DOI:
10.1039/D3RA00761H
(Paper)
RSC Adv., 2023,
13, 19455-19463
Simultaneous determination of phenols in the four main original plants of the famous traditional Chinese medicine Shihu by pressurized capillary electrochromatography
Received
6th February 2023
, Accepted 9th June 2023
First published on 27th June 2023
Abstract
A rapid pressurized capillary electrochromatography (pCEC) method has been established for the simultaneous analysis of 11 phenols in the four main original plants of the famous traditional Chinese medicine (TCM) Shihu. The effects of wavelength, mobile phase, flow rate, pH value, concentration of buffer, and applied voltage were systematically studied. The investigated 11 phenols could be isolated in 35 min on a reversed-phase EP-100-20/45-3-C18 capillary column using the established method. To apply the established pCEC method, all phenols except tristin (11) were detected in the four Dendrobium plants. A total of 10 components were detected in D. huoshanense, 6 components in D. nobile, 3 components in D. chrysotoxum, and 4 components in D. fimbriatum. The consistent evaluation revealed that the similarities among the four original plants of Shihu were 38.2–86.0% based on the 11 polyphenols and 92.5–97.7% based on the pCEC fingerprints. These further suggested that the components of the four original plants of TCM Shihu might be significantly different. Further investigation should be conducted to confirm and evaluate if the four species could be used as the same medicine with the same amount according to Chinese Pharmacopoeia (ChP).
1. Introduction
Dendrobium is one of the biggest genera of the Orchidaceae family with more than 1100 species worldwide, among which 74 species and 2 varieties are distributed in China. Most Dendrobium plants are used as the famous traditional Chinese medicine (TCM) Shihu for a very long time.1 Dendrobium huoshanense C. Z. Tang and S. J. Cheng, Dendrobium nobile Lindl, Dendrobium chrysotoxum Lindl, and Dendrobium fimbriatum Hook are the most important original plants of the TCM Shihu owing to their similar and prominent biological activities.2,3 The stems of the four different herbs are believed to produce the same functions, such as the treatment of stomach, salivary, and ophthalmic disorders, and thus can be used as the same medicine based on Chinese Pharmacopoeia (ChP). The phytochemical documents indicated that Shihu is rich in polysaccharides, polyphenols, and alkaloids.4–6 These metabolites exhibited multiple pharmacological properties, including the immunoenhancement of the body and the special curative effect on the human eye, pharynx, lung, stomach, intestine, kidney, and the cardiovascular system,7–9 and thus were the main bioactive components of Shihu. One medicine from multiple origins was common in herbs besides Shihu. This demands the chemical compositions, at least the main bioactive components from the different original plants are the same, so that they may be used instead of the other in the clinic. The rationality of using the four Dendrobium plants as one medicine needs to be clarified. As the major active constituents of Shihu, polyphenols are also rich in many other natural sources, such as crude extracts of fruits, herbs, vegetables, grains, and nuts. In short, phenols are increasingly attracting considerable interest in most fields, including the food industry, and significant efforts have been spent to develop and enhance the analytical methods for their detection including bioassay,10 ELISA,11,12 and chromatographic methods, such as high-performance liquid chromatography (HPLC), gas chromatography with mass spectrometry (GC-MS), and liquid chromatography with mass spectrometry (LC-MS).13–18 Compared with the previous analytical techniques, pressurized capillary electrochromatography (pCEC)19,20 is a potential separation system in which a mobile phase is driven by both a pressurized flow and an electro-osmotic flow (EOF). This setup is especially compatible with the isolation of highly polar compounds, for example, polyphenols.21 To our knowledge, few references had ever mentioned the application of the pCEC technique for the analysis of polyphenols in natural medicines and food. Zhu used pressurized capillary electrochromatography for determining three isoflavones from Pueraria lobata.23 Derazshamshir A. used capillary electrochromatography to determine catecholamines (CAs) and their metabolites.24 Zhao used pressurized capillary electrochromatography to determine costunolide and dehydrocostus lactone in Maren Runchang Wan.25 The method we established simultaneously determined 11 compounds, which is more than the sample of compounds measured by others using this method.26 The accuracy of the method we established is also better than that of others, and the results showed that the RSD value was less than 1.84%, and the method's accuracy was adequate.27
Based on the above foundation, a fast pCEC method was established for the simultaneous determination of the phenols in the major original plants of Shihu. In order to isolate and simultaneously quantify the phenols in D. huoshanense, D. nobile, D. chrysotoxum and D. fimbriatum, the proposed method was then effectively used and verified. The consistency of the four Dendrobium plants was then evaluated based on the composition of polyphenols and the pCEC fingerprints of their methanol extracts to explore the logic of the use of four different Dendrobium species as the same medicine and thus providing references for other multiple-origin plants.
2. Materials and methods
2.1. Chemicals and reagents
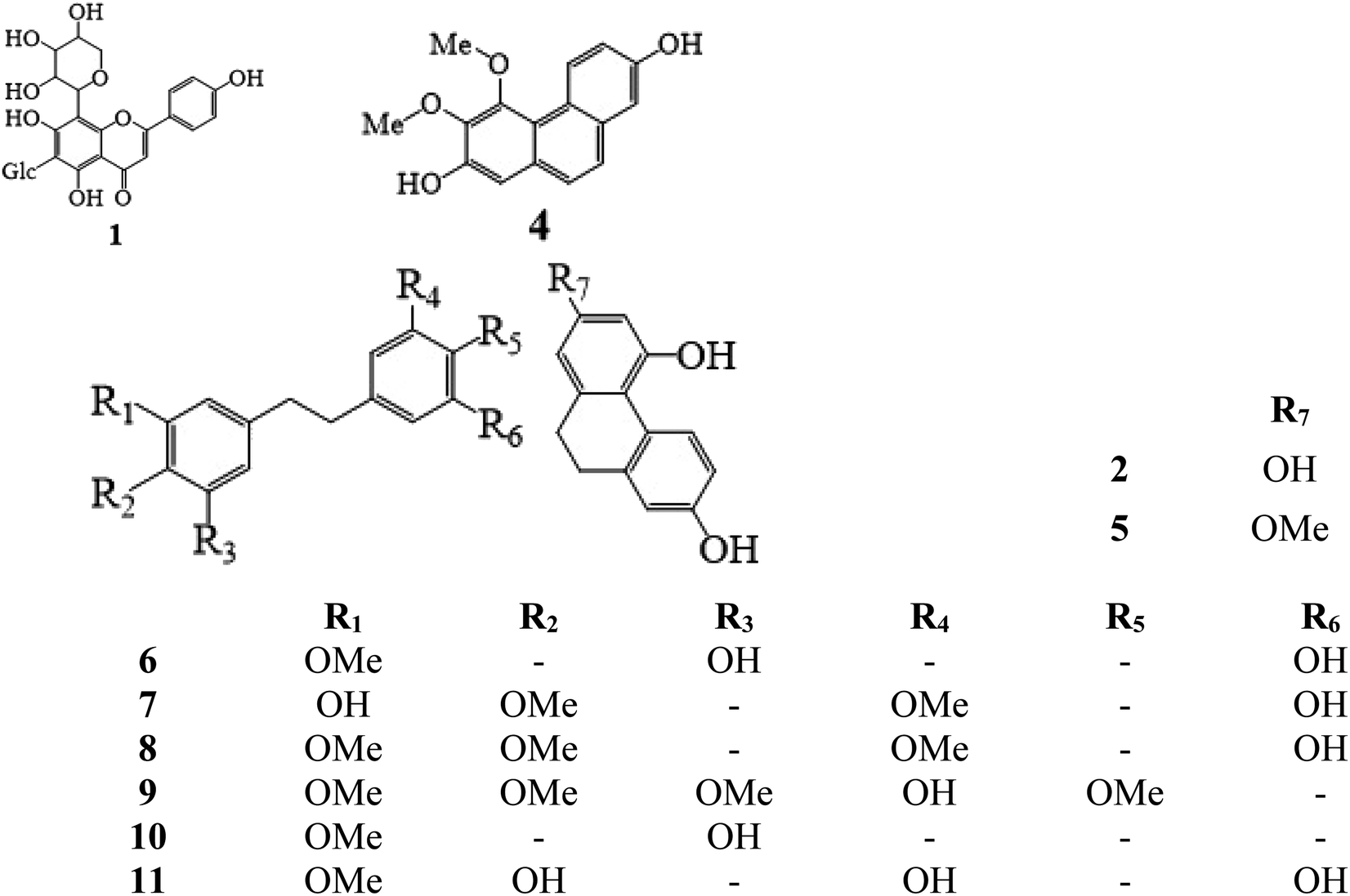
Methanol and acetonitrile (chromatographic grade) were purchased from Tedia High-Purity Solvent Co., Ltd. (USA). Secondary distilled water was made in the laboratory using a Milli-Q Plus purification system (Millipore, Billerica, MA, USA). Ammonium acetate, formic acid, potassium dihydrogen phosphate, borax, and sodium hydroxide were bought from Aladdin Reagent Co., Ltd. (Shanghai, China). Standards of schaftoside (1) 2,4,7-trihydroxy-9,10-dihydrophenanthrene (2), dihydroresveratrol (3), nudol (4), coelonin (5), batatasin III (6), gigantol (7), 3′-hydroxy-3,4,5′-trimethoxybibenzy (8), erianin (9), dihydropinosylvin methyl ether (10), and tristin (11) were obtained from Sichuan Weikeqi Biological Technology Co., Ltd. (Sichuan, Chengdu, China). The structures of these standards are shown in Fig. 1.
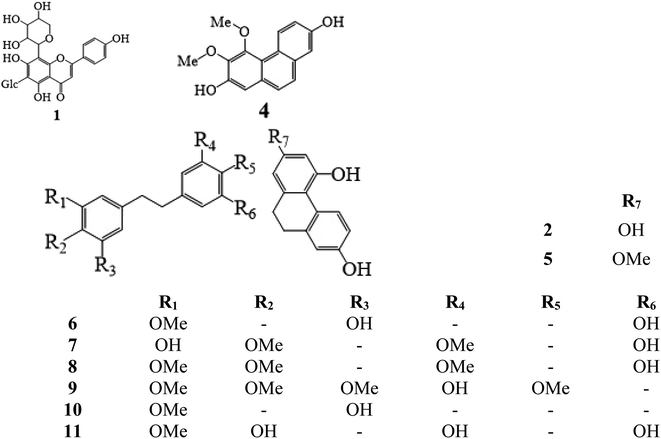 |
| | Fig. 1 Chemical structures of 11 phenolic compounds. Schaftoside (1), 2,4,7-trihydroxy-9,10-dihydrophenanthrene (2), dihydroresveratrol (3), nudol (4), coelonin (5), batatasin III (6), gigantol (7), 3′-hydroxy-3,4,5′-trimethoxybibenzy (8), erianin (9), dihydropinosylvin methyl ether (10), and tristin (11). | |
2.2. Sample preparation and purification procedures
D. huoshanense, D. nobile, D. chrysotoxum, and D. fimbriatum samples were collected on November 14, 2020, in Huoshan County, Anhui Province, China, and identified by Pro. Nai-Fu Chen, West Anhui University, China. The samples were prepared as follows: the fresh stems were freeze-dried at −50 °C to constant weight, then separately powdered using a blender and screened using a 200 mesh sieve. 2.0 g of each powdered sample was extracted with 100 mL 90% methanol 3 times. By decompressing and concentrating the filtrate to dryness, the methanol extracts of 4 Dendrobium plants were produced.
A CNWBOND LC-C18 SPE cartridge (2 g 10 mL; ANPEL Laboratory Technologies Inc., Shanghai, China) was used for solid-phase extraction according to our previous study;2 the detailed steps were as follows: the cartridge was activated using 50 mL of methanol followed by 30 mL of Milli-Q water. An accurate 10 mL of the sample solution was injected into the activated and conditioned SPE cartridge. The elution was performed with 50 mL of 60% methanol solution (the required substance is in the elution), concentrated eluent to 1 mL under reduced pressure to prepare the purified sample.
2.3. Preparation of standard solutions
Appropriate amounts of the substances were mixed and dissolved in methanol. Diluted working standard solutions were prepared as follows: (1) 0.11 mg mL−1, (2) 0.12 mg mL−1, (3) 0.21 mg mL−1, (4) 0.67 mg mL−1, (5) 0.32 mg mL−1, (6) 0.13 mg mL−1, (7) 0.33 mg mL−1, (8) 0.15 mg mL−1, (9) 0.25 mg mL−1, (10) 0.10 mg mL−1, (11) 0.12 mg mL−1. All the solutions were stored at 4 °C.
2.4. Instruments and analytical conditions
The prepared standard phenolic compounds were analyzed using a TriSep™ 2100 pCEC system (Unimicro Technologies Co., Ltd., Pleasanton, CA, USA) comprising two high-pressure infusion modules, a microfluidic module, a high-voltage power module (±30 kV), and a variable-wavelength UV detector (190–800 nm). Chromatographic separation was performed on an EP-100-20/45-3-C18 (Electropak™, C18, ODS, 3 μm, effective length 20 cm × 100 μm i.d.). In this experiment, the wavelength and chromatographic conditions were optimized. The detection wavelength was selected over a range of 190–400 nm. The optimization of chromatographic conditions includes the composition of mobile phases, mobile phase concentration, pH, flow rate, and voltage. In order to better realize the effective separation of chromatographic peaks in the sample, ACN–borax buffer, ACN–phosphate buffer, ACN–ammonium acetate buffer, and ACN–formic acid buffer were selected for comparison. In our experiment, the effect of buffer concentrations on the separation of the 11 analytes was investigated using 0.0 mM, 2.5 mM, 5.0 mM, 7.5 mM, and 10.0 mM, 12.5 mM borax buffer solution in ACN. In pCEC, the pH value of the mobile phase is an important factor to maintain EOF. As such, we compared the resolution of the objective chemicals isolated using the 10.0 mM borax buffer solution in ACN at pH = 7, 8, 9, 10, and 11. The effect of the pCEC flow rate on the resolution of the analytes was investigated by various flow rates of 0.02–0.06 mL min−1. In addition, the influence of applied voltage (from −2.5 kV to +10 kV) was chosen for further optimization so as to shorten the analysis time and prevent peak broadening.
Column EP-100-20/45-3-C18 (Electropak™, C18, ODS, 3 μm, effective length 20 cm × 100 μm i.d.); detection wavelength 280 nm; the voltage applied to the column +5 kV; flow rate 0.03 mL min−1; ACN/10.0 mM borax buffer (containing 0.1 mol L−1 NaOH, pH = 9) as the mobile phase, using gradient elution mode as follows 0 min–10% ACN; 6 min–10% ACN; 7 min–30% ACN; 15 min–30% ACN; 16 min–40% ACN; 20 min–40% ACN; 24 min–80% ACN; 29 min–80% ACN; 31 min–100% ACN; 35 min–100% ACN; the analysis time of each sample was 35 minutes, and fifteen more minutes were needed to equilibrate the column at the end, the injection volume was 3 μL (the volume of the sampling loop was 0.05 μL).
To perform pCEC's response surface methodology optimization, design expert software version 13 was used. Similarity analysis (SA) was performed on the basis of the relative retention time (RRT) and relative peak area (RPA) using the professional software Similarity Evaluation System for Chromatographic Fingerprint of Traditional Chinese Medicine (2004A).
2.5. Method validation
The developed pCEC method was validated by linearity, sensitivity, precision, recovery, stability, and specificity, in accordance with the ICH Q2(R2) guidelines.22
In order to create calibration curves, a minimum of eight calibration concentration levels were used, and the peak area (x) was plotted as a function of the corresponding mass (y, ng) of the working solution. The limit of detection (LOD) and quantification (LOQ) were the lowest concentrations of target analytes that could be detected and measured at a signal-to-noise ratio (S/N) of 3 and 10, respectively. The peak area was determined, a precision indicator known as the relative standard deviation (RSD) was computed and the stability was performed using a single standard solution sampled at 0, 2, 4, 8, 12, and 24 h. To evaluate the accuracy of the method, three different concentration levels (75%, 100%, and 125%) of the standard solutions were added to the samples with known content for the recovery tests. Three parallel samples were extracted for each level. Specificity evaluation was performed by injecting separately 3 μL solution of the mixed working standards, sample and blank into the chromatographic system.
3. Results and discussion
3.1. Optimization of pCEC conditions
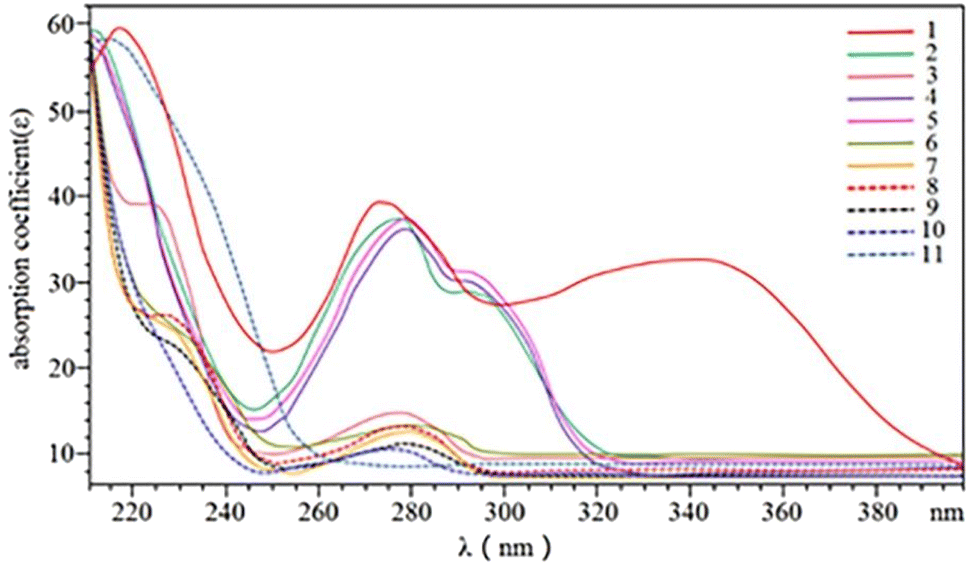
3.1.1. Wavelength. Optimization of the UV-visible spectrum of the 11 phenolics was performed (Fig. 2). Considering the demands on simultaneous detection with higher efficiency, a wavelength of 280 nm was chosen in the following experiments.
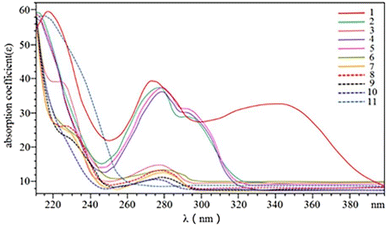 |
| | Fig. 2 UV-visible spectra of 11 phenolic compounds. Schaftoside (1), 2,4,7-trihydroxy-9,10-dihydrophenanthrene (2), dihydroresveratrol (3), nudol (4), coelonin (5), batatasin III (6), gigantol (7), 3′-hydroxy-3,4,5′-trimethoxybibenzy (8), erianin (9), dihydropinosylvin methyl ether (10), and tristin (11). | |
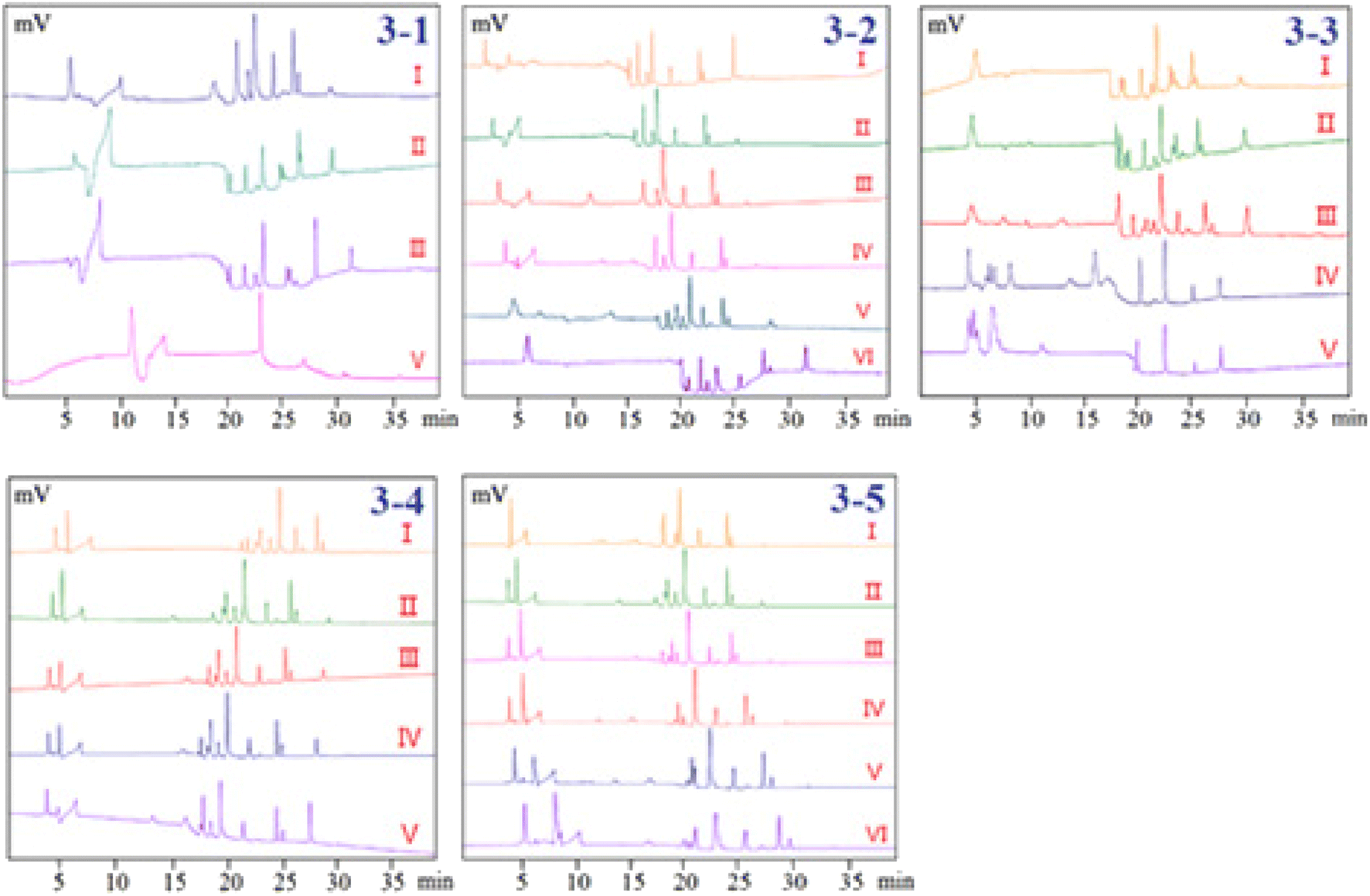
3.1.2. Separation performance. Electroosmotic flow has the same function as a pump in HPLC, driving particles forward, and completing the separation of cations, anions, and neutral particles by adding differences in the electrophoresis speed and direction of different particles. To obtain higher resolution and shorter elution time, the mobile phase composed of acetonitrile (ACN) and different kinds of buffer salts were optimized. Since ACN was used as an organic modifier, it has the ability to modify and lower the viscosity of the mobile phase, lower the system's pressure, and increase the solubility of chemicals in the sample, which was helpful to obtain a quick analysis time, it was often chosen as the initial organic modifier. In the present study, the following mobile phases with different ratios, which succeeded in the analysis of phenolic compounds by LC or LC-MS in pCEC were compared as follows: ACN–formic acid buffer (pH = 3), ACN–ammonium acetate buffer (pH = 7), ACN–phosphate buffer (pH = 7) and ACN–borax buffer (pH = 9). Peak forms and resolution were the major indices in the condition optimization of the pCEC analysis. Poor resolution and serious peak tailing of the mixed standards were displayed when ACN–formic acid or ACN–ammonium acetate buffer was employed as the mobile phase. The resolution of the ACN–phosphate buffer was higher than that of the ACN–formic acid and ACN–ammonium acetate system, however, it was obviously lower than that of the ACN–borax buffer (Fig. 3-1). Thus, the ACN–borax system was chosen as the mobile phase for further optimization. According to the results, the mobile phase which consisted of (A) ACN and (B) Milli-Q water was chosen and its gradient mode was as follows: 0 min–10% ACN; 6 min–10% ACN; 7 min–30% ACN; 15 min–30% ACN; 16 min–40% ACN; 20 min–40% ACN; 24 min–80% ACN; 29 min–80% ACN; 31–35 min–100% ACN. The analysis time of each sample was 35 minutes, and fifteen more minutes were also needed to equilibrate the column at the end.
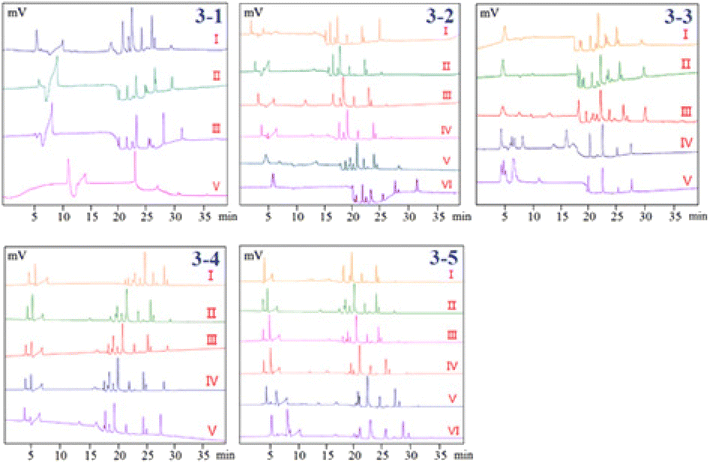 |
| | Fig. 3 Optimization of pCEC conditions. (3-1) Separation performance, I: ACN–borax buffer; II: ACN–phosphate buffer; III: ACN–ammonium acetate buffer; IV: ACN–formic acid buffer; (3-2) effect of borax concentration, I: 0.0 mM; II: 2.5 mM; III: 5.0 mM; IV: 7.5 mM; V: 10.0 mM; VI: 12.5 mM; (3-3) effect of pH values of buffer solution, I: pH = 7; II: pH = 8; III: pH = 9; IV: pH = 10; V: pH = 11; (3-4) effect of flow rate, I: flow rate = 0.02 mL min−1; II: flow rate = 0.03 mL min−1; III: flow rate = 0.04 mL min−1; IV: flow rate = 0.05 mL min−1; V: flow rate = 0.06 mL min−1; (3-5) effect of applied voltage, I: −2.5 kV; II: 0 kV; III: +2.5 kV; IV: +5 kV; V: +7.5 kV; VI: +10 kV. | |
3.1.3. Effect of borax concentration in buffer solution. Capillary electrochromatography (CEC), in which the electroosmotic flow (EOF) created from the electrical double layer was made to act as a pump to drive the mobile phase in a capillary column packed with micro-particulates or coated with a stationary phase. Both neutral and charged species can be resolved by CEC. It has been demonstrated that the efficiency of a separation obtained by electroosmotic propulsion is superior to that obtained by pressure-driven flow (as is the case in HPLC). CEC combines the best features of CE and versatile selectivity and large sample capacity of HPLC, promising high efficiency, high resolution, high selectivity, and high peak capacity. However, in practice, when CEC is used without pressure, often in a commercial CE instrument, there are problems and difficulties associated with bubble formation and column dry-out. These difficulties can be overcome by a pressurized CEC (pCEC) system, in which a supplementary pressure is applied to the column in addition to the EOF. In such a system, pressure can be applied to the capillary column to suppress the bubble formation. Quantitative sample introduction in pCEC can be easily achieved through a rotary-type injector. Most importantly, it is amenable to a solvent gradient mode, similar to that in HPLC, by programming the composition of the mobile phase.The resolution of the investigated phenols will be affected by the change in the buffer concentration since it will alter the EOF of the pCEC system. Therefore, our experiments systematically investigated the effects of the borax buffer concentration (0.0 mM, 2.5 mM, 5.0 mM, 7.5 mM, 10.0 mM, and 12.5 mM of borax in ACN at pH 9.0) on the separation system. The results showed that the resolutions and RTs of the analytes were slightly increased with buffer concentration rising from 0.0 mM to 12.5 mM. Fig. 3-2 shows that the separation resolution became better as the borax concentration increased, but a longer analysis time was needed and the chromatographic column was very easy to be blocked at 12.5 mM borax. In this case, 10 mM was chosen as the optimum concentration of borax for further optimization.
3.1.4. Effect of pH values of buffer solution. In pCEC, the pH of the mobile phase has a strong impact on the resolution, retention time, and detection sensitivity of analytes. Anions moved toward the anode at the capillary sample end in the pCEC system. Greater bulk EOF of buffer through the capillary towards the cathode at the detector end of the capillary caused anions to roam towards the detector. Because the pH of the 10 mM borax buffer solution was about 9, the pH range of 7 to 11 was considered. However, it was shown that when the pH was less than or greater than 9, the resolution of the sample decreased. In order to further upgrade the mobile phase, a pH of 9.0 for the ACN borax buffer was adopted (Fig. 3-3).
3.1.5. Effect of flow rate. The flow rates from 0.02 to 0.06 mL min−1 were adopted to investigate the influence of flow rates on the pCEC separation of the 11 phenolic compounds (Fig. 3-4). It was demonstrated that the retention time of 11 analytes was significantly shortened as the flow rate increased. An excessively high flow rate would cause the loss of packing materials from the cartridge and negatively affect the column efficiency and detection stability. Taking full consideration of the retention time, resolution, and sensitivity of the analytes, a flow rate of 0.03 mL min−1 was used for additional analysis.
3.1.6. Effect of applied voltage. The EOF and electrophoresis of the analytes would be enhanced by an increase in the applied voltage, which would have an impact on the RTs and the baseline of the pCEC separation of the analytes. Therefore, the influence of the applied voltage (from −2.5 kV to 10 kV) was investigated in the present study. As it was shown in Fig. 3-5, the retention time was getting longer and longer as the positive voltage was applied to the outlet of the capillary, but the resolution was improved. The resolution and peaks were both taken into account when the applied voltage was +5 kV. Nevertheless, if the applied voltage were continually rising, the higher applied voltage would cause the pCEC system to become unstable because of the high background current's effect on Joule heating. Therefore, +5 kV of the applied voltage was selected as the optimized voltage.
3.1.7. Optimization design and analysis. In order to maximize the separation between the investigated phenols in less running time, a central composite design was used to examine the influence of each independent chromatographic component as well as the interaction between numerous parameters. Four significant chromatographic parameters were chosen in this study based on preliminary tests and knowledge of earlier literature investigations. The four key factors used for the optimization were A: borax concentration, B: pH, C: flow rate, and D: applied voltage. The levels of each factor were A: borax concentration (7.5–12.5 mM), B: pH (8–10), C: flow rate (0.02–0.04 mL min−1), D: applied voltage (+2.5 to +7.5 kV). The eleven observed responses were the resolution between (1) schaftoside/2,4,7-trihydroxy-9,10-dihydrophenanthrene (R1,2), (2) 2,4,7-trihydroxy-9,10-dihydrophenanthrene/dihydroresveratrol (R2,3), (3) dihydroresveratrol/nudol (R3,4), (4) nudol/coelonin (R4,5), (5) coelonin/batatasin III (R5,6), (6) batatasin III/gigantol (R6,7), (7) gigantol/3′-hydroxy-3,4,5′-trimethoxybibenzy (R7,8), (8) 3′-hydroxy-3,4,5′-trimethoxybibenzy/Erianin (R8,9), (9) erianin/dihydropinosylvin methyl ether (R9,10), (10) dihydropinosylvin methyl ether/tristin (R10,11), and (11) the run time (Table 1). In this experimental design, a total of 30 experiments were performed to optimize the chromatographic conditions. The optimized chromatographic condition was 12.5 mM borax buffer (pH adjusted to 9 with 0.1 mol L−1 NaOH) and ACN was used as the mobile phase at a flow rate 0.03 mL min−1, UV detection wavelength 280 nm, separation voltage +4.3 kV.
Table 1 Analytical parameters of 11 phenolic compoundsa
| Analyte |
Calibration curve |
Liner range (μg mL−1) |
LOD (μg mL−1) |
LOQ (μg mL−1) |
Precision (RSD) |
Recovery (%) |
Recovery (RSD) |
Stability (RSD) |
| Schaftoside (1), 2,4,7-trihydroxy-9,10-dihydrophenanthrene (2), dihydroresveratrol (3), nudol (4), coelonin (5), batatasin III (6), gigantol (7), 3′-hydroxy-3,4,5′-trimethoxybibenzy (8), erianin (9), dihydropinosylvin methyl ether (10), and tristin (11). |
| 1 |
y = 502.44x + 106.33 |
0.19–17.11 |
0.05 |
0.18 |
0.85 |
95.69 |
1.15 |
1.72 |
| 2 |
y = 778.02x + 149.19 |
0.21–18.42 |
0.03 |
0.11 |
0.63 |
90.64 |
1.57 |
0.66 |
| 3 |
y = 652.68x + 252.38 |
0.37–33.16 |
0.04 |
0.14 |
1.62 |
97.85 |
0.74 |
1.52 |
| 4 |
y = 149.55x − 281.58 |
5.30–105.26 |
0.08 |
0.27 |
1.44 |
94.72 |
1.24 |
1.20 |
| 5 |
y = 675.55x + 17.219 |
0.56–50.00 |
0.02 |
0.06 |
1.84 |
96.68 |
1.63 |
1.56 |
| 6 |
y = 875.38x − 232.15 |
0.49–20.72 |
0.02 |
0.05 |
1.22 |
91.37 |
1.38 |
1.67 |
| 7 |
y = 1269.6x − 675.23 |
0.59–52.63 |
0.01 |
0.03 |
0.64 |
96.35 |
0.94 |
0.58 |
| 8 |
y = 768.73x − 101.34 |
0.55–23.16 |
0.02 |
0.06 |
1.78 |
92.14 |
0.86 |
1.47 |
| 9 |
y = 197.19x − 287.03 |
1.99–6.14 |
0.07 |
0.22 |
0.53 |
93.66 |
0.71 |
0.66 |
| 10 |
y = 1640.5x − 123.02 |
0.18–15.79 |
0.01 |
0.03 |
0.69 |
95.42 |
1.52 |
0.76 |
| 11 |
y = 433.83x + 50.197 |
0.44–18.42 |
0.03 |
0.10 |
1.32 |
94.33 |
1.46 |
1.41 |
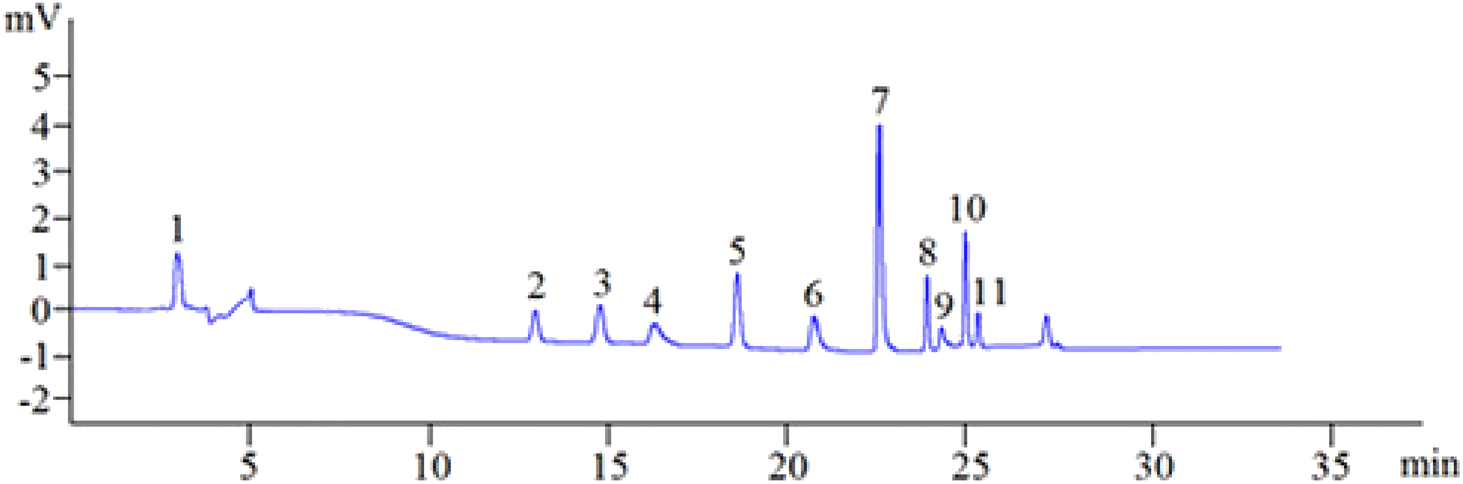
3.1.8. Optimized separation. To sum up, in the aforementioned parameter optimization trials, the best pCEC conditions of simultaneous analysis of the 11 phenolic compounds were as follows: separation voltage +4.3 kV, UV detection wavelength 280 nm, the flow rate of 0.03 mL min−1 with ACN/12.5 mM borax buffer (pH = 9) as the mobile phase, using gradient elution mode as follows: 0 min–10% ACN; 6 min–10% ACN; 7 min–30% ACN; 15 min–30% ACN; 16 min–40% ACN; 20 min–40% ACN; 24 min–80% ACN; 29 min–80% ACN; 31 min–100% ACN; 35 min–100% ACN; the sample analysis time was 35 min, and the column was equilibrated for 15 min. Manual sample injection was used for sample injection with an injection volume of 3 μ L. Under the above-optimized pCEC conditions, the 11 phenolic compounds can be successfully separated and determined in 35 min (Fig. 4).
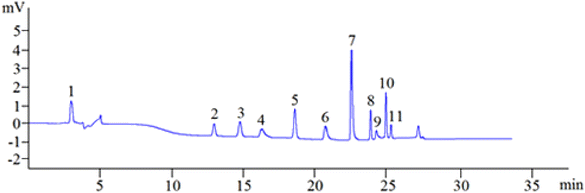 |
| | Fig. 4 A representative chromatogram of the 11 standard phenolic compounds. Schaftoside: 11.51 μg mL−1 (1), 2,4,7-trihydroxy-9,10-dihydrophenanthrene: 12.40 μg mL−1 (2), dihydroresveratrol: 22.32 μg mL−1 (3), nudol: 70.86 μg mL−1 (4), coelonin: 33.66 μg mL−1 (5), batatasin III: 13.95 μg mL−1 (6), gigantol: 35.43 μg mL−1 (7), 3′-hydroxy-3,4,5′-trimethoxybibenzy: 15.59 μg mL−1 (8), erianin: 26.57 μg mL−1 (9), dihydropinosylvin methyl ether: 10.63 μg mL−1 (10), tristin: 12.40 μg mL−1 (11). | |
3.2. Method validation of the established pCEC method
The optimized chromatographic method was validated according to the validation principles stated in the ICH guidelines “validation of analytical procedures: text and methodology Q2 (R2)” (ICH 2022). Key validation parameters evaluated include linearity, sensitivity, precision, recovery, stability, and specificity.
3.2.1. Analytical specificity. Blank solutions, standard mixtures of 11 phenolic compounds, and the 4 Dendrobium plants were analyzed under the conditions of optimized methods. By comparing their RTs with those of reference compounds, researchers are able to decide the specificity of peaks in the four Dendrobium samples. All samples were found to have fewer interferences in the areas with important retention periods. The capacity to differentiate analytes from other Dendrobium plant matrix components was shown by well-resolved peaks, which suggested good analytical specificity of techniques for the established pCEC method.
3.2.2. Linearity. By adopting the optimal pCEC conditions, the analyte concentrations in serially diluted mixed standard working solutions were analyzed. The ratios of each analyte's peak area and concentration were used to establish the calibrations of 11 analytes (Table 1). It was demonstrated that all analytes exhibited excellent linearity within the range of 0.19–17.11 μg mL−1 for schaftoside (1), 0.21–18.42 μg mL−1 for 2,4,7-trihydroxy-9,10-dihydro-phenanthrene (2), 0.37–33.16 μg mL−1 for dihydroresveratrol (3), 5.30–105.26 μg mL−1 for nudol (4), 0.56–50.00 μg mL−1 for coelonin (5), 0.49–20.72 μg mL−1 for batatasin III (6), 0.59–52.63 μg mL−1 for gigantol (7), 0.55–23.16 μg mL−1 for 3′-hydroxy-3,4,5′-trimethoxybibenzy (8), 1.99–6.14 μg mL−1 for erianin (9), 0.18–15.79 μg mL−1 for dihydropinosylvin methyl ether (10) and 0.44–18.42 μg mL−1 for tristin (11). The correlation coefficients were above 0.999 for all 11 analysts.
3.2.3. Sensitivity. LOQ was chosen as the concentration of a phenolic compound-producing S/N = 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 under the optimal pCEC circumstances. The LOD was obtained by the consecutive analysis of spiked matrices with decreasing quantities of each phenolic standard until S/N = 3:1 was attained. Relevant results from Table 1 indicated that the proposed method was highly sensitive.
:1 under the optimal pCEC circumstances. The LOD was obtained by the consecutive analysis of spiked matrices with decreasing quantities of each phenolic standard until S/N = 3:1 was attained. Relevant results from Table 1 indicated that the proposed method was highly sensitive.
3.2.4. Precision. The same mixed standard working solution was continuously injected six times while the peak area was recorded and the RSDs were computed under the optimal pCEC conditions. The results showed that the RSDs were less than 1.84%, and the method's accuracy was adequate (Table 1).
3.2.5. Recoveries. Recovery was conducted by adding the mixed standard solutions of the 11 phenolic compounds into Dendrobium samples with high, intermediate, and low levels. The results showed that the recoveries were in the range of 90.64–97.85% and the detection technique worked accurately and reproducibly (Table 1).
3.2.6. Stability. To assess the stability of the investigated phenolic compounds, the mixed phenolic compounds were tested at different times. The study showed that the RSDs of different times were ≤2.0% for all phenolic compounds in a week under room temperature, suggesting that the analytes were stable throughout the whole sample preparation procedure (Table 1).Overall, the experimental results suggested that the developed pCEC method could be utilized to simultaneously quantify all the investigated 11 phenols and was quick, reliable, sensitive, and accurate.
3.3. Detection of polyphenols in four Dendrobium plants
3.3.1. Qualification of the polyphenols in the investigated plants. In our experiments, the qualification analysis of the polyphenols was performed by the comparison of the retention time between the peaks of standards and those of their possible corresponding samples. To improve the accuracy of the identifications, the internal standard experiments were further carried out.
3.3.1.1. Location of the chromatographic peak of the 11 phenolic compounds. For better qualitative analysis of phenolic compounds, the chromatographic peak positions of each of the 11 phenolic compounds were further investigated by a TriSep™ 2100 pCEC system on an EP-100-20/45-3-C18 column. The chromatographic peak position was identified by the relative retention time (RRT), which was calculated according to the following equation:
where tR,i is the retention time of any of the investigated phenolic compounds, tR,s is the retention time of the internal reference, and ΔtR,is is the variation in the retention time in both. Among them, the chromatographic peak position of each phenolic compound was explicitly identified and designated as the reference peak in the four Dendrobium plants. The results showed that the calculation of the RRT was stable (RSDs < 5%) and could be used for identifying the chromatographic peak locations of the phenolic compounds.
3.3.1.2. The internal standards experiment. For qualitative analysis, an internal standard method was used to analyze the polyphenols in four Dendrobium plants. Specifically, polyphenol compounds as internal standards were added into the sample solution in turn to determine the position of each compound in the sample, comparing the changes in the target peak area and retention time in the sample. The results showed that the peak area corresponding to each compound increased and there was no significant difference in the retention time. The detailed data are shown in Table 2. It is worth noting that the introduction of internal standards in both samples and calibration standards improved the precision and accuracy of the qualitative analysis.
Table 2 The difference in the retention time before and after the introduction of internal standardsa
| Times |
RT1 |
RT2 |
RT3 |
RT4 |
RT5 |
RT6 |
RT7 |
RT8 |
RT9 |
RT10 |
RT11 |
| RT = retention time. |
| 1 |
0.0981 |
0.0879 |
0.0763 |
0.0695 |
0.0901 |
0.0833 |
0.0930 |
0.0865 |
0.0782 |
0.0858 |
0.0923 |
| 2 |
0.0971 |
0.0895 |
0.0814 |
0.0734 |
0.0897 |
0.0849 |
0.0942 |
0.0859 |
0.0813 |
0.0849 |
0.0897 |
| 3 |
0.0902 |
0.0916 |
0.0759 |
0.0725 |
0.0924 |
0.0867 |
0.0915 |
0.0798 |
0.0801 |
0.0835 |
0.0874 |
| 4 |
0.0973 |
0.0883 |
0.0817 |
0.0682 |
0.0896 |
0.0851 |
0.0889 |
0.0834 |
0.0796 |
0.0861 |
0.0916 |
| 5 |
0.0981 |
0.0876 |
0.0815 |
0.0713 |
0.0923 |
0.0865 |
0.0872 |
0.0876 |
0.0853 |
0.0842 |
0.0925 |
| 6 |
0.0986 |
0.0849 |
0.0796 |
0.0688 |
0.0915 |
0.0904 |
0.0856 |
0.0843 |
0.0794 |
0.0797 |
0.0893 |
| Average |
0.0966 |
0.0883 |
0.0794 |
0.0706 |
0.0909 |
0.0862 |
0.0901 |
0.0846 |
0.0807 |
0.0840 |
0.0905 |
| RSD |
3.28% |
2.51% |
3.36% |
2.98% |
1.42% |
2.81% |
3.76% |
3.30% |
3.09% |
2.78% |
2.22% |
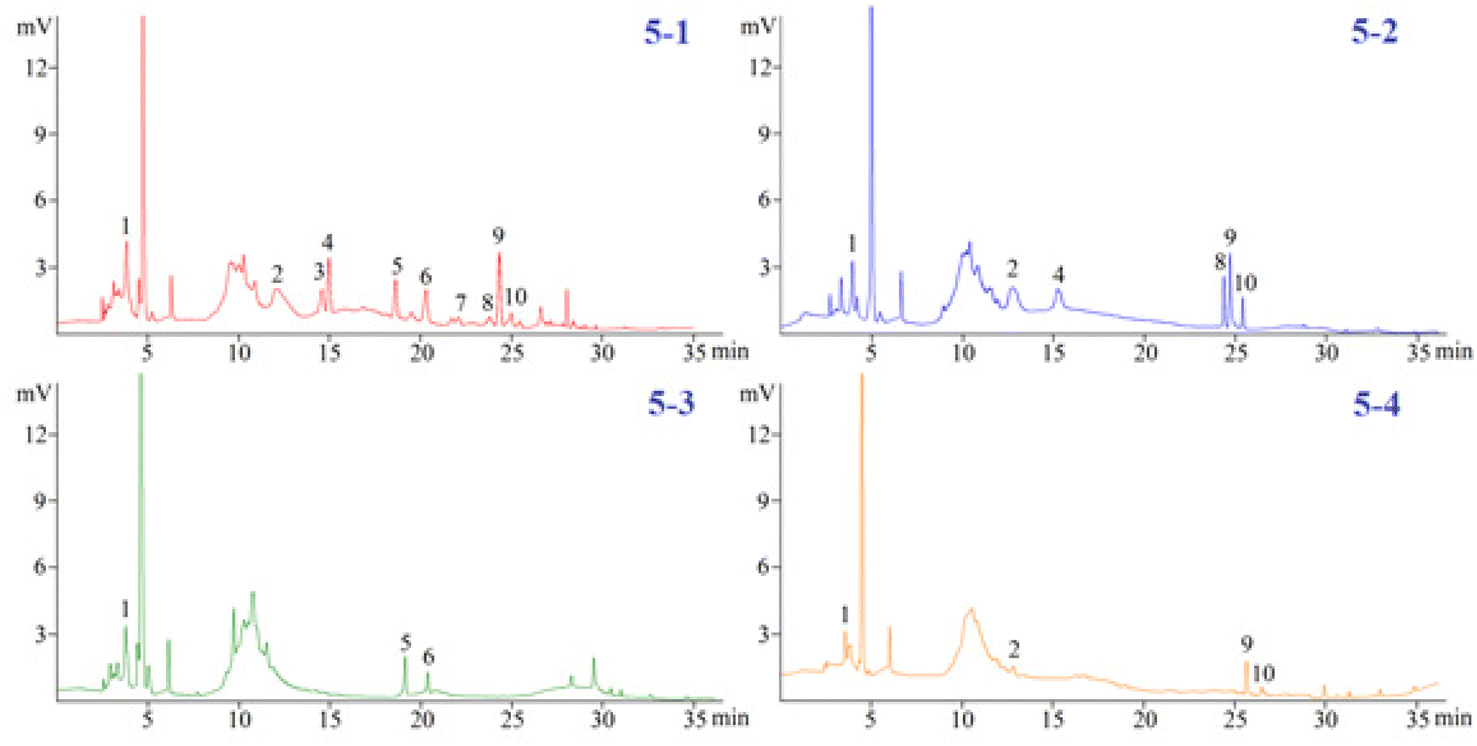
3.3.2. The analysis of the polyphenols in the four Dendrobium plants. The investigated phenols in the 4 Dendrobium plants were subjected to a quantitative examination using the developed pCEC technique. Except for tristin (11), 10 of them were checked and the amounts are summarized in Table 3. In 60% SPE fraction, the content of compounds (1), (2), (4), (5), and (6) was found to be the highest in D. huoshanense; compounds (8) and (10) had the highest content of D. nobile; compounds (3) and (7) had a unique component of D. huoshanense. In our experiment, compound 11 was not found in the 4 Dendrobium samples. Accordingly, the levels of 11 in the 4 Dendrobium samples may be quite low and may change depending on where they are located and when they are harvested (Fig. 5).
Table 3 Content of target components in the 60% solid phase extraction portion of the dry plant (mg kg−1)a
| Compounds |
D. huoshanense |
D. nobile |
D. chrysotoxum |
D. fimbriatum |
| Schaftoside (1), 2,4,7-trihydroxy-9,10-dihydrophenanthrene (2), dihydroresveratrol (3), nudol (4), coelonin (5), batatasin III (6), gigantol (7), 3′-hydroxy-3,4,5′-trimethoxybibenzy (8), erianin (9), dihydropinosylvin methyl ether (10), and tristin (11). |
| 1 |
11.38 ± 0.22 |
8.57 ± 0.38 |
6.16 ± 0.28 |
3.81 ± 0.08 |
| 2 |
12.72 ± 1.10 |
10.46 ± 0.38 |
— |
2.83 ± 0.11 |
| 3 |
5.46 ± 0.50 |
— |
— |
— |
| 4 |
9.06 ± 0.36 |
7.10 ± 0.30 |
— |
— |
| 5 |
6.16 ± 0.66 |
— |
5.13 ± 0.11 |
— |
| 6 |
5.84 ± 0.24 |
— |
2.83 ± 0.13 |
— |
| 7 |
2.48 ± 0.14 |
— |
— |
— |
| 8 |
3.30 ± 0.04 |
6.73 ± 0.24 |
— |
— |
| 9 |
13.73 ± 0.82 |
11.08 ± 1.24 |
— |
7.60 ± 0.45 |
| 10 |
3.70 ± 0.17 |
3.97 ± 0.20 |
— |
1.76 ± 0.01 |
| 11 |
— |
— |
— |
— |
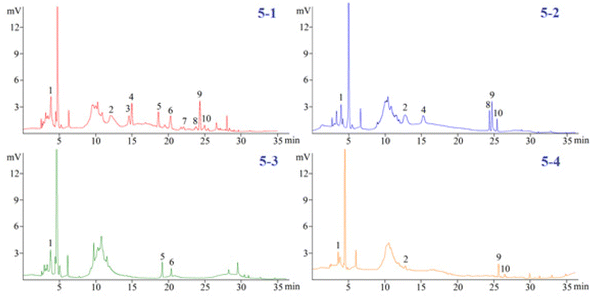 |
| | Fig. 5 Detection of polyphenols in four Dendrobium plants. (5-1) D. Huoshanense, (5-2) D. nobile, (5-3) D. chrysotoxum, and (5-4) D. fimbriatum. | |
3.4. Consistency evaluation
3.4.1. Consistency evaluation based on chemicals. Correlation coefficients (cor) were determined using the cosine angle technique based on the content data of 4 Dendrobium plants and were used as a similarity metric to assess the similarity of the chemical compositions of the 4 Dendrobium plants directly. As can be seen from Table 4, in terms of the content of four species of Dendrobium, there are differences in the similarity between different species of Dendrobium. The similarity between D. huoshanense and D. nobile was the closest (0.823), but the similarity between D. huoshanense and the other two species of D. nobile was the farthest. D. chrysotoxum and the other three species had a low similarity. It is suggested that Dendrobium is one of the medicinal plants with multiple origins, and its rationality needs to be further studied.
Table 4 Similarity analysis based on the 11 polyphenols of four Dendrobium plants
| |
D. huoshanense |
D. nobile |
D. chrysotoxum |
D. fimbriatum |
| D. huoshanense |
1 |
0.823 |
0.456 |
0.795 |
| D. nobile |
|
1 |
0.382 |
0.860 |
| D. chrysotoxum |
|
|
1 |
0.497 |
| D. fimbriatum |
|
|
|
1 |
3.4.2. Consistency evaluation based on pCEC fingerprint. Chromatographic fingerprint methodology, which stresses the systematic characterization of sample compositions, has recently been recognized as a helpful tool for regulating the quality of herbal medicines. In this work, the chromatographic fingerprint of 4 Dendrobium plants was also examined using the established pCEC technique. The similarities between 4 different Dendrobium plants ranged from 92.5% to 97.7%. According to the findings, there were significant differences in 4 different Dendrobium plants (Table 5). The fingerprint method provided a higher similarity between the samples, which may be due to the high sensitivity of chromatographic fingerprint technology to the characteristic peaks of samples, while the compound-based similarity assessment resulted in significant differences due to differences in the main component contents of the samples. More studies would be conducted in the future to evaluate the consistency of Dendrobium plants.
Table 5 Similarity analysis based on the pCEC fingerprints of the four Dendrobium plants
| |
D. huoshanense |
D. nobile |
D. chrysotoxum |
D. fimbriatum |
| D. huoshanense |
1 |
0.977 |
0.951 |
0.959 |
| D. nobile |
|
1 |
0.970 |
0.943 |
| D. chrysotoxum |
|
|
1 |
0.925 |
| D. fimbriatum |
|
|
|
1 |
4. Conclusions
We developed and assessed a pCEC technique for the rapid simultaneous detection of 11 typical phenolic compounds in 35 min. Systematic research was performed on pCEC circumstances. The established approach demonstrated excellent linearity, accuracy, and sensitivity. Compared with the reported studies by Zhu, our proposed method provided a shorter analysis time. This suggested approach made it possible to identify more phenolic compounds, and it offers an accurate and cost-effective alternative for phenolic compound analysis with a wide range of potential applications.2 We employed the conventional approach in the circumstances of our experiment involving a total of 10 phenolic compounds including schaftoside, 2,4,7-trihydroxy-9,10-dihydrophenanthrene, dihydroresveratrol, nudol, coelonin, batatasin III, gigantol, 3′-hydroxy-3,4,5′-trimethoxybibenzy, erianin, and dihydropinosylvin methyl ether were detected in D. huoshanense. This is the first report of the systematic and simultaneous identification and quantification of multiple phenolic compounds in 4 Dendrobium plants using pCEC, and this methodology could provide this plant with a new tool for quick quality control. This thorough examination of the composition of four Dendrobium plants will serve as the foundation for additional studies into the functions of numerous discovered substances and may help in the design and formulation of nutraceutical and cosmetic products in the future.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This study was granted by Natural Science Foundation of Anhui Scientific Research and Innovation Team of Quality Evaluation and Improvement of Traditional Chinese Medicine (No. 2022AH010090) and the Project of Anhui Rural Revitalization of Traditional Chinese Medicine Industry Collaborative Technology Service Center (GXXT-2022-079); The Provincial Level Nature Science Foundation of Anhui Education Department (KJ2019A0628, KJ2019A0617, KJ2019A0626, KJ2021A0957, KJ2020A0634, KJ2020A0632); School Level Projects of West Anhui University (WGKQ2021030); The Open Fund of Anhui Engineering Laboratory for Conservation and Sustainable Utilization of Traditional Chinese Medicine Resources, (TCMRPSU-2022-06); The Project from Anhui Province Rural Revitalization Collaborative Technology Service Center. The authors would like to thank the anonymous reviewers for their invaluable suggestions that helped improve the manuscript.
References
- N. D. Chen, H. Chen, J. Li and H. Yu, J. Mol. Struct., 2015, 1086, 255–265 CrossRef CAS.
- A. L. Zhu, J. W. Hao, L. Liu, N. D. Chen, G. L. Wang, X. Liu, Q. Li, H. M. Xu and W. H. Yang, Front. Nutr., 2021, 8, 771078 CrossRef PubMed.
- National Pharmacopoeia Committee, Pharmacopoeia of the People's Republic of China One, China Medical Science and Technology Press, Beijing, China, 2020, p. 135 Search PubMed.
- Y. J. Fan, T. Jiang and Z. Chun, Plant Physiol. Biochem., 2021, 162, 656–666 CrossRef CAS PubMed.
- M. S. Zhang, H. L. Ling and G. Wang, et al., Nat. Prod. Res., 2022, 36, 5393–5399 CrossRef CAS PubMed.
- X. L. Fu, S. Chen and X. T. Wang, et al., Biochem. Biophys. Rep., 2021, 26, 100877 CAS.
- Z. Niu, J. Pan, Q. Xue, S. Zhu, W. Liu and X. Ding, Acta Pharm. Sin. B, 2018, 8, 466–477 CrossRef PubMed.
- M. Li, Y. He, C. Peng, X. F. Xie and G. Y. Hu, Oncol. Lett., 2018, 16, 5006–5012 Search PubMed.
- Q. Zeng, C. H. Ko, W. S. Siu, K. K. Li, C. W. Wong, X. Q. Han, L. Yang, C. B. Lau, J. M. Hu and P. C. Leung, Chin. J. Nat. Med., 2018, 16, 481–489 CAS.
- Y. Bai, F. Du, Y. Bai and H. Liu, Anal. Methods, 2010, 2, 1867–1873 RSC.
- R. Bari and J. D. G. Jones, Plant Mol. Biol., 2009, 69, 473–488 CrossRef CAS PubMed.
- A. Santner, C. V. Li and M. Estelle, Nat. Chem. Biol., 2009, 5, 301–307 CrossRef CAS PubMed.
- S. D. S. Chiwocha, S. R. Abrams, S. J. Ambrose, A. J. Cutler, M. Loewen, A. R. S. Ross and A. R. Kermode, Plant J., 2003, 35, 405–417 CrossRef CAS PubMed.
- D. Rao, Y. D. Hu and R. X. Zhao, Int. J. Anal. Chem., 2022, 2022, 9510598 Search PubMed.
- C. Wu, S. Gui, Y. Huang, Y. Dai, Q. Shun, K. Huang, S. Tao and G. Wei, Anal. Methods, 2016, 8, 3802–3808 RSC.
- Y. D. Yuan, M. Y. Yu, B. Zhang, X. Liu and J. C. Zhang, PLoS One, 2019, 14, 1–16 Search PubMed.
- X. Y. Huang and Y. I. Yin, Southwest Chin. J. Agric. Sci., 2007, 20, 735–738 Search PubMed.
- J. Liu, T. He and Z. Chun, Zhongguo Zhongyao Zazhi, 2009, 34, 2853–2856 CAS.
- Q. M. Chen, Q. Liu, L. Shen, Y. Wang and C. Yan, Chin. J. Chromatogr., 2018, 36, 388–394 CAS.
- P. Li, S. P. Li, F. Q. Yang and Y. T. Wang, J. Sep. Sci., 2007, 30, 900–905 CrossRef CAS PubMed.
- S. Liu, W. Chen, L. Qu, Y. Gai and X. Jiang, Anal. Bioanal. Chem., 2013, 405, 1257–1266 CrossRef CAS PubMed.
- ICH Q2(R2) Validation of Analytical Procedures, https://www.ema.europa.eu/en/ich-q2r2-validation-analytical-procedures, accessed on 18 August 2022 Search PubMed.
- J. Q. Zhu, H. Cheng and M. M. Zhou, et al., J. Pharm. Biomed. Anal., 2022, 212, 114592 CrossRef CAS PubMed.
- A. Derazshamshir and S. I. Aşir, et al., J. Chromatogr. Sci., 2019, 57, 758–765 CAS.
- Y. Y. Zhao, L. J. Li and W. Y. Huang, et al., Chin. J. Anal. Lab., 2016, 35, 150–152 CAS.
- L. S. Yan, Z. H. Wang and G. A. Luo, et al., Chem. J. Chin. Univ., 2004, 25, 827–830 CAS.
- W. L. Zhou, W. B. Kan and Y. H. Wang, et al., J. Instrum. Anal., 2015, 34, 321–327 CAS.
Footnote |
| † This author contribution equal. |
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *abc,
Jing-Wen Hao*ab,
Shi-Tang Maab,
Min Zhangab,
Yu Songd,
Yun-Jiang Minab,
Ya-Qin Buac and
Sheng Liac
*abc,
Jing-Wen Hao*ab,
Shi-Tang Maab,
Min Zhangab,
Yu Songd,
Yun-Jiang Minab,
Ya-Qin Buac and
Sheng Liac