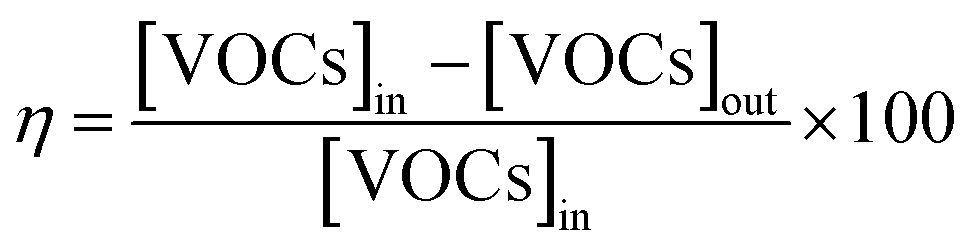DOI:
10.1039/D3RA00957B
(Paper)
RSC Adv., 2023,
13, 13354-13364
A glucose-assisted redox hydrothermal route to prepare a Mn-doped CeO2 catalyst for the total catalytic oxidation of VOCs†
Received
12th February 2023
, Accepted 17th April 2023
First published on 2nd May 2023
Abstract
In this study, a novel glucose-assisted redox hydrothermal method has been presented to prepare an Mn-doped CeO2 catalyst (denoted as Mn-CeO2-R) for the first time. The obtained catalyst contains uniform nanoparticles with a small crystallite size, a large mesopore volume, and rich active surface oxygen species. Such features collectively contribute to improving the catalytic activity for the total catalytic oxidation of methanol (CH3OH) and formaldehyde (HCHO). Interestingly, the large mesopore volume feature of the Mn-CeO2-R samples could be considered an essential factor to eliminate the diffusion limit, favoring the total oxidation of toluene (C7H8) at high conversion. Therefore, the Mn-CeO2-R catalyst outperforms both bare CeO2 and conventional Mn-CeO2 catalysts with T90 values of 150 °C and 178 °C for HCHO and CH3OH, respectively, and 315 °C for C7H8, at a high GHSV of 60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 mL g−1 h−1. Such robust catalytic activities signify a potential utilization of Mn-CeO2-R for the catalytic oxidation of volatile organic compounds (VOCs).
000 mL g−1 h−1. Such robust catalytic activities signify a potential utilization of Mn-CeO2-R for the catalytic oxidation of volatile organic compounds (VOCs).
1. Introduction
Volatile organic compounds (VOCs) emitted from industrial activities have caused concern as the root cause of well-known serious human health and environmental issues.1 Indeed, methanol (CH3OH), formaldehyde (HCHO), and toluene (C7H8), the three most typical harmful VOC pollutants, cause various health problems (e.g., headache, nausea, irritation of the skin, nose, eyes, and throat).2,3 In addition, HCHO molecules stimulate discomfort at low levels and are classified as a group 1 carcinogen by The International Agency for Research on Cancer (IARC). Therefore, catalytic oxidation-driven VOC-to-CO2 conversion has arisen as an efficient platform to completely remove those mentioned pollutants.4 To this end, noble metal-based catalysts (e.g., Pt, Au, Pd) have been proven effective with high performance.5,6 However, high cost and water vapor density restrict the utilization of precious metals for the catalytic oxidation of VOCs.
Recent attempts have pointed out the enormous potential of cerium oxide (CeOx)-based materials as robust total oxidation catalysts for the oxidation of VOCs. Indeed, CeOx catalysts, possessing abundant oxygen storage and reversible Ce4+/Ce3+ redox pairs, can change the oxygen release/uptake behavior during the action of oxidation catalysts, benefiting electron-involving chemical reactions.7,8 However, bare CeO2 materials exhibit limited catalytic activity, which can be described through two major issues: (i) low pore volume and large particle size, which restricts the access of reactants onto active centers, and/or (ii) the availability of active oxygen species that can participate in electron transfer processes during the catalytic oxidation reaction.
Heteroatom doping-driven defect engineering into the CeO2 structure has emerged as a promising method to address the mentioned issues and enhance VOC catalytic oxidation.9,10 To this end, manganese (Mn) species have been considered viable dopants that can generate numerous deficient sites.11,12 Therefore, the formation of active oxygen species on the catalyst surface can be feasible.10 Thus, attempts have been devoted to developing cerium (Ce)–Mn binary oxide catalysts through various synthetic methods (e.g., impregnation, sol–gel, co-precipitation, redox-precipitation, urea-based combustion, and hydrothermal) directed for a wide range of applications (e.g., VOCs removal,9,10,12–16 organic synthesis,17 electrocatalytic ammonia synthesis,18 and CO oxidation).19 However, the prepared catalysts were unsuccessful in simultaneously addressing the two challenges posed by CeO2-based catalysts (i.e., low pore volume and active oxygen species). Combining hydrothermal and redox precipitation methods has attracted increasing attention for nanosynthesis because of the following features: (i) the formation of uniform CeOx/MnOx interfacial contact can induce an efficient doping effect, and (ii) rich surface adsorbed oxygen species that facilitate catalytic oxidation performance can be achieved.20,21 This approach involves the participation of a reducing agent (e.g., citric acid, hydrochloric acid, or organic compounds). To this end, glucose, a natural, non-toxic, and inexpensive substance, has been employed as an electron donor in the redox-precipitation method. Glucose can function as a capping agent, tailoring the growth of uniform and small-sized metal oxide nanoparticles during the synthesis.22
Herein, we attempt to incorporate Mn atoms into CeO2 catalysts (Ce to Mn molar ratio of 1:1) through the glucose-assisted redox hydrothermal method followed by annealing. The characterizations reveal an interesting Mn-doped CeO2 nanocatalyst (denoted as Mn-CeO2-R) that possesses a large pore volume and rich surface oxygen species, which are rarely reported elsewhere. Consequently, the as-prepared Mn-CeO2-R nanocatalyst possesses a high amount of surface oxygen species and large pore volume exhibiting outstanding performance in the total catalytic oxidation of CH3OH, HCHO, and C7H8.
2. Experimental
2.1. Materials
Cerium(III) nitrate hexahydrate (>99%, Sigma Aldrich), manganese(II) nitrate tetrahydrate (>99%, Sigma Aldrich) urea (>99%, Xilong Scientific), D-glucose anhydrous (99%, Scharlau), and KMnO4 (>99%, Merck) were purchased and utilized for synthesis without further purification.
2.2. Material synthesis
For the glucose-assisted redox hydrothermal method, 4.35 g of Ce(NO3)3·6H2O, 1.6 g of KMnO4, and 2.4 g of glucose were added into 50 mL of double distilled water. The obtained mixture was transferred into a Teflon-line stainless steel autoclave and then sealed. The hydrothermal reaction was carried out at 150 °C for 10 hours. After the reaction, the autoclave was naturally cooled to room temperature. The obtained solid was filtered, washed with 1 L of distilled water, and then dried at 110 °C for 12 hours. The dried solid was then annealed at 400 °C under ambient conditions for 4 hours. The final Ce–Mn oxide catalyst was denoted as Mn-CeO2-R. A conventional hydrothermal method was executed with the same procedure by using urea as a precipitation agent with molar Ce–Mn–urea to 1–1–2 (4.34 g of Ce(NO3)3·6H2O, 2.5 g of Mn(NO3)2·4H2O, and 1.2 g of urea are mixed in 50 mL). The solid was denoted as Mn–CeO2–C. The bare CeO2 catalyst was also synthesized with the same procedure without using Mn precursors.
2.3. Characterization
The phase and crystal structure of bare CeO2 and Ce–Mn oxide catalysts were analyzed by a Smartlab X-ray diffractometer (XRD, Rigaku) equipped with a CuKα anode. Morphologies of the catalysts were observed by JSM-6010 Plus/LV scanning electron microscope (SEM, Jeol) and a high-resolution transmission electron microscope (HRTEM, JEM 2100, Joel). The BET (Brunauer–Emmett–Teller) surface area, micropore surface area (Smp), micropore volume (Vmp), and pore distribution of the catalysts, which were previously degassed at 150 °C for 2 hours, were measured by the Asap2020 analyzer (Micromeritics). The Smp and Vmp values were calculated by the t-plot method. The hydrogen temperature-programmed reduction (H2-TPR) experiments were conducted on 0.04 g of the Ce–Mn oxide catalysts by the same procedure previously published.23 Thermogravimetry (TGA) analyses were performed in nitrogen (99.999%) and air atmosphere by STA6000 analyzer (PerkinElmer). Raman spectra were recorded by a Raman microscope (Xplora plus, Horiba) with an excitation laser of 532 nm.
2.4. Catalytic test
Catalytic total oxidation of CH3OH, HCHO, or C7H8 over the Ce–Mn catalysts was conducted in a continuous-flow stainless steel BTRS microreactor (9 mm-inner diameters, Parker). 0.1 g of each catalyst was loaded into the reactor, degassed at 250 °C for 2 hours in a dried air flow, and then cooled to 30 °C. VOC vapor was generated by passing a nitrogen stream with a suitable flow rate into a thermostat bubble system containing C7H8 or formalin liquid. The VOC-containing air stream (300 ppm HCHO, 40 ppm CH3OH, or 1000 ppm C7H8) was fed into the microreactor at a flow rate of 100 mL min−1 and a gas hourly space velocity (GHSV) of 60000 mL g−1 h−1. The catalyst temperature was controlled from 30–400 °C with a heating ramp of 1 °C min−1. The feed and reaction products were analyzed by an online gas chromatograph (7890B, Agilent) equipped with a flame ionization detector (FID) and a thermal conductivity detector (TCD). The conversion of CH3OH, HCHO, and C7H8 (η) was calculated by the formula (1).| |
 | (1) |
where [VOCs]in and [VOCs]out are representative of the concentration of CH3OH, HCHO, or C7H8 in the inlet and outlet stream, respectively.
3. Result and discussion
The incorporation of Mn species into the CeOx parent structure is conducted through a glucose-assisted redox hydrothermal route, followed by calcination at 400 °C for 4 hours, as shown in Scheme 1 (see Section 2.2 for details). In this circumstance, KMnO4 is used as a Mn precursor and a strong oxidant, while Ce3+ and glucose act as Ce precursor and reductant, respectively, to generate homogeneous dispersion of MnO2 and Ce(OH)4/CeO2. CeO2 nanoparticles are hydrothermally formed through the hydrolysis reactions of Ce3+/Ce4+ cations. The as-synthesized nano-species (MnO2, CeO2) are transformed into Mn-doped CeO2 catalysts during the calcination step. The synthesis reactions could be executed as follows:| | |
8MnO4− + C6H12O6 → 8MnO2 + 6CO2 + 8OH− + 2H2O
| (2.1) |
| | |
3Ce3+ + MnO4− + 6H2O → MnO2 + 3Ce4+ + 4OH−
| (2.2) |
| | |
Ce4+ + 4OH− → Ce(OH)4
| (2.3) |
| | |
Ce(NO3)4 + 4H2O → Ce(OH)4 + 4NO3− + 4H+
| (2.4) |
| | |
Ce(NO3)3 + 3H2O → Ce(OH)3 + 3NO3− + 3H+
| (2.5) |
| | |
2Ce(OH)3 + 1/2O2 → 2CeO2 + 3H2O
| (2.6) |
| | |
Ce(OH)4 → CeO2 + 2H2O
| (2.7) |
 |
| | Scheme 1 The illustration of the glucose-assisted redox hydrothermal synthesis of Mn-doped CeO2. | |
In addition, the excess glucose is hydrothermally transformed to poly-carbohydrates and gluconic acid, which could act as stabilizers. The presence of glucose helps to inhibit the growth of nuclei during hydrothermal-derived recrystallization, favoring the formation of Ce–Mn nanoparticles.
3.1. Morphology
Scanning electron microscopy (SEM) was employed to investigate the morphology of the Mn-doped CeOx material as depicted in Fig. 1. The conventional Mn-doped CeO2 (denoted as Mn-CeO2-R) and bare CeO2 samples, prepared without the addition of KMnO4 (see Section 2.2 for the synthesis details), were also prepared for comparison. Various-sized microparticles with different forms and shapes can be observed in the Mn-CeO2-C (Fig. 1A) and bare CeO2 (Fig. 1B) samples. In contrast, the as-synthesized Mn-CeO2-R sample contained uniform nanoparticles (Fig. 1C and D). Fig. 1E exhibits the transmission electron microscopy (TEM) images of the prepared Mn-CeO2-R sample, confirming the existence of Mn-doped CeOx nanoparticles. In addition, the HRTEM image shows the lattice spacing of 0.31 nm and 0.27 nm corresponding to the (111) and (200) planes of the CeO2 phase, respectively (Fig. 1F). Such observation implies a strong impact of the preparation methods using Ce3+, glucose, and KMnO4 as precursors on the formation and stabilization of the prepared catalyst with uniform nanoparticles. Decreasing material size into the nanoscale could encourage reducing the diffusion resistance of reactant molecules into active sites.
 |
| | Fig. 1 SEM images of (A) bare CeO2, and Mn-doped ceria catalysts: (B) Mn-CeO2-C, (C) and (D) Mn-CeO2-R; (E) and (F) TEM and HRTEM images of Mn-CeO2-R. | |
3.2. Phase and crystal structure
The X-ray diffraction (XRD) characterization was conducted to investigate the crystallography of the as-prepared samples as shown in Fig. 2. The XRD pattern of the bare CeO2 (line a) sample exhibits five characteristic peaks centering at 2θ of 28.6°, 33.1°, 47.4°, 56.5°, and 59.4°, which could be attributed to the (111), (200), (220), (311), and (222) crystallographic planes of the CeO2 cubic fluorite structure (JCPDS card no. 00-043-1002). The Mn-CeO2-R catalyst (line b) reveals a similar XRD pattern as the bare CeO2 sample without the existence of typical MnOx peaks. Furthermore, peak broadening and slight shifting could be observed. These findings suggest the insertion of Mn cations into the CeO2 lattice structure.24 The XRD pattern of Mn-CeO2-R witnesses a remarkable distortion within the CeO2 structure. Indeed, the intensity of all the characteristic diffraction peaks significantly decreases in the absence of MnO2-associated peaks. The observation could be rooted in the high degree of Ce4+ substitution by Mn dopants.25 Additionally, the crystallite sizes (Dc) calculated by Scherrer equations are given in Table 1. The measured Dc value of Mn-CeO2-R is found to be the smallest crystallite size (i.e., 4 nm). To this end, it can be said that the prepared Mn-CeO2-R catalyst possesses a significantly distorted structure that could offer unique surface properties (e.g., active oxygen species). In addition, its small crystal size benefits the easy access of reactant and oxygen molecules into the active sites for the catalytic oxidation of VOCs.21
 |
| | Fig. 2 XRD pattern of (a) bare CeO2 and Mn-doped ceria catalysts: (b) Mn-CeO2-C and (c) Mn-CeO2-R. | |
Table 1 Physicochemical characteristics of the Mn-doped ceria catalystsa
| Samples |
SBET (m2 g−1) |
Smp (m2 g−1) |
Vmp (cm3 g−1) |
Vp (cm3 g−1) |
Dc (nm) |
Weight lossb (%) |
ΔWL (%) |
H2 consumption (mmol g−1) |
| Smp: micropore surface area, Vmp: micropore volume, Vp: pore volume, Dc: crystallite size. Weight loss corresponding to the removal of surface lattice oxygen; ΔWL: difference in weight loss at 400 °C under both nitrogen and air atmosphere. |
| CeO2 |
89 |
59 |
0.03 |
0.03 |
21.0 |
1.5 |
0.06 |
1.1 |
| Mn-CeO2-C |
93 |
50 |
0.02 |
0.04 |
10.9 |
1.7 |
0.36 |
1.7 |
| Mn-CeO2-R |
58 |
0.7 |
0 |
0.18 |
4.0 |
2.2 |
0.46 |
1.8 |
Raman spectroscopy was then employed to investigate the doping effect of Mn ions within the CeO2 lattice, as depicted in Fig. 3. The spectrum of the bare CeO2 sample (line a) exhibits a dominant band located at 451 cm−1, which could be attributed to the F2g symmetric stretching mode rooted in oxygen atoms surrounding Ce cations.17 Minor bands centering at ∼240 cm−1 and ∼594 cm−1 could be assigned to double acoustic (2TA) mode and defect-induced mode (D), respectively.26,27 Incorporating Mn into the ceria structure through the conventional method (i.e., the Mn-CeO2-C sample) induces a light modification within the CeO2 lattice despite a decrease in the intensity and broadening of the F2g band. The Mn-CeO2-R catalyst (line a) signifies a remarkable decrease in intensity and unambiguous red shift of the F2g band toward a low Raman shift region, which could be ascribed to the replacement of Ce atoms by Mn atoms.15,27 Furthermore, the disappearance of the D band, which is assigned to the distortion of the anionic lattice due to the formation of oxygen vacancy defects within the cubic fluorite structure, could be noticed. This feature indicates that oxygen defects do not occur in Mn-CeO2-R.
 |
| | Fig. 3 Raman spectra of general (A) and extended regions (B) of (a) bare CeO2, and Mn-doped ceria catalysts, (b) Mn-CeO2-C, and (c) Mn-CeO2-R. | |
3.3. Textural properties
Fig. 4 shows nitrogen adsorption–desorption isotherms and pore size distribution of bare CeO2 and Ce–Mn catalysts obtained by physisorption of nitrogen at 77 K. All curves show type IV isotherm associated with H3 hysteresis loop of slit-like pores-containing porous materials.28 BJH pore size distribution curves (Fig. 4A–D) show peaks centering at 35 Å for bare CeO2 and Mn–CeO2 catalysts and 39 Å for Mn-CeO2-R catalysts, respectively. The measured BET surface area (SBET), micropore surface area (Smp), micropore volume (Vmp), and pore volume (Vp) calculated by the desorption branch are given in Table 1. The SBET, Smp, and Vp values of the bare CeO2 samples were found to be 89 m2 g−1, 59 m2 g−1, and 0.03 cm3 g−1, respectively. The micropore volume of the CeO2 sample is almost equal to its total pore volume, indicating that a majority of micropores having a diameter of less than 2 nm occupy its porous volume. The incorporation of Mn into the CeO2 structure of Mn-CeO2-C via the conventional method slightly changes these values. Interestingly, the Mn-CeO2-R sample exhibits a significant reduction in the SBET (58 m2 g−1), Smp (0.7 m2 g−1), and Vmp (0 cm3 g−1), whereas Vp remarkably increases to 0.18 cm3 g−1, which is 9 and 4.5 times higher than that of bare CeO2 and Mn-CeO2-C, respectively. The suppression of micropores and enlargement of pore volume benefit the intraparticle diffusion of reactants and products during catalytic reactions at high temperatures. To this end, it can be said that the outstanding textural properties of the as-prepared Mn-CeO2-R, which is difficult to be obtained through conventional methods, could promote the oxidation of VOCs.
 |
| | Fig. 4 (A and B) Nitrogen-isotherm adsorption curves, and (C and D) pore size distribution of (a) bare CeO2 and Mn-doped ceria catalysts, (b) Mn-CeO2-C, and (c) Mn-CeO2-R. | |
3.4. Thermogravimetric/differential thermal analysis
Thermogravimetric (TG) analysis in a nitrogen atmosphere was carried out to explore the evolution of surface lattice oxygen of bare CeO2 and Mn-doped ceria catalysts. As shown in Fig. 5A, the TG and corresponding derivative thermogravimetry (DTG) curve of bare CeO2 shows three weight loss steps: (i) at T < 160 °C corresponding to the removal of water that had been physicochemically adsorbed on the catalyst; (ii) from 160–400 °C along with one peak at 214 °C and a shoulder at 280 °C on the corresponding DTG curve is attributed to the loss of chemically adsorbed oxygen and carbonate at the surface; (iii) from 400–780 °C with an equivalent DTG peak at 470 °C (Fig. 5A), which is assigned to the removal of surface lattice oxygen atoms, which are much weaker than that of bulk oxygen.29 At 900 °C, the phase composition of the sample is CeO2, as confirmed by the XRD pattern of the samples obtained from the TGA analysis (Fig. S1†). The thermogravimetric profiles of the Mn-CeO2-C catalyst signify a peak at 478 °C, which can be ascribed to the removal of surface lattice oxygen (Fig. 5B). Two additional DTG peaks at 600–675 °C and 870 °C have been attributed to the decomposition of Mn(IV,III) oxides into Mn3O4 at the surface and in bulk structure, respectively, which can be confirmed by the XRD measurements of the obtained sample after performing TGA analysis, as shown in Fig. S1A and B.† Regarding the Mn-CeO2-R sample, the removal of the surface lattice oxygen peak is broad and shifts to 545 °C (Fig. 5C). The amount of surface lattice oxygen available on the Ce–Mn catalyst, which is determined from the weight loss values during this second step, can be ordered as Mn-CeO2-R (2.2%) > Mn-CeO2-C (1.7%) > CeO2 (1.5%) (Table 1). This indicates that Mn doping enhances the active lattice oxygen for the catalytic oxidation process. The Mn-CeO2-R could release the highest content of surface oxygen species for the total oxidation of VOCs. It has been proven that the oxygen molecules located at the surface contribute to the catalytic cycle of the oxidation process.4,12,16
 |
| | Fig. 5 Thermogravimetry (TGA) and differential thermogravimetry (DTG) curves of (A) bare CeO2 and Mn-doped ceria catalysts: (B) Mn-CeO2-C and (C) Mn-CeO2-R under nitrogen atmosphere. | |
The TGA analyses are also performed in the presence of air to characterize the available oxygen vacancies in the catalysts by comparing them with those obtained under a nitrogen atmosphere. Fig. 6A–C show TGA profiles of the bare CeO2 and Ce–Mn oxide catalysts carried out in both nitrogen and air atmosphere. At T < 800 °C, the weight loss curves evolve in nearly the same trends. The lower weight loss under an oxygen atmosphere indicates the penetration of oxygen molecules from the gas phase into the position of oxygen vacancies of ceria surface and structure to balance the weight loss due to the release of surface and bulk oxygen molecules with temperature. The weight loss difference (ΔWL), which is calculated by the formula ΔWL = Δ2 − Δ1, where Δ1 and Δ2 represent the difference in weight loss in an oxygen and nitrogen atmosphere at 200 °C and 440 °C, respectively, is employed as a means of the semi-quantitative determination of surface oxygen vacancies available on the Ce–Mn oxide catalysts. The ΔWL values can be arranged as CeO2 (0.46%) > Mn-CeO2-C (0.36%) > Mn-CeO2-R (0.06%) (Table 1). Hence, the Mn-CeO2-R catalyst contains fewer oxygen vacancies than CeO2, which agrees with the Raman interpretation.
 |
| | Fig. 6 Thermogravimetry (TGA) curves of (A) bare CeO2 and Mn-doped ceria catalysts: (B) Mn-CeO2-C; (C) and (D) Mn-CeO2-R under nitrogen and air atmosphere. | |
3.5. H2-TPR measurement
H2-TPR experiments are conducted to explore the amount of active surface oxygen in the bare CeO2 and prepared Mn-doped CeO2 catalysts as depicted in Fig. 7. The H2-TPR signal of bare CeO2 (line a) reveals two large peaks located at 300–500 °C and 600–800 °C, which are attributed to the reduction of surface lattice oxygen species on the surface and bulk lattice oxygen in CeO2, respectively.30 These peaks appear at lower temperatures in the Mn-CeO2-C (line b) sample, suggesting the higher mobility of lattice oxygen species at both surfaces and in the bulk structure.31 Additionally, two peaks arising at 244 °C and 333 °C are attributed to the reduction of adsorbed oxygen species at the catalyst surface. Such oxygen species may be due to the mutual interaction between Ce and Mn in the fluorite structure.12 This phenomenon could be observed at 200–350 °C in the H2-TPR signal of the Mn-CeO2-R sample. In this circumstance, a high amount of adsorbed oxygen and lattice oxygen species at the surface are crucial factors that contribute to high catalytic activity in total oxidation reactions.
 |
| | Fig. 7 H2-TPR profiles of (a) bare CeO2, and Mn-doped ceria catalysts: (a) bare CeO2, (b) Mn-CeO2-C, and (c) Mn-CeO2-R. | |
The low temperature-hydrogen consumption (T < 500 °C) could convey active oxygen species as presented in Table 1. The oxygen reactivity of the prepared materials can be classified as Mn-CeO2-R (1.8 mmol g−1) > Mn-CeO2-C (1.7 mmol g−1) > CeO2 (1.1 mmol g−1), respectively. Accordingly, the Mn-CeO2-R catalyst offers the largest amount of active oxygen at the catalyst surface, which is extremely reactive and energetic for reduction at relatively low temperatures during the catalytic total oxidation reaction.
3.6. Catalytic performance of total oxidation of VOCs
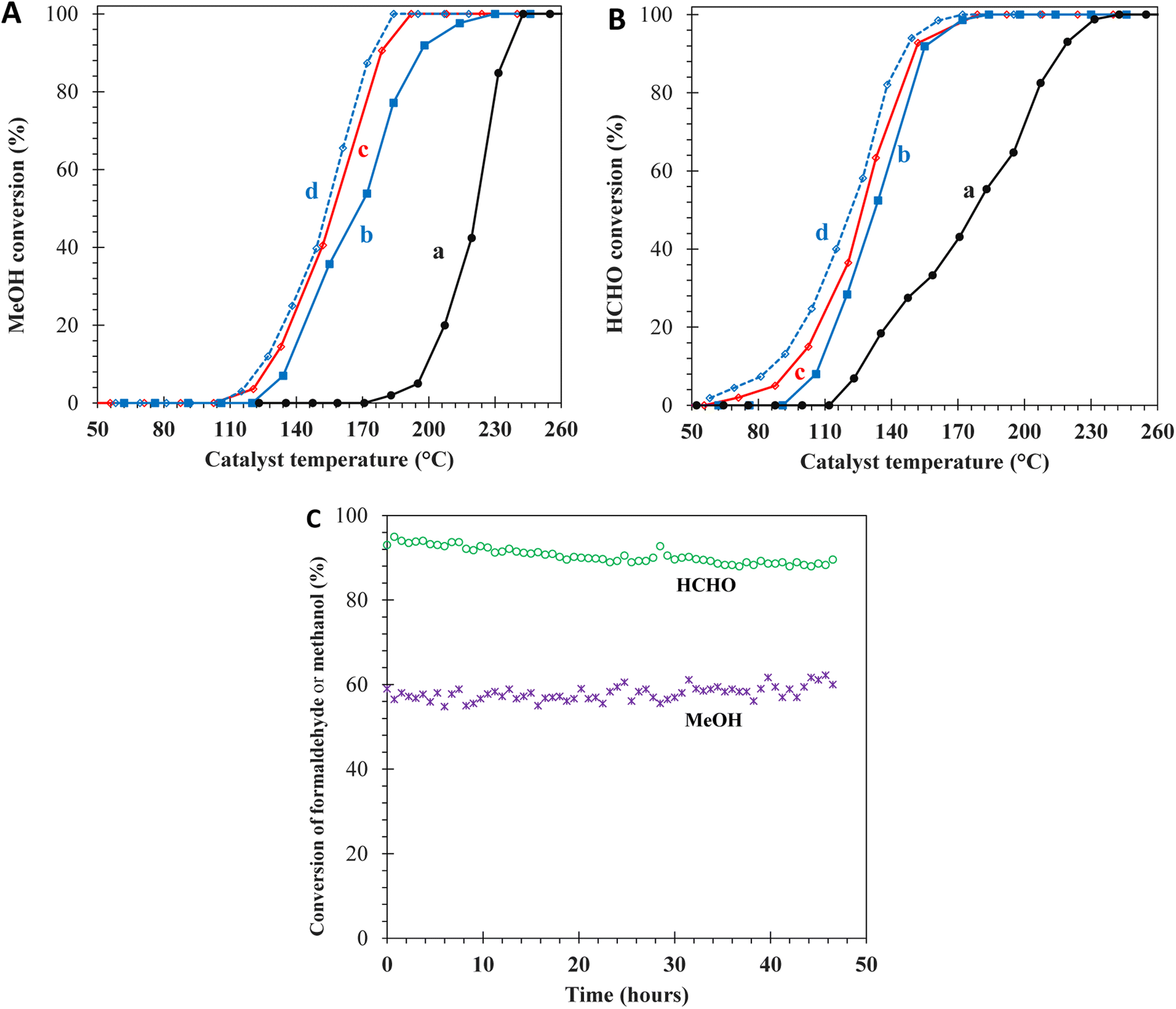
The utilized characterizations have pointed out three features of the prepared Mn-CeO2-R catalysts: (i) small nanoparticle size, (ii) enhanced textural properties, and (iii) rich surface oxygen species, which could significantly promote the total catalytic oxidation reactions. Motivated by these properties, two small molecules, CH3OH (kinetics diameter; d = 3.8 Å) and HCHO (d = 3.7 Å), were employed as model VOCs to evaluate the catalytic performance. Fig. 8A and B show the light-off curves representing the conversion of CH3OH and HCHO versus temperature, respectively, which reach 100% at 180–240 °C, obtained over the Ce–Mn catalysts. The conversion curves of CH3OH and HCHO over Mn-doped ceria catalysts are located at much lower temperatures than that of CeO2, indicating the strong enhancement of catalytic activities. At maximum conversion, 100% of the reactant molecules are converted into CO2. The T50 and T90 values, i.e., the temperatures at which 50% and 90% of CH3OH or HCHO are converted, are interpolated from the light-off curves to evaluate the catalytic activity of the total oxidation of CH3OH and HCHO as summarized in Table 2. The lower T50 and T90 values indicate better catalytic activity. The catalytic performance of the prepared catalysts for HCHO oxidation is arranged as Mn-CeO2-R (T50 = 127 °C; T90 = 150 °C) > Mn-CeO2-C (133 °C; 154 °C) > CeO2 (178 °C; 216 °C). A similar trend is observed in the catalytic CH3OH oxidation reaction. Therefore, it can be said that the Mn-CeO2-R sample significantly outperforms the remaining materials in the catalytic oxidation of HCHO and CH3OH compounds. Furthermore, the Mn-CeO2-R catalyst delivers reaction rates of 0.75 mmol g−1 h−1 and 0.04 mmol g−1 h−1 at GHSV of 60000 mL g−1 h−1 in CH3OH and HCHO oxidation at 150 °C, respectively. The prepared Mn-CeO2-R catalyst maintains its crystalline structure during these reactions at 155 °C, corresponding to 88% and 58% conversion in the oxidation of HCHO and CH3OH for 46 hours, as depicted in Fig. S2.† At low GHSV of 30000 mL g−1 h−1, the T90 values reduce to 145 °C for HCHO and 175 °C for CH3OH.
 |
| | Fig. 8 Light-off curves of (A) MeOH conversion; (B) HCHO conversion over the catalysts: (a) bare CeO2, (b) Mn-CeO2-C, (c) and (d) Mn-CeO2-R at GHSV = 60000 mL g−1 h−1, and 30000 mL g−1 h−1, respectively; (C) stability of HCHO and CH3OH at 155 °C, GHSV = 60000 mL g−1 h−1. | |
Table 2 Catalytic performances of the catalysts toward oxidation of methanol, formaldehyde, and toluene
| Catalysts |
Methanol |
Formaldehyde |
Toluene |
| T50 (°C) |
T90 (°C) |
Reaction rate at 150 °C (mmol g−1 h−1) |
T50 (°C) |
T90 (°C) |
Reaction rate at 150 °C (mmol g−1 h−1) |
T50 (°C) |
T90 (°C) |
Reaction rate at 350 °C (mmol g−1 h−1) |
Ea (kJ mol−1) |
| Catalytic test experiment conducted at GHSV of 30000 mL g−1 h−1. |
| CeO2 |
222 |
235 |
0 |
178 |
216 |
0.25 |
232 |
390 |
2.30 |
10.9 |
| Mn-CeO2-C |
168 |
196 |
0.03 |
133 |
154 |
0.72 |
259 |
360 |
2.36 |
23.7 |
| Mn-CeO2-R |
157 |
178 |
0.04 |
127 |
150 |
0.75 |
278 |
315 |
2.54 |
27.7 |
| Mn-CeO2-Ra |
154 |
175 |
0.04 |
122 |
145 |
0.76 |
|
|
|
|
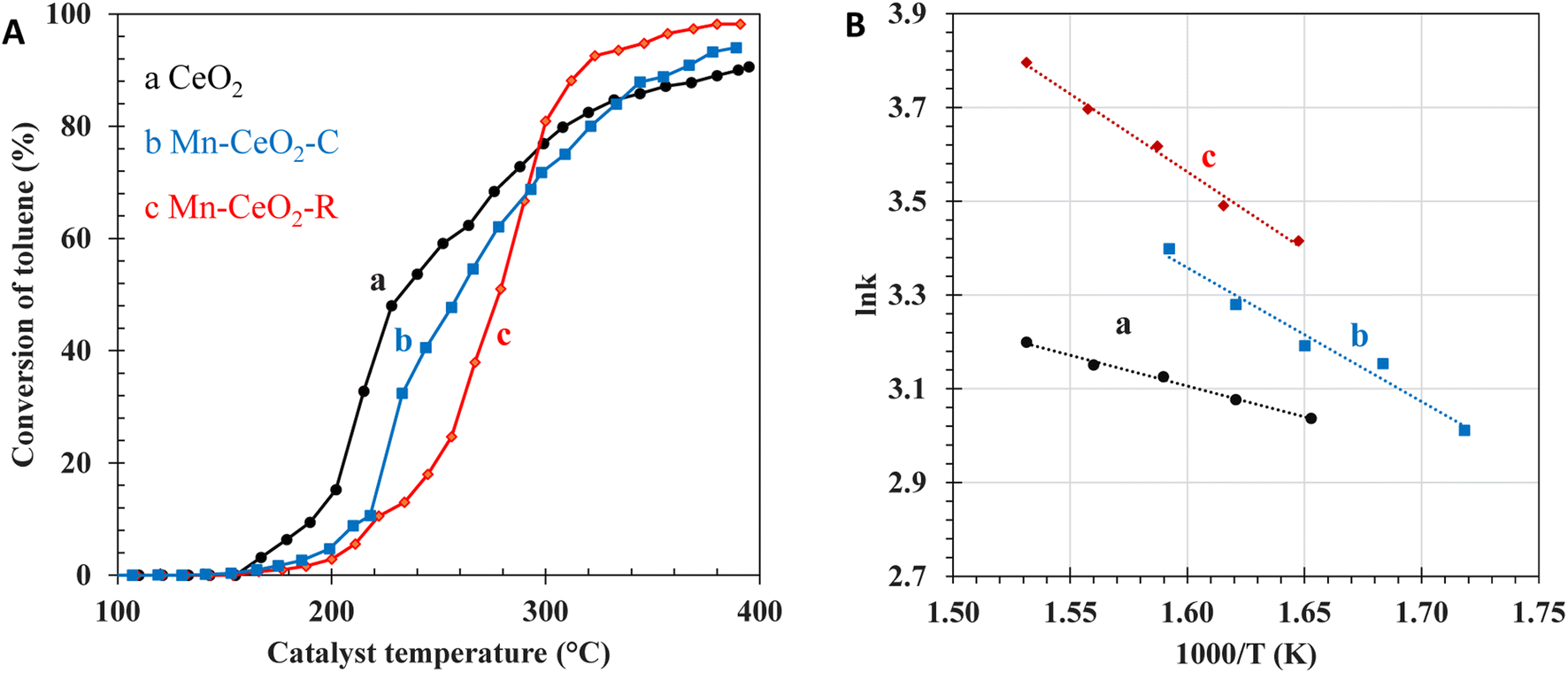
The next attempt is to evaluate the catalytic performance toward the total oxidation of larger molecules (e.g., C7H8, d = 5.9 Å). Fig. 9 shows the light-off curve, which is representative of the conversion of C7H8 versus the catalyst temperature. No by-product is detected by TCD or FID detectors. The selectivity of CO2 is nearly 100%. Once again, the catalytic performance of the Mn-CeO2-R catalyst is significantly enhanced in comparison to the bare CeO2 and Mn-CeO2-C catalysts, when the temperature is above 300 °C. The obtained T90 values can be arranged as Mn-CeO2-R (315 °C) < Mn-CeO2-R (360 °C) < bare CeO2 (390 °C). This finding is consistent with the number of surface oxygen species, crystallite size, nanoparticles sizes, and pore volume. At high temperatures, the Mn-CeO2-R catalyst exhibits the best catalytic performance toward the total oxidation of C7H8, offering a reaction rate of 2.54 mmol g−1 h−1 at 350 °C.
 |
| | Fig. 9 Light-off curves of (A) C7H8 conversion, (B) Arrhenius plots over the various catalysts: (a) bare CeO2, (b) Mn-CeO2-C, and (c) Mn-CeO2-R. | |
At T < 300 °C, the T50-derived C7H8 conversion of the Mn-CeO2-R catalyst is lower than that of Mn-CeO2-C and bare CeO2 catalysts, which is consistent with their BET surface area and amount of oxygen vacancies. Indeed, the bare CeO2 material possessing rich oxygen vacancies, as revealed by TGA and Raman, displays better oxidation of C7H8 at low C7H8 conversion and low temperatures. However, the abundance of micropores and very low pore volume of CeO2 and the Mn-CeO2-C samples restrict the conversion of C7H8 at high temperatures, which could originate from the limitation of inner diffusion of reactants/products into/from the active sites. The Arrhenius plots are representative of the lnk as a function of 1000/T, where T and k are the temperatures of the catalysts and the rate constant of the C7H8 oxidation reaction, respectively. The plots are constructed at high C7H8 conversion greater than 80% and temperature above 300 °C. It is assumed that the total oxidation of C7H8 follows a first-order reaction kinetics with respect to C7H8. Moreover, the obtained energy activation has been highlighted to understand the intra and extra-particle diffusion of the Ce–Mn oxide catalysts, as shown in Fig. 8B. The obtained activation energies (Ea) of CeO2, Mn-CeO2-C, and Mn-CeO2-R are 10.9 kJ mol−1, 23.7 kJ mol−1, and 27.7 kJ mol−1, respectively (Table 2). As a result, the Ea value observed for the CeO2 catalyst is close to that of the diffusion-controlled step (∼10 kJ mol−1). This indicates that diffusion significantly impacts the mass transfer limitation over CeO2 at high C7H8 conversion.32,33 This is consistent with the abundant micropores, large particle size, and lowest catalytic activity of the CeO2 catalyst at high conversion. Accordingly, the Mn-CeO2-R catalyst containing uniform nanoparticles and large pore volume favors the access of reactants into the reactive site and the expulsion of pore diffusion products out of the inner catalyst, leading to the higher catalytic conversion of C7H8.
In general, the Mn-CeO2-R catalyst with the smallest crystallite size, largest pore volume, and the highest amount of active surface oxygen, exhibits the best catalytic performance toward the total oxidation of CH3OH, HCHO, and C7H8. Mars–Van Krevelen mechanism could be employed to explain the enhanced VOC oxidation activity of the prepared Mn-CeO2-R catalyst based on structural characterizations – activity correlation. The adsorbed VOC molecules are activated by active surface oxygen species and subsequently converted to CO2 and water. This process leaves oxygen vacancies at the catalyst surface, which are later replenished by gas-phase oxygen molecules. The high amount of surface oxygen vacancies of ceria increases the reaction rate, which is valid for the total oxidation of C7H8 at low temperatures (T < 300 °C) in our study. However, the catalytic performance at the high conversion of C7H8 is restricted by the presence of micropores. The Mn-CeO2-R catalyst, containing a trace amount of surface oxygen vacancies, shows superior catalytic performance for the total oxidation of HCHO and CH3OH. Accordingly, the abundance of active surface oxygen, created by the incorporation of Mn into ceria catalysts, controls the total oxidation of HCHO and CH3OH. Nevertheless, the effects of porosity and nanoparticles sizes are important in the total oxidation of C7H8 at high conversion. The high proportion of mesopores and large pore volume of Mn-doped ceria nanoparticles, created by the doping of Mn into ceria by glucose-assisted redox hydrothermal method using KMnO4 and glucose as precursors, favors the intraparticle and extra-particle diffusion.
4. Conclusion
This work offers a novel synthetic approach to prepare Mn-doped CeO2 nanoparticle catalysts through a glucose-assisted redox hydrothermal method using cerium(III) nitrate, KMnO4 and glucose as precursors. The resulting catalysts, consisting of Mn doping into ceria, are characterized by uniform nanoparticles with a small crystallite size, a large mesopore volume, and a high active surface oxygen amount. These characteristics lead to superior catalytic conversion of CH3OH and HCHO as compared to bare CeO2 and conventional Mn-doped CeO2 materials. Additionally, the enhanced pore volume and micropore suppression in the Mn-doped CeO2 nanoparticles eliminate diffusion limits and improve the catalytic activity with high C7H8 conversion (T > 300 °C). The findings demonstrate the potential of the glucose-assisted redox hydrothermal method for preparing Mn-doped ceria catalysts for low-temperature oxidation of CH3OH, HCHO, and C7H8.
Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could influence the work reported in this paper.
Acknowledgements
This research is funded by the Ministry of Education and Training, Vietnam, under grant number B2022-DNA-07.
References
- C. He, J. Cheng, X. Zhang, M. Douthwaite, S. Pattisson and Z. Hao, Chem. Rev., 2019, 119, 4471–4568 CrossRef CAS PubMed.
- A. Mellouki, T. J. Wallington and J. Chen, Chem. Rev., 2015, 115, 3984–4014 CrossRef CAS PubMed.
- A. Krishnamurthy, B. Adebayo, T. Gelles, A. Rownaghi and F. Rezaei, Catal. Today, 2020, 350, 100–119 CrossRef CAS.
- Y. Lyu, C. Li, X. Du, Y. Zhu, Y. Zhang and S. Li, Environ. Sci. Pollut. Res., 2020, 27, 2482–2501 CrossRef CAS PubMed.
- H. Huang, Y. Xu, Q. Feng and D. Y. C. Leung, Catal. Sci. Technol., 2015, 5, 2649–2669 RSC.
- R. Liu, H. Wu, J. Shi, X. Xu, D. Zhao, Y. H. Ng, M. Zhang, S. Liu and H. Ding, Catal. Sci. Technol., 2022, 12, 6945–6991 RSC.
- A. M. D'Angelo, A. C. Y. Liu and A. L. Chaffee, J. Phys. Chem. C, 2016, 120, 14382–14389 CrossRef.
- S. Scirè, S. Minicò, C. Crisafulli, C. Satriano and A. Pistone, Appl. Catal., B, 2003, 40, 43–49 CrossRef.
- J. Quiroz, J.-M. Giraudon, A. Gervasini, C. Dujardin, C. Lancelot, M. Trentesaux and J.-F. Lamonier, ACS Catal., 2015, 5, 2260–2269 CrossRef CAS.
- S. Kurajica, K. Mužina, G. Dražić, G. Matijašić, M. Duplančić, V. Mandić, M. Župančić and I. K. Munda, Mater. Chem. Phys., 2020, 244, 122689 CrossRef CAS.
- Y. Zheng, J. Zhou, X. Zeng, D. Hu, F. Wang and Y. Cui, RSC Adv., 2022, 12, 25898–25905 RSC.
- X. Zhang, J. Zhao, Z. Song, W. Liu, H. Zhao, M. Zhao, Y. Xing, Z. Ma and H. Du, J. Colloid Interface Sci., 2020, 562, 170–181 CrossRef CAS PubMed.
- D. Delimaris and T. Ioannides, Appl. Catal., B, 2008, 84, 303–312 CrossRef CAS.
- M. T. N. Dinh, J.-M. Giraudon, A. M. Vandenbroucke, R. Morent, N. De Geyter and J.-F. Lamonier, Appl. Catal., B, 2015, 172–173, 65–72 CrossRef CAS.
- S. Guan, Q. Huang, J. Ma, W. Li, A. T. Ogunbiyi, Z. Zhou, K. Chen and Q. Zhang, Ind. Eng. Chem. Res., 2020, 59, 596–608 CrossRef CAS.
- X. Tang, Y. Li, X. Huang, Y. Xu, H. Zhu, J. Wang and W. Shen, Appl. Catal., B, 2006, 62, 265–273 CrossRef CAS.
- R. Zhang, L. Guo, C. Chen, J. Chen, A. Chen, X. Zhao, X. Liu, Y. Xiu and Z. Hou, Catal. Sci. Technol., 2015, 5, 2959–2972 RSC.
- Y. Ji, W. Cheng, C. Li and X. Liu, Inorg. Chem., 2022, 61, 28–31 CrossRef CAS PubMed.
- K. Kim and J. W. Han, Catal. Today, 2017, 293–294, 82–88 CrossRef CAS.
- J. Du, Z. Qu, C. Dong, L. Song, Y. Qin and N. Huang, Appl. Surf. Sci., 2018, 433, 1025–1035 CrossRef CAS.
- B. de Rivas, R. López-Fonseca, C. Jiménez-González and J. I. Gutiérrez-Ortiz, J. Catal., 2011, 281, 88–97 CrossRef CAS.
- X. Sun, C. Zheng, F. Zhang, Y. Yang, G. Wu, A. Yu and N. Guan, J. Phys. Chem. C, 2009, 113, 16002–16008 CrossRef CAS.
- M. T. Nguyen Dinh, C. C. Nguyen, T. L. Truong Vu, V. T. Ho and Q. D. Truong, Appl. Catal., A, 2020, 595, 117473 CrossRef.
- B. Murugan, A. V. Ramaswamy, D. Srinivas, C. S. Gopinath and V. Ramaswamy, Chem. Mater., 2005, 17, 3983–3993 CrossRef CAS.
- Y. Zhang, F. Yang, R. Gao and W.-L. Dai, Appl. Surf. Sci., 2019, 471, 767–775 CrossRef CAS.
- Y. Lee, G. He, A. J. Akey, R. Si, M. Flytzani-Stephanopoulos and I. P. Herman, J. Am. Chem. Soc., 2011, 133, 12952–12955 CrossRef CAS PubMed.
- B. M. Reddy, L. Katta and G. Thrimurthulu, Chem. Mater., 2010, 22, 467–475 CrossRef CAS.
- S. Brunauer, L. S. Deming, W. E. Deming and E. Teller, J. Am. Chem. Soc., 1940, 62, 1723–1732 CrossRef CAS.
- M. V. Grabchenko, G. V. Mamontov, V. I. Zaikovskii, V. La Parola, L. F. Liotta and O. V. Vodyankina, Appl. Catal., B, 2020, 260, 118148 CrossRef CAS.
- H. Zhu, J. Xu, Y. Yichuan, Z. Wang, Y. Gao, W. Liu and H. Yin, J. Colloid Interface Sci., 2017, 508, 1–13 CrossRef CAS PubMed.
- E. Gao, G. Sun, W. Zhang, M. T. Bernards, Y. He, H. Pan and Y. Shi, Chem. Eng. J., 2020, 380, 122397 CrossRef CAS.
- R. J. Farrauto, in Handbook of Industrial Chemistry and Biotechnology, Springer International Publishing, Cham, 2017, pp. 1995–2035 Search PubMed.
- V. J. Inglezakis and A. A. Zorpas, Desalin. Water Treat., 2012, 39, 149–157 CAS.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *d
*d