DOI:
10.1039/D3RA00852E
(Paper)
RSC Adv., 2023,
13, 11472-11479
Strategy for oxygen vacancy enriched CoMn spinel oxide catalyst activated peroxodisulfate for tetracycline degradation: process, mechanism, and toxicity analysis†
Received
8th February 2023
, Accepted 29th March 2023
First published on 12th April 2023
Abstract
Antibiotic-like organic pollutants are harmful to aquatic ecosystems and seriously disrupt the ecological balance. Herein, we propose a simple and versatile method to prepare cobalt–manganese oxides with high specific surface area and abundant oxygen vacancies using low-temperature reduction crystallization, which greatly facilitates the adsorption and electron transfer between the catalyst, PDS, and TC, thus accelerating the degradation of tetracycline (TC). Among them, the degradation efficiency of TC in the CoMn2O4(C)/PDS system was 99.8% in 60 min and the degradation rate remained above 90% after four cycles. The possible degradation mechanism is also discussed, where Co is the main metal active center of the catalyst and Mn plays an auxiliary catalytic role to promote the generation of reactive radicals in PDS through redox interactions between Co and Mn, where SO4−˙ is the main active species for TC degradation. Finally, the possible degradation pathways of TC are proposed and the toxicity of the intermediates is evaluated. Findings from this work will shed light on the rational design of bimetallic oxide catalysts.
Introduction
As one of the greatest scientific achievements of the 20th century, antibiotics play a key role in the treatment of human and animal diseases.1–4 Tetracycline, a typical antibiotic, ranks second in the global production and consumption of antibiotics. It is reported that annually about 210![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 tons of TC is being discharged into the aquatic environment together with wastewater during the last few decades.5–9 Due to the excessive and irrational use of antibiotics coupled with the fact that humans and animals can only metabolize a small percentage of tetracycline, almost most of it is excreted directly out of the body through urine. Moreover, some studies have reported that TC has been found in groundwater, surface water, and even drinking water, posing a serious threat to the environment and humans.10 Therefore, there is an urgent need to find fast, efficient and recyclable methods to remove TC from water.
000 tons of TC is being discharged into the aquatic environment together with wastewater during the last few decades.5–9 Due to the excessive and irrational use of antibiotics coupled with the fact that humans and animals can only metabolize a small percentage of tetracycline, almost most of it is excreted directly out of the body through urine. Moreover, some studies have reported that TC has been found in groundwater, surface water, and even drinking water, posing a serious threat to the environment and humans.10 Therefore, there is an urgent need to find fast, efficient and recyclable methods to remove TC from water.
To date, various methods have been developed to remove TC from water, such as adsorption,11 microbial degradation,12 membrane bioreactors,13 and advanced oxidation processes (AOPs).14 Among them, AOPs can produce highly reactive oxygen species (ROS), such as sulfate (SO4−˙), hydroxyl (·OH), singlet oxygen (1O2), and superoxide (·O2−), which are capable of converting various pollutants, especially the kind that are difficult to degrade, into mostly harmless compounds or even completely mineralized into water, carbon dioxide, and inorganic ions.15–21 In recent decades, sulfate radical-based advanced oxidation processes (SR-AOPs) have been increasingly investigated for their excellent performance and prevalence in degrading emerging contaminants. SR-AOPs have many advantages: (a) the redox potential of SO4−˙ (E0 = 2.5–3.1 V) is higher than that of ·OH (E0 = 1.8–2.7 V);22 (b) SO4−˙ has a longer half-life (τ1/2 = 30–40 μs) than ·OH (τ1/2 < 1 μs);23 (c) SO4−˙ can remain active over a wide pH range (2.0–8.0);24 (d) SO4−˙ exhibits good selectivity and effectiveness due to the transfer of electrons between organic pollutants through unsaturated bonds or aromatic functional groups with π electrons.25 Up to now, persulfate can be activated by various activation methods to produce SO4−˙, including thermal activation,26 UV light,27 ultrasound,28 and transition metals.29 Ömür Gökkuş30 reported that Ru-coated graphite is an inexpensive and effective anode material in the electrochemical oxidation treatment of tetracycline. Ledjeri et al.31 combined the electro/Fe3+/peroxydisulfate (PDS) process with biological treatment for the degradation of tetracycline and found that under optimal conditions, TC could be completely removed within 40 min and the mineralization rate was 98% after 3 h of electrolysis. Wang et al.32 introduced Fe into nickel-based metal–organic framework materials (FeNi-MOFs) and activated PDS to degrade tetracycline under visible light irradiation. Guo et al.33 prepared Fe–Co/BC catalysts for the activation of PDS to degrade TC. The high activity of Fe–Co/BC is mainly due to the synergistic effect between Fe and Co. Among them, activation using transition metals would be the simplest because it would be the most cost-effective method compared to other methods, especially those based on energy, such as electrolysis, heating, and ultrasound. In transition metal catalysts, the cobalt ion (Co2+) was found to be the most efficient ion for activating PMS/PDS.34 Recent reports have shown that the combination of cobalt oxides with manganese (Mn) can promote redox reactions and improve catalytic performance.35–37 Compared to monometallic oxides, bimetallic oxides have higher oxygen reduction activity, stability, and catalytic activity.
In this work, we have successfully prepared three catalysts, CoMn2O4(P), CoMn2O4(S), and CoMn2O4(C) for the activation of PDS to degrade TC using the reduction crystallization method for the first time. The above three catalysts were characterized by various surface analysis techniques, and the effects of reaction temperature, reaction time, and reductant concentration on the degradation of TC were systematically investigated. The possible degradation pathways of TC were inferred from the detection of intermediates by high-performance liquid chromatography-mass spectrometry (HPLC-MS). Finally, the catalytic stability of Co–Mn spinel was evaluated. Our work may provide a valuable reference for the development of more efficient multiphase catalysts for pollutant control applications.
Experimental
Materials
All chemicals used in this study were at least of analytical grade (AR). We used the following chemicals: manganese acetate tetrahydrate (Mn(CH3COO)2·4H2O), cobalt chloride hexahydrate (CoCl2·6H2O), potassium permanganate (KMnO4), sodium borohydride (NaBH4), sodium hypophosphite (NaH2PO2), hydrazine hydrate (N2H4·H2O), sodium hydroxide (NaOH), methanol (MeOH), tert-butanol (TBA), peroxydisulfate (PDS), tetracycline (TC), and 5,5-dimethyl-1-pyrrolidine N-oxide (DMPO). Ultrapure water (resistivity > 18.2 MΩ cm−1) was used to prepare all solutions. All the chemicals were used as received without further purification.
Synthesis of the catalyst
In this work, CoMn2O4−δ was synthesized by the reduction crystallization method.
Preparation of amorphous MnO2. Mn(CH3COO)2 (100 mL, 0.03 mol L−1) was slowly added to KMnO4 solution (50 mL, 0.04 mol L−1, pH = 12) at room temperature, and then a brown precipitate of MnO2 was formed under constant stirring, and the precipitate was filtered and dried overnight, and the dried solid was amorphous MnO2 nanoparticles.
Preparation of CoMn2O4−δ. Then in the nanocrystal CoMn2O4−δ synthesis, amorphous MnO2 (0.696 g) and CoCl2·6H2O (0.952 g) were mixed in 20 mL of water. An excess of the ready-made NaBH4 solution (NaBH4 solution dissolved in NaOH solution with pH = 11–12) was added dropwise to the mixture under vigorous magnetic stirring and controlled temperature. The purplish-red solution immediately turned colorless and bubbling violently. The resulting precipitate was collected by centrifugation, washed repeatedly with distilled water to remove residual ions, and dried at 80 ± 8 °C for 12 h. It was recorded as CoMn2O4−δ(P). When NaH2PO2 and N2H4·H2O were used as the reducing agents, the samples were noted as CoMn2O4−δ(C) and CoMn2O4−δ(S), respectively, and the procedure was the same as described above.
Characterization of catalyst
The identification of the catalyst phase and crystal structure was carried out by X-ray diffraction (XRD, Bruker D8 Advance A25, Germany). The morphological details of the solid products were observed with scanning electron microscopy (SEM, ZEISS Gemini 300, Germany) and transmission electron microscopy (TEM), respectively. The specific surface area and pore structure of different catalysts were analyzed by the Brunauer–Emmett–Teller analyzer (BET, ASAP 2460, USA). Defect analysis was detected using an electron paramagnetic resonance spectrometer (EPR, Bruker A300-10/12, Germany). The chemical composition and chemical state of elements were determined by X-ray photoelectron spectroscopy (XPS, Thermo Scientific K-Alpha, USA). Finally, degradation products were analyzed by the UPLC-MS method to identify the intermediate products of TC degradation.
Experimental procedure and analysis
TC degradation experiments were conducted in 500 mL conical flasks containing 250 mL of TC solution (50 mg L−1) and a certain quantity of the catalyst (0.25 g) at 30 °C to maintain the same reacting temperature and mixing rate. All batch experiments were implemented in an air bath shaker at 180 rpm. At a given time interval, 0.5 mL of the reaction solution was taken, and then the TC concentration was detected with a 0.22 μm cellulose acetate membrane. The concentration of TC was analyzed by high performance liquid chromatography (HPLC, Shimadzu LC-20AT, Japan) with Agilent HC-C18 column (5 μm, 150 × 4.6 mm) at λ = 356 nm. The mobile phase was the organic phase (acetonitrile:methanol = 2:1, v = 0.28 mL min−1) and the aqueous phase (0.01 M, oxalic acid, v = 0.56 mL min−1) at an isocratic flow rate of 0.84 mL min−1. After a cycle of TC adsorption and degradation, the used catalyst was collected by centrifugation, washed with deionized water, and vacuum dried at 60 °C.
Results and discussion
Characterization of catalysts
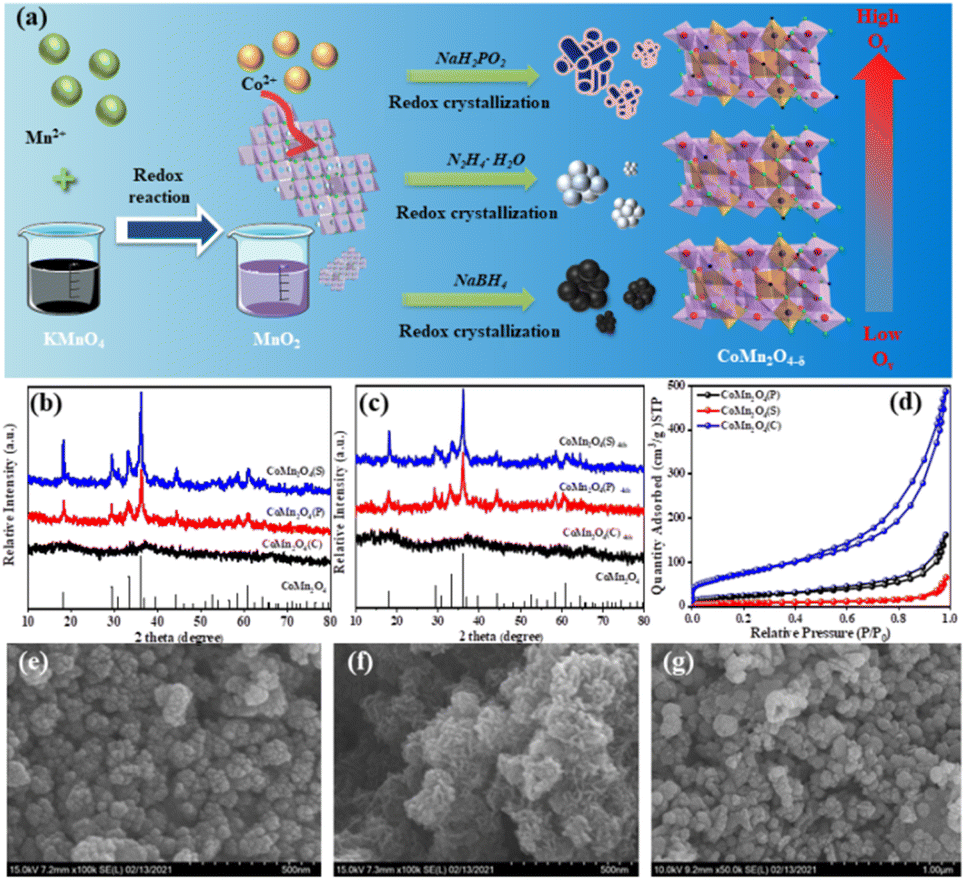
The XRD patterns of the synthesized catalysts are shown in Fig. S1.† Firstly, the strong reducing agent NaBH4 was selected as the reducing agent for the screening of each preparation condition, and the corresponding XRD patterns are shown in Fig. S1a–c.† Fig. S1a† shows the preparation of cobalt–manganese spinel by reduction of NaBH4 at different temperatures, from which it can be seen that the reaction at 20 °C and 40 °C for 15 min did not form the crystalline form corresponding to CoMn2O4 spinel. The peaks at 18.1°, 29.3°, 30.9°, 33.3°, 36.1°, 44.2°, 58.4°, 60.9°, and 64.4° are in good agreement with the CoMn2O4 standard card (PDF #77-0471) and correspond to (101), (112), (200), (103), (211), (200), (321), (224) and (400) crystal planes,38 indicating that the cobalt–manganese spinel, CoMn2O4 was successfully synthesized, and by further increasing the temperature to 80 °C it can be seen that the crystalline strength did not change significantly, laying a certain foundation for the subsequent preparation experiments. Fig. S1b and c,† respectively, show the effect of different concentrations and reaction times of NaBH4 on the crystalline shape of spinel. It was found that the increase of reducing agent concentration did not have a significant effect on the crystalline strength of spinel; however, the crystalline shape of spinel appeared to be strengthened after 12 h of the reaction, so the subsequent preparation of spinel was performed by extending the reaction time to prepare spinel with better crystalline shape. Fig. S1d† shows the preparation of spinel by reduction with the weak reducing agent NaH2PO4, and it was found that no characteristic peaks of CoMn2O4 were formed at different reaction times, temperatures, and reducing agent concentrations. In addition, another reductant N2H4·H2O was used for the preparation of spinel.
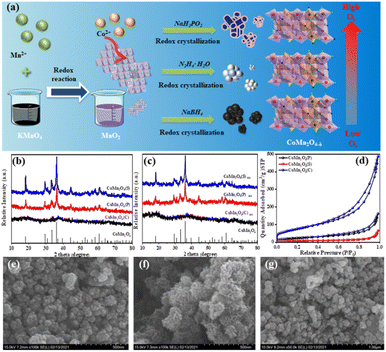
The preparation process of the catalysts is shown in Fig. 1a. Fig. 1b compares the crystalline shapes of the spinels prepared with different reducing agents, and it can be seen from the XRD that the strongest crystalline shape of the spinels formed by N2H4·H2O under the same conditions, followed by NaBH4, and the use of NaH2PO2 did not show the corresponding CoMn2O4 spinels in the XRD patterns. Also, XRD characterization of the three different spinels was performed again after four uses and no significant change in the crystalline shape occurred (Fig. 1c).
 |
| | Fig. 1 (a) Schematic diagram of the catalysts preparation process. (b and c) XRD patterns of before and after catalytic oxidation. (d) N2 isotherms adsorption–desorption curves; (e–g) SEM images of CoMn2O4(P), CoMn2O4(C) and CoMn2O4(S) catalysts. | |
The texture parameters including the BET surface area and pore volume of the prepared catalysts were measured from N2 physical adsorption–desorption. The N2 adsorption–desorption isotherms of the three catalysts were typical type IV with long and narrow hysteresis lines at relative pressures (P/P0) of 0.6 to 1.0, indicating the mesoporous structure (Fig. 1d) and the three catalysts demonstrated H3 type hysteresis loops, indicating the presence of slit-shaped pores. The pore properties including Brunauer–Emmett–Teller (BET) specific surface area, total pore volume, and pore size are summarized in Table S1.† The BET-specific surface area was 79.40, 24.24, and 242.52 m2 g−1 and the pore volume was 0.248, 0.098, and 0.694 cm3 g−1 for CoMn2O4(P), CoMn2O4(S), and CoMn2O4(C), respectively. This is consistent with the degradation test results of tetracycline, where the larger the specific surface area and the more active sites of the catalyst, the better the degradation effect.
Fig. 1e–g shows the SEM images of CoMn2O4(P), CoMn2O4(S) and CoMn2O4(C). All three catalysts have irregular granular morphology and rough surface of the samples, indicating a high degree of defects that can act as active centers of the catalysts and improve the catalytic performance. The TEM magnification (Fig. S2†) shows that CoMn2O4(P) and CoMn2O4(S) possess the tetragonal morphology of spinel with an average size of about 50–100 nm. The TEM of CoMn2O4(C) shows a lamellar structure.39
Defect characteristics
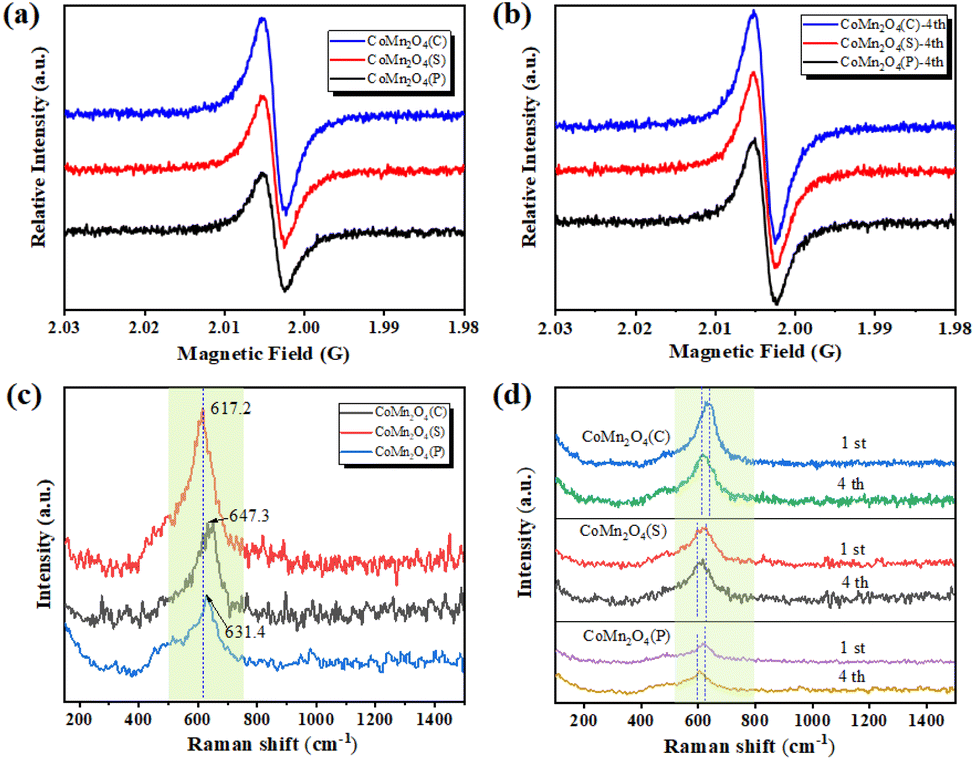
The oxygen vacancies of the three catalysts were further confirmed using an electron paramagnetic resonance spectrometer (EPR). As shown in Fig. 2a, the signal of CoMn2O4(C) was the strongest with a g factor of 2.004, indicating the abundant oxygen vacancies in CoMn2O4(C). Sufficient oxygen vacancies could provide more adsorption sites and active sites, exhibiting excellent catalytic performance.40 After the catalyst was used 4th for drying, another EPR test was conducted. The test results (Fig. 2b) showed that the defects of the catalysts appeared to increase to some extent after use, which means that the crystalline shape was damaged to some extent during the use, and also this is the reason why the removal rate did not significantly lower during the cyclic experiments of tetracycline catalytic degradation, the crystalline shape was damaged, the defects increased, and the removal effect remained the same.
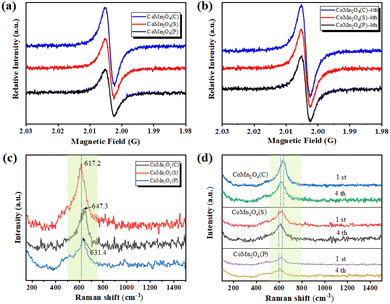 |
| | Fig. 2 (a and b) EPR and (c and d) Raman spectra of CoMn2O4(P), CoMn2O4(S), and CoMn2O4(C) before and after use. | |
The Raman spectrum of CoMn2O4(C) has a significantly higher front intensity at 617.2 cm−1 than CoMn2O4(S) and CoMn2O4(P) with an obvious red-shift, indicating an inversion of the spinel of CoMn2O4(C), which should be due to the effect of Mn on the Co–O bond at the tetrahedral site (Fig. 2c). Furthermore, the red-shift also indicates more oxygen vacancies, which is beneficial to the activation of oxygen species and promotes catalytic performance.41 On the other hand, these phenomena can also be clarified as the simultaneous co-existence of Co and Mn cations with different valence states at the tetrahedral and octahedral sites, which facilitates the synergistic redox reaction between the redox-coupled ion pairs, thus promoting charge transfer and oxygen vacancy generation.42 Finally, the catalysts were subjected to another Raman test after four uses and it was found that the Raman spectra of all three catalysts were significantly red-shifted, which was consistent with the EPR results (Fig. 2d).
Surface valences and PDS activation
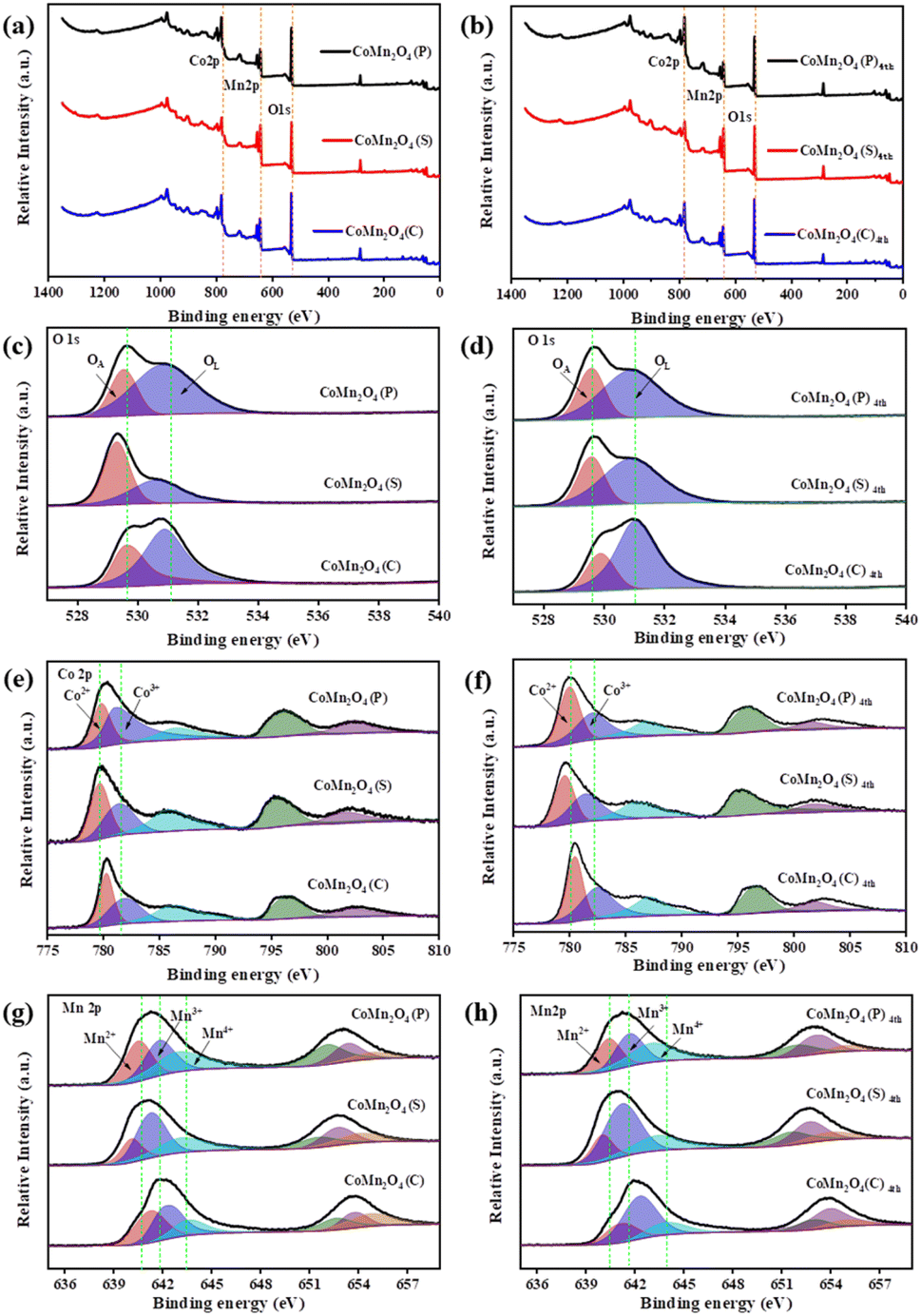
The full XPS survey scan spectrum of the as-prepared materials in Fig. 3 showed the existence of all elemental components (O, Co, and Mn) consistent with their chemical structures. As shown in Fig. 3c and d, O 1s peaks included two distinct oxygen components. The peak at ∼529 eV and 530 eV (marked as OL and OA),43 ascribed to lattice oxygen in a coordinately saturated environment, surface-adsorbed oxygen species, and oxygen vacancies in the unsaturated coordination modes, respectively,44 demonstrating the coexistence of OA adsorbed on oxygen vacancies and OL in the three catalysts. As shown in Tables S2 and S3,† the CoMn2O4(C) exhibits more adsorbed oxygen (OA) which means it has more oxygen vacancies before and after use. The OA/OL ratio is consistent with the performance ranking of the catalyst, which indicates that oxygen vacancies play an important part in TCs and PDS activation. In addition, the increase in adsorbed oxygen concentration might be ascribed to the formation of Co–OH or Mn–OH groups or O2 adsorbed on the catalyst surface and these oxygen species play a key role in PDS activation.45
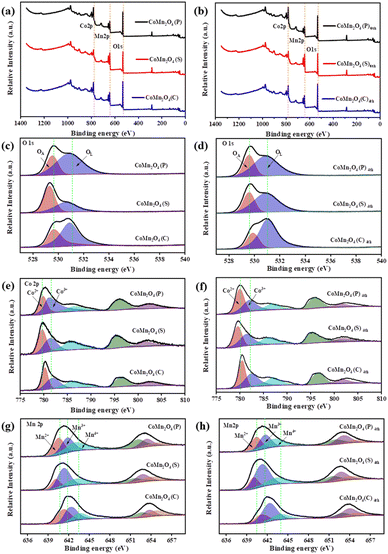 |
| | Fig. 3 XPS spectra of the synthesized catalysts before and after use: (a and b) survey spectra of the samples; (c and d) O 1s (e and f) Co 2p and (g and h) Mn 2p for CoMn2O4(P), CoMn2O4(S), CoMn2O4(C). | |
The XPS spectrum of Co 2p shows (Fig. 3e and f) that there are two different valence peaks in CoMn2O4(P), CoMn2O4(S), and CoMn2O4(C), and the corresponding valence states are Co2+ and Co3+. As shown in Fig. 3f, Co2+ in all catalysts increased to some extent after use, indicating that valence conversion between metals occurred during the catalytic process, which promoted the catalytic reaction. In the XPS spectrum of Mn 2p (Fig. 3g and h), a corresponding increase of Mn3+ and Mn4+ also occurred. However, compared with Co2+, the rise of Mn3+ and Mn4+ was in a smaller range, indicating that the main metal-active neutral in the catalytic process was Co. The defects in the spinel structure are considered to be Schottky defects and occur in pairs with oxygen vacancies and cation vacancies. In addition, it has been shown that Co cations can occupy tetrahedral and octahedral positions in CoMn2O4, while Mn cations are located only in octahedral positions. Therefore, it is further demonstrated that Co is the key metal for the activation of PDS.
Based on these results, a possible mechanism is proposed to effectively activate PDS through CoMn2O4. It is assumed that metal ions act as Lewis sites and combine with H2O resulting in lots of hydroxyls forming activated oxygen species on the surface of CoMn2O4. Meanwhile, S2O82− is bonded in the form of CoMn2O4–O–H–S2O82− through a hydrogen bond in the solution.46 In this case, Co2+, Mn2+ and Mn3+ undergo redox reactions with S2O82− to produce SO4−˙ (eqn (1)–(3)). SO4−˙ can be partially converted by the equation conversion to ·OH (4). In addition, the reduction potential required for the reduction of Co3+ and Mn3+ by PDS to generate Co2+ and Mn2+ is low, so the process is thermodynamically easy to carry out as in eqn (5) and (6).
| | |
Co2+ + S2O82− → Co3+ + SO4−˙ + SO42−
| (1) |
| | |
Mn2+ + S2O82− → Mn3+ + SO4−˙ + SO42−
| (2) |
| | |
Mn3+ + S2O82− → Mn4+ + SO4−˙ + SO42−
| (3) |
| | |
H2O + SO4−˙ → ·OH + SO4−˙ + H+
| (4) |
| | |
Mn3+ + Co3+ → Mn4+ + Co2+
| (5) |
| | |
Mn2+ + Co3+ → Mn3+ + Co2+
| (6) |
Catalytic performance
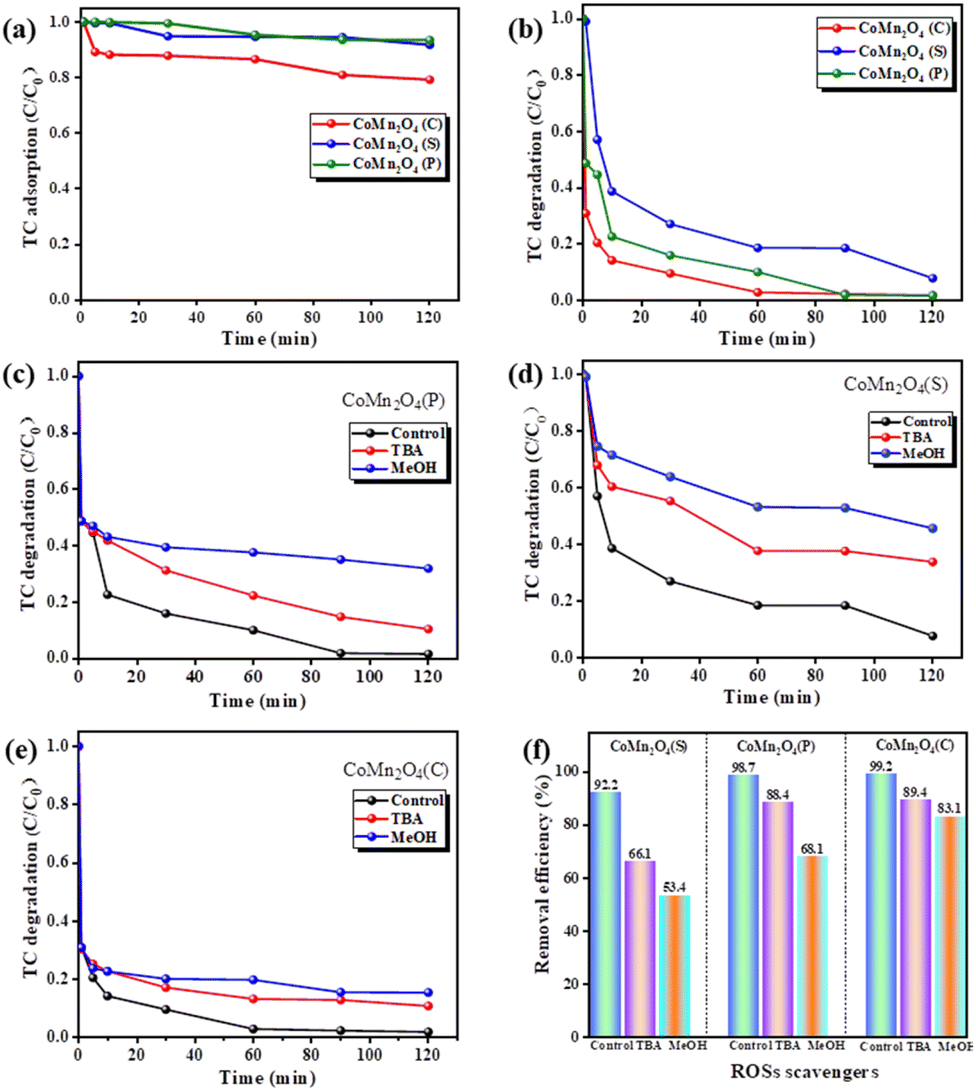
Fig. 4 shows a comparative test of the catalytic degradation of tetracycline by PDS activated by different catalysts. As shown in Fig. 4a, CoMn2O4(P), CoMn2O4(C), and CoMn2O4(S) removed no more than 20% of TC in 120 min, indicating that the degradation of TC depends mainly on catalysis rather than adsorption. CoMn2O4(C)/PDS exhibited the highest TC degradation efficiency of 99.8% within 120 min, because of strong molecular-level interactions between Co/Mn and PDS that accelerated charge transfer on the surface of CoMn2O4(C).47,48 Guo et al.49 reported that the interplay between Co and Mn in CoMn2O4 resulted in increasing Mn–O covalence, accelerating electron mobility between Mn and O species, and facilitating the activation of PMS by the CoMn2O4 activator.
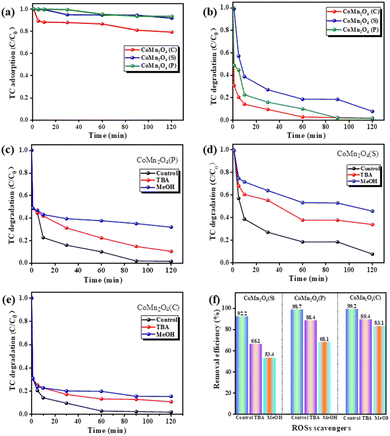 |
| | Fig. 4 The catalytic performance of various catalysts (a) adsorption, (b) degradation. (c–f) Effect of different quenchers on TC degradation in CoMn2O4−δ/PDS system. Reaction conditions: [catalyst] = 0.25 g, [PMS] = 10 mL, 0.1 mol L−1, [TC] = 250 mL, 50 mg L−1, [temperature] = 30 °C. | |
For the purpose of this experiment, TC was selected as the representative organic pollutant, and the removal rate of TC by NaBH4 under different conditions is shown in Fig. S3.† As shown in Fig. S3a,† the reduction preparation of spinel at 70 °C has the best degradation effect on tetracycline under four temperature conditions, which can reach 99.8%. Fig. S3b† shows that the concentration of the reducing agent during the preparation of spinel has no significant effect on the degradation effect of tetracycline, probably because the reducing agent is added in excess during the formation of spinel, so the change of concentration is not the main factor affecting the process of spinel preparation. Fig. S3c† shows the catalytic degradation test of cobalt–manganese spinel on tetracycline at different reaction times, from which it can be seen that the degradation effect decreases as the reaction time increases, especially the catalytic degradation effect of 1 mol L−1 70 °C 15 min (P) in the first ten minutes is significantly higher than that of 1 mol L−1 70 °C 12 h (P), while it can be inferred with reference to the XRD results that better is the crystal shape, worse is the catalytic degradation effect of tetracycline.
Since activation of PDS can generate both free radicals and non-free radicals, we performed quenching experiments on the above three catalysts using methanol (MeOH) and tert-butanol (TBA) to determine the main types of free radicals generated during PDS activation. As it is well-known, MeOH is usually used to scavenge both ·OH and SO4−˙ due to its high reactivity for both oxidation species. Additionally, TBA mainly is used as a quenching agent for ·OH because of its high reactivity, but not for SO4−˙ radicals.50 The results of the quenching experiments (Fig. 4c–f) showed that the removal of tetracycline by CoMn2O4(S), CoMn2O4(P), and CoMn2O4(C) decreased to 66.1%, 88.4%, 89.4%, and 53.4%, 68.1%, 83.1% after the addition of the quenching agents tert-butanol (TBA) and methanol (MeOH) for the quenching of ·OH and SO4−˙, respectively. Methanol exhibits a clear quenching effect, unlike TBA, implying that the addition of MeOH can reduce the reaction rate more than TBA, so the main reaction species for TC degradation is SO4−˙.
Stability and reusability of CoMn2O4−δ
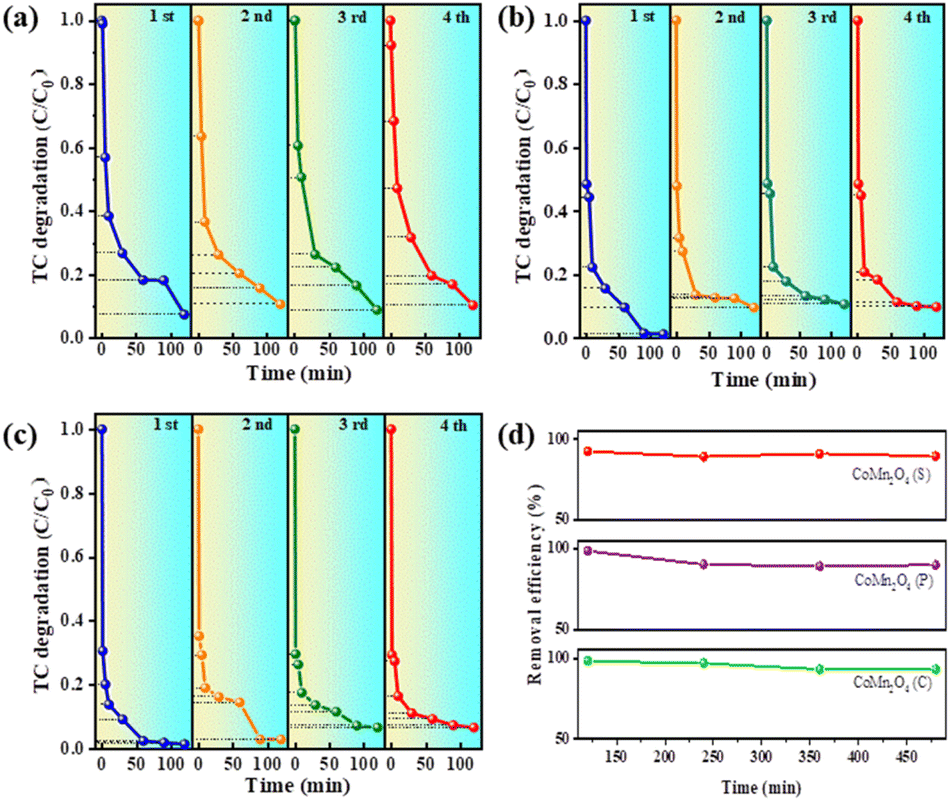
The reusability and stability of a catalyst are essential factors in determining its practical applications. Therefore, the catalytic performance of the three catalysts was investigated by four consecutive TC removal tests under identical reaction conditions. More specifically, after the first adsorption/degradation process, the used catalyst was collected by centrifugation, washed with water, and dried. After re-dispersing the sample into a fresh TC solution, a subsequent cycle was conducted. Fig. 5a–c shows four cycles of CoMn2O4(S), CoMn2O4(P), and CoMn2O4(C), and the experimental results show that the catalytic degradation of tetracycline by CoMn2O4(P) and CoMn2O4(C) showed a 5–10% decrease in the process of cycling. In contrast, CoMn2O4(S) did not show a decrease in the degradation of tetracycline after four cycles. Fig. 5d shows a summary plot of CoMn2O4−δ in four cycles with the time taken at 120 min after each cycle for the plot. In summary, it can be concluded that the stronger the crystalline shape of the cobalt–manganese spinel in the activation of PDS for the degradation of tetracycline, the more stable the lattice formed between the crystalline shape of the spinel will be and it will not be easily destroyed, so it can maintain a stable catalytic activity.
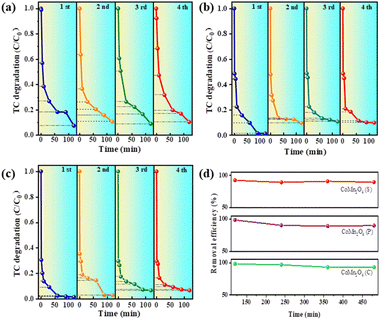 |
| | Fig. 5 Cycling experiments of tetracycline degradation by (a) CoMn2O4(S), (b) CoMn2O4(P), (c) CoMn2O4(C), and (d) CoMn2O4−δ. Reaction conditions: [catalyst] = 0.25 g, [PMS] = 10 mL, 0.1 mol L−1, [TC] = 250 mL, 50 mg L−1, [temperature] = 30 °C. | |
Proposed degradation pathway of TC
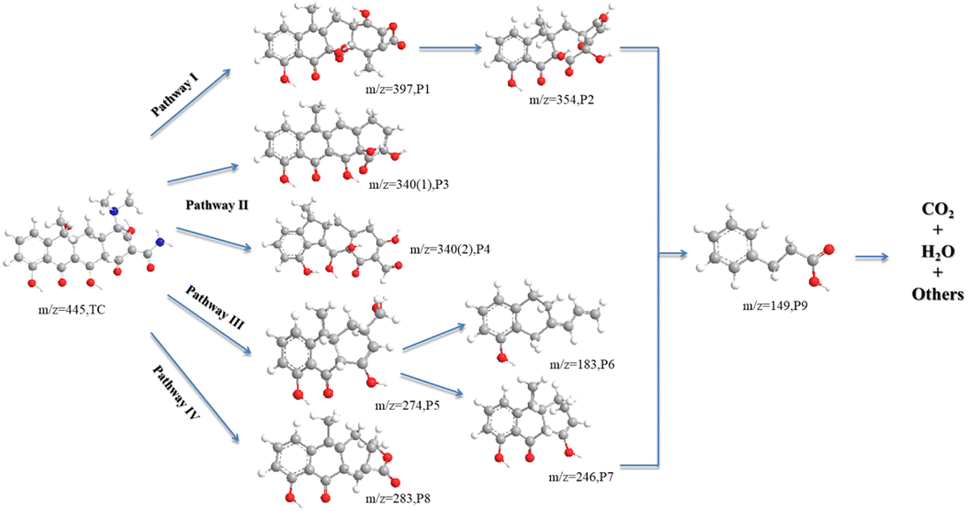
The LC-MS technique was used to study the degradation process of TC in CoMn2O4(S), CoMn2O4(P), and CoMn2O4(C)–PDS systems. The molecular structure of TC has three functional groups, namely a double bond, an amino group, and a phenolic hydroxyl group. The LC-MS analysis of the degradation products of CoMn2O4(S), CoMn2O4(P), and CoMn2O4(C) in the catalytic degradation of tetracycline at 30 min and 120 min showed that there were some differences in the m/z of the three products at 30 min sampling and testing, but the m/z values of the reaction products corresponding to the three catalysts were identical when the reaction products at 120 min were taken for testing, and it was tentatively judged that the degradation mechanism of tetracycline by the three catalysts was the same. Fig. S4–S9† shows the mass spectra of TC degradation products there at different times, and the details of the intermediates are shown in Table S4.† A total of 9 intermediates were detected, indicating that the tetracycline molecule was degraded mainly through functional group deletion and ring-opening reactions, such as hydroxylation, dehydroxylation, demethylation, and amide bond breakage.
The oxidative degradation of TC consisted of four possible degradation pathways, as shown in Fig. 6. In pathway I, TC (m/z = 445) takes off an amino group to form P1 (m/z = 397), which in turn loses CO2 to obtain P2 (m/z = 354). In pathway II, P3 (m/z = 340(1)) is the action of electrophilic addition, dehydroxylation, and dimethylamine on the corresponding groups on C2, C3, C4, and C6. The P4 (m/z = 340(2)) is formed by the removal of dimethylamine, hydroxyl, carbonyl, amino, and hydroxylation from tetracycline. In pathway III, the methyl group is shed due to the lower N–C bond energy of the dimethylamine at C4, the product 1 in C1 is dehydrated and the methanol group in C2 substitutes the acylamine, which in turn breaks the methanol group in C2 and the carbon atom undergoes cleavage to produce the product with P5 (m/z = 274), which is further dehydroxymethylated to form the product with P7 (m/z = 246),51 and also undergo ring opening to form a product with P6 (m/z = 183). In pathway IV, P8 (m/z = 283) was obtained by dehydration of TC, dehydration of amide group, demethylation, and cleavage followed by cleavage of carbon atom ring and then successive dehydroxylation reactions. The intermediate product with P9 (m/z = 149) was seen in the analysis of all samples by LC-MS, which was the compound with the smallest m/z value detected during the degradation of tetracycline. By increasing the reaction time, oxidative decomposition and ring-opening reactions occur further, and the aromatic compounds are finally decomposed into H2O, CO2, and inorganic ions.
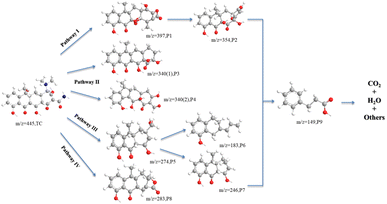 |
| | Fig. 6 Possible degradation pathways of TC in CoMn2O4−δ/PDS systems. | |
Toxicity evaluation of intermediate products
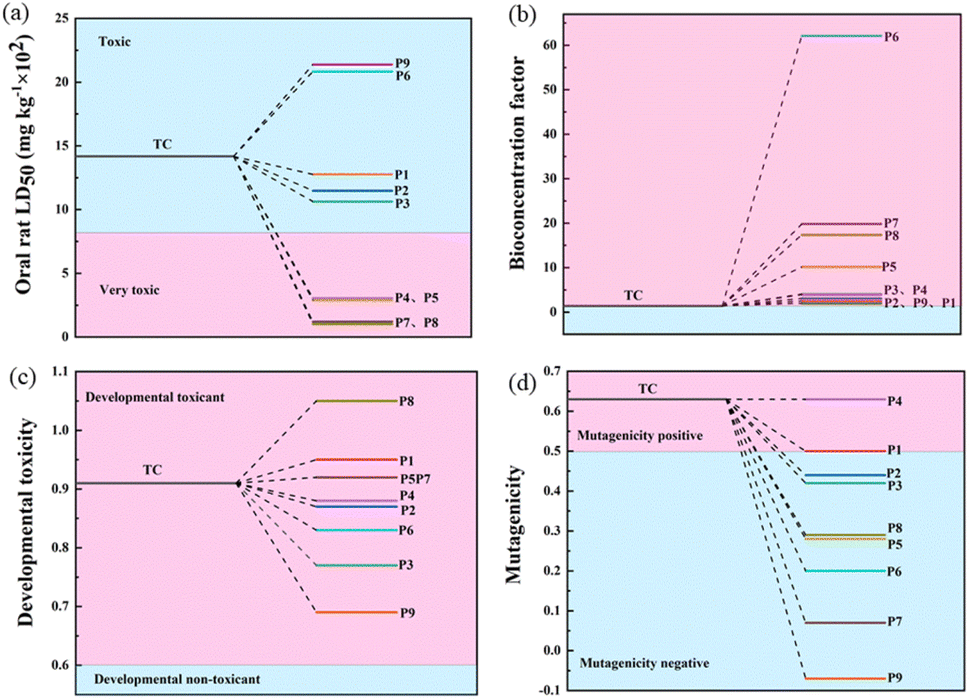
The acute toxicity, bioaccumulation factors, developmental toxicity, and mutagenicity of TC and its degradation intermediates were evaluated using the Toxicity Evaluation Software Tool (TEST). The LD50 of TC in rats was 807 mg kg−1, which is considered very toxic.41 As shown in Fig. 7a, the LD50 of most of the degradation intermediates of TC was above 1000 mg kg−1, indicating that the acute toxicity was effectively controlled. However, the products P1, P2, P3, P6, and P9 were still considered “toxic”. Fig. 7b shows that the bioaccumulation factors of all intermediates increased, with the highest bioaccumulation factor for product P6. Fig. 7c shows that the developmental toxicity of TC is also significantly reduced during the oxidation process. Specifically, TC was developmentally toxic, and almost all intermediates except P8, P1, P5, and P7 had lower developmental toxicity than TC. In addition, TC was positively mutated (Fig. 7d) and all intermediate products except P4 and P1 were negatively mutated. From the calculations, the CoMn2O4−δ/PDS system can mitigate the toxicity of TC and effectively suppress environmental hazards even if some intermediates are still toxic.
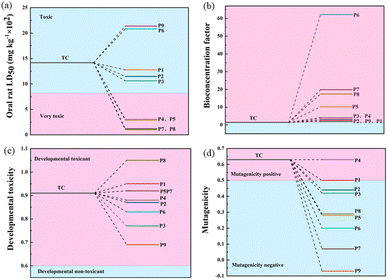 |
| | Fig. 7 Acute toxicity LD50 (a), bioaccumulation factor (b), developmental toxicity (c), and mutagenicity (d) of TC and degradation intermediates. | |
Conclusions
In this study, we have successfully prepared CoMn2O4−δ by low-temperature reduction crystallization method, which has a high specific surface area and abundant oxygen vacancies, compared with other spinel synthesis methods; this method is energy-saving, simple, and efficient, and is an excellent activator for TC degradation by activated persulfate (PDS). The bimetallic oxide catalysts were thoroughly characterized for pertinent surface properties, and tested for the catalytic performance in PDS activation for the removal of TC in water. In TC degradation experiments, CoMn2O4(C) was found to exhibit good degradation and cycling stability, which was attributed to the irregular structure of CoMn2O4(C), thus exposing more active sites. In addition, the redox interaction between oxygen vacancies and Co–Mn promoted the generation of reactive radicals (SO4−˙, ·OH) by PDS, which accelerated the degradation of tetracycline. Finally, the pathway of TC degradation by CoMn2O4−δ was proposed in combination with LC-MS and toxicity assessment, and the biological toxicity was significantly reduced after the reaction. The results of this study may expand the further development of transition metal oxide catalysts for the activation of persulfates.
Author contributions
Jingdan Gao and Ruijia Xiong conducted the experiments. Lei Wang and Song Qiao edited the program and performed the data analysis. Yonggang Sun and Yulong Ma analyzed the experimental results. Juan Zhang, Wenxin Ji, and Yuanyuan Li revised the manuscript. Yonggang Sun and Yulong Ma directed the research, acquired funding, and reviewed the manuscript. All authors contributed to the revision and final discussion of the manuscript.
Conflicts of interest
The authors have no relevant financial or non-financial interests to disclose.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (22006079), The West Light Talent Program of the Chinese Academy of Sciences, the Youth Talent Training Program of the Ningxia, and the Scientific and Technological Innovation Team of Industrial Water Treatment and Recycling, Ningxia.
Notes and references
- M. M. Mullis, I. M. Rambo, B. J. Baker and B. K. Reese, Front. Microbiol., 2019, 10, 2518 CrossRef PubMed
 .
. - M. Kumar, S. Jaiswal, K. K. Sodhi, P. Shree, D. K. Singh, P. K. Agrawal and P. Shukla, Environ. Int., 2019, 124, 448–461 CrossRef CAS PubMed .
- X. Wang, R. Yin, L. Zeng and M. Zhu, Environ. Pollut., 2019, 253, 100–110 CrossRef CAS PubMed .
- M. J. F. Calvete, G. Piccirillo, C. S. Vinagreiro and M. M. Pereira, Coord. Chem. Rev., 2019, 395, 63–85 CrossRef CAS .
- Y. Ben, C. Fu, M. Hu, L. Liu, M. H. Wong and C. Zheng, Environ. Res., 2019, 169, 483–493 CrossRef CAS PubMed .
- F. Saadati, N. Keramati and M. M. Ghazi, Crit. Rev. Environ. Sci. Technol., 2016, 46, 757–782 CrossRef CAS .
- F. Chen, Q. Yang, X. Li, G. Zeng, D. Wang, C. Niu, J. Zhao, H. An, T. Xie and Y. Deng, Appl. Catal., B, 2017, 200, 330–342 CrossRef CAS .
- D. Huang, X. Wang, C. Zhang, G. Zeng, Z. Peng, J. Zhou, M. Cheng, R. Wang, Z. Hu and X. Qin, Chemosphere, 2017, 186, 414–421 CrossRef CAS PubMed .
- X. Peng, Z. Yang, F. Hu, C. Tan, Q. Pan and H. Dai, Sep. Purif. Technol., 2022, 287, 120525 CrossRef CAS .
- J. Deng, C. Ya, Y. Ge, Y. Cheng, Y. Chen, M. Xu and H. Wang, RSC Adv., 2018, 8, 2338–2349 RSC .
- V. Nguyen, T. Nguyen, C. Chen, C. Hung, T. Vo, J. Chang and C. Dong, Bioresour. Technol., 2019, 284, 197–203 CrossRef CAS PubMed .
- J. Tang, M. Yang, M. Yang, J. Wang, W. Dong and G. Wang, New J. Chem., 2015, 39, 4919–4923 RSC .
- N. Prado, J. Ochoa and A. Amrane, Bioresour. Technol., 2009, 100, 3769–3774 CrossRef CAS PubMed .
- W. H. M. Abdelraheem, M. N. Nadagouda and D. D. Dionysiou, Appl. Catal., B, 2020, 269, 118807 CrossRef CAS .
- L. Lai, J. Yan, J. Li and B. Lai, Chem. Eng. J., 2018, 343, 676–688 CrossRef CAS .
- J. Rodríguez-Chueca, S. Giannakis, M. Marjanovic, M. Kohantorabi, M. R. Gholami, D. Grandjean, L. F. de Alencastro and C. Pulgarín, Appl. Catal., B, 2019, 248, 62–72 CrossRef .
- F. Ghanbari and M. Moradi, Chem. Eng. J., 2017, 310, 41–62 CrossRef CAS .
- J. Sharma, I. M. Mishra, D. D. Dionysiou and V. Kumar, Chem. Eng. J., 2015, 276, 193–204 CrossRef CAS .
- C. Nie, Z. Dai, H. Meng, X. Duan, Y. Qin, Y. Zhou, Z. Ao, S. Wang and T. An, Water Res., 2019, 166, 115043 CrossRef CAS PubMed .
- X. Chen, W. Wang, H. Xiao, C. Hong, F. Zhu, Y. Yao and Z. Xue, Chem. Eng. J., 2012, 193–194, 290–295 CrossRef CAS .
- Q. Zhao, Q. Mao, Y. Zhou, J. Wei, X. Liu, J. Yang, L. Luo, J. Zhang, H. Chen, H. Chen and L. Tang, Chemosphere, 2017, 189, 224–238 CrossRef CAS PubMed .
- J. Rodríguez-Chueca, C. García-Cañibano, R. J. Lepistö, Á. Encinas, J. Pellinen and J. Marugán, J. Hazard. Mater., 2019, 372, 94–102 CrossRef PubMed .
- J. C. Espinosa, P. Manickam-Periyaraman, F. Bernat-Quesada, S. Sivanesan, M. Álvaro, H. García and S. Navalón, Appl. Catal., B, 2019, 249, 42–53 CrossRef CAS .
- W. Li, S. Li, Y. Tang, X. Yang, W. Zhang, X. Zhang, H. Chai and Y. Huang, J. Hazard. Mater., 2020, 389, 121856 CrossRef CAS PubMed .
- J. Zhang, C. Zhai, W. Zhao, Y. Chen, R. Yin, L. Zeng and M. Zhu, Chem. Eng. J., 2020, 397, 125310 CrossRef CAS .
- Y. Qian, X. Liu, K. Li, P. Gao, J. Chen, Z. Liu, X. Zhou, Y. Zhang, H. Chen, X. Li and G. Xue, Chem. Eng. J., 2020, 384, 123332 CrossRef CAS .
- Y. Zhu, M. Wei, Z. Pan, L. Li, J. Liang, K. Yu and Y. Zhang, Sci. Total Environ., 2020, 705, 135960 CrossRef CAS PubMed .
- L. Yang, J. Xue, L. He, L. Wu, Y. Ma, H. Chen, H. Li, P. Peng and Z. Zhang, Chem. Eng. J., 2019, 378, 122146 CrossRef CAS .
- W. Oh, Z. Dong and T. Lim, Appl. Catal., B, 2016, 194, 169–201 CrossRef CAS .
- I. Köktaş, Ö. Gökkuş, I. Kariper and A. Othmani, Chemosphere, 2023, 315, 137758 CrossRef PubMed .
- A. Ledjeri, I. Yahiaoui and F. Aissani-Benissad, J. Environ. Manage., 2016, 184, 249–254 CrossRef CAS PubMed .
- D. Wang, M. Suo, S. Lai, L. Deng, J. Liu, J. Yang, S. Chen, M. Wu and J. Zou, Appl. Catal., B, 2023, 321, 122054 CrossRef CAS .
- L. Guo, L. Zhao, Y. Tang, J. Zhou and B. Shi, Chem. Eng. J., 2023, 452, 139381 CrossRef CAS .
- C. Cai, S. Kang, X. Xie, C. Liao, X. Duan and D. D. Dionysiou, J. Hazard. Mater., 2020, 399, 122979 CrossRef CAS PubMed .
- Y. Aoki, K. Takase, H. Kiuchi, D. Kowalski, Y. Sato, H. Toriumi, S. Kitano and H. Habazaki, J. Am. Chem. Soc., 2021, 143, 6505–6515 CrossRef CAS PubMed .
- A. Khan, K. Zhang, A. Taraqqi-A-Kamal, X. Wang, Y. Chen and Y. Zhang, J. Colloid Interface Sci., 2021, 599, 805–818 CrossRef CAS PubMed .
- N. S. Shah, J. Ali Khan, M. Sayed, Z. Ul Haq Khan, H. Sajid Ali, B. Murtaza, H. M. Khan, M. Imran and N. Muhammad, Chem. Eng. J., 2019, 356, 199–209 CrossRef CAS .
- Y. Wang, W. Guo and X. Li, RSC Adv., 2018, 8, 36477–36483 RSC .
- C. Ma and Y. Zhang, Sep. Purif. Technol., 2021, 258, 118024 CrossRef CAS .
- Y. Liu, C. Wang, J. Veder, M. Saunders, M. Tade, S. Wang and Z. Shao, J. Colloid Interface Sci., 2018, 530, 556–566 CrossRef CAS PubMed .
- G. Li, N. Li, Y. Sun, Y. Qu, Z. Jiang, Z. Zhao, Z. Zhang, J. Cheng and Z. Hao, Appl. Catal., B, 2021, 282, 119512 CrossRef CAS .
- W. Gu, C. Li, J. Qiu and J. Yao, J. Alloys Compd., 2022, 892, 162185 CrossRef CAS .
- K. Li, R. Zhang, R. Gao, G. Shen, L. Pan, Y. Yao, K. Yu, X. Zhang and J. Zou, Appl. Catal., B, 2019, 244, 536–545 CrossRef CAS .
- H. Zhao, Z. Qu and H. Sun, Appl. Surf. Sci., 2020, 529, 147044 CrossRef CAS .
- Y. Sun, X. Zhang, N. Li, X. Xing, H. Yang, F. Zhang, J. Cheng, Z. Zhang and Z. Hao, Appl. Catal., B, 2019, 251, 295–304 CrossRef CAS .
- P. Wang, J. Wang, X. An, J. Shi, W. Shangguan, X. Hao, G. Xu, B. Tang, A. Abudula and G. Guan, Appl. Catal., B, 2021, 282, 119560 CrossRef CAS .
- Y. Wang, H. Sun, H. M. Ang, M. O. Tadé and S. Wang, Appl. Catal., B, 2015, 164, 159–167 CrossRef CAS .
- C. Li, C. Chen, J. Lu, S. Cui, J. Li, H. Liu, W. Li and F. Zhang, Chem. Eng. J., 2018, 337, 101–109 CrossRef CAS .
- Z. Y. Guo, C. X. Li, M. Gao, X. Han, Y. J. Zhang, W. J. Zhang and W. W. Li, Angew. Chem., Int. Ed., 2021, 60, 2 CrossRef CAS .
- T. Zhang, H. Zhu and J. Croué, Environ. Sci. Technol., 2013, 47, 2784–2791 CrossRef CAS PubMed .
- Q. Wang, H. Zhu and B. Li, Chem. Eng. J., 2019, 378, 122072 CrossRef CAS .
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *,
Lei Wang,
Song Qiao,
Juan Zhang,
Wenxin Ji and
Yuanyuan Li
*,
Lei Wang,
Song Qiao,
Juan Zhang,
Wenxin Ji and
Yuanyuan Li