
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEvaluation of the anti-proliferative activity of 2-oxo-pyridine and 1′H-spiro-pyridine derivatives as a new class of EGFRWt and VEGFR-2 inhibitors with apoptotic inducers†
Reham R. Raslan a,
Yousry A. Ammar*b,
Sawsan A. Fouada,
Sadia A. Hesseina,
Nadia A. M. Shmiessa and
Ahmed Ragab*b
a,
Yousry A. Ammar*b,
Sawsan A. Fouada,
Sadia A. Hesseina,
Nadia A. M. Shmiessa and
Ahmed Ragab*b
aDepartment of Chemistry, Faculty of Science (Girls), Al-Azhar University, Nasr City, Cairo, Egypt. E-mail: rehamsieam.5919@azhar.edu.eg; sawsan_ahmedfouad@azhar.edu.eg; sawsan_ahmedfouad@yahoo.com
bDepartment of Chemistry, Faculty of Science (Boys), Al-Azhar University, Nasr City, 11884, Cairo, Egypt. E-mail: ahmed_ragab@azhar.edu.eg; ahmed_ragab7@ymail.com
First published on 3rd April 2023
Abstract
Developing new agents for cancer treatment remains a top priority because it is one of the deadliest worldwide. A new series of 2-oxo-pyridine and 1′H-spiro-pyridine derivatives were designed and synthesized based on an N-(ethyl benzoate) moiety. The structure of the designed derivatives was confirmed by different spectroscopic techniques (FT-IR and NMR) and elemental analysis and then evaluated as antiproliferative against HepG-2 and Caco-2 cell lines compared with Doxorubicin. The spiro-pyridine derivatives 5, 7, and 8 exhibited a remarkably higher activity against Caco-2 cell lines than that of other derivatives. Additionally, these derivatives exhibited activation in the Bax and suppressed Bcl-2 expression with variable degrees. Interestingly, compound 7 showed the lowest cytotoxicity value on Caco-2 cells (IC50 = 7.83 ± 0.50 μM) compared with Doxorubicin (IC50 = 12.49 ± 1.10 μM). Additionally, this compound showed activation of the Bax gene (7.508-fold) and suppressed Bcl-2 (0.194-fold) compared to untreated Caco-2 cells, as revealed by the qRT-PCR technique. Moreover, compound 7 could inhibit EGFR and VEGFR-2 with sub-micromole values of 0.124 μM and 0.221 μM compared with Erlotinib (IC50 = 0.033 μM) and Sorafenib (IC50 = 0.043 μM), respectively. Further, cell cycle and apoptosis analysis demonstrated that compound 7 promoted apoptosis by increasing the apoptosis rate from 1.92 to 42.35% and the S cell accumulation ratio from 31.18 to 42.07% compared to untreated Caco-2 cells. Finally, the most active compound 7 showed good drug-likeness and toxicity profiles. Besides, molecular docking studies were performed to determine the binding mode, which is in agreement with the in vitro results.
1. Introduction
Cancer is one of the most difficult and complex diseases to treat and is considered a global health problem that affects much of the world population due to uncontrolled cell growth.1 A cancer cell can use multiple compensatory pathways for survival, which is why it is a multifactorial genetic and/or epigenetic disease with complicated signaling networks.2 According to predictions, in 2023, 15 million deaths per year, also cancer represents a real crisis for public health and health systems worldwide.3 The most common form of cancer worldwide is colorectal cancer (CRC), the second leading cause of death among cancer patients.4 As a result, current drugs developed based on the single-target single-drug design tend to be less effective in treating heterogeneous, complex, multigenic cancers.5 One or more cures such as surgery, radiotherapy, and systemic therapy were introduced for cancer therapy.6,7 Traditionally, chemotherapeutics have been the first choice in cancer treatment.8 During chemotherapy, programmed cell death (apoptosis) is induced.9 Hence, during carcinogenesis, the inhibition of apoptosis will play an important role in cancer progression; also, the cancer cells use different molecular pathways to inhibit apoptosis.10 BAX and Bcl-2 are apoptotic and anti-apoptotic proteins, respectively, and their ratio determines the frequency of apoptosis in cells.11 To avoid the cytotoxic side effects of standard chemotherapy, cancer therapies have been developed with a specific target.12The protein tyrosine kinase inhibitors (PTKIs) are the first targeted drug therapy to be discovered and developed, which compete with the ATP binding site of the kinases to start reducing substrate protein phosphorylation and inhibiting cell proliferation cascades.13 Under normal and pathological conditions, protein tyrosine kinases (PTKs) control most cellular functions including metabolism, transcription, apoptosis, cytoskeleton rearrangement, cell proliferation, and differentiation.14 The epidermal growth factor receptor (EGFR) plays a critical role in cell proliferation and migration, and there are many FDA-approved drugs such as Erlotinib and Gefitinib (first generation), Afatinib and Dacomitinib (second generation) and Rociletinib and Nazartinib (third generation) using this mechanism.15,16 The expression of vascular endothelial growth factors (VEGF) rises under pathological conditions, where EGFR is activated. The process of angiogenesis is initiated when VEGF binds to the receptor of vascular endothelial growth factor (VEGFR), specifically VEGFR-2.17 As a member of the tyrosine kinase family that promotes the angiogenesis mechanism, VEGFR is an excellent therapeutic target for cancer. Consequentially, therapeutic drugs inhibiting EGFR and VEGFR-2 can improve cancer therapy efficiency and overcome resistance problems.18 Cancer drug resistance and undesired toxicity make it challenging to develop anticancer agents with desired clinical outcomes.19
The pyridine nucleus is an important heterocyclic scaffold that is found in many vitamins (B3 and B6), alkaloids (trigonelline), co-enzymes (nicotinamide), and natural antibiotics (pyridomycin).20,21 Moreover, pyridine nuclei have the ability to alter the target drug's aqueous solubility, metabolic stability, and lipophilicity.22 Therefore, the synthesis of novel pyridine-based derivatives has attracted much attention in the cancer therapy field.23,24 Compared to pyridines, 2-oxo-pyridone acts as both a donor and an acceptor of the hydrogen bonds. Additionally, it is highlighted as a kinase inhibitor due to attractive binding to kinases at their ATP binding clefts.25,26 Furthermore, pyridine derivatives in cancer research are promising anticancer agents since they have been shown to cure a number of vicious illnesses, such as idiopathic respiratory fibrosis, malignant breast cancer, and myeloid leukemia.27 Many approved drugs such as Crizotinib, Regorafenib, Sorafenib, and Vismodegi contain pyridine nuclei as the main pharmacophore (Fig. 1). Several studies have demonstrated the effectiveness of Sorafenib in treating various cancer types including pancreatic, colon, breast, and thyroid cancers.28 Moreover, Regorafenib inhibits tumor angiogenesis, cell proliferation, and other features of tumors by targeting VEGFR-1/2/3 and BRAF as multi-kinase inhibitors.29 Vismodegib treats patients with basal cell carcinoma that has spread locally or metastatically using the hedgehog pathway inhibitor.30 In recent years, oxindole-based scaffolds have attracted much attention because of their broad pharmacological significance.31 Additionally, Sunitinib and Nintedanib are clinically approved oxindole-containing drugs recognized as multi-kinase inhibitors for treating renal cell carcinoma and adenocarcinoma, respectively.32 Furthermore, the other rings incorporated at C-3 of the indole ring can vary greatly. This opens enormous possibilities for synthesizing novel pharmacodynamic indole heterocycles (Fig. 1). Moreover, spirooxindole is present in many alkaloids with therapeutic values, such as mitraphylline, strychnofoline, and spirotryprostatin A and B that have excellent anticancer activity.33,34
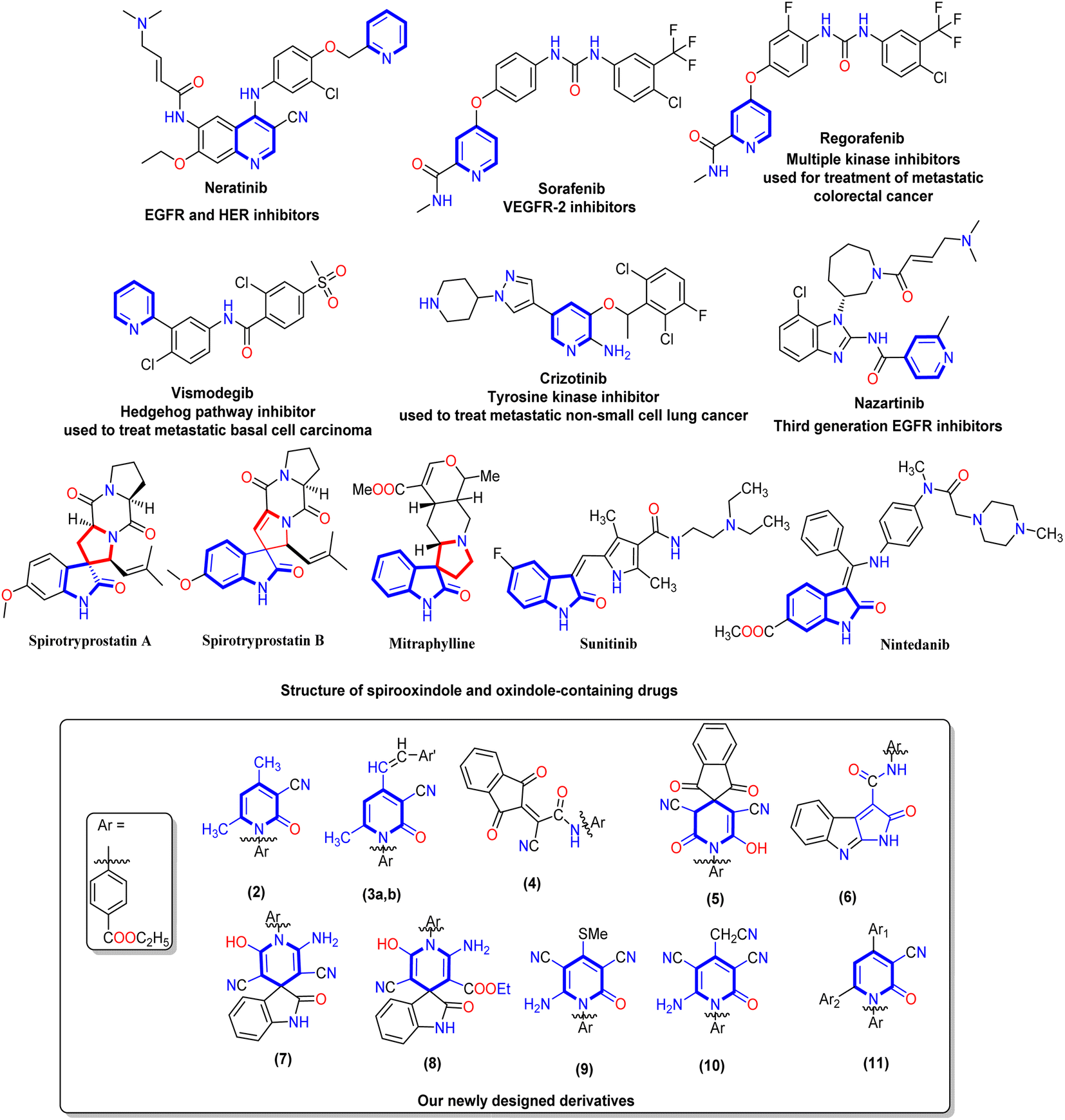 | ||
| Fig. 1 Represented FDA-approved drugs containing a pyridine nucleus and a chemical structure of spirooxindole and oxindole represented by blue cores, and our newly designed derivatives 2–11. | ||
Finally, one of the most efficient and interesting methods for discovering and designing a new bioactive agent is the molecular hybridization approach that combines two pharmacophores to form a new bioactive core.35–40 Additionally, forming new pyridine derivatives with different substituents on its main core and the presence of N-(ethyl benzoate) at N1 of pyridine or adding pyridine heterocycles to the spirooxindole core is expected to increase the potency of anticancer therapeutics in medicinal chemistry. Our work was designed to synthesize new 2-oxo-pyridine and spirooxindole derivatives and evaluate their activity on HepG-2 and Caco-2 cell lines. Mitochondrial apoptosis pathway proteins (BAX and Bcl-2) were also tested for the most active derivatives. Moreover, the cell cycle and apoptosis analysis, EGFR, and VEGFR-2 were evaluated for the most active member. Finally, the in silico ADMET and docking simulations were performed compared to positive controls.
2. Results and discussion
2.1. Chemistry
The key starting material ethyl 4-(2-cyanoacetamido)benzoate (1) was prepared by reacting ethyl 4-aminobenzoate with ethyl cyanoacetate according to the previously reported method.20 Our work planned to synthesize 3-cyanopyrid-2-one and spiro-pyridine derivatives via the reaction of ethyl 4-(2-cyanoacetamido)benzoate (1) with different reagents, as described in Schemes 1–3. Treatment of ethyl 4-(2-cyanoacetamido)benzoate (1) with acetylacetone in ethanol and the presence of piperidine as a basic catalyst furnished the corresponding 4,6-dimethyl-2-oxo-pyridine derivative 2 via intramolecular hetero-cyclization and loss of water molecules. The analytical analysis and spectral data evidenced the structure of compound 2. The IR spectra of compound 2 revealed an absorption band at ν 2219 cm−1 due to the cyano group and bands at ν 1723 and 1660 cm−1 corresponding to two carbonyl groups. Its 1H NMR spectrum showed three singlet signals at δ 1.36, 4.38, 1.97, and 2.40 ppm corresponding to ethoxy and two methyl groups, and a singlet signal at δ 6.49 ppm corresponding to pyridine-CH, as well as four aromatic protons ranging from δ 7.50 to 8.12 ppm with the coupling constant J = 8.2 Hz. Additionally, its 13C NMR afforded new characteristic signals at δ 14.15, 20.69, 21.38, and 61.14 ppm attributed to two methyl carbons and ethoxy group, besides the carbon of cyano group (CN) and carbon of pyridine-C5 appeared at δ 115.75 and 100.04 ppm, besides, signals at δ 160.32, and 165.05 ppm were ascribed to two carbonyl groups (Scheme 1). Moreover, the reaction of the 4,6-dimethyl-2-oxo-pyridine derivative 2 with various aromatic aldehydes such as p-methoxy benzaldehyde and p-ethoxy benzaldehyde in an ethanolic solution containing piperidine as a catalyst afforded the corresponding 4-(alkoxystyryl)-6-methyl-2-oxopyridine derivatives 3a,b. The IR spectra of compound 3a, revealed absorption bands at ν 2216, 1718, and 1663 cm−1 due to cyano and two carbonyl groups, respectively. Furthermore, its 1H NMR spectrum indicated the lack of the methyl signal at δ 1.97 ppm and two signals with integration three at δ 2.02 and 3.83 ppm attributable to methyl (CH3) and methoxy (OCH3) protons. Besides, there were two new singlet signals at δ 6.11 and 6.72 ppm for the styryl moiety (CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH), and one at δ 6.49 ppm for pyridine-H5. Moreover, its 13C NMR spectrum of compound 3b displayed signals at δ 14.11, 14.52, 61.08, and 63.36 ppm related to two ethoxy groups, signals at δ 102.68 and 115.79 ppm corresponding to pyridine-C5 and carbon of cyano group, and a singlet signal at δ 21.62 ppm for the methyl group at C6 of pyridine. The downfield region also exhibited three signals at δ 160.43, 160.84, and 165.00 ppm related to carbon attached to ethoxy and the two carbonyl groups.
CH), and one at δ 6.49 ppm for pyridine-H5. Moreover, its 13C NMR spectrum of compound 3b displayed signals at δ 14.11, 14.52, 61.08, and 63.36 ppm related to two ethoxy groups, signals at δ 102.68 and 115.79 ppm corresponding to pyridine-C5 and carbon of cyano group, and a singlet signal at δ 21.62 ppm for the methyl group at C6 of pyridine. The downfield region also exhibited three signals at δ 160.43, 160.84, and 165.00 ppm related to carbon attached to ethoxy and the two carbonyl groups.
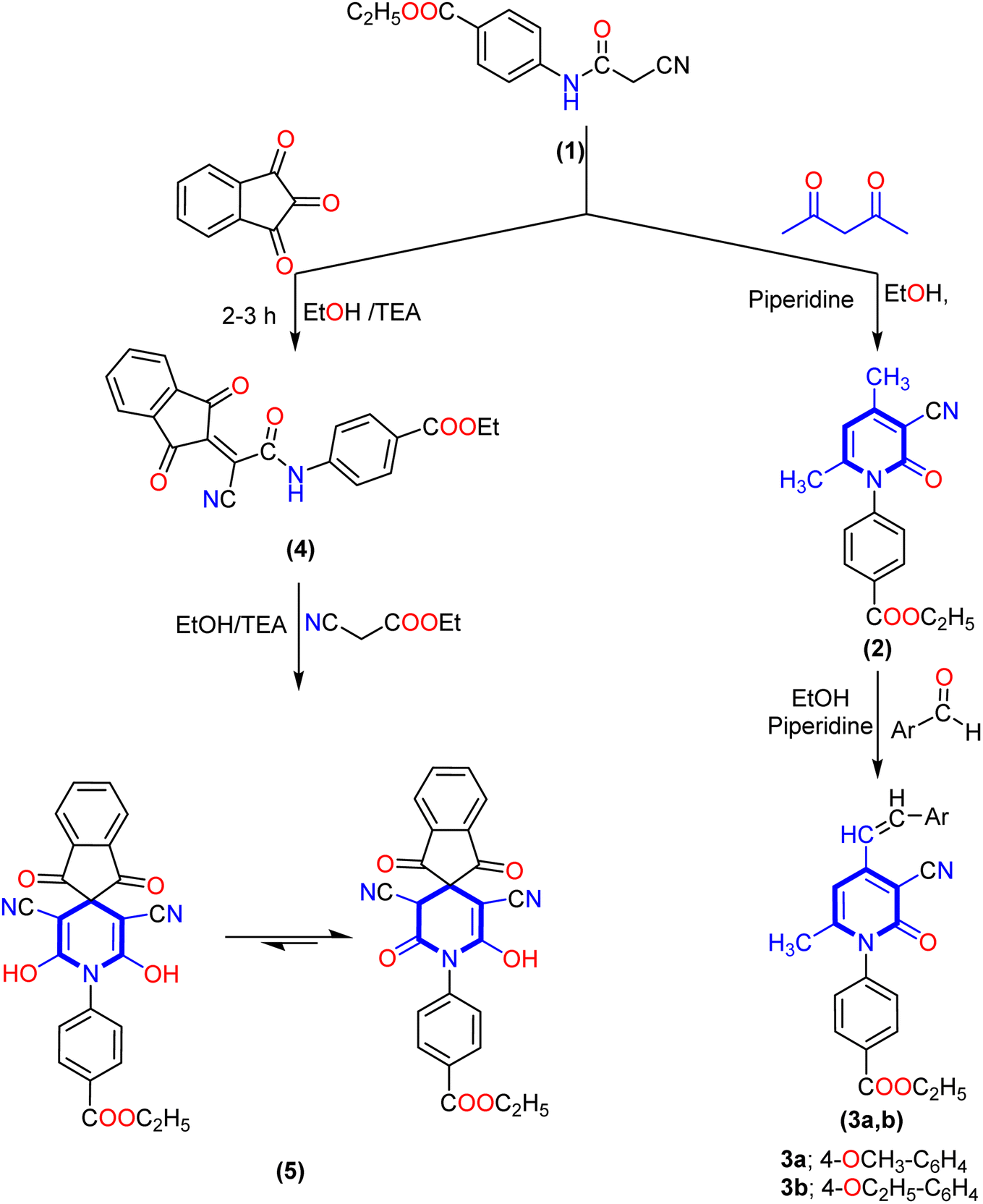 | ||
| Scheme 1 Synthesis of the new 2-oxo-pyridine derivatives (2 and 3a,b) and 1′H-spiro-indene-2,4′-pyridine derivative 5. | ||
Interestingly, our work was extended to study the synthetic accessibility of 2-cyano acetamide derivative 1 with some reagents for producing spiro compounds containing a pyridine moiety. The reactivity of 2-cyanoacetamide towards ninhydrin and isatin was investigated. First, the reaction of cyano acetamide derivative 1 with ninhydrin produced the 2-(1,3-dioxo-1,3-dihydro-2H-inden-2-ylidene)acetamide derivative 4. Meanwhile, the spiropyridine derivative 5 was obtained via the reaction of arylidene derivative 4 with ethyl cyanoacetate by Michael's addition of the active methylene group of ethyl cyanoacetate to the activated double bond of arylidene 4, followed by intramolecular cyclization via nucleophile attack of the lone pair of the amino group of amide to the carbonyl group of ethyl cyanoacetate, and therefore, the product tautomerized to create the 1′H-spiro-indene-2,4′-pyridine derivative 5.
Furthermore, the formation of spiro-pyridine derivative 5 was confirmed chemically by one-pot multicomponent reactions between cyanoacetamide derivative 1, ninhydrin, and ethyl cyanoacetate in molar ratio and in the presence of ethanolic solution with a few drops of triethylamine (TEA) as described in the experimental section. The IR spectrum of 1′H-spiro-indene-2,4′-pyridine derivative 5 revealed broad absorption bands at ν 3373, 2212, and 1713 cm−1 for the hydroxyl, cyano (CN), and carbonyl groups, respectively. At the same time, 1H NMR spectra displayed two signals as triplet and quartet at δ 1.32 and 4.29 ppm assignable to the protons of the ethoxy group and eight aromatic protons ranging between δ 7.62 and 7.95 ppm, as well as the presence of a new downfield singlet signal at δ 10.65 ppm attributed to two hydroxyl groups (exchangeable with D2O). Its 13C NMR spectra afforded a new characteristic signal at δ 59.40 ppm for the spiro carbon, singlet signals at δ 115.63 and 115.97 ppm for the two cyano groups, and three signals at δ 164.28, 165.15 and 165.80 ppm due to two carbons attached with OH and carbonyl groups of ninhydrin and ester group, respectively. In addition, the aromatic carbons ranged from δ 118.22 to 142.58 ppm.
Similarly, the treatment of 2-cyanoacetamide derivative 1 with isatin in an ethanolic solution catalyzed with piperidine produced the non-isolable arylidene derivative intermediate (A), which was then subjected to intramolecular cyclization via nucleophilic addition of the hydroxyl group (produced from tautomerism of the amide group) to the cyano group, followed by the Dimroth rearrangement as described previously41 to produce pyrrolo[2,3-b]indole derivative 6. The IR spectrum of compound 6 demonstrated the lack of a cyano group (CN) and presented a typical absorption band at ν 3278 and 1714 cm−1, referred to the NH group and two carbonyl groups (ester and amide). The 1H NMR spectra showed triplet and quartet signals at δ 1.31 and 4.32 ppm assigned to aliphatic protons of the ethoxy group and downfield singlet signals at δ 10.88 and 11.03 ppm associated with two NH groups, as well as the eight aromatic protons displayed from δ 6.83 to 7.98 ppm. Its 13C NMR spectrum showed two signals at δ 14.20 and 60.56 ppm for ethyl group carbons and distinctive three signals at δ 165.55, 165.64, and 168.98 ppm for two carbonyl groups of the amide (CO–NH) and ester (COOEt) groups, respectively (Scheme 2).
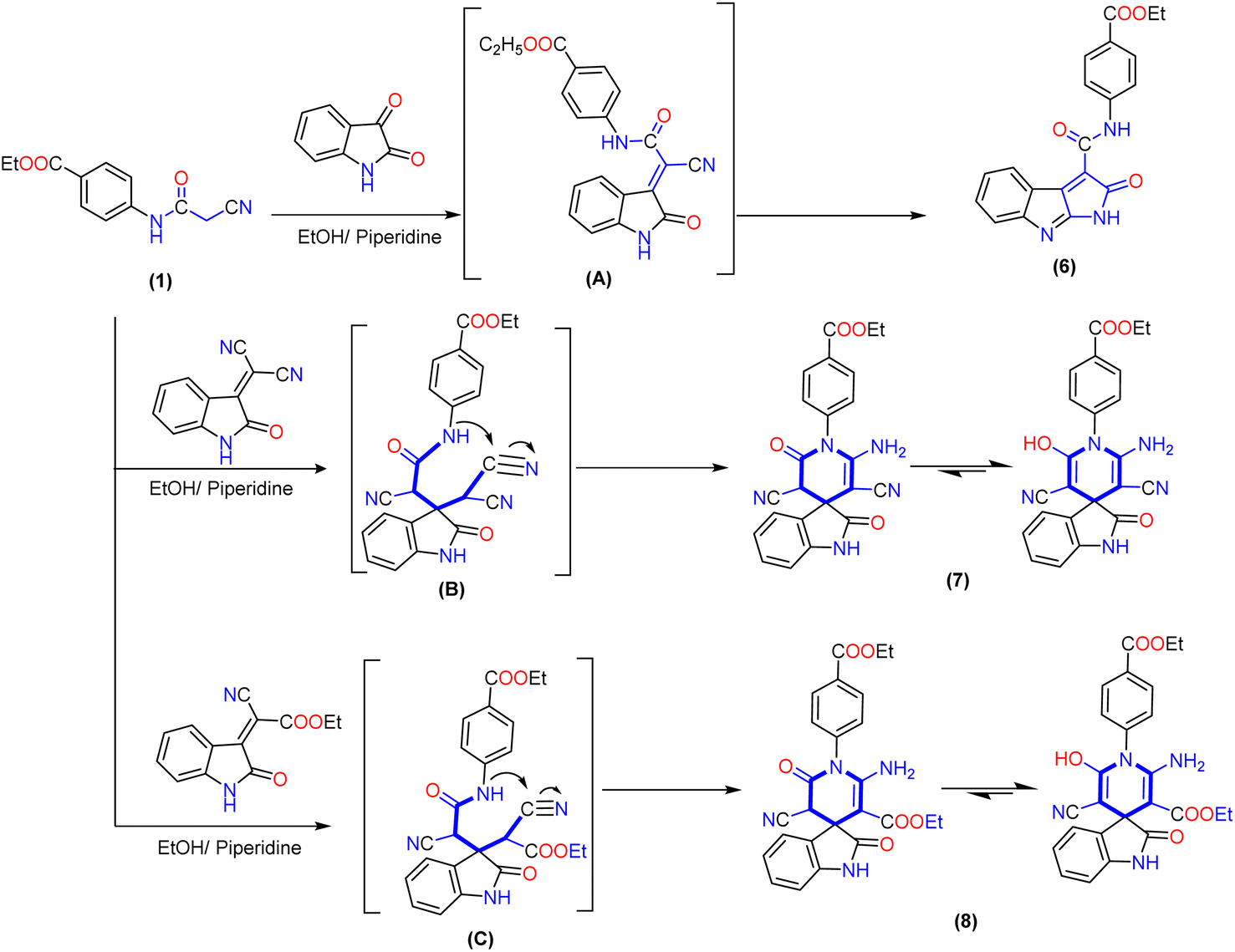 | ||
| Scheme 2 Synthetic pathway to produce fused pyrrolo[2,3-b]indole derivative 6 and 1′H-spiro-indoline-3,4′-pyridine derivatives 7 and 8. | ||
Interestingly, 1′H-spiro-indoline-3,4′-pyridine derivatives 7 and 8 are accomplished by the Michael addition reaction between cyanoacetamide derivative 1 and activated double bond of 2-(2-oxoindolin-3-ylidene)malononitrile or 2-cyano-2-(2-oxoindolin-3-ylidene)acetate in an ethanolic solution catalyzed with piperidine. Additionally, the reaction proceeds through intramolecular cyclization of non-isolable intermediate (B and C) by nucleophilic addition of the NH group to the cyano group, and the product obtained tautomerizes to yield 1′H-spiropyridoindoline derivatives 7 and 8. To evidence the structure of isolated product 7, the IR exhibited strong stretching frequencies in the region of ν 3316, 3210, 2188, and 1713 cm−1 attributable to hydroxy, amino (NH2), cyano (CN), and carbonyl groups, respectively. Moreover, its 1H NMR spectrum was distinguished by triplet and quartet signals at δ 1.26 and 4.20 ppm for the aliphatic proton of the ethoxy group. Moreover, eight aromatic protons ranged from δ 6.55 to 7.62 ppm with a coupling constant (J) ranging from 6.6 to 9.2 Hz, a broad band with integration two at δ 8.00 ppm related to the amino group, and two singlet signals at δ 9.05 and 10.86 ppm due to protons of hydroxyl group and NH of isatin. Its 13C NMR spectrum of compound 8 afforded new characteristic signals at δ 47.70, 103.24, and 109.61 ppm for the (C-spiro) and carbons of pyridine ascribed to CN and COOEt, respectively. In addition, five deshielded signals appeared at δ 153.46, 165.25, 165.94, 169.04, and 169.41 ppm attributed to two carbons attached with amino and OH groups and three carbonyl groups, respectively. Besides, the aromatic carbons ranged from δ 116.08 to 144.13 ppm (Scheme 2).
Furthermore, the reaction of 2-cyanoacetamide derivative 1 with some reagents containing an activated double bond, such as 3,3-bis(methylsulfanyl)methylene malononitrile or 2-aminoprop-1-ene-1,1,3-tricarbonitrile afforded the corresponding 4-(thiomethyl)-2-oxopyridine derivative 9 and 3,5-dicyano-4-(cyanomethyl)-2-oxopyridine derivative 10, respectively. The structure of the produced compounds was confirmed by elemental analysis and spectrum data. The IR spectrum of 4-(thiomethyl)-2-oxopyridine derivative 9 exhibited absorption bands at ν 3365, 3318, 2214, 1707, and 1681 cm−1 related to amino, cyano (CN) and two carbonyl groups (CO), respectively. Additionally, 1H NMR spectrum of compound 9 displayed two signals at δ 1.36 and 4.39 ppm assigned to the aliphatic protons of the ethoxy group, a singlet signal at δ 2.50 attributed to the thiomethyl group, two doublet signals at δ 7.52 and 8.13 ppm corresponding to four aromatic CH protons, and a broad singlet signal at δ 7.88 ppm related to the NH2 group. Similarly, 1H NMR spectrum of compound 10 displayed three signals at δ 1.31, 4.29, and 3.94 ppm attributed to aliphatic protons of the ethoxy group and methylene group of the acetonitrile moiety in position four at the pyridine ring. In addition to two doublet signals at δ 7.68 and 7.93 ppm corresponding to four aromatic protons with coupling constant J = 8.8 and 8.6 Hz, a singlet signal appeared at δ 6.76 ppm assigned to the amino (NH2) group. Moreover, the 13C NMR spectrum of compound 10 showed signals at δ 14.18, 60.50, and 26.97 ppm due to the ethoxy group and carbon of methylene group, and three signals deshielded at δ 152.55, 161.69, and 165.20 ppm for the carbon of pyridine bearing amino and two carbonyl groups, respectively.
Finally, treatment of 2-cyanoacetamide derivative 1 and 3-(4-hydroxyphenyl)-1-(p-tolyl)prop-2-en-1-one as the α,β-unsaturated carbonyl compound in the presence of a catalytic quantity of triethylamine (TEA) gave 1,4,6-triaryl-2-oxo-pyridine derivative 11. The IR spectra of compound 11 showed absorption bands at ν 3350, 2210, and 1693 cm−1 for the hydroxy, cyano, and two carbonyl groups, respectively. The 1H NMR spectrum of compound 11 revealed three singlet signals at δ 1.30, 4.29, and 2.12 ppm attributed to the ethoxy group and methyl of the tolyl moiety, respectively. In addition, a singlet signal at δ 6.52 ppm for pyridine-H5 and twelve aromatic protons appeared as six doublet signals with coupling constant (J) ranging from 8.0 to 8.6 Hz, as well as one exchangeable singlet signal at δ 10.60 ppm assigned to the hydroxyl group. The 13C NMR spectra displayed signals at δ 97.64, 101,89, and 116.36 ppm due to the carbon of pyridine attached to the nitrile group, pyridine-C5, and cyano group, as well as three deshielded signals at δ 161.64, 162.18 and 165.30 ppm attributed to carbon attached with OH and two carbonyl groups of pyridine and ester, respectively (Scheme 3).
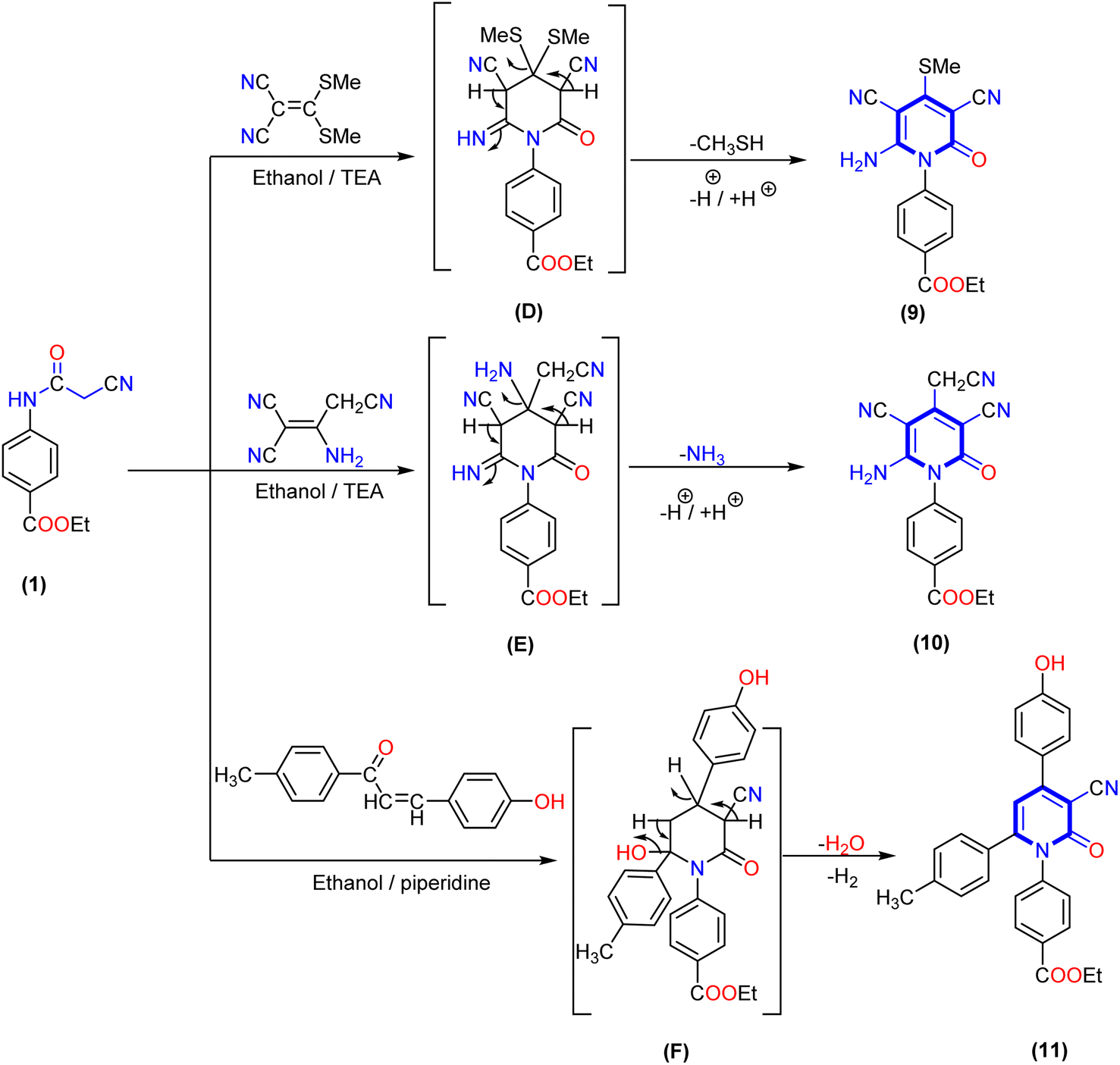 | ||
| Scheme 3 Synthesis of 3-cyano 2-oxo-pyridine derivatives (9–11). | ||
2.2. Biological activity
| Comp. no. | In vitro cytotoxic activity IC50 (μM) | |
|---|---|---|
| HepG-2 | Caco-2 | |
| 2 | 51.59 ± 2.90 | 41.49 ± 2.50 |
| 3a | 78.17 ± 3.80 | 84.43 ± 4.0 |
| 3b | 63.11 ± 3.40 | 75.68 ± 3.50 |
| 4 | 28.09 ± 2.10 | 37.32 ± 2.40 |
| 5 | 10.58 ± 0.80 | 9.78 ± 0.70 |
| 6 | 39.30 ± 2.50 | 45.37 ± 2.70 |
| 7 | 8.90 ± 0.60 | 7.83 ± 0.50 |
| 8 | 8.42 ± 0.70 | 13.61 ± 1.20 |
| 9 | 34.91 ± 2.20 | 56.19 ± 3.10 |
| 10 | 25.25 ± 1.90 | 37.78 ± 2.30 |
| 11 | 14.87 ± 1.20 | 19.23 ± 1.40 |
| DOX | 4.50 ± 0.20 | 12.49 ± 1.10 |
According to Table 1, the synthesized derivatives 2–11 exhibited moderate to promising potency with IC50 values ranging between (8.42 ± 0.70 to 78.17 ± 3.80 μM) and (7.83 ± 0.50 to 84.43 ± 4.0 μM) against HepG-2 and Caco-2 cell lines compared with Doxorubicin (IC50 HepG-2 = 4.50 ± 0.20 μM and IC50 Caco-2 = 12.49 ± 1.10 μM). Among the synthesized derivatives, the spiro-pyridine derivatives 5, 7, and 8 revealed a remarkably higher activity against HepG-2 and Caco-2 cell lines. Generally, the result indicated that the spiro-pyridine derivatives 5 (IC50 = 10.58 ± 0.8 and 9.78 ± 0.7 μM), 7 (IC50 = 8.90 ± 0.6 and 7.83 ± 0.5 μM), and 8 (IC50 = 8.42 ± 0.7 and 13.61 ± 1.2 μM) represented the highest anticancer potential against all tested human cancer cells compared to other compounds and Doxorubicin, which has IC50 values of 4.50 ± 0.2 and 12.49 ± 1.1 μM against HepG-2 and Caco-2, respectively. The spiro-pyridine derivatives 5 and 7 demonstrated a superior activity against Caco-2 cell lines with IC50 values of 7.83 ± 0.50 and 9.78 ± 0.70 μM.
In terms of structure–activity relationship (SAR), it was found that 1′H-spiro-indene-2,4′-pyridine derivative 5 with an indene moiety showed IC50 values of 10.58 ± 0.80 and 9.78 ± 0.70 μM compared with Doxorubicin (IC50 = 4.50 ± 0.20 and 12.49 ± 1.10 μM) against HepG-2 and Caco-2, respectively. Moreover, replacing the indene moiety with an indoline scaffold, as represented in compounds 7 and 8, improved the IC50 values against the HepG-2 cell line with IC50 values of 8.90 ± 0.60 and 8.42 ± 0.70 μM, indicating that the presence of ethyl carboxylate groups in position three in the pyridine nucleus decreases the cytotoxicity and have more antiproliferative activity than the cyano group against HepG-2. However, the presence of a cyano group at C3 of pyridine in 1′H-spiro-indoline-3,4′-pyridine derivative 7 enhanced the antiproliferative activity against the Caco-2 cell line with IC50 values of 7.83 ± 0.50 μM compared with 3'-(ethyl carboxylate)-2-oxo-1′H spiro-indoline-3,4′-pyridine 8 (IC50 = 13.61 ± 1.20 μM) and Doxorubicin (IC50 = 12.49 ± 1.10 μM).
Furthermore, the 4,6-dimethyl-2-oxo-pyridine derivative 2 exhibited a moderate cytotoxic activity with IC50 values of 51.59 ± 2.90 and 41.49 ± 2.50 μM against HepG-2 and Caco-2 cell lines. Additionally, the reaction of a methyl group at C4 of the pyridine nucleus to afford 4-(alkoxystyryl)-6-methyl-2-oxopyridine derivatives 3a and 3b decreases the activity by increasing the cytotoxic values with IC50 values of 78.17 ± 3.80 and 63.11 ± 3.40 μM against HepG-2 and IC50 = 84.43 ± 4.0 and 75.68 ± 3.50 μM against the Caco-2 cell line and that may be related to these derivatives 3a,b possessing one extra phenyl group. The cytotoxicity results also indicated that the ethoxy group is more advantageous than the methoxy group. Introducing the indene moiety to 2-cyanacetamide derivative 2 to form a new arylidine derivative 4 does not enhance the activity. Considering the results in Table 1 and Fig. 2, it was found that introducing three aryl groups (as hydrophobic tails) to the pyridine nucleus as designated in 1,4,6-triaryl-2-oxo-pyridine derivative 11 caused enhancement in cytotoxic activity against HepG-2 and Caco-2 cell lines with IC50 values of 14.87 ± 1.20 and 19.23 ± 1.40 μM, but still higher than Doxorubicin. Moreover, modification of a 3,5-dicyano-2-oxopyridine derivative by insertion of thiomethyl 9 and cyanomethyl 10 at C4 of pyridine nucleus gave a mild change in activity with IC50 values of 34.91 ± 2.20 and 25.25 ± 1.90 μM and 56.19 ± 3.10 and 37.78 ± 2.30 μM against HepG-2 and Caco-2 cell lines, respectively.
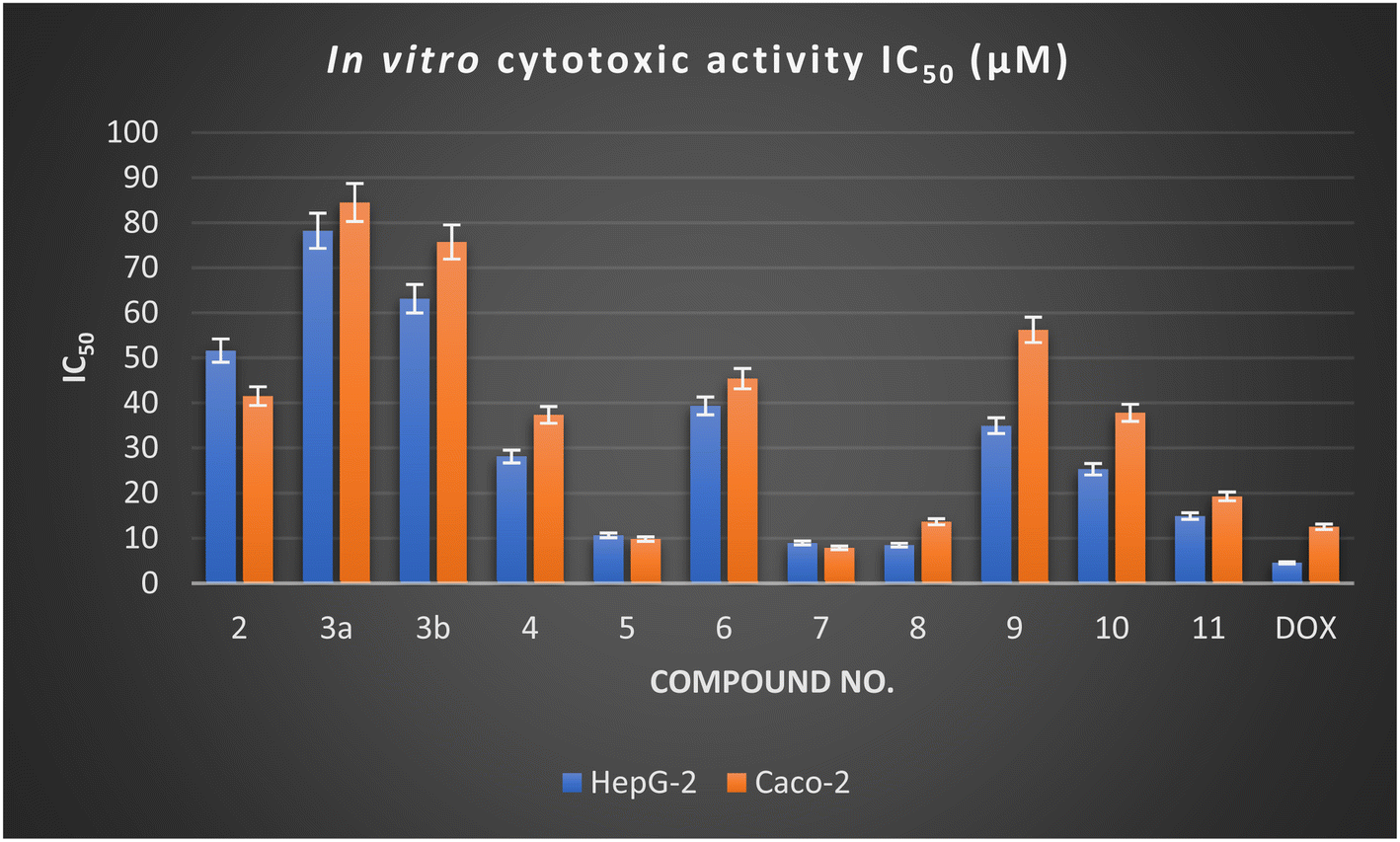 | ||
| Fig. 2 In vitro cytotoxic activity of the newly designed derivatives against the HepG-2 and Caco-2 cell lines. | ||
Finally, the antiproliferative activity of the designed pyridine and spiro-pyridine derivatives showed moderate to good activity against HepG-2 and Caco-2 cell lines. Among the designed compound, the spiro-pyridine derivatives 5, 7, and 8 showed promising results with IC50 values ranging from 7.83 ± 0.50 to 13.61 ± 1.20 μM against the Caco-2 cell line compared with Doxorubicin (IC50 = 12.49 ± 1.10 μM). In contrast, these derivatives showed potency against HepG-2 with an IC50 value lower than 10 μM.
2.2.2.1. Mitochondrial apoptosis pathway proteins (BAX and Bcl-2). The Bcl-2 family of genes plays a crucial role in regulating apoptosis, particularly Bax and Bcl-2, associated with the apoptotic pathway. Additionally, the Bax and Bcl-2 genes play a role in downstream caspase protein initialization.44,45 The effects of most active derivatives 5, 7, and 8 at a concentration of their IC50 values 9.78, 7.83, and 13.61 μM against Bax (pro-apoptotic) and Bcl-2 (anti-apoptotic) genes on Caco-2 cells were studied. The expression of Bax and Bcl-2 was evaluated using specific primers and antibodies according to the manufacturer's instructions using the qRT-PCR technique, as described previously.46,47 Generally, the results revealed a remarkable change in the expression of apoptotic genes Bax (increased) and Bcl-2 (decreased) compared to the untreated control cells (Caco-2).
As depicted in Table 2, the most active compounds 5, 7, and 8 exhibited significant activation in the Bax expression by 4.508, 7.508, and 4.188 folds, respectively. Meanwhile, these derivatives displayed suppression of Bcl-2 expression with values ranging from 0.194 to 0.436. Among these derivatives, compound 7 significantly increased Bax expression in Caco-2 by 7.508 folds and caused down-regulation to the Bcl-2 gene by 0.194 folds. These changes in expression profiles significantly reduced the growth rate of Caco-2 cells when treated with the most active derivatives.
| Tested cpd. | qRT-PCR results fold change | |
|---|---|---|
| Pro-apoptotic | Anti-apoptotic | |
| Bax | Bcl-2 | |
| 5/Caco-2 | 4.508 | 0.436 |
| 7/Caco-2 | 7.508 | 0.194 |
| 8/Caco-2 | 4.188 | 0.275 |
| Cont. Caco-2 | 1 | 1 |
Finally, it can be concluded that the most active derivatives 5, 7, and 8 can be represented as kinase inhibitors by activation of Bax and deactivation of Bcl-2 genes by variable degree, and the explanation for these results may be related to the imbalance of Bax and Bcl-2 by promoting proapoptotic and antiapoptotic factors.
2.2.2.2. Cell cycle analysis and apoptosis induction by annexin-V assay for the most active compound 7. The antiproliferative agents often inhibit the proliferation of cells by stopping the cell cycle at specific points. The cell cycle analysis was performed on the most active compound 7 over the colorectal adenocarcinoma (Caco-2) cell line. Generally, the flow cytometry analysis demonstrated that 2-oxo-1′H-spiro-indoline-3,4′-pyridine derivative 7 induced cell cycle arrest at the S phase. Moreover, the effect of the most potent compound 7 on the Caco-2 cell line at its IC50 value (7.83 μM) markedly increased the S cell accumulation ratio from 31.18 to 42.07%. Moreover, the percentage of G0-G1 and G2/M cells decreased simultaneously, with values of 51.08 and 6.85%, compared with untreated Caco-2 cancer cells with 54.69 and 14.13%, respectively Fig. 3.
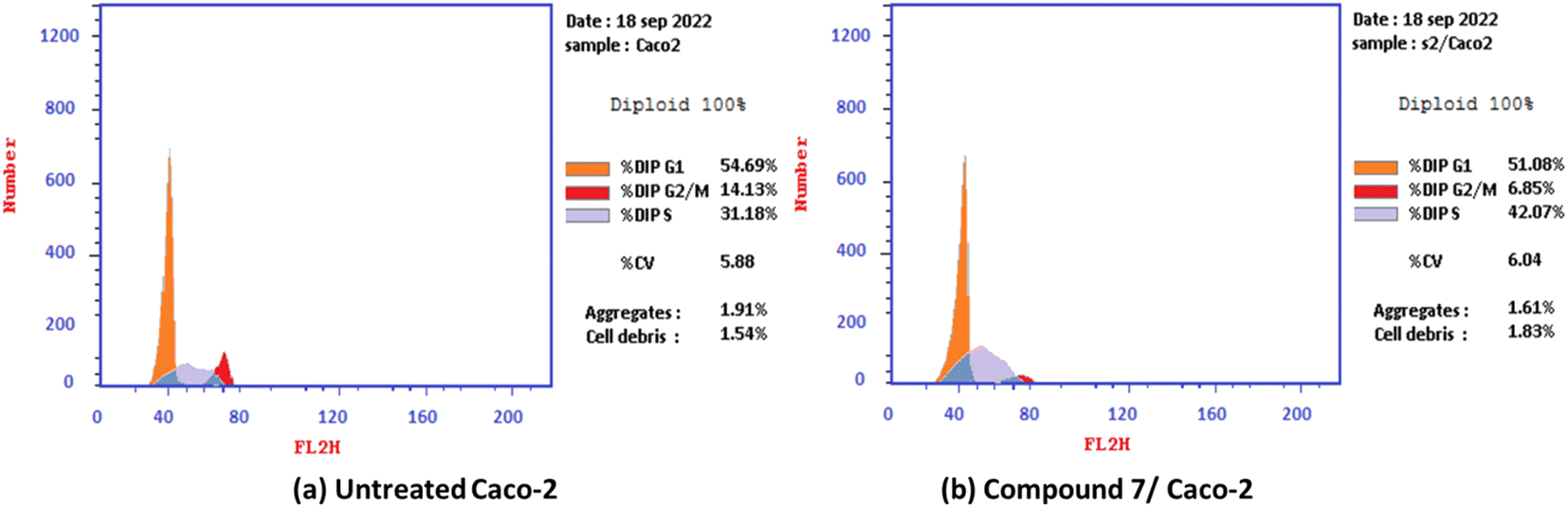 | ||
| Fig. 3 Cell cycle distribution% assessment using FACS analysis: (a) untreated Caco-2 cells and (b) compound 7 treated with Caco-2 at IC50 = 7.83 μM. | ||
Additionally, the ability of the most active derivative to induce apoptosis was confirmed by applying double staining of annexin-V/propidium iodide (PI) to stain DNA and, therefore, stated the dead cells. The most active 2-oxo-1′H-spiro-indoline-3,4′-pyridine derivative 7 exhibited a significantly higher apoptotic effect in Caco-2 cells with a value of 42.35% compared to that of control cells of 1.92%. In addition, this compound displayed a necrosis value of 3.02 compared with 1.42% for control cells. In addition, the 2-oxo-1′H-spiro-indoline-3,4′-pyridine derivative 7 presented a remarkable increase in the DNA content in early apoptosis with a value of 22.31 (67.60-folds) and late apoptosis with a value of 17.02 (100.11-folds) compared to the control as described in Table 3 and Fig. 4.
| Cpd. no. | Apoptosis | Necrosis | ||
|---|---|---|---|---|
| Total | Early | Late | ||
| 7/MCF-7 | 42.35 | 22.31 | 17.02 | 3.02 |
| Cont. Caco-2 | 1.92 | 0.33 | 0.17 | 1.42 |
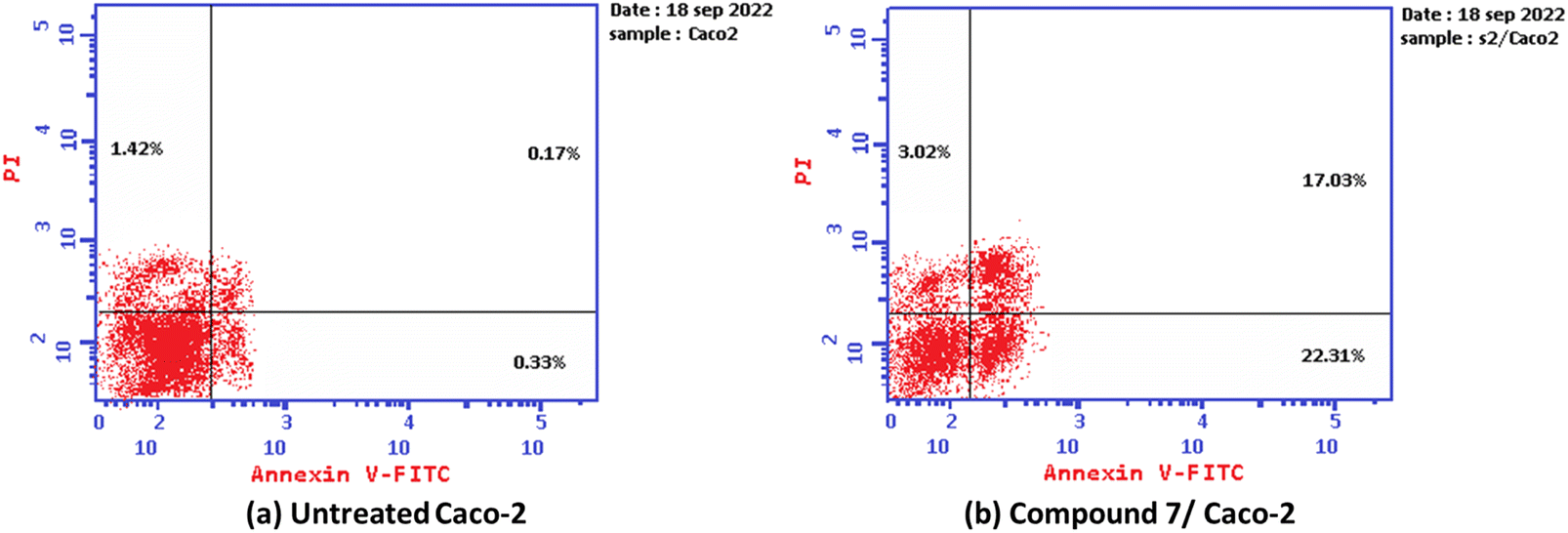 | ||
| Fig. 4 Graph showing the apoptosis-inducing effects of the most active derivative 7 in Caco-2 cells when treated at its IC50 for 24 h: (a) untreated cell and (b) compound 7/Caco-2. | ||
2.2.2.3. Evaluate the receptor tyrosine kinase proteins (EGFR and VEGFR). To investigate the plausible mode of action of the anticancer activity, the in vitro inhibitory activity for most active derivative 7 against the epidermal growth factor EGFR (wide) and vascular epidermal growth factor receptor VEGFR-2 was evaluated using ELISA analysis. Erlotinib was used as an EGFR positive control, while Sorafenib was used for VEGFR-2. The results of inhibitory activity for the target compound 7 and positive controls against receptor tyrosine kinase proteins expressed by IC50 (μM) and the inhibitory percentage at 10 μM are summarized in Table 4. All the results of the target 2-oxo-1′H-spiro-indoline-3,4′-pyridine derivative 7 against tested proteins displayed values with sub-micromolar units (<0.25 μM).
| Cpd. no. | Enzyme inhibitory activity IC50a (μM) | |||
|---|---|---|---|---|
| EGFRWt | Inhibitory%b | VEGFR-2 | Inhibitory%b | |
| a The data are an average of three independent tests.b Inhibitory% at 10 μM. | ||||
| 7 | 0.124 ± 0.009 | 88.41 | 0.221 ± 0.009 | 87.03 |
| Doxorubicin | 0.349 ± 0.016 | 77.59 | — | — |
| Erlotinib | 0.033 ± 0.002 | 92.85 | — | — |
| Sorafenib | — | — | 0.043 ± 0.002 | 92.08 |
According to the above-mentioned results, 2-oxo-1′H-spiro-indoline-3,4′-pyridine derivative 7 significantly reduced the activity of EGFR with an IC50 value of 0.124 ± 0.009 and an inhibitory percentage (IP) of 88.41% compared with Doxorubicin (IC50 = 0.349 ± 0.016 and IP = 77.59%) and Erlotinib (IC50 = 0.033 ± 0.002 and IP = 92.85%). Simultaneously, 2-oxo-1′H-spiro-indoline-3,4′-pyridine derivative 7 efficiently suppressed VEGFR-2 activity with an IC50 of 0.221 ± 0.009 μM compared to Sorafenib (IC50 = 0.043 ± 0.002 μM). Moreover, compound 7 revealed an inhibitory percentage against VEGFR-2 activity of 87.03%, which was nearly equipotent to Sorafenib (IP = 92.08%).
Finally, it can be concluded that the evaluation findings suggest that the 2-oxo-1′H-spiro-indoline-3,4′-pyridine derivative 7 may benefit EGFR and VEGFR-2 inhibitory actions with a higher inhibitory percentage to EGFR wide type than VEGFR-2 due to its sub-micromolar level of inhibition.
2.3. In silico ADME and toxicity predictions
| Test items | Most active 2-oxo-1′H-spiro-indoline-3,4′-pyridine derivative 7 and positive controls | ||||
|---|---|---|---|---|---|
| 7 | Dox.a | Erl.a | Sor.a | ||
| a DOX. = Doxorubicin, Erl. = Erlotinib, and Sor. = Sorafenib. | |||||
| SwissADME | Molecular properties | ||||
| MLog P | 0.36 | −2.10 | 1.89 | 2.91 | |
| XLogP3 | 2.59 | 1.27 | 4.11 | 4.07 | |
| Fraction Csp3 | 0.13 | 0.44 | 0.27 | 0.10 | |
| TPSA (Å2) | 152.47 | 206.07 | 74.73 | 92.35 | |
| M. wt. | 427.41 | 543.52 | 393.44 | 464.82 | |
| nHBA (NO) | 6 | 12 | 6 | 7 | |
| nHBD (OHNH) | 3 | 6 | 1 | 3 | |
| NRB | 4 | 5 | 10 | 9 | |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|||||
| Pharmacokinetics | |||||
| GI absorption | Low | Low | High | Low | |
| BBB permeant | No | No | Yes | No | |
| P-gp substrate | Yes | Yes | No | No | |
| Skin permeation (logKp), cm s−1 |
−7.07 | −8.71 | −6.35 | −6.25 | |
|
|||||
| Drug likeness and medicinal chemistry | |||||
| LogS (ESOL) |
−4.14 | −3.91 | −4.11 | −5.11 | |
| Solubility | Mod. Soluble | Soluble | Mod. soluble | Mod. soluble | |
| PAINS | 0 | 1 (quinone) | 0 | 0 | |
| Synthetic accessibility | 4.52 | 5.81 | 3.19 | 2.87 | |
| Bioavailability score | 0.55 | 0.17 | 0.55 | 0.55 | |
| Lipinski rule (violation) | Yes (0) | No (3) | Yes (0) | Yes (0) | |
| Veber rule (violation) | No (1) | No (1) | Yes (0) | Yes (0) | |
The result represented that 2-oxo-1′H-spiro-indoline-3,4′-pyridine derivative 7, Erlotinib, and Sorafenib obey the Lipinski rule of five without any violation, except Doxorubicin that exhibited unfollow Lipinski rule due to three violations including M. wt over 500 Dalton, hydrogen bond acceptors = 12 (two higher than standard), and hydrogen bond donor = 6 (one more than standard). Additionally, the 2-oxo-1′H-spiro-indoline-3,4′-pyridine derivative 7 and Doxorubicin do not follow the Veber rule due to the topological polar surface area (TPSA) being higher than 140. Moreover, all the tested compounds exhibited soluble to moderately soluble behaviours with Log S (ESOL) values ranging from −5.11 to −3.91. Additionally, the most active 2-oxo-1′H-spiro-indoline-3,4′-pyridine derivative 7, Erlotinib, and Sorafenib demonstrated no Pan-assay interference compound (PAIN) alarms in their structure, while doxorubicin showed only one PAIN alarm due to the quinone ring.
Furthermore, the tested derivative and positive controls revealed acceptable bioavailability scores with values of 0.55, while Doxorubicin displayed 0.17. Additionally, compound 7 showed easy synthetic accessibility = 4.52, Erlotinib (3.19) and Sorafenib (2.87) less than Doxorubicin (5.81). Moreover, for pharmacokinetic prediction, all tested derivatives displayed low gastrointestinal tract and did not pass the BBB, except Erlotinib, which showed high GI and passed the BBB. As well, compound 7 and Doxorubicin were revealed to be substrates for P-gp, which does not cause problems with drug excretion. In contrast, Erlotinib and Sorafenib are not substrates for P-gp. Based on these data, 2-oxo-1′H-spiro-indoline-3,4′-pyridine derivative 7 appears to be a promising drug candidate for further research and development.
Fig. 5 shows the calculation of radar charts obtained from the SwissADME web tool to predict the accessibility of the tested derivatives to be oral bioavailability. These charts involve six parameters as insaturation (INSATU), polarity (POLAR), insolubility (INSOLU), flexibility (FLEX), lipophilicity (LIPO), and size, and the tested compound is represented by a red line integrated into the pink area. The optimal range for each property in the radar charts can be represented as follows: INSATU (insaturation), 0.25 < fraction of Csp3 < 1; POLAR (polarity), 20 Å2 < topological surface area (TPSA) < 130 Å2; INSOLU (insolubility), 0 < logS (ESOL) < 6; FLEX (flexibility), 0 < number of rotatable bonds < 9; lipophilicity (LIPO), −0.7 < XLOGP 3 < + 5.0; and SIZE, 150 g mol−1 < MW < 500 g mol−1.51 Moreover, the lipophilicity in our work is measured in two ways: Moriguchi octanol–water partition coefficient (MLOGP), which is employed in the Lipinski rule and based on quantitative structure–logP relationships using topological indexes.52 Simultaneously, XLOGP3 predicts a given compound's partition coefficient (logP) value by starting with the known logP value of a reference compound and is used in radar charts.53
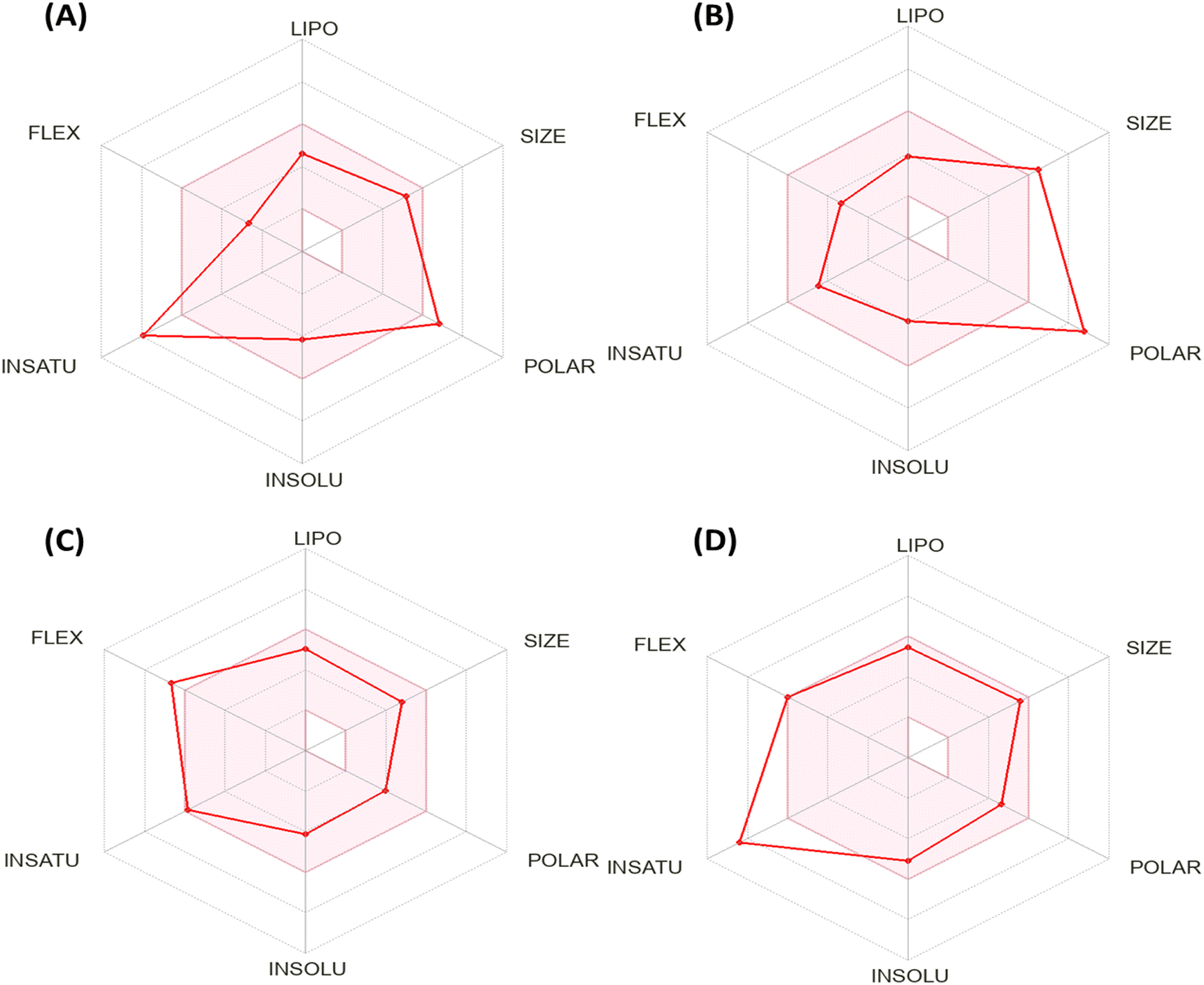 | ||
| Fig. 5 Bioavailability radar chart generated by Swiss-ADME for (A) the most active compound 7, (B) Doxorubicin, (C) Erlotinib, and (D) Sorafenib. | ||
Based on the above-mentioned criteria, the molecules that fall within the pink region of the radar are considered drug-like. The 2-oxo-1′H-spiro-indoline-3,4′-pyridine derivative 7 exhibited four of six rules with two violations as insaturation (INSATU), which refers to the ratio of hybridized sp3 atoms to the total number of C atoms (fraction of Csp3 = 0.13) and the second violation related to the polarity (POLAR) (TPSA = 152.47). At the same time, Doxorubicin demonstrated a violation in polarity that related to a number of hydrogen bond donors and acceptors, as well as TPSA > 140, while it showed a slight violation to flexibility and that related to the number of rotatable bonds. Moreover, Sorafenib demonstrated insaturation (INSATU) violation with a fraction of Csp3 value of 0.10.
However, the positive controls showed activity to immunotoxin, mutagenic, and cytotoxicity, except Sorafenib, which displayed inactivity to mutagenicity. Additionally, all the tested derivatives exhibited inactivity to heat shock factor response element (HSE) (performing important functions in the folding and unfolding or translocation of proteins, as well as in the assembly and disassembly of protein complexes) with probability values ranging between 0.88 and 0.96 and inactivity to mitochondrial membrane potential (MMP) (playing a vital role in energy storage during oxidative phosphorylation) with probability values ranging between 0.56 and 0.70, except Sorafenib that displayed activity with a probability of 0.79. In addition, compound 7 and Erlotinib depicted inactive tumor suppressor phosphoprotein (p53) with probabilities of 0.78 and 0.89, respectively, while showing activity for Doxorubicin and Sorafenib (Table 6).
| Oral toxicity prediction | Most active 2-oxo-1′H-spiro-indoline-3,4′-pyridine derivative 7 and positive controls | ||||
|---|---|---|---|---|---|
| 7 | Dox.a | Erl.a | Sor.a | ||
| a DOX. = Doxorubicin, Erl. = Erlotinib, and Sor. = Sorafenib. | |||||
| ProTox-II prediction | LD50 mg kg−1 | 1000 | 205 | 125 | 800 |
| Toxicity class | IV | III | III | IV | |
| Hepatotoxicity | Active | Inactive | Active | Active | |
| 0.50 | 0.86 | 0.78 | 0.82 | ||
| Carcinogenicity | Inactive | Inactive | Inactive | Inactive | |
| 0.56 | 0.90 | 0.51 | 0.50 | ||
| Immunotoxicity | Inactive | Active | Active | Active | |
| 0.99 | 0.99 | 0.91 | 0.92 | ||
| Mutagenicity | Inactive | Active | Active | Inactive | |
| 0.61 | 0.98 | 0.55 | 0.79 | ||
| Cytotoxicity | Inactive | Active | Active | Active | |
| 0.59 | 0.94 | 0.75 | 0.77 | ||
| Heat shock factor response element (HSE) | Inactive | Inactive | Inactive | Inactive | |
| 0.88 | 0.98 | 0.96 | 0.96 | ||
| Mitochondrial membrane potential (MMP) | Inactive | Inactive | Inactive | Active | |
| 0.70 | 0.56 | 0.68 | 0.79 | ||
| Phosphoprotein (tumor suppressor) p53 | Inactive | Active | Inactive | Active | |
| 0.78 | 0.52 | 0.89 | 0.57 | ||
| pkCSM prediction | AMES toxicity | No | Yes | No | No |
| Skin sensitisation | No | No | No | No | |
| hERG I inhibitor | No | No | No | No | |
| hERG II inhibitor | No | No | Yes | Yes | |
| Max. tolerated dose (human) (log mg per kg per day) | 0.047 | 0.654 | 0.654 | 0.677 | |
| Oral rat chronic toxicity (LOAEL) (log mg per kg_bw per day) | 1.722 | 3.296 | 1.404 | 1.054 | |
| Oral rat acute toxicity (LD50) (mol kg−1) | 2.185 | 3.978 | 2.757 | 2.595 | |
For pkCSM prediction, all tested derivatives featured non-AMES (except Doxorubicin), non-skin sensitivity (unable to elicit an allergic response), and non-inhibitors for hERG I and II (except Erlotinib and Sorafenib that displayed inhibitor to hERG II). Moreover, the most promising compound 7 expressed the lowest maximum tolerated dose (human) (0.047 log mg per kg per day) and oral rat chronic toxicity (LOAEL = 1.722 log mg per kg_bw per day). Besides, compound 7 showed oral rat acute toxicity (LD50 = 2.185 mol kg−1) lower than that of Erlotinib (LD50 = 2.757 mol kg−1), Sorafenib (LD50 = 2.595 mol kg−1), and Doxorubicin (LD50 = 3.978 mol kg−1).
Finally, based on the previous toxicity profile, it can be concluded that the most active 2-oxo-1′H-spiro-indoline-3,4′-pyridine derivative 7 exhibited a safety profile that appeared through non-carcinogenic, non-immunotoxin, non-mutagenic, and non-cytotoxic activity with a good LD50 value.
2.4. Molecular docking simulations
To determine the suitable mechanism of anticancer activity and to explain the experimental result obtained previously, the molecular docking simulation for the most active 2-oxo-1′H-spiro-indoline-3,4′-pyridine 7 was performed inside the active sites of Bcl-2 (PDB: 4AQ3), EGFR (PDB:1M17), and VEGFR-2 (PDB: 4ASD). All these proteins were downloaded from the protein data bank (https://www.rcsb.org/; access 1/2/2023).The docking pose for the most active 2-oxo-1′H-spiro-indoline-3,4′-pyridine 7 exhibited binding energy S = −17.10 kcal mol−1 through two hydrogen bond sidechain acceptors between Arg105 and nitrogen of the cyano group at C5 of pyridine with bond lengths of 2.31 and 2.59 Å and strengths of 16 and 10%, respectively. In addition, the arene–cation interaction takes place between Arg105 and phenyl of isatin derivative. The most active 2-oxo-1′H-spiro-indoline-3,4′-pyridine 7 exhibited hydrophobic interaction with the pocket through the isatin nucleus, pyridine core, and phenyl of ethyl benzoate group with the residues Leu96, Ala108, Met74, Tyr67, Phe63, and Phe71 (Fig. 6).
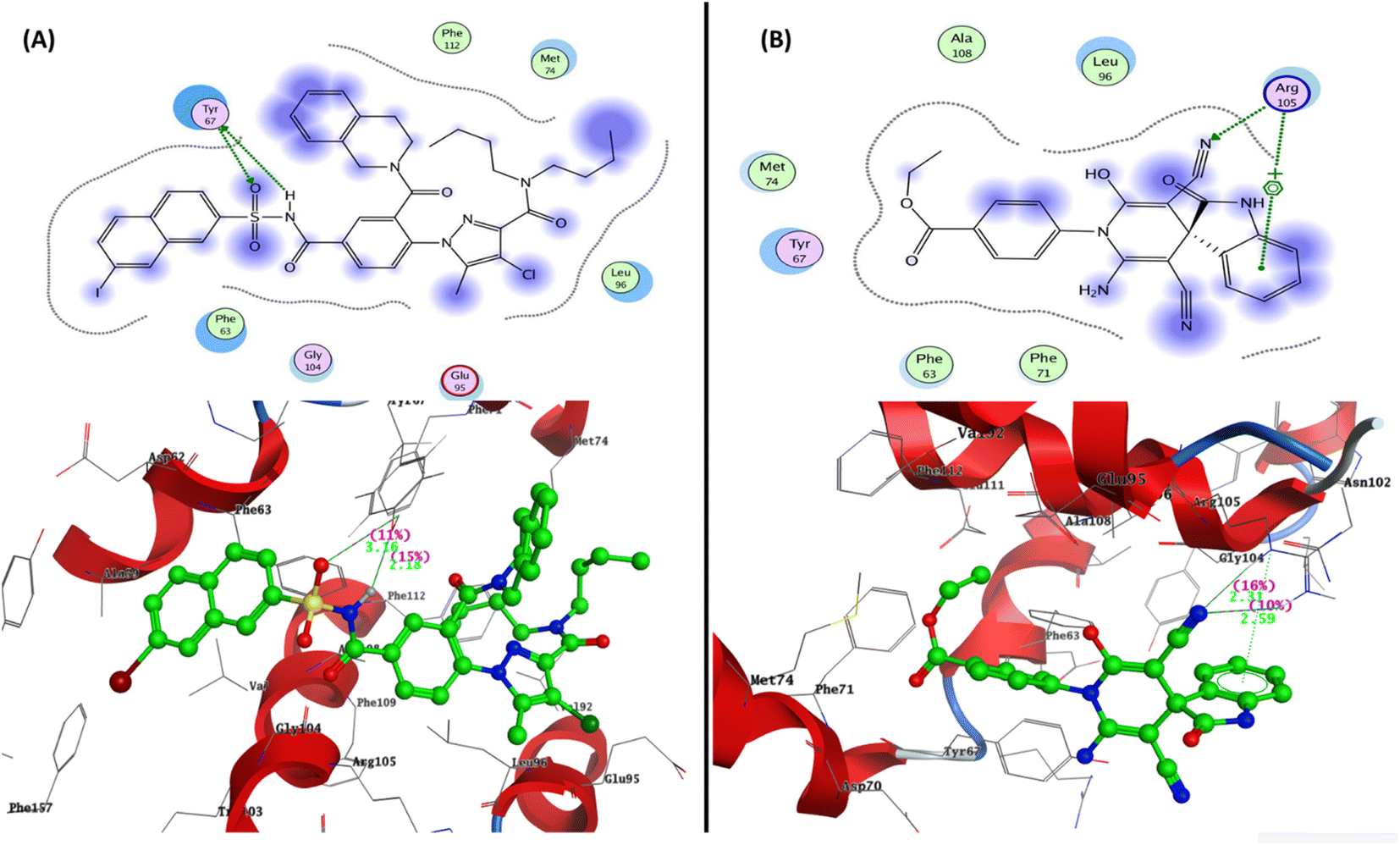 | ||
| Fig. 6 Representation of the 2D and 3D binding modes of (A) co-crystallized ligand and (B) 2-oxo-1′H-spiro-indoline-3,4′-pyridine 7 inside Bcl-2 binding pocket (PDB: 4AQ3). | ||
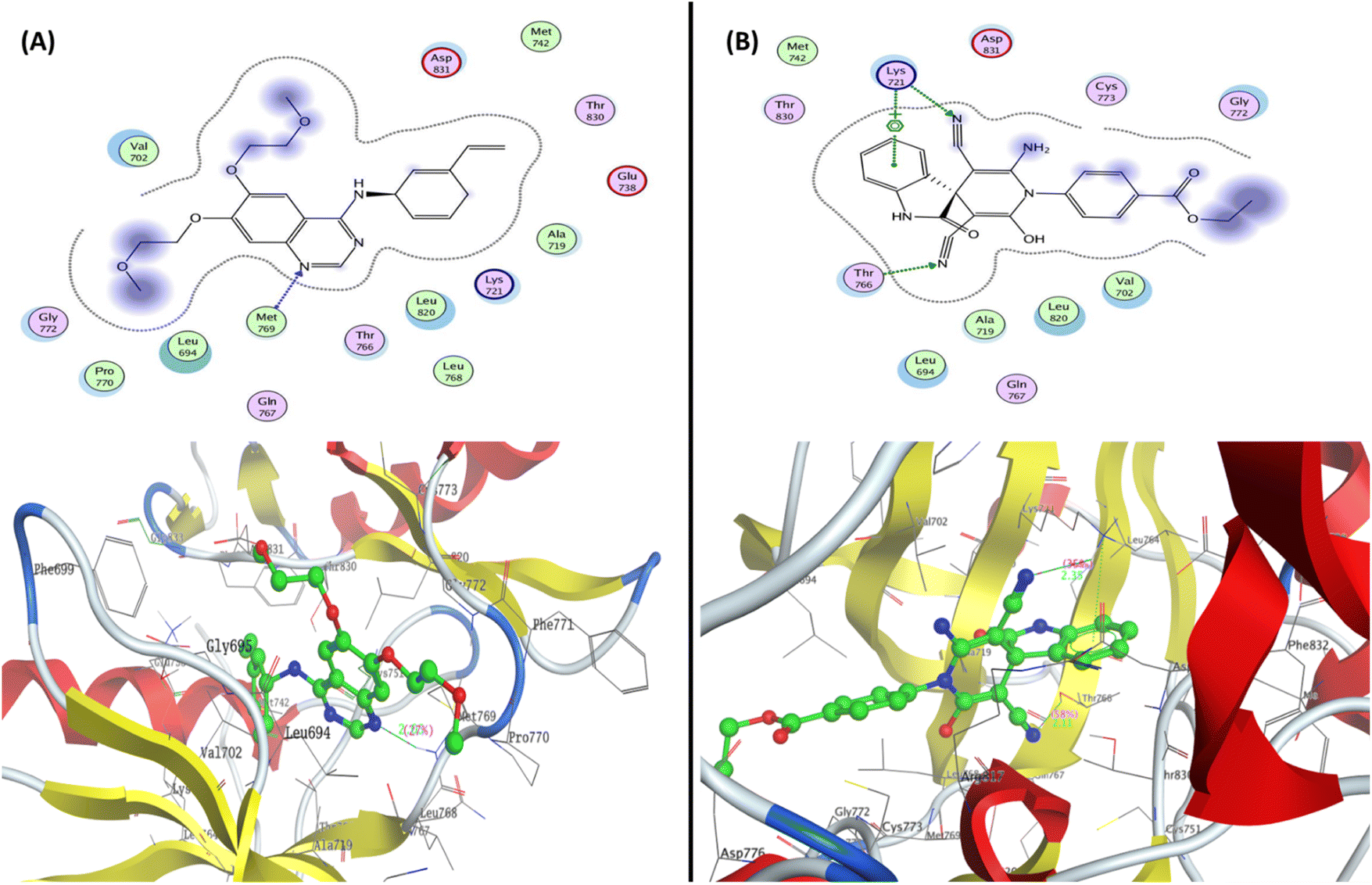 | ||
| Fig. 7 Representation of the 2D and 3D binding mode of (A) co-crystallized ligand and (B) 2-oxo-1′H-spiro-indoline-3,4′-pyridine 7 inside the EGFR binding pocket (PDB: 1M17). | ||
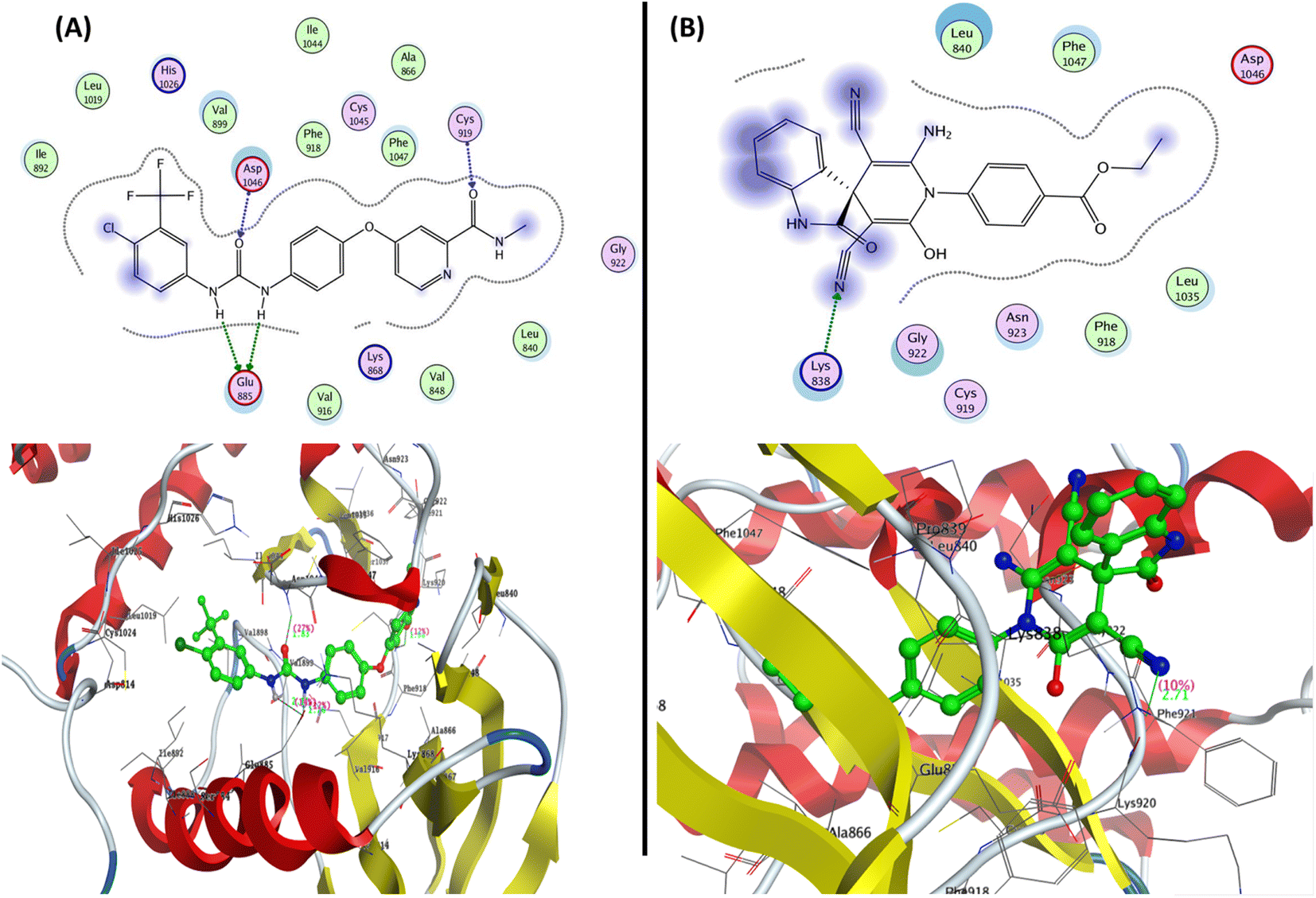 | ||
| Fig. 8 Representation of the 2D and 3D binding modes of (A) co-crystallized ligands and (B) 2-oxo-1′H-spiro-indoline-3,4′-pyridine 7 inside the VEGFR-2 binding pocket (PDB: 4ASD). | ||
3. Conclusion
In this work, reaction of 4-(3-cyano-4,6-dimethyl-2-oxopyridin-1(2H)-yl)benzoate (1) with different reagents afforded 2-oxo-pyridine and spirooxindole, as well as arylidine of 2-cyanoacetamide and pyrrolo[2,3-b]indole derivatives. The analytical analysis and spectral data evidenced the structure of newly designed derivatives. The in vitro cytotoxic activity of all synthesized derivatives was evaluated against HepG-2 and Caco-2 cell lines. The synthesized derivatives 2–11 exhibited moderate to promising potency with IC50 values ranging between (8.42 ± 0.70 to 78.17 ± 3.80 μM) and (7.83 ± 0.50 to 84.43 ± 4.0 μM) against HepG-2 and Caco-2 cell lines compared with Doxorubicin (IC50 HepG-2 = 4.50 ± 0.20 μM and IC50 Caco-2 = 12.49 ± 1.10 μM). The SAR study showed that spirooxindole displayed better IC50 values in both cell lines with more potency to Caco-2 cells than other pyridine derivatives. The most active derivatives 5, 7, and 8 were evaluated on Bax and Bcl-2 gene expression. The results exhibited activation in the Bax expression by 4.508, 7.508, and 4.188 folds and suppressed Bcl-2 expression with values of 0.436, 0.194, and 0.275, respectively. Compound 7 exhibited the highest Bax and Bcl-2 inhibition at its IC50 value on the Caco-2 cell line. Additionally, the cell cycle and apoptosis analysis using annexin PI were evaluated. The results indicated accumulation of cells at the S phase with a ratio of 42.07% and induced total apoptosis with 42.35% compared to control untreated cells. Furthermore, 2-oxo-1′H-spiro-indoline-3,4′-pyridine derivative 7 reduced the activity of EGFR and VEGFR-2 with IC50 values of 0.124 ± 0.009 and 0.221 ± 0.009 μM, compared with Erlotinib (IC50 = 0.033 ± 0.002) and Sorafenib (IC50 = 0.043 ± 0.002 μM), respectively. The in silico ADMET prediction of compound 7 exhibited good drug-likeness, medicinal chemistry, and safety profile that appeared through non-carcinogenic, non-immunotoxic, non-mutagenic, and non-cytotoxic activity with a good LD50 value and appeared to be a promising drug candidate for further research and development. Finally, the molecular docking studies were performed, and the results indicated a negative binding energy with a similar or very close to binding mode as described for the co-crystallized ligand inside the active sites of Bcl-2 (PDB: 4AQ3), EGFR (PDB: 1M17), and VEGFR-2 (PDB: 4ASD).4. Experimental
4.1. Material and instrument
All melting points were measured using an Electrothermal LA 9000 SERIS, Digital Melting Point Apparatus and are uncorrected. IR spectra were obtained by the KBr disc method using a Nikolet IR 200 FT IR Spectrophotometer. The 1H NMR spectra were recorded at 400 MHz on Mercury and the 13C NMR spectra at 101 MHz using an NMR Spectrometer with DMSO-d6 as a solvent; chemical shifts were quantified in ppm relative to TMS as an internal standard. TLC on silica gel aluminum sheets was used to monitor the reactions and assess the purity of the compounds, and the spots were found by briefly exposing the sheets to a UV analysis lamp at 254/366 nm. Elemental microanalyses were performed at the microanalytical facility and were determined to be within 0.5% of the theoretical values. The information of the chemicals used in this study is provided in a ESI.†4.2. Chemistry
Off-white solid: yield (84%); mp = 240–242 °C; IR (KBr): ν/cm−1 3064 (arom. CH), 2929, 2906 (aliph. CH), 2219 (CN), 1723 (CO, ester), 1660 (CO, pyridine); 1H NMR (400 MHz, DMSO-d6); δ/ppm 1.36 (t, 3H, CH3), 1.97 (s, 3H, CH3), 2.40 (s, 3H, CH3), 4.38 (q, 2H, OCH2), 6.49 (s, 1H, pyridine-H5), 7.52 (d, J = 8.2 Hz, 2H, Ar-CH), 8.12 (d, J = 8.2 Hz, 2H, Ar-CH), 13C NMR (101 MHz, DMSO-d6) δ/ppm 14.15, 20.69, 21.38 (3CH3), 61.14 (OCH2), 100.04 (CH-pyridine), 109.13 (C–CN), 115.75 (CN), 128.67, 130.48, 130.61, 141.46, 151.70 (Ar-Cs), 159.99 (C–CH3), 160.33 (CO-pyridine), 165.03 (CO-ester); anal. calcd for C17H16N2O3 (296.33): calcd (%): C, 68.91; H, 5.44; N, 9.45. Found (%): C, 68.74; H, 5.51; N, 9.67.
:5) catalyzed with piperidine was heated under reflux for 5–7 h. The reaction mixture was cooled to ambient temperature, poured into crushed ice, filtrated out, and recrystallized from ethanol to afford the desired product 3.
4.2.2.1 Ethyl (E)-4-(3-cyano-4-(4-methoxystyryl)-6-methyl-2-oxopyridin-1(2H)-yl)benzoate (3a). Canary yellow powder: yield (72%); mp = 170 °C; IR (KBr): ν/cm−1 3065 (arom. CH), 2978, 2934, 2837 (aliph. CH), 2216 (CN), 1718 (CO, ester), 1663 (CO, pyridine); 1H NMR (400 MHz, DMSO-d6); δ/ppm 1.35 (t, 3H, CH3), 2.02 (s, 3H, CH3), 3.83 (s, 3H, OCH3), 4.36 (q, 2H, OCH2), 6.11 (s, 1H, methylinic-CH), 6.49 (s, 1H, pyridine-H5), 6.72 (s, 1H, methylinic-CH), 7.32 (d, J = 8.8 Hz, 2H, Ar-CH), 7.54 (d, J = 6.8 Hz, 2H, Ar-CH), 7.56 (d, J = 6.8 Hz, 2H, Ar-CH), 7.69 (d, J = 8.4 Hz, 2H, Ar-CH); 13C NMR (101 MHz, DMSO-d6) δ/ppm 14.38, 21.88 (2CH3), 55.69 (OCH3), 61.49 (OCH2), 97.23 (C–CN), 103.12 (CH-pyridine), 114.95, 115.01 (CN), 127.65, 128.87, 129.00, 130.08, 130.80, 130.90, 130.99, 141.78, 142.04, 151.32 (Ar-Cs), 161.10 (C–OCH3), 161.45 (CO. pyridine), 165.42 (CO. ester); anal. calcd for C25H22N2O4 (414.46): calcd (%): C, 72.45; H, 5.35; N, 6.76; found (%): C, 72.37; H, 5.51; N, 6.70.
4.2.2.2 Ethyl (E)-4-(3-cyano-4-(4-ethoxystyryl)-6-methyl-2-oxopyridin-1(2H)-yl)benzoate (3b). Yellow powder: yield (78%); mp = 180–182 °C; IR (KBr): ν/cm−1 3066 (arom. CH), 2980, 2933 (aliph. CH), 2215 (CN), 1718 (CO, ester), 1658 (CO, pyridine); 1H NMR (400 MHz, DMSO-d6) δ/ppm 1.31–1.36 (m, 6H, 2CH3), 2.00 (s, 3H, CH3), 4.10 (q, 2H, OCH2), 4.37 (q, 2H, OCH2), 6.80, 6.90 (2s, 2H, 2 methylinic- CH), 7.05 (d, J = 7.8 Hz, 2H, Ar-CH), 7.09 (s, 1H, pyridine-H5), 7.54 (d, J = 8.4 Hz, 2H, Ar-CH), 7.66 (d, J = 8.4 Hz, 2H, Ar-CH), 8.11 (d, J = 8.6 Hz, 2H, Ar-CH); 13C NMR (101 MHz, DMSO-d6): δ/ppm 14.11, 14.52, 21.62 (3CH3), 61.08, 63.36 (2 OCH2), 97.03 (C–CN), 102.68 (CH-pyridine), 115.79 (CN), 118.96, 127.42, 128.76, 129.13, 129.25, 129.74, 130.36, 140.07, 141.56, 150.82 (Ar-Cs), 154.24 (C–CH3), 160.43 (C-OC2H5), 160.84 (CO-pyridine), 165.00 (CO-ester); anal. calcd for C26H24N2O4 (428.49): calcd (%): C, 72.88; H, 5.65; N, 6.54. Found (%): C, 72.45; H, 5.84; N, 6.75.
Brown crystals: yield (89%); mp = 98–100 °C; IR (KBr): ν/cm−1 3304 (NH), 3071 (arom. CH), 2981, 2937 (aliph. CH), 2210 (CN), 1714 (br, 4CO); 1H NMR (400 MHz, DMSO-d6); δ/ppm: 1.32 (t, 3H, CH3), 4.30 (q, 2H, OCH2), 7.62 (d, J = 8.6 Hz, 2H, Ar-CH), 7.80 (d, J = 8.0 Hz, 1H, Ar-CH), 7.84 (d, J = 8.8 Hz, 2H, Ar-CH), 7.91(d, J = 8.4 Hz, 1H, Ar-CH), 7.96 (d, J = 6.6 Hz, 1H, Ar-CH), 8.01 (d, J = 7.8 Hz, 1H, Ar-CH), 11.07 (s, 1H, NH exchangeable by D2O); 13C NMR (101 MHz, DMSO-d6) δ/ppm 14.18 (CH3), 60.17 (OCH2), 112.62 (C–CN), 116.03 (CN), 118.23, 122.23, 126.37, 129.85, 131.97, 132.90, 135.52, 145.62 (Ar-Cs), 154.27 (C–NH), 163.36 (CO-amide), 165.56 (![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) C–CN), 165.87(CO-ester), 191.20 (2CO-ninhydrin); anal. calcd for C21H14N2O5 (374.35): calcd (%): C, 67.38; H, 3.77; N, 7.48. Found (%): C, 67.50; H, 3.80; N, 7.50.
C–CN), 165.87(CO-ester), 191.20 (2CO-ninhydrin); anal. calcd for C21H14N2O5 (374.35): calcd (%): C, 67.38; H, 3.77; N, 7.48. Found (%): C, 67.50; H, 3.80; N, 7.50.
4.2.4.1 Method A. A mixture of arylidene 4 (0.01 mol) and ethyl cyanoacetate (0.01 mol) in an ethanolic solution containing a few drops of triethylamine (TEA) was prepared and then heated under reflux for 5 h. The reaction mixture was cooled to ambient temperature, poured into crushed ice, filtrated out, and recrystallized from ethanol to afford the spiro derivative 5.
4.2.4.2 Method B. A mixture of cyanoacetamide derivative 1 (0.01 mol), ninhydrin (0.01 mol), and ethyl cyanoacetate (0.01 mol) in an ethanolic solution was catalysed with a few drops of triethylamine (TEA) via a one-pot reaction. The reaction mixture was refluxed for 10 h, cooled, and poured into crushed ice. The resulting solid product was filtered and crystallized from ethanol to afford the desired product 5.
Black powder: yield (85%); mp = 120–122 °C; IR (KBr): ν/cm−1 3373 (br, 2OH), 3066 (arom. CH), 2979, 2932, 2868 (aliph. CH), 2212 (CN), 1713 (br, 3CO); 1H NMR (400 MHz, DMSO-d6) δ/ppm 1.32 (t, 3H, CH3), 4.29 (q, 2H, OCH2), 7.64 (d, J = 8.6 Hz, 2H, Ar-CH), 7.70 (d, J = 8.6 Hz, 1H, Ar-CH), 7.83 (d, J = 8.0 Hz, 1H, Ar-CH), 7.95 (m, 4H, Ar-CH), 10.65 (br s, 2H, 2OH exchangeable by D2O); 13C NMR (101 MHz, DMSO-d6) δ/ppm 14.13 (CH3), 59.40 (C-Spiro), 60.45 (OCH2), 112.56 (2–CN), 115.63, 115.97 (2CN), 118.22, 118.58, 124.81, 129.78, 130.29, 130.95, 142.58 (Ar-Cs), 164.28 (2–OH), 165.15 (CO. ester), 165.80 (2CO); anal. calcd for C24H15N3O6 (441.40): calcd (%): C, 65.31; H, 3.43; N, 9.52; found (%): C, 65.50; H, 3.15; N, 9.60.
Brownish red crystals: yield (91%); mp = 150 °C; IR (KBr): ν/cm−1 3278 (2NH), 2982, 2935 (aliph. CH), 1714 (br, 3CO); 1H NMR (400 MHz, DMSO-d6); δ/ppm 1.31 (t, 3H, CH3), 4.32 (q, 2H, CH2), 6.85 (d, J = 8.4 Hz, 1H, Ar-CH), 6.92 (d, J = 7.8 Hz, 1H, Ar-CH), 7.06 (t, 1H, Ar-CH), 7.33 (d, J = 6.4 Hz, 1H, Ar-CH), 7.62 (d, J = 9.4 Hz, 1H, Ar-CH), 7.83 (d, J = 8.0 Hz, 1H, Ar-CH), 7.98 (d, J = 7.6 Hz, 2H, Ar-CH), 10.88, 11.03 (2s, 2H, 2NH exchangeable by D2O); 13C NMR (101 MHz, DMSO-d6) δ/ppm 14.20 (CH3), 60.56 (OCH2), 112.19, 112.61, 117.82, 121.16, 121.69, 122.76, 124.68, 129.85, 131.01, 132.64, 138.37, 144.09 (Ar-Cs), 150.71 (C–NH), 159.36 (CN), 165.55, 165.64, 168.98 (3CO); anal. calcd for C20H15N3O4 (361.36): calcd (%): C, 66.48; H, 4.18; N, 11.63. Found (%): C, 66.72; H, 4.00; N, 11.55.
4.2.6.1 Ethyl 4-(2′-amino-3′,5′-dicyano-6′-hydroxy-2-oxo-1′H-spiro[indoline-3,4′-pyridin]-1′-yl)benzoate (7). Black crystals: yield (80%); mp = 170–172 °C; IR (KBr): ν/cm−1 3316, 3210 (br, OH, OH, and NH2), 2978, 2934, 2869 (aliph. CH), 2188 (CN), 1713 (br, 2CO); 1H NMR (400 MHz, DMSO-d6) δ/ppm: 1.26 (t, 3H, CH3), 4.20 (q, 2H, CH2), 6.55 (d, J = 8.8 Hz, 1H, Ar-CH), 6.83 (d, J = 7.8 Hz, 1H, Ar-CH), 6.96 (d, J = 8.0 Hz, 1H, Ar-CH), 7.12 (d, J = 7.2 Hz, 2H, Ar-CH), 7.32 (d, J = 6.6 Hz, 1H, Ar-CH), 7.62 (d, J = 9.2 Hz, 2H, Ar-CH), 8.00 (br s, 2H, NH2 exchangeable by D2O), 9.05 (s, 1H, NH exchangeable by D2O), 10.86 (s, 1H, OH exchangeable by D2O); 13C NMR (101 MHz, DMSO-d6) δ/ppm 14.65 (CH3), 49.07 (C-spiro), 59.94 (OCH2), 103.68, 110.02 (2
–CN), 115.77, 116.51 (2CN), 118.71, 121.63, 122.18, 125.49, 129.79, 131.49, 133.11, 133.82, 144.57 (Ar-Cs), 153.90 (–NH2), 165.62 (–OH), 166.35, 169.47 (2CO); anal. calcd for C23H17N5O4 (427.42): calcd (%): C, 64.63; H, 4.01; N, 16.39; found (%): C, 64.50; H, 4.15; N, 16.45.
4.2.6.2 Ethyl 2′-amino-5′-cyano-1′-(4-(ethoxycarbonyl)phenyl)-6′-hydroxy-2-oxo-1′H spiro[indoline-3,4′-pyridine]-3′-carboxylate (8). Brown crystals: yield (82%); mp = 143–145 °C; IR (KBr): ν/cm−1 3288 (br, OH, NH, and NH2), 2938, 2867 (aliph. CH), 2208 (CN), 1712 (br, 2CO); 1H NMR (400 MHz, DMSO-d6) δ/ppm 0.87 (t, 3H, CH3), 1.28 (t, 3H, CH3), 4.21 (q, 2H, CH2), 4.30 (q, 2H, CH2), 6.57 (d, J = 9.4 Hz, 2H, Ar-CH), 6.85 (d, J = 7.6 Hz, 2H, Ar-CH), 7.14 (d, J = 7.0 Hz, 2H, Ar-CH), 7.64 (d, J = 8.0 Hz, 2H, Ar-CH), 7.98 (s, 2H, NH2), 10.61 (s, 1H, NH exchangeable by D2O), 10.88 (s, 1H, OH exchangeable by D2O); 13C NMR (101 MHz, DMSO-d6) δ/ppm 14.21, 14.40 (2CH3), 47.70 (C-spiro), 59.52, 61.67 (2OCH2), 103.24 (
–CN), 109.61 (–COOEt), 112.68 (CN), 116.08, 121.21, 122.83, 123.03, 124.73, 125.68, 133.11, 131.06, 138.44, 142.10, 144.13 (Ar-Cs), 153.46 (–NH2), 165.25 (–OH), 165.94, 169.04, 169.41 (3CO); anal. calcd for C25H22N4O6 (474.47): calcd (%): C, 63.29; H, 4.67; N, 11.81. Found (%): C, 63.75; H, 4.38; N, 11.65.
Yellow crystal: yield (77%); mp = 290 °C; IR (KBr): ν/cm−1 3365, 3318 (NH2), 3086 (arom. CH), 2978, 2903 (aliph. CH), 2214 (CN), 1707 (ester CO), 1681 (CO); 1H NMR (400 MHz, DMSO-d6); δ/ppm: 1.36 (t, 3H, CH3), 2.50 (s, 3H, H3S–), 4.39 (q, 2H, CH2), 7.52 (d, J = 12.0 Hz, 2H, Ar-CH), 7.88 (br s, 2H, NH2 exchangeable by D2O), 8.13 (d, J = 8.6 Hz, 2H, Ar-CH); 13C NMR (101 MHz, DMSO-d6) δ/ppm 14.17 (CH3), 17.47 (H3S–), 61.06 (OCH2), 76.75, 89.27 (2–CN), 115.16, 115.91 (2CN), 129.32, 131.15, 131.31, 138.02 (Ar-Cs), 156.35 (–NH2), 158.78 (CO-pyridine), 160.80 (CO-ester), 165.21 (–SMe); anal. calcd. for C17H14N4O3S (354.38): calcd (%): C, 57.62; H, 3.98; N, 15.81; found (%): C, 57.84; H, 3.60; N, 15.97.
Yellow powder: yield (65%); mp = 130–131 °C; IR (KBr): ν/cm−1 3336 (br, NH2), 3065 (arom. CH), 2987, 2924 (aliph. CH), 2257 (aliph. CN), 2206 (CN), 1707 (ester CO), 1691 (CO); 1H NMR (400 MHz, DMSO-d6) δ/ppm 1.31 (t, 3H, CH3), 3.94 (s, 2H, CH2CN), 4.29 (q, 2H, CH2), 6.76 (s, 2H, NH2 exchangeable by D2O), 7.68 (d, J = 8.8 Hz, 2H, Ar-CH), 7.93 (d, J = 8.6 Hz, 2H, Ar-CH); 13C NMR (101 MHz, DMSO-d6) δ/ppm 14.18 (CH3), 26.97 (H2CN), 60.50 (OCH2), 91.72, 100.59 (2–CN), 115.67, 118.62 (2CN), 124.86, 126.88, 130.34, 134.79, 142.63 (Ar-Cs), 152.55 (–NH2), 161.69 (CO-pyridine), 165.20 (CO-ester); anal. calcd for C18H13N5O3 (347.33) Calcd. (%): C, 62.24; H, 3.77; N, 20.16. Found (%): C, 62.35; H, 3.40; N, 20.41.
Yellowish white crystals: yield (70%); mp = 180–182 °C; IR (KBr) ν/cm−1 3350 (OH), 3063 (arom. CH), 2990, 2973 (aliph. CH), 2210 (CN), 1693 (CO); 1H NMR (400 MHz, DMSO-d6) δ/ppm 1.30 (t, 3H, CH3), 2.12 (s, 3H, CH3), 4.29 (q, 2H, CH2), 6.52 (s, 1H, pyridine-H5), 7.14 (d, J = 8.6 Hz, 2H, Ar-CH), 7.50 (d, J = 8.4 Hz, 2H, Ar-CH), 7.67 (d, J = 8.6 Hz, 2H, Ar-CH), 7.93 (d, J = 8.6 Hz, 2H, Ar-CH), 8.10 (d, J = 8.0 Hz, 2H, Ar-CH), 8.14 (d, J = 8.2 Hz, 2H, Ar-CH), 10.60 (s, 1H, OH exchangeable by D2O); 13C NMR (101 MHz, DMSO-d6) δ/ppm 14.21, 21.72 (2CH3), 60.53 (OCH2), 97.64 (–CN), 112.61, 116.36 (2CN), 116.90, 119.79, 122.78, 125.02, 127.33, 130.11, 131.03, 133.17, 134.00, 138.91, 142.91, 151.24 (Ar-Cs), 161.64 (–OH), 162.18 (CO-pyridine), 165.30 (CO-ester); anal. calcd for C28H22N2O4 (450.49): calcd (%): C, 74.65; H, 4.92; N, 6.22; found (%): C, 74.39; H, 5.05; N, 6.46.
4.3. Biological activities
The in vitro cytotoxicity screening of the newly synthesized 2-oxopyridine, spirooxindole, and other derivatives was done against two human cancer cell lines (HepG2 and Caco-2) by an MTT assay for an incubation period of 24 h, as described previously.42,43 The cell lines were obtained from ATCC via Holding company for biological products and vaccines (VACSERA), Cairo, Egypt. Moreover, the in vitro flow cytometry cell cycle analysis and apoptosis analysis using an Annexin V-FITC Apoptosis Detection BioVision Kit were performed at VACSERA Tissue Culture Unit, Cairo, Egypt, as described previously.11 The effect of most active derivatives 5, 7, and 8 on the gene expression of Bcl-2 and Bax was determined by qRT-PCR using the Bio-Rad Laboratories iScript TM One-Step RT-PCR Kit with SYBR® Green according to the manufacturer's instructions and as described previously.46,47 Additionally, the in vitro EGFR wt and VEGFR for the most active 2-oxo-1′H-spiro-indoline-3,4′-pyridine 7 was performed using the BPS-Bioscience EGFR Kinase Assay Kit Catalog # 40321 and VEGFR-2(KDR) Kinase Assay Kit Catalog # 40325, respectively according to the manufacturer's instructions.4.4. Molecular docking simulations
The molecular docking studies for the most active 2-oxo-1′H-spiro-indoline-3,4′-pyridine derivative 7 inside the active sites of Bcl-2 (PDB: 4AQ3), EGFR (PDB: 1M17), and VEGFR-2 (PDB: 4ASD) were performed using Molecular Operating Environmental (MOE)58–60 version 2009.10. All these proteins were downloaded from the protein data bank (https://www.rcsb.org/; access 1/2/2023). The structure of compound 7 was constructed in 2D using ChemBioDraw. 2014 and exported to MOE. Additionally, the structure of compound 7 was protonated and then the structure was minimized using forcefield MMFF94x, as described previously.61–64 For Bcl-2 (PDB: 4AQ3), the validation process was carried out by selecting only one chain and deleting all other chains. Moreover, the redocking process was performed, and the co-crystallized ligand (sulphonamide molecule) exhibited binding energy S = −23.39 kcal mol−1 with RMSD = 1.49 Å, where the alpha triangle placement and London dG as rescoring functions were selected. For EGFR (PDB: 1M17), the validation process reported that Erlotinib (co-crystallized ligand) showed binding energy S = −17.84 kcal mol−1 with RMSD = 1.73 Å, where the triangle matcher placement and London dG as rescoring functions were selected. Moreover, for VEGFR-2 (PDB: 4ASD), the redocking process of the co-crystallized ligand (Sorafenib) showed binding energy S = −16.169 kcal mol−1 with RMSD = 1.484 Å via four hydrogen bonds, where the triangle matcher placement and London dG as rescoring functions were selected.Data availability
All data that support the finding of this study are available in the ESI data of this article.†Conflicts of interest
The authors declare no conflict of interest.References
- G. Stazi, R. Fioravanti, A. Mai, A. Mattevi and S. Valente, Curr. Opin. Chem. Biol., 2019, 50, 89–100 CrossRef CAS PubMed.
- G. Housman, S. Byler, S. Heerboth, K. Lapinska, M. Longacre, N. Snyder and S. Sarkar, Cancers, 2014, 6, 1769–1792 CrossRef PubMed.
- A. K. A. Bass, E.-S. M. Nageeb, M. S. El-Zoghbi, M. F. A. Mohamed, M. Badr and G. E.-D. A. Abuo-Rahma, Bioorg. Chem., 2022, 119, 105564 CrossRef CAS PubMed.
- H. A. Bakshi, G. A. Quinn, M. M. Nasef, V. Mishra, A. A. A. Aljabali, M. El-Tanani, Á. Serrano-Aroca, M. Webba Da Silva, P. A. McCarron and M. M. Tambuwala, Cells, 2022, 11, 1502 CrossRef CAS PubMed.
- A. Thakur, G. J. Tawa, M. J. Henderson, C. Danchik, S. Liu, P. Shah, A. Q. Wang, G. Dunn, M. Kabir, E. C. Padilha, X. Xu, A. Simeonov, S. Kharbanda, R. Stone and G. Grewal, J. Med. Chem., 2020, 63, 4256–4292 CrossRef CAS PubMed.
- M. İ. Han, Ö. D. Yeşil Baysal, G. Ş. Başaran, G. Sezer, D. Telci and Ş. G. Küçükgüzel, Tetrahedron, 2022, 115, 132797 CrossRef CAS.
- E. Dickens and S. Ahmed, Surgery, 2018, 36, 134–138 Search PubMed.
- B. A. Chabner and T. G. Roberts, Nat. Rev. Cancer, 2005, 5, 65–72 CrossRef CAS PubMed.
- K. Cole, K. Pravoverov and J. E. Talmadge, Cancer Metastasis Rev., 2021, 40, 391–411 CrossRef PubMed.
- R. M. Mohammad, I. Muqbil, L. Lowe, C. Yedjou, H.-Y. Hsu, L.-T. Lin, M. D. Siegelin, C. Fimognari, N. B. Kumar, Q. P. Dou, H. Yang, A. K. Samadi, G. L. Russo, C. Spagnuolo, S. K. Ray, M. Chakrabarti, J. D. Morre, H. M. Coley, K. Honoki, H. Fujii, A. G. Georgakilas, A. Amedei, E. Niccolai, A. Amin, S. S. Ashraf, W. G. Helferich, X. Yang, C. S. Boosani, G. Guha, D. Bhakta, M. R. Ciriolo, K. Aquilano, S. Chen, S. I. Mohammed, W. N. Keith, A. Bilsland, D. Halicka, S. Nowsheen and A. S. Azmi, Semin. Cancer Biol., 2015, 35, S78–S103 CrossRef PubMed.
- M. M. S. Wassel, Y. A. Ammar, G. A. M. Elhag Ali, A. Belal, A. B. M. Mehany and A. Ragab, Bioorg. Chem., 2021, 110, 104794 CrossRef CAS PubMed.
- Y. A. Ammar, A. M. Sh El-Sharief, A. Belal, S. Y. Abbas, Y. A. Mohamed, A. B. M. Mehany and A. Ragab, Eur. J. Med. Chem., 2018, 156, 918–932 CrossRef CAS PubMed.
- K. S. Bhullar, N. O. Lagarón, E. M. McGowan, I. Parmar, A. Jha, B. P. Hubbard and H. P. V. Rupasinghe, Mol. Cancer, 2018, 17, 48 CrossRef PubMed.
- J. J. Cui, M. Tran-Dubé, H. Shen, M. Nambu, P.-P. Kung, M. Pairish, L. Jia, J. Meng, L. Funk, I. Botrous, M. McTigue, N. Grodsky, K. Ryan, E. Padrique, G. Alton, S. Timofeevski, S. Yamazaki, Q. Li, H. Zou, J. Christensen, B. Mroczkowski, S. Bender, R. S. Kania and M. P. Edwards, J. Med. Chem., 2011, 54, 6342–6363 CrossRef CAS PubMed.
- S. Antar, A. A. Al-Karmalawy, A. Mourad, M. Mourad, M. Elbadry, S. Saber and A. Khodir, Pharm. Sci., 2022, 28, 525–540 CAS.
- E. A. Abdelsalam, A. A. Abd El-Hafeez, W. M. Eldehna, M. A. El Hassab, H. M. M. Marzouk, M. M. Elaasser, N. A. Abou Taleb, K. M. Amin, H. A. Abdel-Aziz, P. Ghosh and S. F. Hammad, J. Enzyme Inhib. Med. Chem., 2022, 37, 2265–2282 CrossRef CAS PubMed.
- N. A. Pennell and T. J. Lynch Jr, Oncologist, 2009, 14, 399–411 CrossRef CAS PubMed.
- J. Tabernero, Mol. Cancer Res., 2007, 5, 203–220 CrossRef CAS PubMed.
- S. Sarkar, A. Mazumdar, R. Dash, D. Sarkar, P. B. Fisher and M. Mandal, Cancer Biol. Ther., 2010, 9, 592–603 CrossRef CAS PubMed.
- K. E. Saadon, N. M. H. Taha, N. A. Mahmoud, G. A. M. Elhagali and A. Ragab, J. Iran. Chem. Soc., 2022, 19, 3899–3917 CrossRef CAS.
- A. Ragab, S. A. Fouad, O. A. A. Ali, E. M. Ahmed, A. M. Ali, A. A. Askar and Y. A. Ammar, Antibiotics, 2021, 10, 162 CrossRef CAS PubMed.
- R. Matsa, P. Makam, G. Sethi, A. A. Thottasseri, A. R. Kizhakkandiyil, K. Ramadas, V. Mariappan, A. B. Pillai and T. Kannan, RSC Adv., 2022, 12, 18333–18346 RSC.
- T. Al-Warhi, A.-A. M. Sallam, L. R. Hemeda, M. A. El Hassab, N. Aljaeed, O. J. Alotaibi, A. S. Doghish, M. Noshy, W. M. Eldehna and M. H. Ibrahim, Pharmaceuticals, 2022, 15(10), 1262 CrossRef CAS PubMed.
- A. E. Abdallah, R. R. Mabrouk, M. M. S. Al Ward, S. I. Eissa, E. B. Elkaeed, A. B. M. Mehany, M. A. Abo-Saif, O. A. El-Feky, M. S. Alesawy and M. A. El-Zahabi, J. Enzyme Inhib. Med. Chem., 2022, 37, 573–591 CrossRef CAS PubMed.
- Y. Wan, J. Shang, R. Graham, R. S. Baric and F. Li, J. Virol., 2020, 94, 014855 Search PubMed.
- J. Bach, P. Eastwood, J. González, E. Gómez, J. A. Alonso, S. Fonquerna, E. Lozoya, A. Orellana, M. Maldonado, E. Calaf, J. Albertí, J. Pérez, A. Andrés, N. Prats, C. Carreño, E. Calama, J. De Alba, M. Calbet, M. Miralpeix and I. Ramis, J. Med. Chem., 2019, 62, 9045–9060 CrossRef CAS PubMed.
- M. Albratty and H. A. Alhazmi, Arabian J. Chem., 2022, 15, 103846 CrossRef CAS.
- S. M. Wilhelm, C. Carter, L. Tang, D. Wilkie, A. McNabola, H. Rong, C. Chen, X. Zhang, P. Vincent, M. McHugh, Y. Cao, J. Shujath, S. Gawlak, D. Eveleigh, B. Rowley, L. Liu, L. Adnane, M. Lynch, D. Auclair, I. Taylor, R. Gedrich, A. Voznesensky, B. Riedl, L. E. Post, G. Bollag and P. A. Trail, Cancer Res., 2004, 64, 7099–7109 CrossRef CAS PubMed.
- S. M. Wilhelm, J. Dumas, L. Adnane, M. Lynch, C. A. Carter, G. Schütz, K.-H. Thierauch and D. Zopf, Int. J. Cancer, 2011, 129, 245–255 CrossRef CAS PubMed.
- A. Sekulic, M. R. Migden, A. E. Oro, L. Dirix, K. D. Lewis, J. D. Hainsworth, J. A. Solomon, S. Yoo, S. T. Arron, P. A. Friedlander, E. Marmur, C. M. Rudin, A. L. S. Chang, J. A. Low, H. M. Mackey, R. L. Yauch, R. A. Graham, J. C. Reddy and A. Hauschild, N. Engl. J. Med., 2012, 366, 2171–2179 CrossRef CAS PubMed.
- P. Dhokne, A. P. Sakla and N. Shankaraiah, Eur. J. Med. Chem., 2021, 216, 113334 CrossRef CAS PubMed.
- G. J. Roth, R. Binder, F. Colbatzky, C. Dallinger, R. Schlenker-Herceg, F. Hilberg, S.-L. Wollin and R. Kaiser, J. Med. Chem., 2015, 58, 1053–1063 CrossRef CAS PubMed.
- Y. B. Sharma, R. Singh, C. P. Singh, Y. P. Bharitkar and A. Hazra, ChemistrySelect, 2022, 7, e202200707 CAS.
- D. G. Giménez, G. P. Elena, S. R. Teresa, A. Fernández Arche and R. De la Puerta, Planta Med., 2010, 76, 133–136 CrossRef PubMed.
- R. Ayman, M. S. Abusaif, A. M. Radwan, A. M. Elmetwally and A. Ragab, Eur. J. Med. Chem., 2023, 249, 115138 CrossRef CAS PubMed.
- A. Ragab, D. M. Elsisi, O. A. Abu Ali, M. S. Abusaif, A. A. Askar, A. A. Farag and Y. A. Ammar, Arabian J. Chem., 2022, 15, 103497 CrossRef CAS.
- D. M. Elsisi, A. Ragab, A. A. Elhenawy, A. A. Farag, A. M. Ali and Y. A. Ammar, J. Mol. Struct., 2022, 1247, 131314 CrossRef CAS.
- A. Ragab, M. S. Abusaif, N. A. Gohar, D. S. Aboul-Magd, E. A. Fayed and Y. A. Ammar, Bioorg. Chem., 2023, 131, 106307 CrossRef CAS PubMed.
- A. Ragab, M. S. Abusaif, D. S. Aboul-Magd, M. M. S. Wassel, G. A. M. Elhagali and Y. A. Ammar, Drug Dev. Res., 2022, 1305–1330 CrossRef CAS PubMed.
- Y. A. Ammar, J. A. Micky, D. S. Aboul-Magd, S. M. A. Abd El-Hafez, S. A. Hessein, A. M. Ali and A. Ragab, Chem. Biol. Drug Des., 2023, 101, 245–270 CrossRef CAS PubMed.
- A. M. S. El-sharief, Y. A. Ammar, A. Belal, M. A. M. S. El-sharief, Y. A. Mohamed, A. B. M. Mehany, G. A. M. Elhag and A. Ragab, Bioorg. Chem., 2019, 85, 399–412 CrossRef CAS PubMed.
- E. A. Fayed, Y. A. Ammar, M. A. Saleh, A. H. Bayoumi, A. Belal, A. B. M. Mehany and A. Ragab, J. Mol. Struct., 2021, 1236, 130317 CrossRef CAS.
- S. A. Ibrahim, A. Ragab and H. A. El-Ghamry, Appl. Organomet. Chem., 2022, 36, 1–17 Search PubMed.
- M. M. S. Wassel, A. Ragab, G. A. M. Elhag Ali, A. B. M. Mehany and Y. A. Ammar, J. Mol. Struct., 2021, 1223, 128966 CrossRef CAS.
- M. Eldeeb, E. F. Sanad, A. Ragab, Y. A. Ammar, K. Mahmoud, M. M. Ali and N. M. Hamdy, Biomedicines, 2022, 10, 722 CrossRef CAS PubMed.
- R. Z. Batran, S. M. El-Daly, W. A. El-Kashak and E. Y. Ahmed, Chem. Biol. Drug Des., 2022, 99, 470–482 CrossRef CAS PubMed.
- A. E. Azab, M. S. Alesawy, W. M. Eldehna, A. Elwan and I. H. Eissa, Arch. Pharm., 2022, 355, 2200133 CrossRef CAS PubMed.
- A. Ragab, Y. A. Ammar, A. Ezzat, A. M. Mahmoud, M. Basseem, I. Mohamed, A. S. El-tabl and R. S. Farag, Comput. Biol. Med., 2022, 145, 105473 CrossRef CAS PubMed.
- A. S. Hassan, N. M. Morsy, W. M. Aboulthana and A. Ragab, Drug Dev. Res., 2023, 84(1), 3–24 CrossRef CAS PubMed.
- A. Y. Alzahrani, Y. A. Ammar, M. Abu-Elghait, M. A. Salem, M. A. Assiri, T. E. Ali and A. Ragab, Bioorg. Chem., 2022, 119, 105571 CrossRef CAS PubMed.
- A. Kadri and K. Aouadi, J. Appl. Pharm. Sci., 2020, 10, 107–115 CrossRef CAS.
- R. Estrada-Tejedor, S. Noemí, B. Francesc and N. Santi, Afinidad. J. Chem. Eng. Theor. Appl. Chem., 2013, 70, 564 Search PubMed.
- T. Cheng, Y. Zhao, X. Li, F. Lin, Y. Xu, X. Zhang, Y. Li, R. Wang and L. Lai, J. Chem. Inf. Model., 2007, 47, 2140–2148 CrossRef CAS PubMed.
- R. Ayman, A. M. Radwan, A. M. Elmetwally, Y. A. Ammar and A. Ragab, Arch. Pharm., 2023, 356, e2200395 CrossRef PubMed.
- M. M. Abdelgalil, Y. A. Ammar, G. A. M. Elhag Ali, A. K. Ali and A. Ragab, J. Mol. Struct., 2023, 1274, 134443 CrossRef CAS.
- A. Ragab, S. A. Fouad, Y. A. Ammar, D. S. Aboul-magd and M. S. Abusaif, Antibiotics, 2023, 12, 128 CrossRef CAS PubMed.
- E. A. Fayed, M. Mohsen, S. M. A. El-Gilil, D. S. Aboul-Magd and A. Ragab, J. Mol. Struct., 2022, 1262, 133028 CrossRef CAS.
- E. S. A. E. H. Khattab, A. Ragab, M. A. Abol-Ftouh and A. A. Elhenawy, J. Biomol. Struct. Dyn., 2021, 40, 13291–13309 CrossRef PubMed.
- S. A. El-Kalyoubi, A. Ragab, O. A. Abu Ali, Y. A. Ammar, M. G. Seadawy, A. Ahmed and E. A. Fayed, Pharmaceuticals, 2022, 15, 376 CrossRef CAS PubMed.
- H. F. Rizk, M. A. El-Borai, A. Ragab, S. A. Ibrahim and M. E. Sadek, Polycyclic Aromat. Compd., 2023, 43, 500–522 CrossRef CAS.
- A. Ezzat, M. B. I. Mohamed, A. M. Mahmoud, R. S. Farag, A. S. El-Tabl and A. Ragab, J. Mol. Struct., 2022, 1251, 132004 CrossRef CAS.
- A. Y. Alzahrani, Y. A. Ammar, M. A. Salem, M. Abu-Elghait and A. Ragab, Arch. Pharm., 2022, 355, e2100266 CrossRef PubMed.
- H. Ali Mohamed, Y. A. Ammar, G. A. m. Elhagali, H. A. Eyada, D. S. Aboul-Magd and A. Ragab, ACS Omega, 2022, 7, 4970–4990 CrossRef CAS PubMed.
- A. S. Hassan, N. M. Morsy, W. M. Aboulthanab and A. Ragab, RSC Adv., 2023, 13, 9281–9303 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra00887h |
| This journal is © The Royal Society of Chemistry 2023 |