DOI:
10.1039/D3RA00713H
(Paper)
RSC Adv., 2023,
13, 13540-13546
Synthesis and identification of new sacubitril derivatives as lead compounds for antibacterial, antifungal and antitubercular (TB) activities against dormant tuberculosis†
Received
2nd February 2023
, Accepted 13th April 2023
First published on 3rd May 2023
Abstract
We identified twenty-two new sacubitril derivatives (5a–v) as lead compounds for various biologically active targets. These compounds were synthesized by reacting an intermediate compound (2R,4S)-5-([1,1′-biphenyl]-4-yl)-4-(amino)-2-methylpentanoic acid ethyl ester hydrochloride with respective carboxylic acid (RCOOH). The molecular structures of all the newly synthesized compounds were determined by 1H and 13C NMR, ESI mass spectrometry, FTIR spectroscopy, and CHN analysis. Moreover, compound 5n was characterized by a single-crystal X-ray diffraction (SXRD) study to confirm the structure obtained from spectral data. All these compounds were screened for various biological functions such as antifungal, antibacterial, and anti-TB activities. Among these twenty-two compounds (5a–v), some exhibited good to moderate anti-bacterial properties. Similarly, some compounds showed moderate anti-TB and antifungal activities. In addition, the anti-TB activity of compound 5q was estimated against M. tuberculosis in a nutrient starvation model (NSM). Similarly, toxicity was examined against RAW 264.7 cells. These biological activity studies were also correlated with molecular docking studies.
Introduction
A sacubitril derivative, also known as AHU-377 is α-methyl-c-amino-d-biphenyl valeric acid and contains two stereocenters.1 Sacubitril derivatives are crucial and active pharmaceutical ingredients mainly used for the treatment of heart failure along with the combination of valsartan.2 The combination of sacubitril/valsartan in a single crystalline supramolecular complex is composed of two molecular moieties and is called LCZ696 and is marketed as Entresto by Novartis.3,4 In fact, this supramolecular hybrid LCZ696 complex has been identified to reduce the risk of death in patients due to cardiovascular arrest or chronic heart failure. Moreover, the FDA has recently approved the LCZ696 complex as the first dual inhibitor of neutral endopeptidase (NEP) and AT1 receptors for angiotensin II.5
Furthermore, amide and ester-based heterocyclic compounds are also known to sustain various biological functions such as anti-mycobacterial,6–12 anticancer,13,14 antifungal,15,16 and antibacterial activities.17 As reported in the literature, several derivatives of the above-mentioned combinations containing scaffold compounds have been identified to possess potent antituberculosis activities.18–21 In fact, a pyrazinamide amide derivative-based first-line anti-TB drug has already been made commercially available.22 One of the recent and very successful approaches for drug design and discovery is the mimicking of multiple biologically active scaffolds in a single hybrid framework that could exhibit unique and enhanced biological properties.23 In continuation of our ongoing research10–14,16,17 on the design and synthesis of a variety of novel heterocyclic hybrid scaffolds to exhibit the desirable biological activity, the present study is focused on sacubitril derivatives attached to various organic moieties of donor and acceptor abilities, and various lengths of alkyl chains were also synthesized. These derivatives were screened for various biological activities.
In this study, twenty-two new sacubitril derivatives (5a–v) were synthesized by reacting ethyl ester hydrochloride derivatives of (2R,4S)-5-([1,1′-biphenyl]-4-yl)-4-(amino)-2-methylpentanoic acid with corresponding carboxylic acids. The molecular structures of all the newly synthesized compounds were determined by 1H and 13C NMR, 19F NMR, ESI mass spectrometry, FTIR spectroscopy, and CHN analysis. All the newly synthesized compounds were screened for various biological functions such as anti-fungal, anti-bacterial, and anti-TB activities. These biological activity studies were also correlated with molecular docking studies.
Results and discussion
The synthetic pathway of the titled twenty-two new sacubitril derivatives (5a–v) is projected in Scheme 1. The intermediate compounds, were synthesized according to our previously reported procedure.12 In the first step, (3R,5S)-5-([1,1′-biphenyl]-4-ylmethyl)-3-methylpyrrolidin-2-one (1) was reacted with acetic acid, ethyl acetate and HCl to form (2R,4S)-5-([1,1′-biphenyl]-4-yl)-4-amino-2-methylpentanoic acid (2) with a 94% yield. In the 2nd step, compound 2 was reacted with the Boc anhydride reagent to yield the corresponding (2R,4S)-5-([1,1′-biphenyl]-4-yl)-4-((tert-butoxycarbonyl)amino)-2-methylpentanoic acid, 3. In the 3rd step, compound 3 was reacted with SOCl2 under refluxed conditions in EtOH to afford ethyl (2R,4S)-5-([1,1′-biphenyl]-4-yl)-4-amino-2-methylpentanoate hydrochloride 4. In the final step, the coupling reactions of 4 with different carboxylic acids furnished respective amides (5a–v) in excellent yields (Scheme 1).
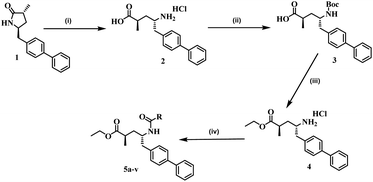 |
| | Scheme 1 Reagents and conditions for the synthesis of the title compounds (5a–v): (i) acetic acid, EtOAc, HCl, 15 h, 90 °C; (ii) DIPEA, Boc-anhydride, DCM, RT, 6 h; (iii) SOCl2, EtOH, reflux, 18 h; and (iv) RCOOH, HATU, DIPEA, DCM, 12 h, RT, N2. | |
In addition, in this work, a total of twenty-two new compounds were synthesized via various substitutions at R positions in order to exhibit enhanced biological activity (Scheme 1). However, the desirable compounds (5a–v) were accomplished in 12 h, with yields ranging between 79 and 97%. The newly synthesized twenty-two derivatives for different substituted (R) analogues (5a–v), yields and their melting points (MPs) are listed in Table 1. 1H NMR, 13C NMR, mass spectrometry, and elemental analysis were used to determine the structures of the newly synthesised compounds (5a–v). All the spectral data for the title compounds (5a–v) are presented in the Experimental section and (S.I, Fig. S1 to S67).†
Table 1 Physical data for the titled compounds (5a–v)
| Compound code |
R |
Yield (%) |
M.P. (°C) |
| 5a |
2-Bromo-5-fluorophenyl |
88 |
205 |
| 5b |
2-Chlorobenzyl |
92 |
184 |
| 5c |
2,6-Dimethoxyphenyl |
82 |
217 |
| 5d |
2-Hydroxyphenyl |
91 |
232 |
| 5e |
3-Trifluoromethylphenyl |
94 |
201 |
| 5f |
4-Trifluoromethylphenyl |
89 |
187 |
| 5g |
2-Fluorophenyl |
88 |
179 |
| 5h |
3,4-Dichlorophenyl |
93 |
214 |
| 5i |
2,4-Dichlorobenzyl |
86 |
198 |
| 5j |
2,4,5-Trifluorobenzyl |
79 |
238 |
| 5k |
3,4,5-Trimethoxybenzyl |
90 |
264 |
| 5l |
4-Fluorobenzyl |
86 |
196 |
| 5m |
Isonicotinoyl |
79 |
223 |
| 5n |
4-Nitrophenyl |
87 |
228 |
| 5o |
Tetra decanoyl |
97 |
176 |
| 5p |
1-Boc-piperidine-4-yl |
92 |
204 |
| 5q |
Cyclopropyl |
88 |
187 |
| 5r |
Cyclopentyl |
91 |
197 |
| 5s |
2-Chloro-5-nitrophenyl |
83 |
235 |
| 5t |
2-Methyl-4-nitrophenyl |
92 |
217 |
| 5u |
4-(Cyanomethyl)phenyl |
95 |
184 |
| 5v |
2-Amino-4-chlorophenyl |
87 |
195 |
X-ray crystal structure
Further, the structures of these newly synthesized compounds determined by spectral data and elemental analysis were confirmed by single-crystal X-ray diffraction (SXRD) analysis of 5n. The single crystals of 5n were grown by diffusion method using a DCM/n-hexane solvent combination. The ORTEP diagram for 5n is shown in Fig. 1 (SXRD data table provided in Table S1, S.I, CCDC 2168484),† which is in agreement with the structure determined by spectral data. Compound 5n crystallized in an orthorhombic crystal system with a space group of P21 (no. 4). In the SXRD structure, the asymmetric unit contains one formula unit (C27H28N2O5), without any solvent molecules in the crystallization.
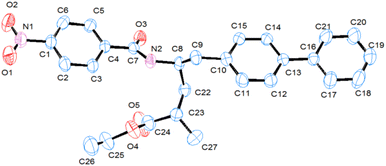 |
| | Fig. 1 Single-crystal X-ray diffraction (SXRD) structure ORTEP (50% probability and hydrogen atoms are omitted for clarity) of compound, 5n. | |
Biological activity study
In vitro anti-mycobacterial activity. For in vitro anti-tuberculosis activity, the Microplate Alamar Blue Assay (MABA) method against Mycobacterium tuberculosis (Mtb) H37Rv (ATCC27294) was employed on all the new twenty-two derivatives (5a–v). The detailed MABA assay24–26 is described in the Experimental section of S.I.† In this assay, ethambutol, rifampicin, and isoniazid were clinically used as reference drugs for the comparison of minimum inhibitory concentration (MIC) values, and these values were determined as duplicates for each compound and expressed in μg mL−1, and the results are summarised in Table 2. As shown in Table 2, the screened compounds displayed good to poor anti-TB activity with MIC values in the range of 6.25 to >25 μg mL−1. Among all the twenty-two new sacubitril-based derivatives (5a–v), compound 5q exhibited potent anti-TB activity with a MIC value of 6.25 μg mL−1. When compared to the standard drugs ethambutol (1.56 μg mL−1), rifampicin, (0.78 μg mL−1) and isoniazid (0.39 μg mL−1), and the compounds 5k and 5p exhibited significant anti-TB activity with the same MIC values of 12.50 μg mL−1. The remaining compounds showed poor anti-TB activity.
Table 2 Anti-TB activity data (MIC and cytotoxicity % of inhibition against RAW 264.7) of selected compounds (5a–v)a
| Compound code |
Anti-TB activity MIC (μg mL−1) |
Cytotoxicity (% of inhibition) against RAW 264.7 |
| 10 μg mL−1 |
25 μg mL−1 |
50 μg mL−1 |
100 μg mL−1 |
| Compounds 5a–5j, 5l–5o and 5r–5v exhibited anti-TB activity with MIC > 25 μg mL−1. |
| 5k |
12.5 |
8.93 ± 0.87 |
22.40 ± 0.90 |
37.93 ± 1.37 |
69.88 ± 0.90 |
| 5p |
12.5 |
6.36 ± 0.91 |
14.93 ± 0.60 |
28.88 ± 1.24 |
51.78 ± 0.91 |
| 5q |
6.25 |
6.40 ± 0.72 |
17.79 ± 0.71 |
30.61 ± 0.71 |
55.77 ± 1.09 |
| Ethambutol |
1.56 |
— |
— |
— |
— |
| Rifampicin |
0.78 |
— |
— |
— |
— |
| Isoniazid |
0.19 |
— |
— |
— |
— |
Antituberculosis nutrient starved model (NSM) assay. For the NSM assay, the Mtb (H37Rv genome) culture was successfully undernourished in phosphate-buffered saline (PBS) for 42 days.30 The test sample 5q, at a concentration of 10 μg mL−1, was evaluated for its activity for 7 days relative to the standard dormant stage captured bacteria. The sample plates were incubated at 37 °C for 28 days. For the analysis, wells with hallucinatory bacterial gathering were subsumed as positive. The MPN values were determined using standard statistical methods (SSMs), as reported previously.31 In addition, the selected 5q sample was screened against the NSM dormant TB model. In this method, surprisingly, compound 5q exhibited 6.4 log lower bacterial counts compared with the two standard drugs, rifampicin and isoniazid (Fig. 2). From these results, we can conclude that compound 5q showed good frail active stages along with the dormant forms of Mtb. Indeed, this behaviour of sample 5q is very imperative for reducing the therapy period for anti-TB drugs.
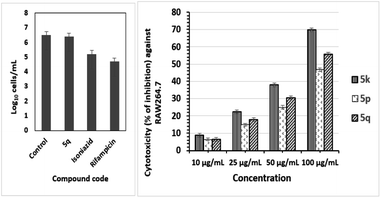 |
| | Fig. 2 Active Mtb compound NSM values for compound 5q, bacterial count estimated (mean ± S.D., n = 3) using the MPN assay (two-way ANOVA “GraphPad” prism software was used). Compound 5q exhibited positive inhibition of growth compared to the control growth (p < 0.0001; left). The percentage of inhibition on the RAW 264.7 cell line at four different concentrations of three compounds 5k, 5p and 5q (right) indicated promising anti-TB activities. | |
Toxicity assay. Three compounds, namely, 5k, 5p, and 5q showed promising anti-TB activity, were examined for their safety profiles using a cytotoxicity study on normal human RAW 264.7 cells at the concentration levels of 10, 25, 50, and 100 μg mL−1.32 The % cytotoxicity inhibition values of 5k, 5p, and 5q are projected in Fig. 2 (right). The observed IC50 values for the three compounds 5k, 5p, and 5q are 69.25, 95.09, and 87.97 respectively, as shown in Table 3. In fact, the cytotoxic inhibition percentages of the three compounds are low, indicating that these three compounds do not affect the normal human immune system, and hence, they could be potential candidates as anti-Tb drugs.
Table 3 Anti-TB activity with IC50 values
| Compound code |
IC50 μM |
| 5k |
69.25 |
| 5p |
95.09 |
| 5q |
87.97 |
| Ethambutol |
— |
| Rifampicin |
— |
| Isoniazid |
— |
Antifungal activity. Candidiasis is a fungal infection caused by Candida albicans.27,28 It is an acute or chronic, superficial or deep infection with a very broad clinical spectrum. All the test compounds were screened for antifungal activities against Candida albicans (ATCC 10231), and the activity was evaluated by measuring the inhibition zone diameter around the well and was recorded in mm. Among all these twenty-two compounds (5a–v), two compounds, namely, 5b and 5n showed moderate activity in the concentration (μg mL−1) range of 75 and 100, by using potato dextrose agar media (Hi-media), as shown in Table 4 (S.I, Fig. S68†). The activity was compared with that of standard antifungal agents fluconazole, griseofulvin and nystatin.
Table 4 Anti-fungal activity of the titled compounds (5a–v)a
| Compounds |
Zone of inhibition (mm) Candida albicans |
| 75 (μg mL−1) |
100 (μg mL−1) |
| Data are presented as mean ± standard deviation (n = 3). |
| 5b |
10.66 ± 0.57 |
12.66 ± 0.57 |
| 5n |
12.33 ± 0.57 |
14.33 ± 0.57 |
| Fluconazole |
15.66 ± 0.57 |
21.0 ± 1.0 |
| Griseofulvin |
20.66 ± 0.57 |
23.33 ± 0.57 |
| Nystatin |
21.33 ± 0.57 |
25.0 ± 1.0 |
Anti-bacterial activity. Antibacterial activity was measured directly by an agar diffusion method.17–19 Sterile Petri plates with 90 mm diameter and 15 mm height were taken and filled with 25 mL of freshly prepared sterile nutrient agar medium (Hi-media) and kept for solidification for 15–20 min. The plates were inoculated with 1 × 108 CFU mL−1 of E. coli (MTCC 9721), P. aeruginosa (MTCC 9800), S. aureus (MTCC 11949), and B. subtilis (MTCC 10010) cultures. The inoculated agar medium was punched with 6 mm-diameter wells, filled with 100 μg mL−1 of test compound and incubated at 37 °C for 24 h in an incubator. The activity was compared with positive (tetracycline 30 μg per disc, ampicillin 10 μg per disc, gentamycin 10 μg per disc) and negative (DMSO) controls. The activity was determined by measuring the zone of inhibition in mm. Among these twenty-two compounds, nine showed good antibacterial activity, and the data are presented as mean ± standard deviation (n = 3) in Table 5.
Table 5 Antibacterial activity of the titled compounds (5a–v)a
| Compound (100 μg mL−1) |
Zone of inhibition (mm) |
| Compounds |
E. coli (MTCC 9721) |
P. aeruginosa (MTCC 9800) |
S. aureus (MTCC 11949) |
B. subtilis (MTCC 9800) |
| Data are presented as mean ± standard deviation (n = 3). |
| 5b |
14.33 ± 0.57 |
22.66 ± 0.57 |
21.33 ± 0.57 |
18.66 ± 0.57 |
| 5c |
11.66 ± 0.57 |
15.33 ± 0.57 |
15.66 ± 0.57 |
11.66 ± 0.57 |
| 5g |
13.66 ± 0.57 |
17.66 ± 0.57 |
19.33 ± 0.57 |
15.33 ± 0.57 |
| 5i |
10.66 ± 0.57 |
16.66 ± 1.15 |
16.66 ± 0.57 |
12.33 ± 0.57 |
| 5l |
13.33 ± 0.57 |
16.66 ± 0.57 |
15.66 ± 1.52 |
12.66 ± 0.57 |
| 5m |
11.33 ± 0.57 |
18.33 ± 0.57 |
19.33 ± 0.57 |
14.33 ± 0.57 |
| 5n |
17.66 ± 0.57 |
25.0 ± 1.0 |
21.0 ± 1.0 |
18.33 ± 0.57 |
| 5p |
13.66 ± 0.57 |
19.33 ± 0.57 |
19.66 ± 0.57 |
13.66 ± 0.57 |
| 5u |
— |
13.33 ± 0.57 |
12.66 ± 0.57 |
12.66 ± 0.57 |
| Tetracycline (30 μg mL−1) |
25.0 ± 1.0 |
32.33 ± 0.57 |
29.66 ± 0.57 |
22.33 ± 0.57 |
| Ampicillin (10 μg mL−1) |
14.33 ± 0.57 |
11.33 ± 0.57 |
13.33 ± 0.57 |
12.66 ± 0.57 |
| Gentamicin (10 μg mL−1) |
21.33 ± 0.57 |
18.33 ± 0.57 |
20.66 ± 0.57 |
19.66 ± 0.57 |
Similarly, minimum inhibitory concentration (MIC) values were determined by a microbroth dilution method using a resazurin indicator.29 For this, in the first step, a test solution of 100 μL (1% w/v) was added to the first row of wells, and other wells were filled with 50 μL of sterile nutrient broth. In the second step, 50 μL of test solution was transferred from the first well to the next well and so on for serial dilutions. Later, respective bacterial suspension (10 μL) and resazurin (0.02%) of 30 μL were added. At the final step, the test plates were covered and incubated at 37 °C for 24 h. All the MIC values are summarized in Table 6 (S.I, Fig. S69 and S70†).
Table 6 Minimum inhibitory concentration of the titled compounds (5a–v)
| Compound (100 μg mL−1) |
Minimum inhibitory concentration (μg mL−1) |
| Compounds |
E. coli (MTCC 9721) |
P. aeruginosa (MTCC 9800) |
S. aureus (MTCC 11949) |
B. subtilis (MTCC 9800) |
| 5b |
12.5 |
3.125 |
3.125 |
6.25 |
| 5c |
25 |
12.5 |
12.5 |
25 |
| 5g |
25 |
6.25 |
6.25 |
12.5 |
| 5i |
25 |
12.5 |
12.5 |
25 |
| 5l |
25 |
12.5 |
12.5 |
25 |
| 5m |
25 |
6.25 |
6.25 |
12.5 |
| 5n |
6.25 |
3.125 |
3.125 |
6.25 |
| 5p |
25 |
6.25 |
6.25 |
25 |
| 5u |
50 |
25 |
25 |
25 |
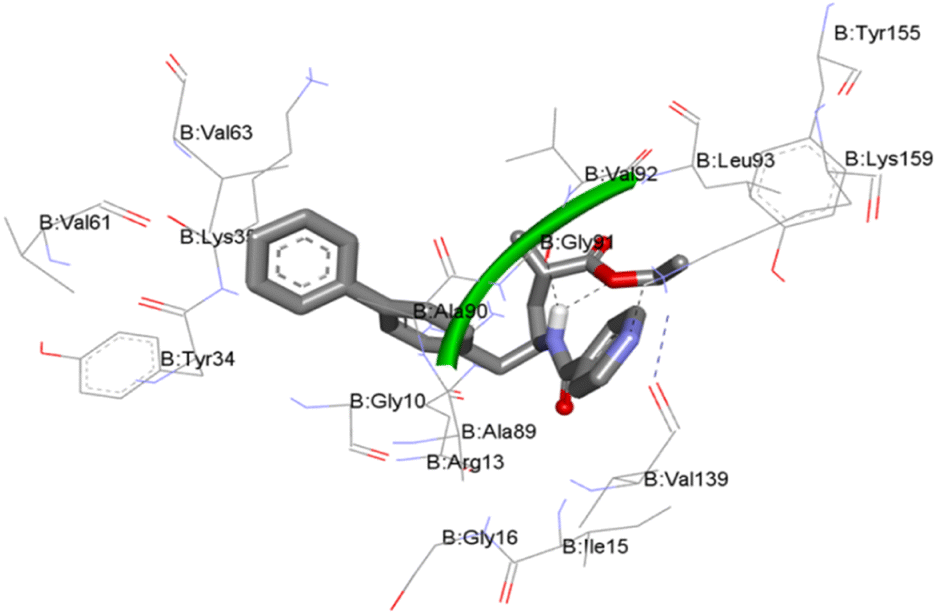
Molecular docking study. For the structure-based drug design, the molecular docking method is one of the most important and frequently used tools. Usually in this study, the binding affinities of small drug molecules were calculated with respect to the target binding sites of the biological receptors.17–19 The molecular docking in silico studies of all twenty-two sacubitril derivatives (5a–v) were performed against the enzyme receptor target oxidoreductase enzyme nicotinamide adenine dinucleotide phosphate (NADPH) using AutoDock Tools (ADT) version 1.5.6 and AutoDock version 4.2.5.1. This group of enzyme receptors usually employ nicotinamide adenine dinucleotide phosphate (NADP, NADP+ or NADPH) as a cofactor. This enzyme receptor performs several enzymatic electron transfer reactions along with the generation of free radicals in immune cells. In fact, these radicals could be useful for the destruction of the pathogens termed respiratory bursts. Moreover, NADPH also furnishes the reducing equivalents for biosynthetic reactions and is antagonistic against the toxicity of reactive oxygen species (ROS), which allows the regeneration of GSH.In this context, the NADPH target receptor was selected for our docking study protocol. The molecular docking binding energies of all twenty-two (5a–v), derivatives against NADPH protein receptors are presented in Table 7. Most of the compounds exhibit good binding energies, and compounds 5m and 5n showed the highest binding energies of −10.19 and −10.18 kcal mol−1, respectively. The ligand interaction with protein amino acid residues of compound 5m is illustrated in Fig. 3. These docking studies have shown a good correlation with antibacterial studies.
Table 7 Binding energies of sacubitril derivative (5a–v) inhibitors against receptor oxidoreductase enzyme nicotinamide adenine dinucleotide phosphate (NADPH) protein amino acid residues
| Compound |
Binding energies (kcal mol−1) |
| NADPH (PDB ID 5B1Y) |
| Binding energy |
No. of H bonds |
Residues involved in bonding |
| 5a |
−8.38 |
03 |
GLY91, ILE15, LEU93 |
| 5b |
−9.85 |
03 |
GLY91(2), LYS159 |
| 5c |
−8.14 |
01 |
VAL188 |
| 5d |
−8.65 |
03 |
SER36, LYS35, GLY91 |
| 5e |
−7.75 |
03 |
TRY155, LYS159, GLY91 |
| 5f |
−8.89 |
03 |
GLY91, ILE15, VAL63 |
| 5g |
−9.59 |
03 |
ARG13, SER12, ARG37 |
| 5h |
−8.29 |
03 |
LYS159(2), ALA89 |
| 5i |
−8.75 |
01 |
LYS159 |
| 5j |
−9.25 |
02 |
TYR155, LYS159 |
| 5k |
−7.34 |
02 |
TYR155, LEU93 |
| 5l |
−8.27 |
01 |
LYS159 |
| 5m |
−10.19 |
02 |
LYS159, GLY91 |
| 5n |
−10.18 |
03 |
SER141, PRO185, LEU93 |
| 5o |
−9.53 |
02 |
LYS35, SER12 |
| 5p |
−9.34 |
02 |
LYS159, ARG13 |
| 5q |
−7.40 |
03 |
LYS159, TYR155 |
| 5r |
−8.09 |
03 |
GLY91, LYS159, ILE15 |
| 5s |
−8.63 |
03 |
LYS159(2), ILE15 |
| 5t |
−8.52 |
03 |
GLY10, LYS35, SER36 |
| 5u |
−8.71 |
03 |
GLY91, ALA89, LYS35 |
| 5v |
−8.48 |
03 |
ILE15, THR190, VAL188 |
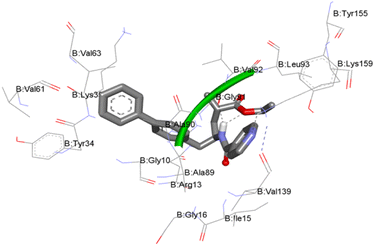 |
| | Fig. 3 Molecular docking (MD) interactive profile of compound 5m (ball-and-stick model) showing its interactive H-bonding map (black dotted lines) with the receptor NADPH protein amino acid residues (stick model). | |
The structure–activity correlation (SAC) of all the twenty-two newly synthesized compounds (5a–v), is described as follows. Overall, the sacubitril derivatives with donor groups such as 5k, 5p and 5q showed anti-Tb activity. Moreover, our cytotoxicity determination of lead compounds such as 5k, 5p, and 5q on normal human RAW 264.7 cells illustrated that these compounds are non-toxic to human cells and could be potential candidates as new anti-TB drugs. The compounds with acceptor groups such as 5b and 5n exhibit anti-fungal activities. Further, more compounds (5b, 5c, 5g, 5i, 5l, 5m, 5n, 5p and 5u) exhibited good to moderate antibacterial activities compared with the selected references. Among the nine compounds, 5m (isonicotinoyl) has the best antibacterial activity performance, and the values are close to the reference. These results also correlated with the docking studies using the oxidoreductase enzyme nicotinamide adenine dinucleotide phosphate (NADPH), where 5m, showed the highest binding of −10.19 kcal mol−1.
Conclusions
We have identified twenty-two new sacubitril derivatives (5a–v) as lead compounds for different biologically active molecules. These compounds were synthesized from (2R,4S)-5-([1,1′-biphenyl]-4-yl)-4-(amino)-2-methylpentanoic acid ethyl ester hydrochloride with the corresponding carboxylic acid. The molecular structures of all the newly synthesized compounds were determined by 1H and 13C NMR, ESI mass spectrometry, FTIR spectroscopy and CHN analysis. Moreover, compound 5n was characterized by a single-crystal X-ray diffraction study, which confirmed the structure obtained by spectral data. All these compounds were screened for various biological functions such as anti-fungal, anti-bacterial and anti-TB activities. Among these twenty-two compounds (5a–v), some compounds exhibited good to moderate anti-bacterial properties. Similarly, some compounds showed moderate antifungal and anti-TB activities. Further, anti-TB active compound 5q was examined against M. tuberculosis in the NSM, and the toxicity of compounds 5k, 5p, and 5q was evaluated against normal human RAW 264.7 cells, finding that they are non-toxic to human cells. These biological activity studies were also correlated with molecular docking studies. This study could provide a roadmap for the synthesis and identification of new sacubitril derivatives as lead compounds for antibacterial, antifungal, and antitubercular (TB) activities against dormant tuberculosis.
Experimental section
The melting points (MPs) for all the compounds used in this study were recorded in capillary tubes using an Elchem lab melting point apparatus and the reported values were uncorrected. The 1H and 13C NMR spectra were recorded using a Bruker FT-NMR spectrometer at 400 MHz and 75 MHz, respectively in CDCl3 and DMSO solvents with TMS as an internal reference. The chemical shift values were reported in parts per million (δ ppm) from internal standard TMS. FT-IR spectra were recorded using a Cary 630 FT-IR Spectrometer with Diamond ATR (Agilent Technologies, USA). All the reagents used in this work were purchased from Sigma-Aldrich and used as received without any further purification. Silica gel 60F254 coated aluminium sheets were used for thin-layer chromatography (TLC) plates and the spots on TLC plates were visualized by UV illumination, heating the plates dipped in KMnO4 stain and exposure to iodine vapour. The organic extracts were dried over anhydrous Na2SO4 and purification was carried out by column chromatography using silica gel (230–400 mesh).
General procedure and spectral data for compound preparation of (5a–v)
Under an N2 atmosphere, a stirred solution of compound (4) in dry dichloromethane at room temperature was added simultaneously to Hexafluorophosphate Azabenzotriazole Tetramethyl Uronium (HATU) and DIPEA, and after five minutes, the respective carboxylic acids were added to the mixture. Later, the reaction mixture was stirred for 12 hours. After the completion of the reaction, the reaction mixture was diluted with an excess of DCM and washed with a saturated bicarbonate solution followed by a brine solution. Later, the organic layer was dried with a sodium sulphate filter and concentrated completely under vacuum. The titled compounds (5a–v) were obtained in good yields by purifying the crude product by column chromatography with an ethyl acetate: hexane solvent mixture.
In silico molecular docking study. Molecular docking was performed using the AutoDock Vina software.29 The crystal structures of the applied proteins were extracted from the RCSB Protein Data Bank. All the proteins were first prepared and cleaned using BIOVIA Discovery Studio 2020 (ref. 30) and AutoDock tools.31 All the ligand structures were optimized employing the Gaussian09 software package. The docked poses were analyzed and visualized in BIOVIA Discovery Studio 2020.32
Author contributions
Dodda Bhargavi, Srihari Konduri and Koya Prabhakara Rao conceived, designed and carried out the experiments. Jyothi Prashanth carried out DFT and other computational analysis. Dodda Bhargavi and Sowjanya Pulipati carried out biological activity. All the authors contributed in analyzing the data and also in writing the paper.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
K. P. R thanks DST-SERB, for the financial support, external project for the core research grant project no. CRG/2019/002217. D. B. thanks VFSTR for the research avenues and HTRA fellowship.
Notes and references
- A. Halama and M. Zapadlo, Org. Process Res. Dev., 2019, 23, 102 CrossRef CAS.
- G. M. Ksander, R. D. Ghai, R. DeJesus, C. G. Diefenbacher, A. Yuan, C. Berry, Y. Sakane and A. Trapani, J. Med. Chem., 1995, 38, 1689 CrossRef CAS PubMed.
- J. J. V. McMurray, M. Packer, A. S. Desai, J. Gong, M. P. Lefkowitz, A. R. Rizkala, J. L. Rouleau, V. C. Shi, S. D. Solomon, K. Swedberg and M. R. Zile, N. Engl. J. Med., 2014, 371, 993 CrossRef PubMed.
- S. H. Lau, S. L. Bourne, B. Martin, B. Schenkel, G. Penn and S. V. Ley, Org. Lett., 2015, 17, 5436 CrossRef CAS PubMed.
- US Food and Drug Administration, Development Approval Process/Drug Innovation, Novel Drugs Summary, 2015, http://www.fda.gov/Drugs/DevelopmentApprovalProcess/DrugInnovation/ucm474696.htm Search PubMed.
- I. V. Daele, H. Munier-Lehmann, M. Froeyen, J. Balzarini and S. V. Calenbergh, J. Med. Chem., 2007, 50, 5281 CrossRef PubMed.
- V. Bhowruth, A. K. Brown, R. C. Reynolds, G. D. Coxon, S. P. Mackay, D. E. Minnikin and G. S. Besra, Bioorg. Med. Chem. Lett., 2006, 16, 4743 CrossRef CAS PubMed.
- R. S. Upadhayaya, G. M. Kulkarni, N. R. Vasireddy, J. K. Vandavasi, S. S. Dixit, V. Sharma and J. Chattopadhyaya, Bioorg. Med. Chem., 2009, 17, 4681 CrossRef CAS PubMed.
- I. Kucukguzel, E. S. Tatar, G. Kucukguzel, S. Rollas and E. de. Clercq, Eur. J. Med. Chem., 2008, 43, 381 CrossRef PubMed.
- S. Konduri, J. Prashanth, V. S. Krishna, D. Sriram, J. N. Behera, D. Siegel and K. P. Rao, Bioorg. Med. Chem. Lett., 2020, 30, 127512 CrossRef CAS PubMed.
- S. Konduri, D. Bhargavi, J. Prashanth, V. S. Krishna, D. Sriram and K. P. Rao, ACS Omega, 2021, 6, 1657 CrossRef CAS PubMed.
- S. Konduri, V. Pogaku, J. Prashanth, V. S. Krishna, D. Sriram, S. Basavoju, J. N. Behera and K. P. Rao, ChemistrySelect, 2021, 6, 3869 CrossRef CAS.
- S. Kancharla, P. Jyothi, K. P. Rao, S. Madala, A. Vejendla and M. V. B. Rao, Lett. Drug Des. Discovery, 2020, 17, 929 CrossRef.
- M. Subramanyam, R. Sreenivasulu, R. Gundla, M. V. B. Rao and K. P. Rao, Lett. Drug Des. Discovery, 2018, 18, 1299 CrossRef.
- Y. H. Ye, L. Ma, Z. C. Dai, Y. Xiao, Y. Y. Zhang, D. D. Li, J. X. Wang and H. L. Zhu, J. Agric. Food Chem., 2014, 62, 4063 CrossRef CAS PubMed.
- B. Srinivas, J. Suryachandram, Y. K. Devi and K. P. Rao, J. Heterocycl. Chem., 2017, 54, 3730 CrossRef CAS.
- B. Srinivas, P. Jyothi, M. V. B. Rao and K. P. Rao, J. Heterocycl. Chem., 2019, 56, 73 CrossRef.
-
(a) I. Kucukguzel, E. S. Tatar, G. Kucukguzel, S. Rollas and M. Kiraz, Bioorg. Med. Chem. Lett., 2001, 11, 1703 CrossRef CAS PubMed;
(b) H. M. Abdel-Rahman, N. A. El-Koussi and H. Y. Hassan, Arch. Pharm., 2009, 342, 94 CrossRef CAS PubMed.
- S. K. Avraham Liav, A. J. Patrick and M. J. Brennan, Bioorg. Med. Chem. Lett., 2008, 18, 2649 CrossRef PubMed.
- Y. Zhang, W. Shi, W. Zhang and D. Mitchison, Microbiol. Spectrum, 2013, 2, 4 Search PubMed.
- H.-W. Han, H.-Y. Qiu, C. Hu, W.-X. Sun, R.-W. Yang, J.-L. Qi, X.-M. Wang, G.-H. Lu and Y.-H. Yang, Bioorg. Med. Chem. Lett., 2016, 26, 3237 CrossRef CAS PubMed.
- L. A. Collins and S. G. Franzblau, Antimicrob. Agents Chemother., 1997, 41, 1004 CrossRef CAS PubMed.
- V. S. Krishna, S. Zheng, E. M. Rekha, L. W. Guddat and D. Sriram, J. Comput.-Aided Mol. Des., 2019, 33, 357 CrossRef CAS PubMed.
- V. Pogaku, V. S. Krishna, C. Balachandran, D. Sriram, K. Rangan, S. Aoki and S. Basavoju, New J. Chem., 2019, 43, 17511 RSC.
- N. A. R. Gow and B. Yadav, Microbiology, 2017, 163, 1145 CrossRef CAS PubMed.
-
(a) K. K. Alluri, R. S. Reshma, R. Suraparaju, S. Gottapu and D. Sriram, Bioorg. Med. Chem., 2018, 26, 1462 CrossRef CAS PubMed;
(b) P. Malapati, V. S. Krishna, R. Nallangi, R. R. Srilakshmi and D. Sriram, Bioorg. Med. Chem., 2018, 26, 177 CrossRef CAS PubMed;
(c) V. S. Krishna, S. Zheng, E. M. Rekha, R. Nallangi, D. V. Sai Prasad, S. E. George, L. W. Guddat and D. Sriram, Eur. J. Med. Chem., 2020, 193, 112178 CrossRef CAS PubMed.
- J. C. Betts, P. T. Lukey, L. C. Robb, R. A. McAdam and K. Duncan, Mol. Microbiol., 2002, 43, 717 CrossRef CAS PubMed.
- V. Pogaku, V. S. Krishna, D. Sriram, K. Rangan and S. Basavoju, Bioorg. Med. Chem. Lett., 2019, 29, 1682 CrossRef CAS PubMed.
- O. Trott and A. J. Olson, J. Comput. Chem., 2010, 31, 455 CAS.
- BIOVIA, Dassault Systèmes, BIOVIA Discovery Studio, Dassault Systèmes, San Diego, CA, USA, 2020 Search PubMed.
- G. M. Morris, R. Huey, W. Lindstrom, F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, J. Comput. Chem., 2009, 16, 2785 CrossRef PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, et al., Gaussian 09, revision a. 02, 2015 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence c,
Sowjanya Pulipatid,
K. K. Praneeth
c,
Sowjanya Pulipatid,
K. K. Praneeth