DOI:
10.1039/D3RA00707C
(Paper)
RSC Adv., 2023,
13, 8720-8735
FeMoS2 micoroparticles as an excellent catalyst for the activation of peroxymonosulfate toward organic contaminant degradation†
Received
2nd February 2023
, Accepted 6th February 2023
First published on 17th March 2023
Abstract
The FeMoS2 catalyst for activating peroxymonosulfate (PMS) is a promising pathway for removing organic pollutants in wastewater, however, the dominant FeS2 phases and sulfur (S) vacancies in it are little involved. Herein, for the first time, novel bimetallic FeMoS2 microparticles were synthesized by a simple method and then applied for PMS activation for degrading organic pollutants. The catalysts were characterized by several techniques, including X-ray diffraction and X-ray photoelectron spectroscopies. The results revealed that new FeMoS2 microparticles containing S vacancies in the main FeS2 phases were obtained. FeS2 and S vacancies were found to play important roles for activating PMS by radical and nonradical pathways. More Fe2+ and Mo4+ were formed in the presence of S vacancies, which offered a new strategy for exploring novel heterogeneous catalysts in the activation of PMS for environmental remediation.
1. Introduction
Organic pollutants such as dyes are widely applied in various fields, e.g., antibiotics,1 food additives,2 and microplastics,3 etc. However, they are harmful to human health. Consequently, it is extremely necessary to remove them using various advanced oxidation processes.1–10 Among these, the Fenton-like technology involving PMS activated by Fe-based catalysts is a good alternative. Over the past years, various Fe-based catalysts have been extensively developed,5–10 including single6,7 and bimetallic Fe-based catalysts.5,8–10 Bimetallic Fe-based catalysts more easily facilitate PMS decomposition by accelerating the transformation of iron(II) and iron(III) (Fe2+ and Fe3+, respectively) due to the function of another metal with variable valence states relative to single Fe-based states (formula (1)). Clearly, the former has more advantages than the latter in PMS activation.| | |
Fe3+ + Mn+ → Fe2+ + Mn+1
| (1) |
To date, research has predominantly focused on Fe–Mn,8 Fe–Cu,5,9 Fe–Co,10 Fe–Ni,11 Fe–Ag,12 and Fe–Mo.13–18 Among these, they are either highly toxicity, such as cobalt and nickel, or expensive, such as silver. It is possible that molybdenum (Mo) better boosts the transformation cycle of Fe2+ and Fe3+ relative to copper (Cu) and manganese (Mn). This is because the Mo (4d55s1) atom accommodates more electrons in empty orbits than Cu (3d104s1) and Mn (3d54s2) atoms. In addition, Mo has other merits like lower toxicity. Clearly, it is an ideal alternative for Mo as a cocatalyst to promote PMS activation in Fe-based catalysts. Enhancement of the Fe/PMS system has been reported in the assistance of Mo species, such as Mo0 (ref. 13) and MoS2.14–18 Compared to Mo0, the exposed Mo4+ species in MoS2 are more suitable for activating PMS in the presence of unsaturated S species,14–19 thereby inducing greater removal of organic pollutants. Recently, some FeMoS2 heterogeneous catalysts in PMS-based reactions have been extensively explored, including Fe3O4@MoS2,20 FeOOH@MoS2,21 and Fe–MoS2,22 which has to overcome the shortcomings of mixtures of FeMoS2-based homogeneous and heterogeneous catalysts.14–17 However, these processes involve complex procedures. More importantly, most Fe species are deposited on MoS2 surfaces, which results in long distances between Fe and Mo atoms, thereby affecting the Fe2+/Fe3+ cycle. To alter this situation, FeMoS2 has been directly prepared by high-temperature treatment of mixtures of Fe, Mo, S, and I powders.23 The results show that Fe atoms are confined near Mo atoms and thus interatomic distances turn shorter, which is better for the adsorption and decomposition of PMS. However, the Fe content is extremely low inside the catalyst in this case, restricting larger applications. Nevertheless, this situation has provided a good inspiration for the synthesis of a novel FeMoS2 with shorter distances between Fe and Mo atoms using in situ synthesis. Interestingly, researchers have found that small crystal phases of FeS2 appear in FeMoS2 via the above method.24,25 It has been confirmed that FeS2 can effectively activate PMS to remove organic pollutants in wastewater,26,27 even having a higher ability relative to MoS2 alone in Fenton-like reaction.28 It has been expected to be able to synthesize FeS2 as dominant phases in FeMoS2-based materials to enhance catalytic activity. Also, this catalyst would be rich in S atoms. In this case, S vacancies are easily generated in these materials by suitable treatment.19,29 Wu et al.29 have demonstrated that the S vacancies play key role in activating PMS in the Fe(III)/CoS2 system. Thus, it is promising to further strengthen the catalytic performance in FeMoS2. There is no doubt that the development of a new FeMoS2 containing S vacancies with main FeS2 phases is highly desired. To the best of our knowledge, the synthesis of this catalyst and its application for activating PMS to degrade organic pollutants has not been reported so far.
In this study, a new FeMoS2 catalyst, containing S vacancies and FeS2 as the dominant phases, was synthesized by a one-step method, which could efficiently activate PMS to degrade organic contaminants. Important influencing factors were studied in detail. A different reaction mechanism was found in this FeMoS2 for PMS activation. The relationship between the catalyst structure and catalytic performance was established.
2. Experiments
2.1. Chemical reagents and materials
The chemical reagents and materials were supplied by commercial purchase. For instances, peroxymonosulfate (2KHSO5·KHSO4·K2SO4), 5,5-dimethyl-1-pyrro-lineN-oxide (DMPO, 98%), and 2,2,6,6-tetra-methylpiperidine (TEMP, 99%) were purchased from Sigma-Aldrich, Inc. (St Louis, MO, USA). Molybdenum(IV) sulfide (MoS2, ≥98.0%), furfuryl alcohol (C5H6O2, ≥98.0%), ammonium molybdate tetrahydrate ((NH4)6Mo7O24·H2O, ≥99.0%), and rhodamine B (RhB) were purchased from Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China). Tiron (C6H7NaO8S2, >98.0%), sodium sulfide (Na2S·9H2O, >98.0%), and methyl orange (MO) were obtained from Damao Chemical Reagent Factory (Tianjin, China). Molybdenum trioxide (MoO3, ≥99.5%) and methylene blue (MB) were purchased from Kemiou Chemical Reagent Co., Ltd. (Tianjin, China). All the above-mentioned chemical reagents and materials were of analytical grade and employed without further purification.
2.2. Synthesis of FeMoS2 catalysts
(i) In situ synthesis: 0.18 g or 0.94 g or 1.98 g of Fe(NO3)3·9H2O, 2.28 g of CH4N2S, and 0.196 g of (NH4)6Mo7O24·4H2O were mixed in 35 mL of distilled water and then evenly stirred. Afterward, the solutions were then autoclaved at 180 °C for 4 h. Then, they were dried at 60 °C and denoted as FeMoS2-IS-60. Finally, dried samples were calcined at different temperatures (200, 300, 400, or 500 °C) for 4 h with a heating rate of 5 °C min−1 under air atmosphere, thus obtaining the targeted products, respectively denoted as FeMoS2-IS-200, FeMoS2-IS-300, FeMoS2-IS-400, and FeMoS2-IS-500. Herein, the theoretic concentrations of Fe in the above catalysts were 1.0, 5.0 and 10 wt%, respectively. Unless otherwise indicated, FeMoS2-IS-400 was abbreviated as FeMoS2-IS and the concentration of Fe was fixed at 5.0 wt%. The MoS2-IS, Fe–Mo, and Fe–S catalysts were synthesized by the same above procedures for FeMoS2-IS except for one of components. MoS2-300 was obtained by heating commercial MoS2 powders at 300 °C for 4 h. The synthetic steps for FeMoS2-IS microparticles are displayed in Fig. S1.†
(ii) Post treatment: 0.19 g of Fe(NO3)3·9H2O and 0.50 g of MoS2 were weighed and added separately to 50 mL of distilled water. After being evenly stirred, they were autoclaved at 180 °C for 4 h, then cooled to room temperature, and dried at 60 °C. Finally, the dried sample was treated at 400 °C for 4 h with a heating rate of 5 °C min−1 under air atmosphere. The targeted product was denoted as FeMoS2-IE (theoretic Fe concentration, 5.0 wt%).
2.3. Characterization of catalysts
The crystal phases of catalyst were analyzed by using (XRD) spectroscopy with a D8-Advance X-ray Diffractometer (Bruker Corp., Billerica, MA, USA). The specific surface area and pore features of catalyst were measured by liquid nitrogen physisorption using an Autosorb-iQ/ASAP 2460 apparatus (Quantachrome Instruments Corp., Boynton Beach, FL, USA). The morphologies of catalysts were examined by scanning electron microscopy (SEM) on an SU-8100 instrument (Hitachi Instruments, Inc., Tokyo, Japan). Fourier transform-infrared spectroscopy (FT-IR) was measured on a Spectrum 100 (PerkinElmer, Inc., Waltham, MA, USA). Raman Spectroscopy was recorded using an LabRAM HR800 (Horiba Ltd., Tokyo, Japan). The surface elemental information of catalysts was characterized by X-ray photoelectron spectroscopy (XPS) on an EscaLab 250xi instrument (Thermo Fisher Scientific Inc., Pittsburgh, PA, USA). The thermogravimetry (TG) was carried out on the TA SDT650/STA 449 F3 instrument. The vacancies of catalyst and reactive oxygen (O) species were measured by electron paramagnetic resonance (EPR) on a Bruker A300 (Bruker Corp). The zeta potential value was measured on Zetasizer Nano ZS90 (Malvern Instruments Ltd., Malvern, UK).
2.4. Removal of contaminants in different reaction processes
Degradation reactions were conducted under atmospheric pressure and room temperature. First, a 10 mg L−1 RhB solution was prepared using distilled water. Then, the catalyst together with PMS was immediately added into the solution to maintain the suitable concentration of each component. After addition, the removal reaction proceeded and, at intervals, samples collected, with methanol added to the samples to capture reactive oxygen species (ROS). The absorbance of RhB was measured by spectrophotometer and pH recorded using a pH meter before and after reaction. According to experimental requirements, the important reaction parameters and the related chemical reagents were timely adjusted under optimal conditions. The decomposition of PMS was quantified by the potassium iodide method.30 The concentration of Fe2+ was quantified using the 1,10-phenanthroline method, with Fe3+ first reduced by hydroxylamine hydrochloride and then total Fe2+ quantified by the above method.13–15
3. Results and discussion
3.1. Characterization of catalysts
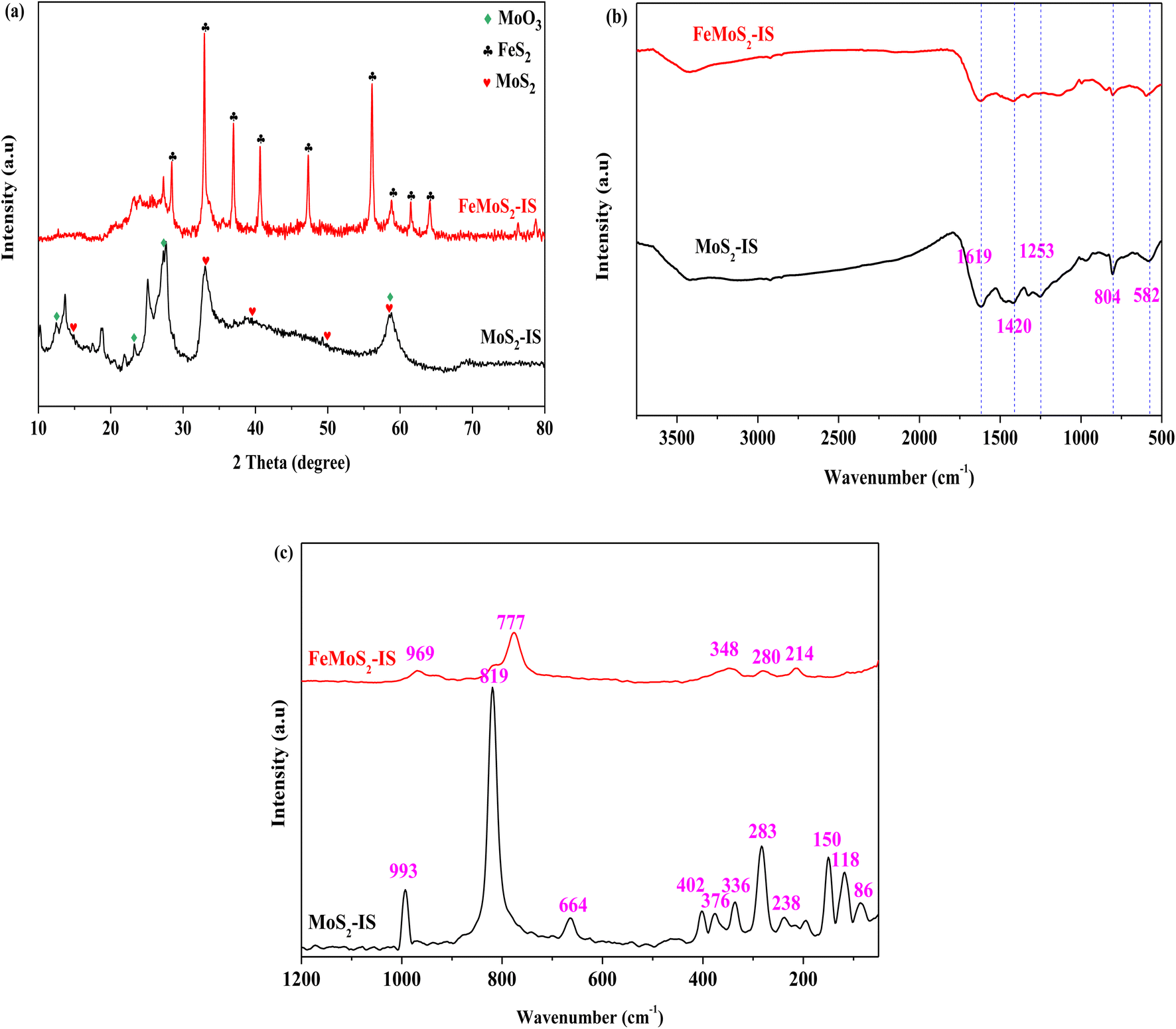
XRD patterns of MoS2-IS and FeMoS2-IS showed that, for MoS2-IS, diffraction characteristic peaks located at 2θ = 14.38°, 32.68°, 39.54°, 49.79° and 58.33°, which were attributed to the crystal phases of MoS2 (PDF-# 37-1492, Fig. 1a). This originated from the reaction between ammonium molybdate and thiourea during hydrothermal treatment.21 Some diffraction characteristic peaks located at 2θ = 12.76°, 23.33°, 27.33° and 58.80° were ascribed to the crystal phases of MoO3 (PDF-# 05-0508). The introduction of O was derived from the oxidation of MoS2 in high-temperature treatment. In addition, introducing O also occurred during hydrothermal treatment.31 These results were further confirmed from the Raman spectra (see Fig. 1c). In general, the widths of diffraction characteristic peaks were large in MoS2-IS, implying that its crystallization was not high, in other words, it is very difficult to identify the crystal phases of MoS2 and MoO3. It was agreement with the literature.20,21 After modification, the diffraction characteristic peaks of FeMoS2-IS were greatly altered relative to those of MoS2-IS, namely, some diffraction characteristic peaks disappeared and others newly appeared. On the one hand, this did not present diffraction characteristic peaks of MoS2. This was most probably due to it being either highly dispersed on the support or having low crystallization. Simultaneously, MoO3 intensity signals were decreased. On the other hand, some clear diffraction characteristic peaks were newly observed. For example, sharp diffraction peaks located at 2θ = 36.96°, 40.64°, 47.30°, 56.12°, 58.82°, 61.48° and 64.12° were observed, which were attributed to the crystal phase of FeS2 (PDF-# 42-1340).24 In other words, the main crystal phases of the catalyst were FeS2. As mentioned above, the crystal phases of FeS2 were little identified in FeMoS2. In fact, FeS2 has been employed to degrade organic pollutants in advanced oxidation processes,26,27,32,33 but it was not FeS2 as dominant phases in the various FeMoS2 catalysts for activating PMS.20–22 Consequently, FeS2 appearance was in favor of the activation of PMS in this reaction.
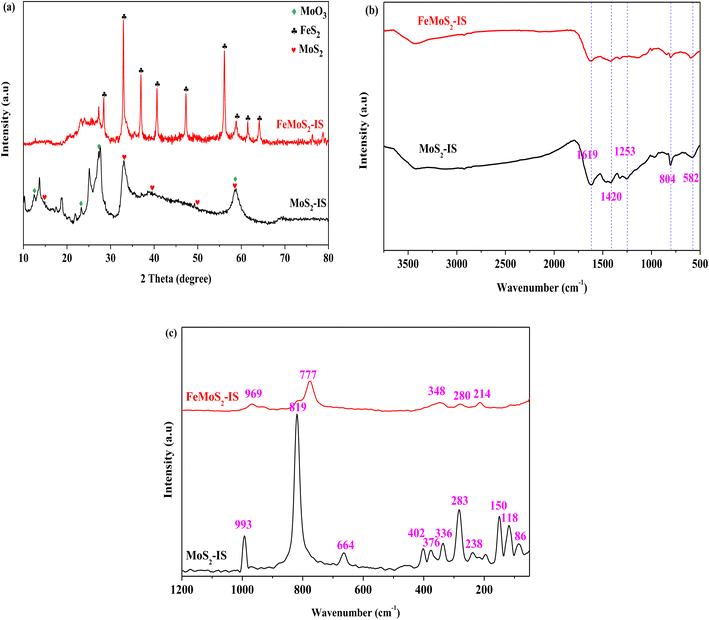 |
| | Fig. 1 (a) XRD patterns of MoS2-IS and FeMoS2-IS; (b) FT-IR spectra of MoS2-IS and FeMoS2-IS; (c) Raman spectra of MoS2-IS and FeMoS2-IS. | |
One wonders whether the synthesis and application of FeS2 occurs without the addition of Mo? The answer is No. As evidenced by XRD characterization, the typical diffraction characteristic peaks of samples was mainly attributed to the structure of FeS2 (Fig. S2a†). Therefore, the Mo existence was no a prerequisite for FeS2 generation but it mainly affected the catalytic performance (see below Fig. 3). In addition, other active sites might have been highly dispersed on the support, such that they were not detected. For instance, with MoS2-300 as an indirect evidence, the reaction between S of MoS2 and O in air occurred. Thus, S vacancies were formed due to the different atomic radius between S and O, as evidenced from EPR characterization (Fig. S2b†). The result indicated that the relative intensity of MoS2-300 was altered relative to those of MoS2, hinting that the S vacancies may generate in the FeMoS2.19
To further explore the differences, SEM was employed and MoS2-IS observed to exhibit a plate-like structure with the partially agglomerated appearance (Fig. S3a and b†). After modification, the morphology of FeMoS2-IS was greatly different and the catalyst shape became large aggregation particles. The pore structure of MoS2-IS and FeMoS2-IS showed that, for MoS2-IS, the SBET, VTotal, and DA of this catalyst were 12.69 m2 g−1, 0.0346 cm3 g−1, and 10.91 nm, respectively (Table 1). After modification to form FeMoS2-IS, the SBET was clearly reduced, which might be attributed to changed morphology, as evidenced from SEM results. In contrast, the VTotal and DA of FeMoS2-IS were increased, which was beneficial for enhancing mass transfer. Combined with the removal of RhB and SBET, the SBET was confirmed not to be an important factor for improving RhB removal.
Table 1 Pore structure of different catalysta
| Catalysts |
SBET (m2 g−1) |
VTotal (cm3 g−1) |
DA (nm) |
| SBET, VTotal and DA were denoted as specific surface area, total pore volume and mean pore sizes, respectively. |
| MoS2-IS |
12.69 |
0.0346 |
10.91 |
| FeMoS2-IS |
7.85 |
0.0375 |
19.12 |
The FT-IR and Raman spectra of MoS2-IS and FeMoS2-IS showed that for the MoS2-IS, five characteristic peaks appeared, located at 1619, 1420, 1253, 804, and 582 cm−1, in the region of 2000–500 cm−1, which belonged to the characteristic peaks of Mo–S (Fig. 1b and c).34,35 After treatment, similar characteristic peaks appeared at the above positions, implying that Mo–S structure in the catalyst remained unchanged. Nevertheless, it was difficult to identify structural differences between them. Therefore, they were characterized by Raman analysis. MoS2-IS exhibited some characteristic peaks located at 445–448, 402, and 376 cm−1, which were attributed to the structure of 2H-MoS2.35 In addition, some characteristic peaks located at 336, 283, and 150 cm−1 were observed, belonging to the structure of 1T-MoS2.35 The results demonstrated the co-existence of 2H–1T MoS2 structure in MoS2-IS. The characteristic peaks located at 993, 819, and 664 cm−1 were observed, which were ascribed to the structure of MoO3.35 Thus, MoS2-IS was concluded to be composed of MoS2 and MoO3. After modification, the above characteristic peaks basically disappeared and some new characteristic peaks located at 969, 777, 348, 280, and 214 cm−1 appeared. This suggested that a new structure in FeMoS2-IS was formed, which was different from MoS2 and MoO3.
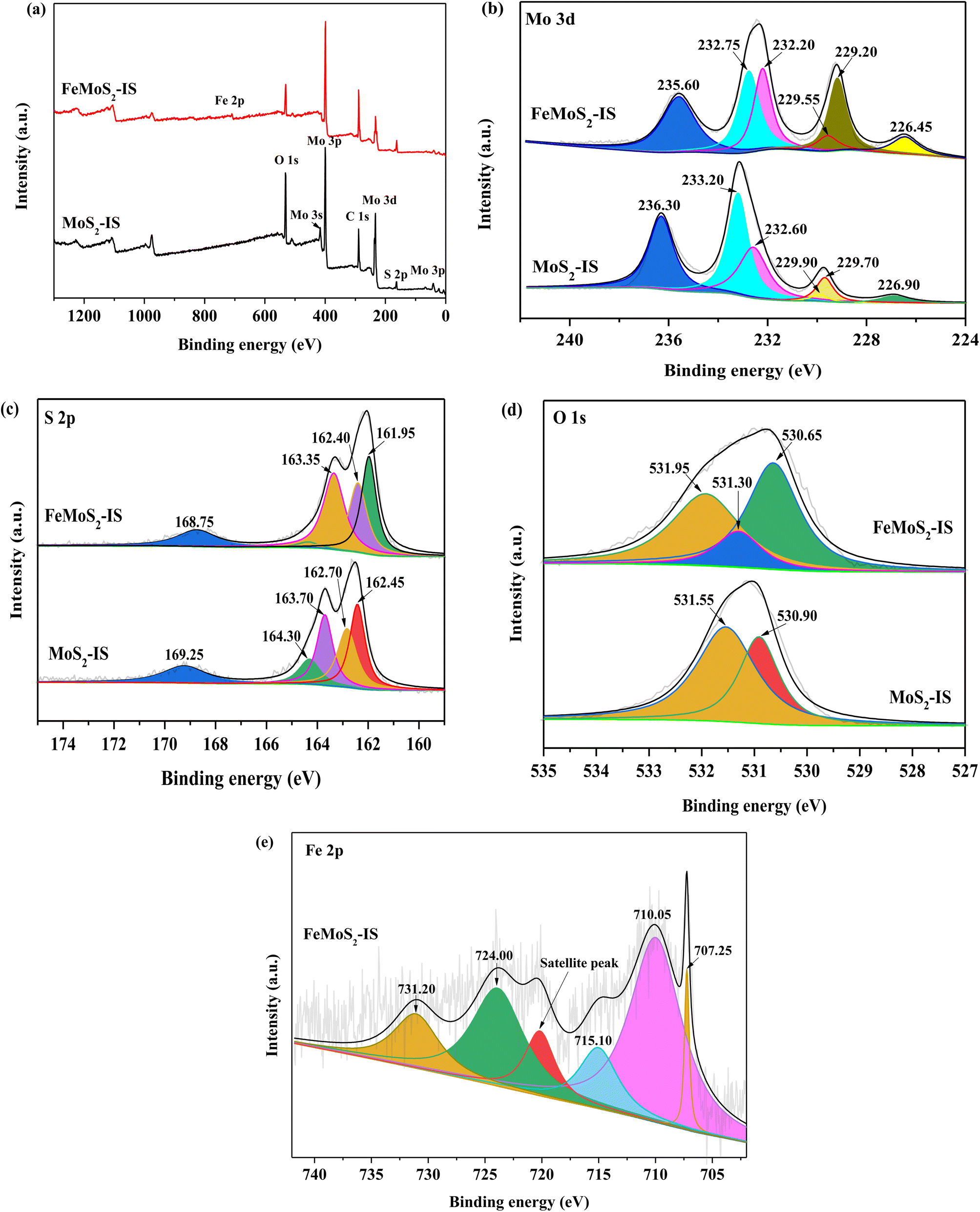
The surface elemental information of MoS2-IS and FeMoS2-IS was confirmed by XPS survey spectra, with the synthesized MoS2-IS and FeMoS2-IS primarily composed of Mo, S, and O (Fig. 2a). In addition, Fe also appeared in FeMoS2-IS. In terms of Mo-3d of MoS2-IS, one weak binding energy peak was located at 226.90 eV, which was attributed to S atoms at the external side of Mo–S (Fig. 2b).34–36 There were also two binding energy (BE) peaks located at 229.75 and 233.05 eV, which respectively corresponded to characteristic peaks of Mo 3d5/2 and Mo 3d3/2, which indicated that Mo4+ was present in MoS2-IS.36 The characteristic BE peak located at 229.75 eV was subdivided into two characteristic peaks located at 229.90 and 229.69 eV, corresponding to the structure of 2H-MoS2 3d5/2 and 1T-MoS2 3d5/2.35 Similarly, the characteristic BE peak located at 232.40 eV was subdivided into two characteristic peaks with the BEs at 233.20 and 232.57 eV, corresponding to the structure of 2H–MoS2 3d3/2 and 1T-MoS2 3d3/2,35 respectively. The above results showed that a mixed structure of 2H- and 1T-MoS2 existed in MoS2-IS. In addition, one clear characteristic BE peak located at 236.30 eV corresponded to a characteristic peak of Mo-based oxides.34–36
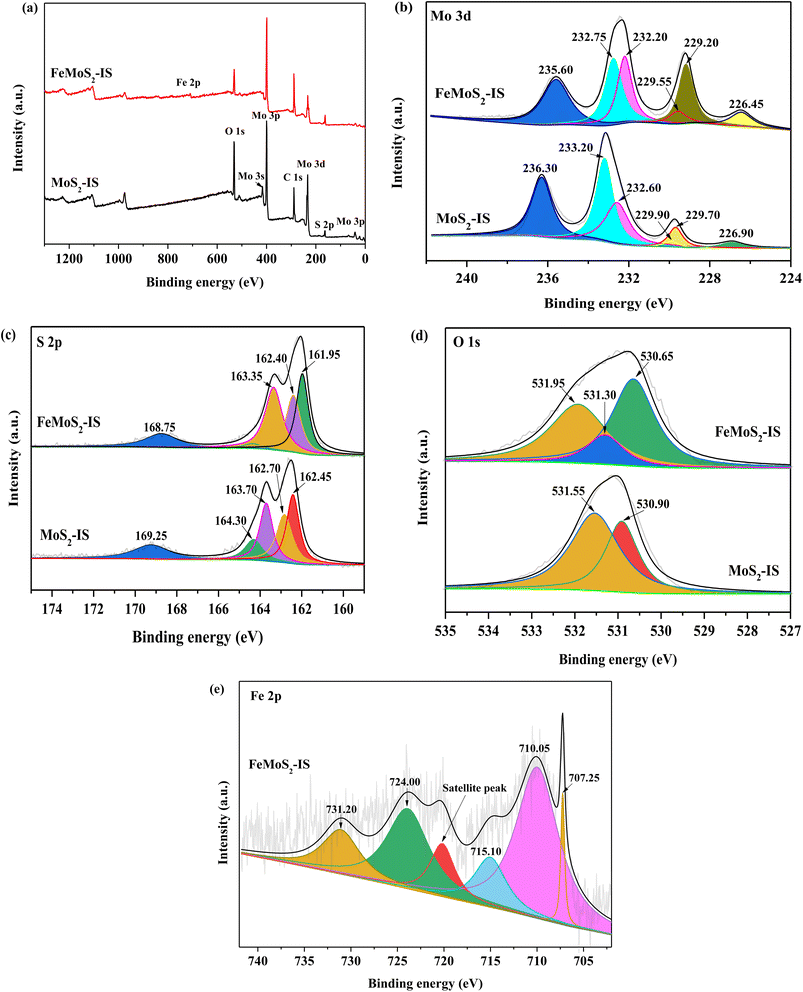 |
| | Fig. 2 XPS spectra of (a) survey spectra, (b) Mo 3d, (c) S 2p and (d) O 1s in the MoS2-IS and FeMoS2-IS, and (e) Fe 2p in the FeMoS2-IS. | |
After treatment, the characteristic peak of Mo 3d was altered. The ratio of Mo4+ to Mo6+ and S atoms from the external of Mo–S were both increased, but the ratio of 2H- and 1T-MoS2 and concentration of Mo6+ were both reduced in FeMoS2-IS, compared to MoS2-IS (Table S1†). This meant that this modification contributed more Mo4+ and external S in Mo–S and/or FeS2. As mentioned above, the decomposition of PMS could be accelerated by enhancing the Fe2+ and Fe3+ cycle in the presence of Mo4+.13–18 Simultaneously, more external S atoms in the catalyst were in better contact H+ in solution such that more active sites, such as Fe2+ and Mo4+, were exposed. Some authors have pointed out that Mo4+, not Mo6+, species were important active sites for decomposing PMS.34–36 Accordingly, more Mo4+ and less Mo6+ species were in favor of strengthening PMS activation. For S 2p in MoS2-IS, there were two distinct characteristic BE peaks located at 163.70 and 162.65 eV,19,35 which was respectively ascribed to the S 2p1/2 and S 2p3/2 (Fig. 2c). In addition, one characteristic peak located at 169.24 eV was attributed to –SOn– due to oxidation during catalyst synthesis.35 After modification, the characteristic peak of S 2p was little changed. Interestingly, the O 1s spectrum could be deconvoluted into two components located at 531.54 and 530.91 eV, corresponding to hydroxyl groups in adsorbed water and O defects in the MoS2-IS (Fig. 2d). After modification, besides the above peaks, there appeared a new peak located at 531.30 eV, which was ascribed to surface adsorbed O species.35 In the case of Fe 2p in FeMoS2-IS (Fig. 2e), a peak located at 707.25 eV was attributed to the existence of FeS2.32 Also, the extra three characteristic peaks, located at 709.99 and 731.07 eV, corresponded to Fe2+.37 Besides, the peak located at 720.18 eV was attributed to the satellite peak. This indicated that Fe2+ was generated in the FeMoS2-IS due to the reduction of Fe3+ by thiourea. There were two characteristic peaks located at 715.02 and 723.90 eV, which corresponded to Fe3+. According to the peak areas of Fe2+ and Fe3+, the Fe2+ concentration and Fe2+/Fe3+ ratio were 0.67 and 1.99, respectively, suggesting that most Fe3+ in ferric nitrate was reduced to Fe2+ during synthesis. The higher Mo4+/Mo6+ and a large number of Fe2+ species were concluded to have been generated after Fe modification.
3.2. Removal of organic pollutants in different catalytic reaction processes
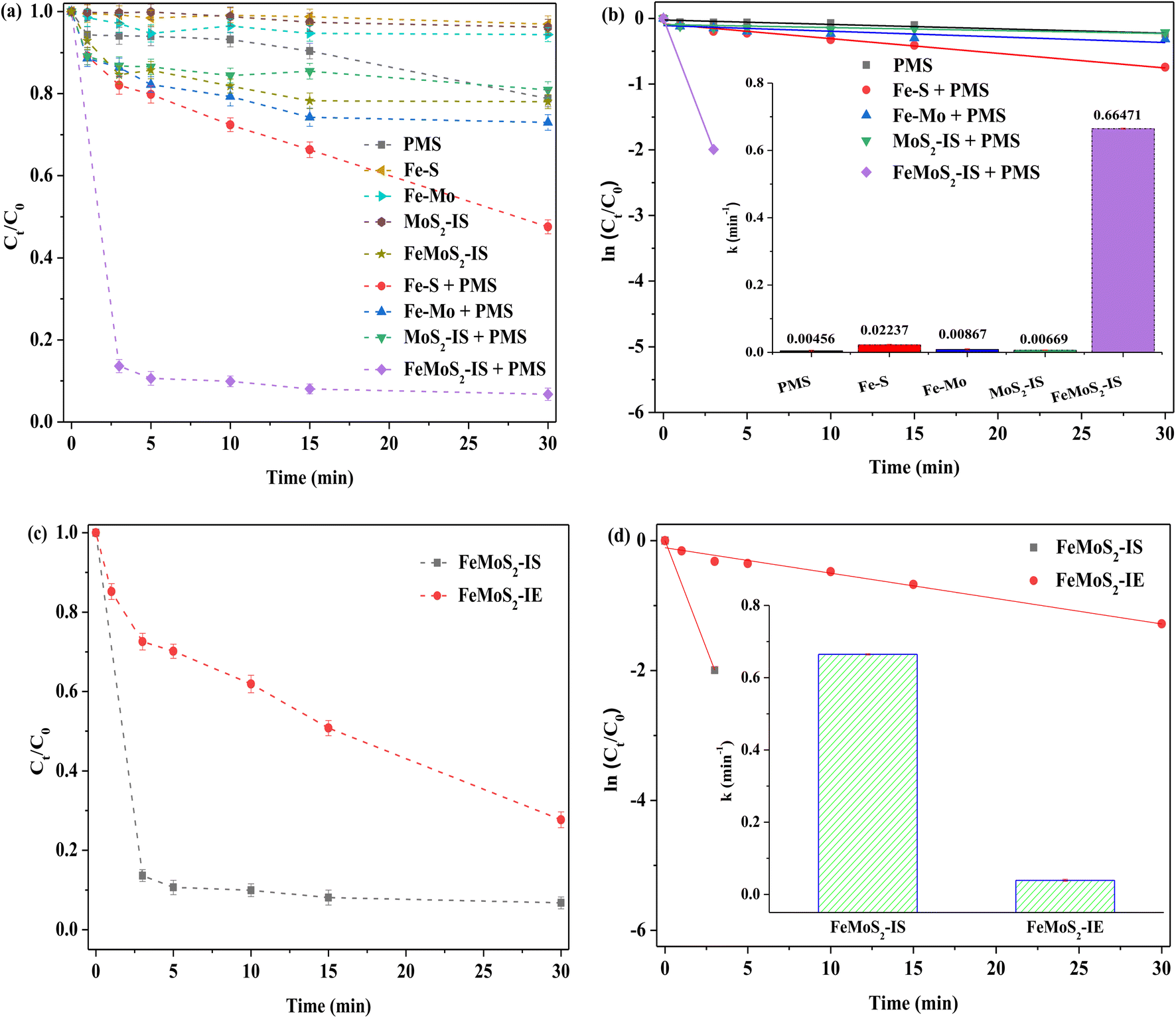
Different catalysts for activating PMS in RhB removal in the darkness were compared. The RhB degradation efficiency was only 21% when PMS was used alone (Fig. 3). Although MoS2-IS was added into the above system, RhB removal hardly increased. According to the pseudo-first order reaction equation, the reaction rate constant (denoted as k) was calculated.20 These values were 0.00456 and 0.00669 min−1 for PMS and PMS + MoS2-IS, meaning that their reaction rates were extremely slow. This was because the MoS2-IS contained a lot of Mo6+ species, such as MoO3, as evidenced from XPS characterization, but it did not act as active sites.19 Also, its low SBET restricted increased catalyst activity and, accordingly, RhB removal was poor. The degradation efficiency of RhB was ∼27% at 0.00867 min−1 for its k using Fe–Mo as a heterogeneous catalyst. This indicated that the bimetallic Fe–Mo catalyst containing no S could activate PMS to remove RhB, which was in agreement with the literature.38 However, this promotion was limited and also illustrated that the S was very important factor for improving RhB removal. If a catalyst without Mo was employed, a positive effect should be achieved. Surprisingly, the results showed that the RhB degradation efficiency over Fe–S was only ∼52%, with k at 0.02237 min−1. This was only due to the generation of FeS2. Clearly, the role of Mo was important in the removal of RhB. In contrast, when FeMoS2-IS was used as a heterogeneous catalyst under identical reaction conditions, ∼86% of RhB was removed, with k at 0.66471 min−1, obtained only in 3.0 min in the presence of PMS. From the above results, FeMoS2-IS possessed many active sites such as FeS2 and S vacancies and their synergism activated PMS to form ROS. Zhou et al.26 have directly used pyrite for activating PMS for the effective degradation and mineralization of diethyl phthalate. Huang et al.39 have considered that S vacancies accelerate electron transfer and reduce Mo6+ to Mo4+ in natural molybdenite/PMS systems. As a result, RhB removal was significantly increased relative to the control experiment.
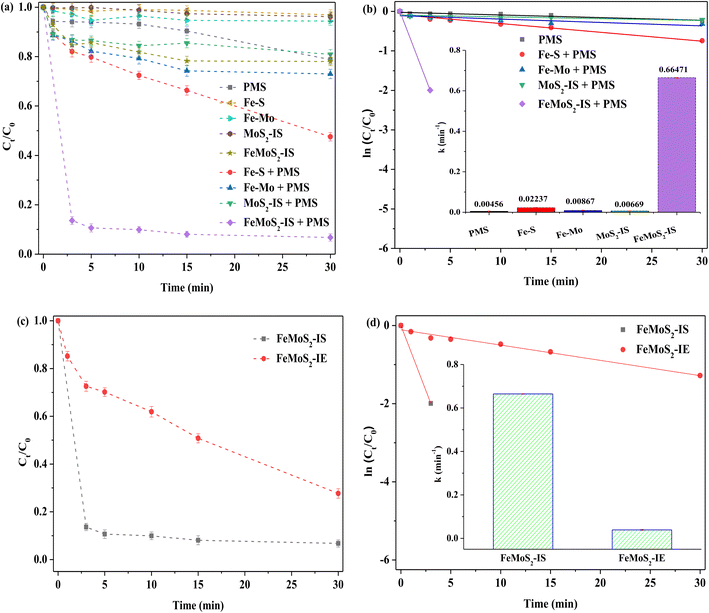 |
| | Fig. 3 (a) RhB degradation of adsorption and different PMS system; (b) pseudo first order kinetics of different catalytic system and reaction rate constant (illustration); (c) removal of RhB activating PMS by FeMoS2 with different methods; (d) pseudo first order kinetics of different FeMoS2 activating PMS system and reaction rate constant (illustration). Reaction conditions: [RhB] = 10 mg L−1, initial pH = 3.0, [Catal. ] = 1.0 g L−1, [PMS] = 1.0 mM and in the darkness. | |
As mentioned earlier, Fe-doped MoS2 was mainly synthesized via a post treatment.20–22 Herein, FeMoS2-IE was obtained by impregnation and high-temperature successive treatments and then it was applied for activating PMS. The results indicated that the RhB degradation efficiency, with k at 0.03914 min−1, reached ∼72% at 30 min. The diffraction characteristic peaks of FeMoS2-IE were mainly ascribed to the mixed structure of MoS2 and MoO3 (Fig. S4†). Meanwhile, Fe species were not detected because they were either highly dispersed on the support or in low crystallinity. On the basis of FeMoS2-IS and FeMoS2-IE, a great difference was found on RhB removal, which was due to different structures between them. As mentioned above, the main phases in the former were MoS2 and MoO3 whereas the dominant phases in the latter were FeS2. As shown in eqn (2)–(5),32,33 the FeS2 to generate dissolved as well as surface Fe2+ was an important reason for promoting RhB degradation. In a few words, this provided a new method for FeS2 synthesis. In addition, this had many merits, e.g., simple operation, low cost, and green. Accordingly, it possessed great potential for industrial application.
| | |
FeS2 + H+ → Fe2+ + H2S
| (2) |
| | |
FeS2 + 3.5O2 + 4H2O → Fe2+ + 2SO42− + 2H+
| (3) |
| | |
2FeS2 + 7O2 + 2H2O → 2Fe2+ + 4SO42− + 4H+
| (4) |
| | |
FeS2 + 14Fe3+ + 8H2O → 15Fe2+ + 2SO42− + 16H+
| (5) |
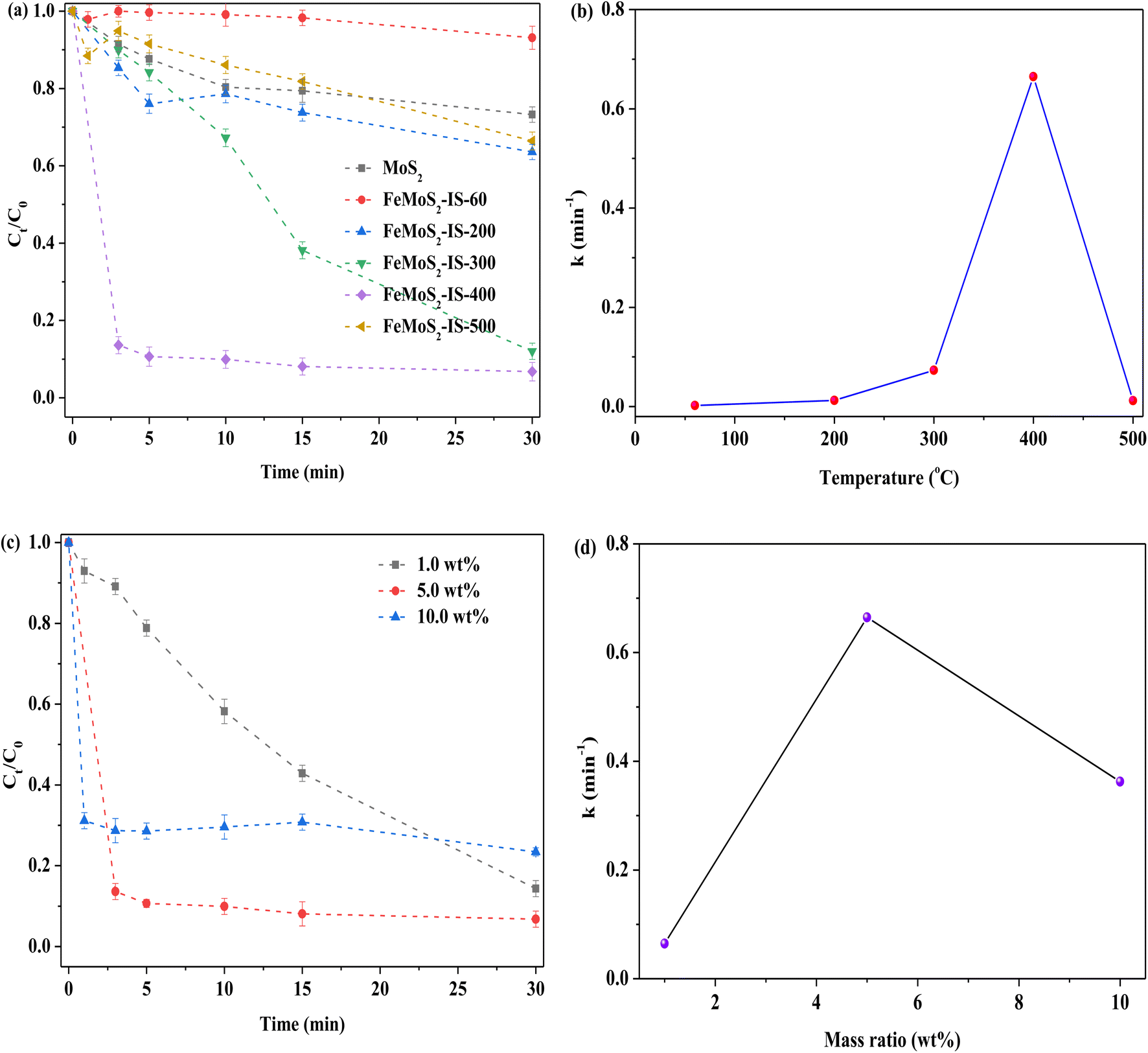
To further explore the roles of FeS2 and S vacancies, the treatment temperature and Fe doping were investigated, particularly how temperature affected the formation of FeS2 and S vacancies (Fig. 4a). When the treatment temperature was 400 °C, RhB removal was the highest, while with further elevation of temperature, RhB removal was clearly reduced. When the catalyst was not heat treatment, besides there being no FeS2 and Mo–S, there was lower Mo4+/Mo6+ and Fe2+/Fe3+ ratios, and higher concentrations of Mo6+ and 2H-/1T-MoS2 obtained in FeMoS2-IS-60 relative to FeMoS2-IS-400 (or FeMoS2-IS), as evidenced by XRD and XPS results (Fig. S5 and S6a–S6e and Table S1†). Accordingly, RhB removal was low. As the temperature was increased, S in the catalyst reacted with atmospheric O to form gaseous SO2 and thus S vacancies generated and more Fe2+ and Mo4+ species exposed. In addition to the activation of PMS by Fe2+ and Mo4+, S vacancies were able to activate PMS to form ROS.19,29 This was due to the ability of defective sites to reduce the adsorption energy of PMS and prolong the bond length of peroxides (–O–O–) in PMS. As the indirect evidence, the comparison of MoS2 and MoS2-300 for activating PMS on RhB removal was conducted. RhB removal over MoS2-300 was remarkably higher than that of MoS2 (Fig. S7†). This result suggested that S vacancies could indeed facilitate RhB removal. Also, more FeS2 was generated with increased calcination temperature and, as a result, RhB removal increased. When the temperature was too high, FeMoS2-IS structure was seriously destroyed, as evidenced from TG-DSC characterization (Fig. S8†). As a consequence, RhB degradation was remarkably reduced.
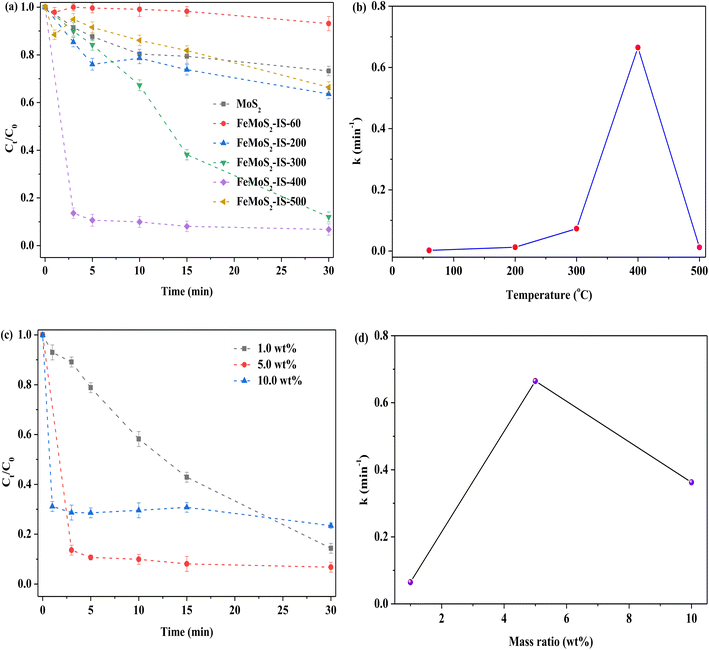 |
| | Fig. 4 (a) Effect of the calcined temperatures of catalyst on the removal of RhB; (b) relationship between calcined temperatures and reaction rate constant; (c) effect of the Fe concentration on the removal of RhB; (d) relationship between Fe concentration and reaction rate constant. Reaction conditions: [RhB] = 10 mg L−1, initial pH = 3.0, [Catal. ] = 1.0 g L−1, [PMS] = 1.0 mM and in the darkness. | |
As mentioned above, Fe modification was in favor of strengthening RhB removal. On the basis of this reason, the influence of Fe concentration was examined in FeMoS2-IS. When the catalyst Fe concentration was increased from 1.0 to 5.0 wt%, RhB degradation efficiency became rapid, at ∼86% in 3.0 min, as compared to that of the former at 30 min (Fig. 4c and d). Further increasing Fe concentration, RhB removal was remarkably reduced. XRD results showed that the main phase was not FeS2 in the 1.0 and 10.0 wt% Fe-modified catalysts (Fig. S9†). Therefore, RhB removal was decreased. In brief, the above results well illustrated that FeS2 and S vacancies were two essential factors for enhancing RhB degradation.
3.3. Effects of operation parameters on RhB removal
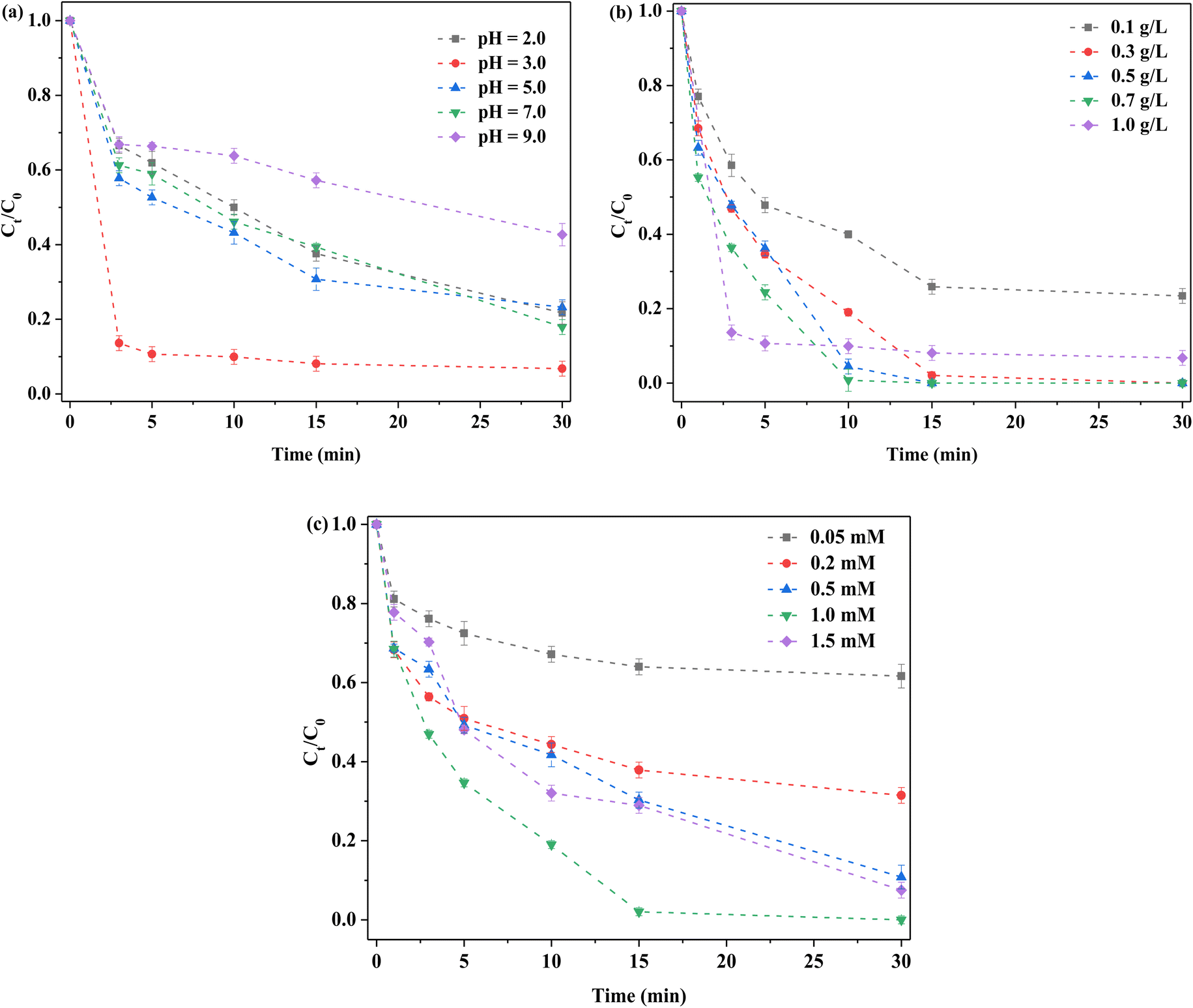
Examination of the effects of initial pH on RhB removal revealed that removal was more favorable under acidic conditions, compared to neutral and alkaline conditions (Fig. 5a). When the initial pH was 3.0, RhB removal was the highest, while at pH 7.0, the obtained result was still satisfactory. Removal was decreased at pH 9.0, at ∼50%. Under an acidic environment, S atoms on the surface of FeMoS2-IS were trapped by H+ in solution to generate H2S,22,23 in which case, more active sites, such as Fe2+ and Mo4+, were exposed and thus more ROS generated, such that the above co-actions promoted RhB removal. In an alkaline environment, this effect was suppressed due to inhibition of S atom capture and, accordingly, RhB degradation decreased. In addition, after PMS was added into the reaction system, the solution became acidity. Except for an initial pH of 2.0, all solutions retained about pH 3.0 after reaction (Fig. S10†). In this case, the above-mentioned effect was to a lesser extent inhibited. The zeta potential value of FeMoS2-IS was −1.66 mV at ∼3.0 pH while the RhB was a cationic dye. Thus, this was in favor of adsorbing RhB. Generally, RhB removal was high over a wide pH range.
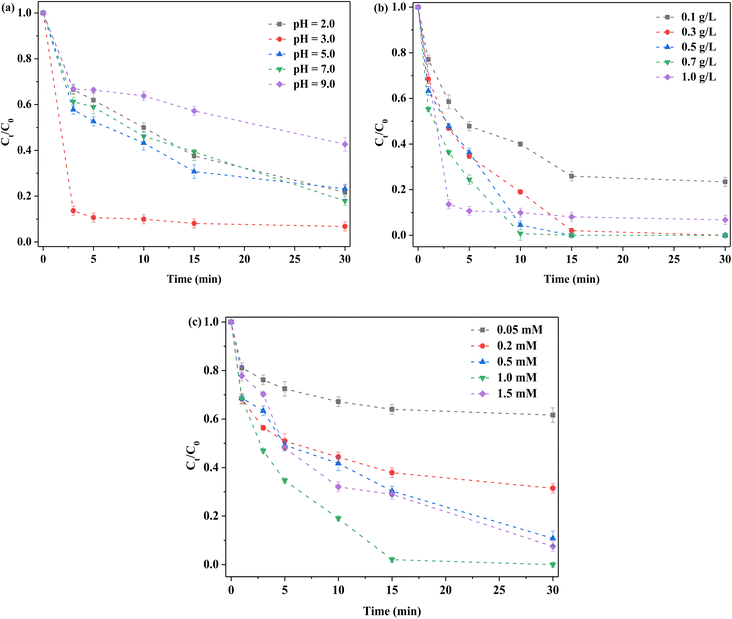 |
| | Fig. 5 Optimized reaction conditions on the removal of 10 mg L−1 RhB in the FeMoS2-IS catalyzed PMS system in the darkness. (a) Initial pH. Reaction conditions: [FeMoS2-IS] = 1.0 g L−1 and [PMS] = 1.0 mM; (b) catalyst dosage. Reaction conditions: initial pH = 3.0 and [PMS] = 1.0 mM; (c) PMS concentration. Reaction conditions: initial pH = 3.0 and [FeMoS2-IS] = 0.3 g L−1. | |
Based on the above results, the effects of FeMoS2-IS dosages were examined (Fig. 5b). When 0.1 g L−1 of catalyst was applied, RhB degradation efficiency was ∼77% in 30 min. With increased dosage, the degradation rate was accelerated. When the catalyst dosage was within 0.3–0.7 g L−1, all degradation efficiency was ∼100% in 15 min. However, when the dosage exceeded 0.7 g L−1, the removal started to decrease. The dosage of catalyst was insufficient, as there was not enough active sites to activate PMS. Therefore, the removal of RhB was low. With increasing catalyst dosage, more ROS were produced and, hence, the removal increased. When the catalyst dosage was too high, agglomeration occurred between catalyst particles, thereby affecting PMS activation. Meanwhile, the repulsion force between catalyst and PMS increased due to their similar charges.40 Similarly, the removal was decreased.
Here, the PMS concentration was optimized. The degradation efficiency of RhB was ∼38% at 30 min when 0.05 mM PMS was employed (Fig. 5c). With increased PMS concentration, RhB removal increased. When the PMS concentration was 1.0 mM, the removal reached the highest and began to decrease with further increased PMS. Increased PMS concentration generated more ROS and thus produced higher RhB removal. When PMS was too high, the removal decreased, which was mainly due to the reaction between partially activated PMS without organic pollutants. In addition, there was newly formed ROS, such as HO2˙−.41 The activity of these species was less than that of the original system. Accordingly, RhB degradation was reduced. In short, optimal reaction conditions were established and therefore the best degradation obtained.
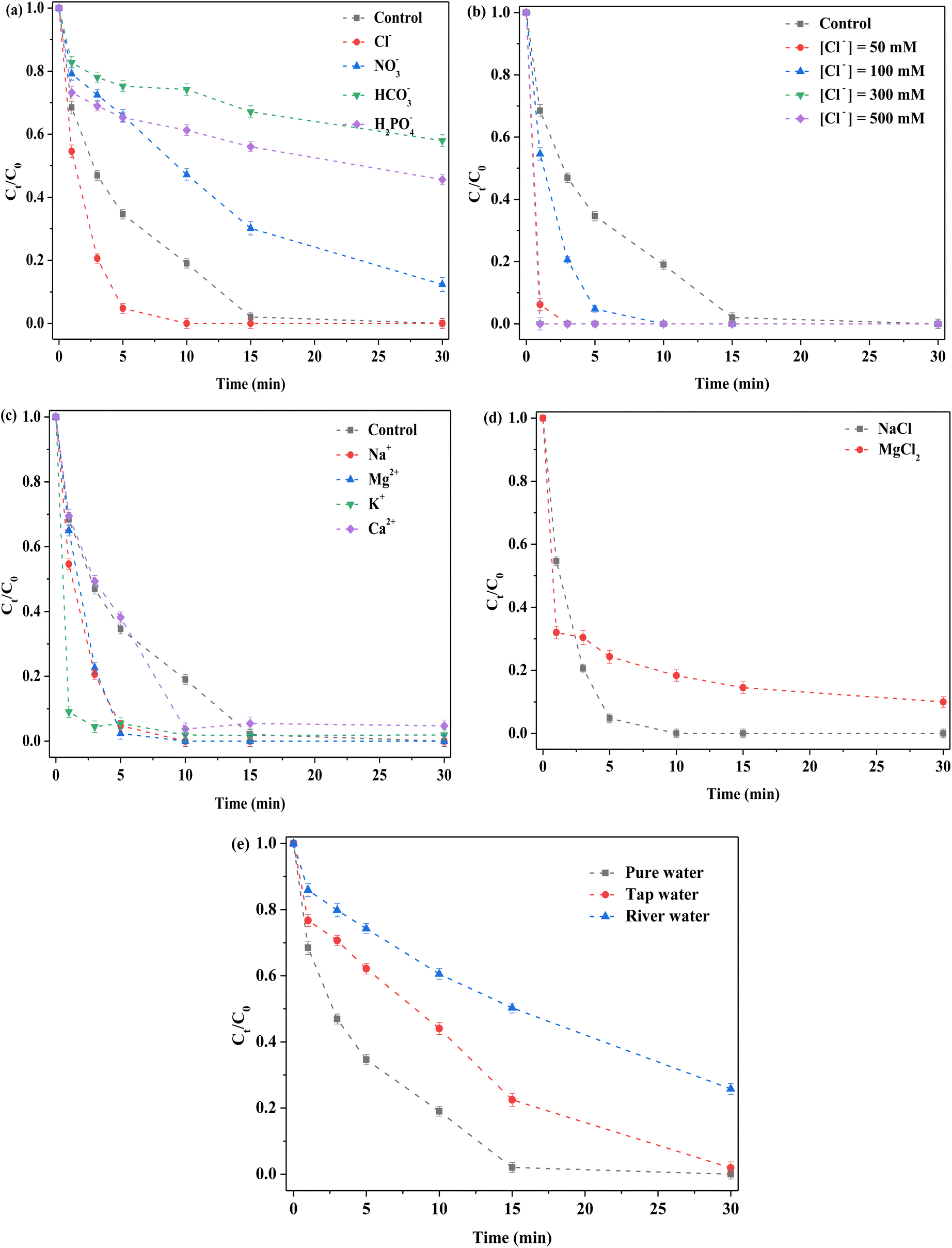
Common anions, including chloride, nitrate and bicarbonate ions (Cl−, NO3−, and HCO3−, respectively) were captured by the highly oxidizing free radical species and the corresponding reaction rate constants considerably varied.22,23,42–44 In this reaction system, different cations and anions were respectively added. RhB degradation efficiency was observed to be close to 100% after 15 min of reaction without any extra anions (Fig. 6a). When different anions (Cl−, NO3−, HCO3−, and H2PO4−) were employed, great changes appeared in RhB degradation. Cl− promoted removal based on the original reaction system, whereas other anions (including NO3−, HCO3−, and H2PO4−) suppressed degradation. After adding Cl−, the degradation efficiency reached 100% in 10 min, which indicated that Cl− played a positive role in RhB removal. To further explore this effect, different concentrations (50, 100, 300 and 500 mM) of NaCl were investigated (Fig. 6b). RhB removal ability was observed to be clearly strengthened by introducing low or high NaCl concentrations, which further confirmed that Cl− was indeed a beneficial additive. This was because Cl− could react with strong free radical species to form other free radical species.41 Accordingly, the promotion might have been attributable to newly formed active species such as Cl˙ and Cl2˙. In addition, other active chlorine species were formed.42,43 For NO3−, RhB removal decreased, which might have been attributable to the competition for reactants. For the HCO3−, the degradation was severely inhibited after HCO3− addition, because solution pH was altered and sharply increased toward 8–9. Under alkaline condition, the binding of H+ with S atoms on the catalyst surface did not occur,22,23 thereby reducing the exposure of active sites, such as Mo4+ and Fe2+ species. In addition, HCO3− reacted with PMS or its related ROS.20 Thus, removal was clearly decreased after HCO3− was added. Similar to HCO3−, the solution pH was remarkably increased after H2PO4− was added to the reaction system, such that it was clear that an inhibition effect occurred due to alkaline condition. In addition, interactions between H2PO4− and Mo on the catalyst could generate a heteropoly acid salt.45 Hence, H2PO4− had a clear negative influence on RhB removal.
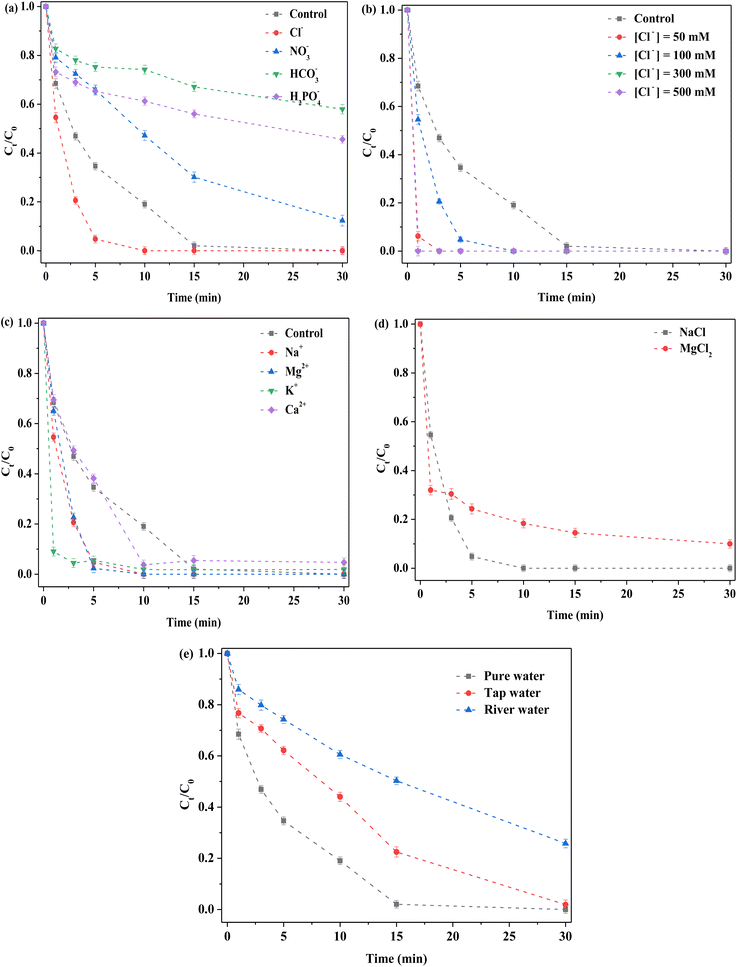 |
| | Fig. 6 Effect of various factors on the removal of RhB. (a) Anions; (b) Cl− concentration; (c) cations; (d) different cations with the same concentration of Cl−; (e) types of water. Reaction conditions: [RhB] = 10 mg L−1, initial pH = 3.0, [FeMoS2-IS] = 0.3 g L−1, [PMS] = 1.0 mM, [anions or cations] = 100 mM and in the darkness. | |
To explore the influence of cations on RhB removal, various cations, including Na+, K+, Mg2+, and Ca2+ with Cl− as the anion were examined in detail. The order of the activity in terms of RhB removal was Na+ > Mg2+ > K+ > Ca2+ > control (Fig. 6c). The results indicated that these cations had no inhibitory effect on RhB degradation. Promotion might have been due to the presence of Cl−. To explore the contribution of Na+ and Cl−, Na+ was replaced by Mg2+ with the same concentration of Cl− (Fig. 6d). When MgCl2 was used as the precursor, RhB removal was significantly lower than that of NaCl as precursor. Thus, the above- mentioned promotion was attributed to the coaction of cations and anions, with the contribution of Cl− greater than that of Na+. To explore the influence of water types, tap water and river water (Xiang-Jiang River, Hunan, China) as solvents were selected. The RhB degradation efficiencies were 100 and 70% in tap and river water at 30 min, respectively (Fig. 6e). In general, the catalyst exhibited satisfactory degradation in different water environments. Using river water as solvent, the low degradation might have been due to the presence of natural organic humic acids and various inorganic salts. In short, the FeMoS2-IS/PMS system had a strong ability to resist external interference. In addition, different organic pollutants were investigated in this system. Most of organic pollutants (RhB, MB, MO, and AOII) were rapidly removed in 30 min, especially AOII (Fig. S11†). These results indicated that the catalytic system possessed excellent ability to remove organic pollutants in wastewater.
The stability of FeMoS2-IS was examined and it was observed that the degradation efficiency of RhB reached 100% with its 1st cycle of reaction (Fig. S12†). After reaction, the catalyst was regenerated by filtering the reaction solution, washing the solid with distilled water and ethanol to separate liquid and solid, and dried. Finally, the dried sample was calcined at 400 °C for 1.5 h. According to the same reaction conditions as the 1st reaction, the regenerated catalyst was added into a 2nd reaction solution. The results indicated that the degradation efficiency of RhB arrived at 100% after 30 min of reaction. Repeating the above same steps, several reactions and regeneration were conducted. After the 5th reaction, the degradation efficiency of RhB was slightly decreased, implying that the catalyst possessed basically retained original activity. Notably, the used catalyst had to be treated at high temperature to recover the initial catalytic activity because it was covered and/or blocked the active sites of catalyst by the intermediates and/or products, which was the main reason for remarkably decreased RhB removal (Fig. S13†). By a simple calcination, the used catalyst was well regenerated.
The structure of fresh and used FeMoS2-IS was characterized by XRD, which showed that used catalyst was greatly changed after the several reaction cycles, compared to that of fresh catalyst, with the diffraction characteristic peaks of FeS2 no longer apparent, which was mainly attributed to the structure of MoO3 (Fig. S14†). This situation mainly originated from the leaching of Fe and oxidation of MoS2 during reaction and regeneration, which were the main reasons for decreasing catalytic activity after the reaction and regeneration. To further confirm this difference, the changes in surface elements on the catalyst after reaction were characterized by XPS. One characteristic peak located at ∼707.25 eV did not appear in Fe 2p, implying that Fe–S had disappeared and, meanwhile, the ratio of Fe2+ and Fe3+ was remarkably reduced, meaning that a lot of Fe2+ were oxidized to Fe3+ (Fig. S15 and Table S1†). This change was further confirmed in S 2p, where the S2− was completely eliminated and the peak was attributed to one characteristic of SOn−. This indicated that Mo–S and Fe–S on the catalyst surface did not indeed exist. For Mo 3d, the ratio of Mo4+ and Mo6+ was reduced whereas the concentration of Mo6+ increased, hinting that great oxidation was occurred. This was confirmed by O 1s XPS characterization; that is, the concentration of lattice O in the catalyst had increased, which was further supported by the fact that the color of the catalyst had turned from black to white. The above results illustrated that the heterogeneous reaction was happened on the surface of catalyst.
3.4. Leaching of various ions during reaction and their effects
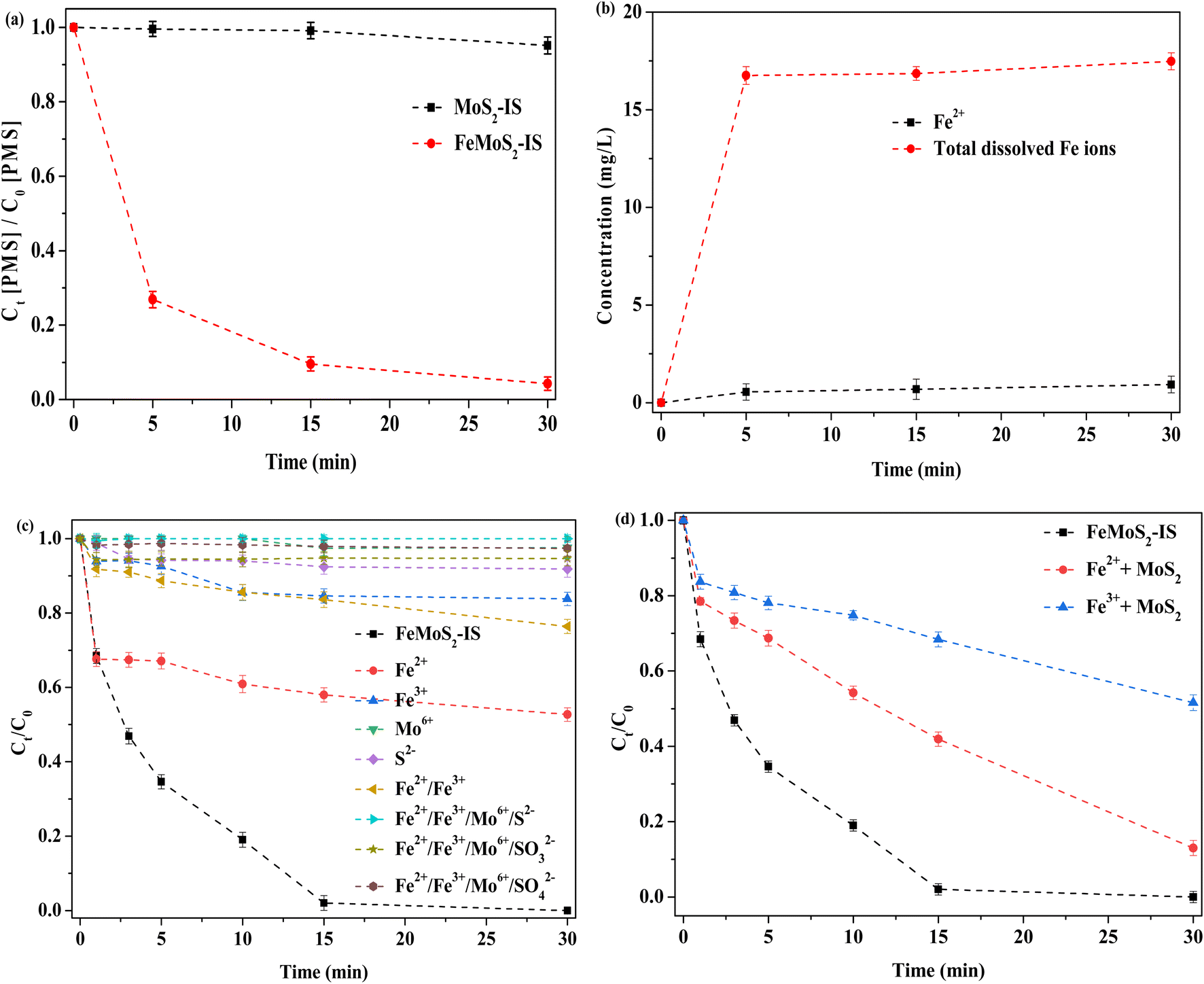
Decomposition of PMS had an important influence on ROS formation and the test was conducted.30 When MoS2-IS acted as catalyst, the decomposition efficiency of PMS was no more than 5% at 30 min (Fig. 7a). This was main reason for the low RhB degradation. In contrast, when FeMoS2-IS was the catalyst, the decomposition efficiency of PMS reached as high as ∼95% in the same time. Thus, FeMoS2-IS had a higher ability to activate PMS and thus removed more RhB in solution. Also, the source of ROS was mainly from PMS decomposition.
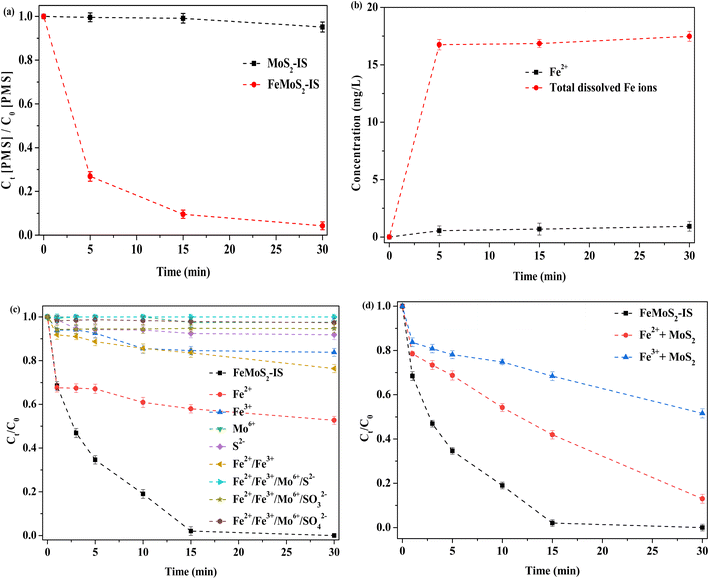 |
| | Fig. 7 (a) The decomposition of PMS; (b) the leaching of Fe2+ and total dissolved Fe ions; (c) removal of RhB in the PMS-based homogeneous reaction system; (d) removal of RhB in the PMS-based heterogeneous reaction system. Reaction conditions: [RhB] = 10 mg L−1, initial pH = 3.0, [FeMoS2-IS and MoS2] = 0.3 g L−1, [PMS] = 1.0 mM, [Fe2+] = 0.55 mg L−1, [Fe3+] = 15.46 mg L−1, [Mo6+] = 1.11 mM, [S2−] = [SO32−] = [SO42−] = 0.14 mM and in the darkness. | |
During reaction, some ions were leached. To examine this, Fe2+ and total Fe ions were evaluated (Fig. 7b). After reaction to 30 min, the concentrations of Fe2+ and total Fe ions were 0.55 and 16.01 mg L−1, respectively. Based on the above eqn (2)–(5), soluble Fe ions were generated. According to the calculation, the loss efficiency of total Fe ions was ∼5.0 wt%, implying that the catalyst had a good stability. Similarly, Mo and S ions moved from the catalyst and entered the reaction solution. Thus, the effect of their activation on PMS in the homogeneous system was investigated (Fig. 7c). When Fe2+ alone was used as homogeneous catalyst, the degradation efficiency of RhB was 47% after 30 min of reaction, which suggested that dissolved Fe2+ made some contributions on RhB removal because Fe2+ itself could directly activate PMS.14–16 The degradation efficiency of RhB was both below 25% when Fe3+ or Fe2+/Fe3+ were employed, which indicated that these Fe ions' ability to activate PMS was poor. When soluble Mo ion ((NH4)6Mo7O24·H2O as precursor) was used, the degradation efficiency of RhB was only ∼2% at 30 min, hinting that dissolved Mo ion had little activation effect on PMS. Similarly, S2− (Na2S as a precursor) alone could not accelerate the removal rate. Surprisingly, the removal did hardly occur when all dissolved ions (Na2SO3 and Na2SO4 as SO32− and SO42− precursors, respectively) were simultaneously added into the reaction system. This result indicated that it contributed the extremely low degradation for RhB even when various ions from the catalyst leached during reaction.
It has been reported that the homogeneous Fe2+ or Fe3+ ions can remarkably enhance RhB removal in the MoS2/PMS system.14–18 To verify this deduction, a series of experiments were designed. The degradation efficiency of RhB was respectively 87 and 48% in Fe2+/MoS2/PMS and Fe3+/MoS2/PMS systems, which meant that Fe ions dissolved by interactions with MoS2 in the FeMoS2-IS could accelerate RhB removal (Fig. 7d). In other words, the heterogeneous catalytic reaction played a vital role on RhB removal. To date, there has been a debate regarding the main active sites in the catalyst FeS2 activating persulfate. Some researchers46,47 have suggested that Fe2+ played a major role. Others48 have pointed out that S species were involved in this reaction in addition to Fe2+. Exploring whether Fe2+ was on the catalyst surface or in solution, different researchers have come to different conclusions: (i) dissolved Fe2+;49 (ii) Fe2+ on the surface of heterogeneous catalyst;50 and (iii) Fe2+ in the both solution and heterogeneous catalysts.51 Clearly, this reaction behavior was different from that of FeS2 catalytic PMS systems in previous studies.
3.5. Identifying ROS in RhB removal
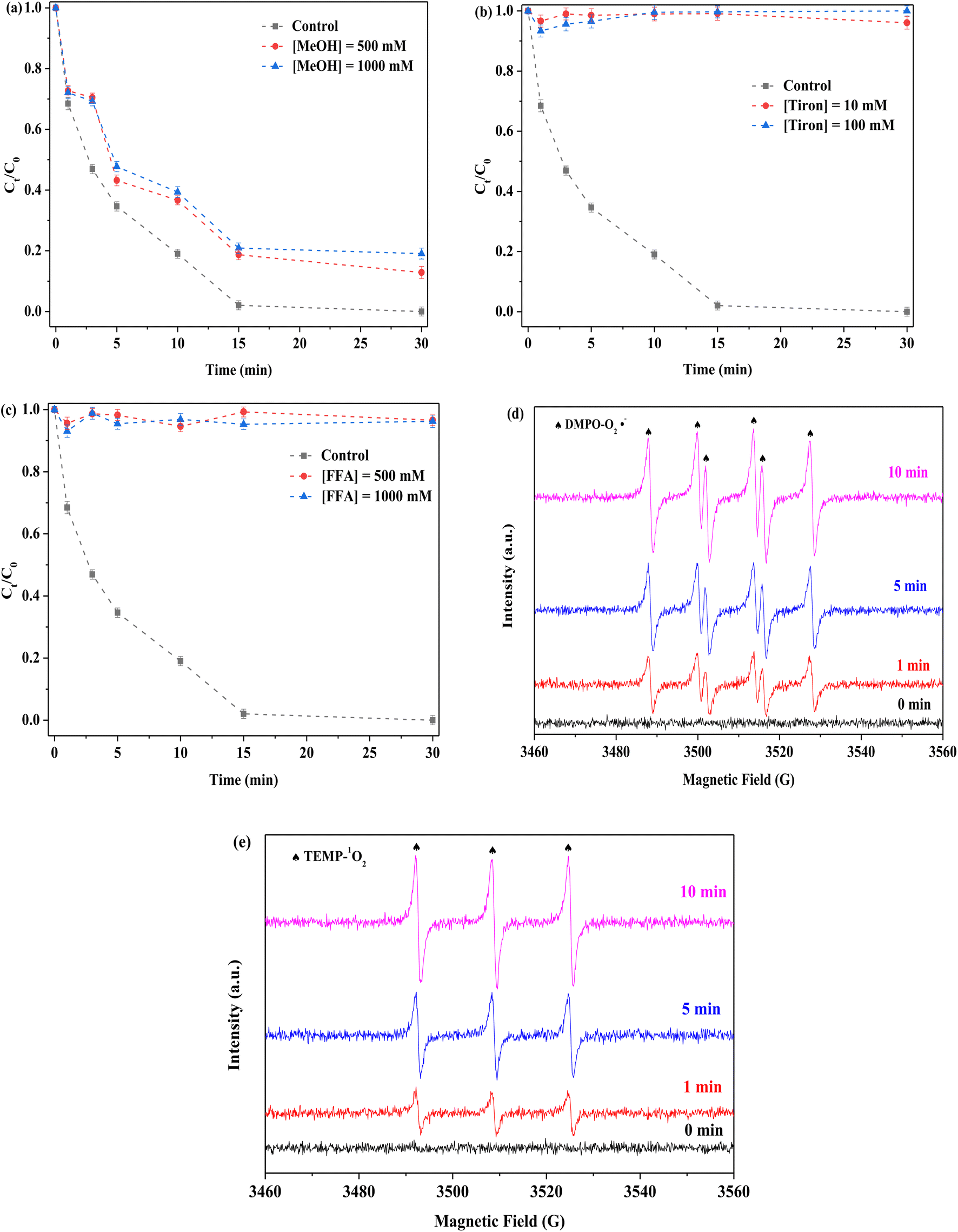
To examine the mechanism of PMS activation, different quenchers were used to detect possible ROS in the FeMoS2-IS/PMS system. In general, free radical species (including SO4˙−, ˙OH, and O2˙−) and non-free radical species, such as singlet oxygen (1O2), usually appeared in the process of PMS activation. In this experiment, methanol (MeOH) was used to identify as SO4˙− and ˙OH,13–15 tiron applied for capturing O2˙−,52,53 and furfuryl alcohol (FFA) employed for capturing 1O2,14 as shown in Fig. 8a–c. FeMoS2-IS was an Fe-based catalyst in the Fenton-like reaction and SO4˙− and ˙OH usually produced during PMS activation. This was verified by the reaction with MeOH. After MeOH in different concentrations (500 and 1000 mM) was employed, RhB degradation efficiency decreased from 100 to 89% and 85% in 30 min, respectively, implying the presence of SO4˙− and ˙OH in the reaction solution but with limited effect. After the addition of phenol with different concentrations, the inhibitions were clearly observed (Fig. S16(a)†), implying that the above two species were appeared on the surface of catalyst. It was further confirmed by the EPR characterization. As shown in Fig. S16(b),† the signals of SO4˙− and ˙OH were appeared and the signal of ˙OH turned stronger with increasing reaction time, implying that the ˙OH was generated from the reaction between SO4˙− and H2O.15 This was mainly attributed to the activation of PMS by Fe2+ and Mo4+.13–23 The above results confirmed that the SO4˙− and ˙OH were not main ROS in this reaction. When tiron at different concentrations (10 and 100 mM) was used, RhB degradation efficiencies were both <5% in 30 min. This indicated that the reaction system contained a lot of O2˙− species and played an important role in degradation. When FFA at different concentrations (500 and 1000 mM) was applied, RhB degradation efficiencies were both no more than 5%, which showed that 1O2 existed in the reaction system. Notably, FFA could react with PMS, resulting in reduced RhB removal.18 To confirm the existence of O2˙− and 1O2 in the reaction system, EPR characterization was applied using DMPO (methanol as solvent) and TEMP, respectively. Some noticeable signals of DMPO-O2˙− adduct were detected in the FeMoS2-IS/PMS system, confirming that the appearance of O2˙− had occurred (Fig. 8d). Similarly, remarkable signals of TEMP-1O2 were observed, implying that many 1O2 were formed in this system (Fig. 8e). This reaction was concluded to indeed contain a mixture of radical and non-radical species, with O2˙− and 1O2 as main ROS.
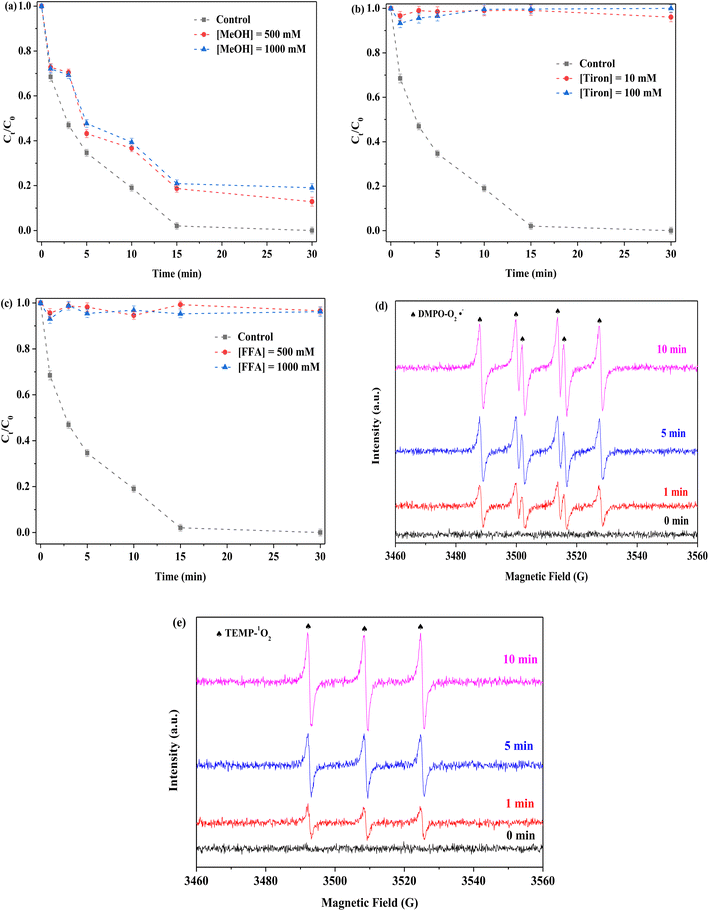 |
| | Fig. 8 Effect of (a) MeOH, (b) Tiron and (c) FFA on the removal of RhB in the FeMoS2-IS/PMS process and signals of EPR (d) and (e). Reaction conditions: [RhB] = 10 mg L−1, initial pH = 3.0, [FeMoS2-IS] = 0.3 g L−1, [PMS] = 1.0 mM and in the darkness. | |
3.6. Feasible synthetic and reaction mechanisms
When all the precursors were added into the water, the solution pH was 1.8. In this environment, the Fe and Mo ions existed in the form of Fe3+ and Mo7O246−. After thiourea introduction, it combined with Fe3+ and Mo7O246−. Under hydrothermal condition, some Fe3+ and Mo6+ were reduced to Fe2+ and Mo4+ by thiourea to form the polymer structure, namely, –Mo–S2–Fe–S2–Mo–.22 This situation allowed a tight connection between FeS2 and MoS2 and they were thus better able to interact with each other. After high temperature treatment, this special structure was partially broken to form FeS2 and MoS2. When the Fe concentration was too low, more MoS2 was formed and, meanwhile, some MoO3 and S vacancies also produced. When the Fe concentration was too high, more FeS2 was generated, but it easily combined with O2 in the air to form iron oxides like α-Fe2O3, due to the lack of Mo protection. The treatment temperature was another important factor for generating FeS2. When the temperature was too low, the above special structure was not decomposed to FeS2. When the temperature was too high, the catalyst was oxidized such that its structure was seriously destroyed. In addition, higher temperature could generate more S vacancies and the existence of S vacancies induced higher Fe2+/Fe3+ and Mo4+/Mo6+.
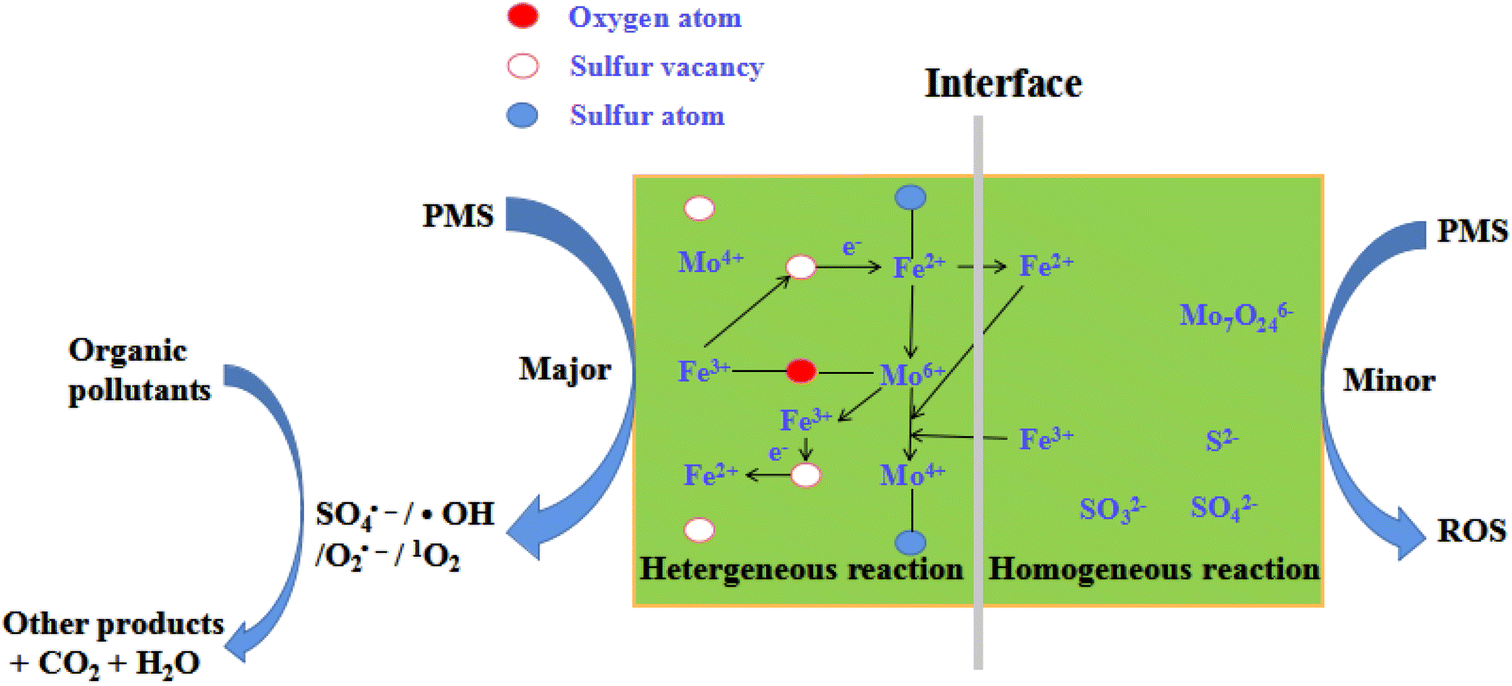
Based on the above results, a feasible reaction mechanism was proposed (Fig. 9). This was a typical heterogeneous catalytic reaction, which was different from that of the literature.20–23,46–51 The solution exhibited acidity during reaction and thus solution H+ corroded FeS2 and MoS2 on the surface of FeMoS2-IS to expose more Fe2+ and Mo4+ to generate SO4˙− and ˙OH by the activation of PMS.13–23. Simultaneously, the catalyst could adsorb these ions by electrostatic interactions. Thus, these were coordination with Mo species to form Fe2+/Fe3+ and Mo4+/Mo6+ cycles, thereby accelerating PMS decomposition toward O2˙− and 1O2. In addition, FeMoS2-IS was rich in S vacancies and these sites had good affinity for PMS, which then reacted with each other to form intermediates.35 At the liquid–solid interface, these intermediates finally evolved into O2˙− and 1O2. Also, O2˙−, as an intermediate, reacted with each other to generate 1O2.20 In addition, the surface Mo6+ on the catalyst could react with O2˙− to produce 1O2.20 Notably, the Mo6+ peroxo complex species could generate 1O2.14 The above pathways were displayed in formulas (6)–(17).
| | |
Fe3+ + Mo4+ → Fe2+ + Mo6+
| (6) |
| | |
Fe2+ + O2 + e− → Fe3+ + O2˙−
| (7) |
| | |
Fe3+/Mo6+ + HSO5− → Fe2+/Mo4+ + SO5˙− + H+
| (8) |
| | |
2SO5˙− + H2O → 2SO4− + 1O2 + H+
| (9) |
| | |
FeMoS2-IS (vacancies) + PMS + e− → PMS*–FeMoS2 (vacancies)
| (10) |
| | |
PMS*–FeMoS2 (vacancies) + H2Obuck → FeMoS2 (vacancies)–H2Ointerface + O2˙−/1O2
| (11) |
| | |
2O2˙− + H2O → H2O2 + OH− + 1O2
| (12) |
| | |
Mo6+ + O2˙− → Mo4+ + 1O2
| (13) |
| | |
HMoO3+ + 2H2O2 → MoO(O2)2 + H+ + H2O
| (15) |
| | |
MoO(O2)2 + H2O → MoO(OH)(O2)2− + H+
| (16) |
| | |
MoO(OH)(O2)2− + H2O → MoO42− + 1O2 + H+
| (17) |
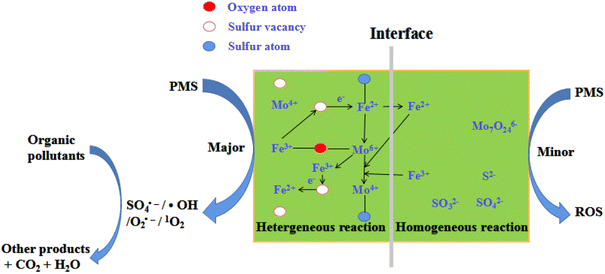 |
| | Fig. 9 Schematic illustration of the removal of organic pollutants in the FeMoS2-IS/PMS system. | |
On all accounts, Fe, Mo, and S played important roles in the structure and catalytic performance of catalyst. First, for Fe ions, Fe2+ acted as an important active site for the formation of FeS2 and the activation PMS/dissolved O2 to generate ROS. Second, the effect of Mo mainly involved in this reaction and promoted RhB removal. Finally, the role of S suppressed the oxidation of low-valent Fe and Mo to high-valent metal ions. In addition, it provided a source of S deficient sites and could directly adsorb and activate PMS.
4. Conclusions
In summary, new Fe–Mo microparticles containing S vacancies with FeS2 as the dominant phases were successfully synthesized by the in situ synthesis. The catalyst effectively removed RhB by PMS activation in wastewater. High-temperature treatment and doping Fe were both beneficial for the formation of active sites, such as FeS2 and S vacancies. This system could effectively eliminate RhB over a wide pH range. All cations had positive effects on the RhB degradation but anions exhibited a dual effect. After operating through several reaction and regeneration cycles, the removal of RhB was still good. It was confirmed that SO4˙−, ˙OH, O2˙− and 1O2 were important ROS in this reaction system, providing a good pathway for degrading organic pollutants in wastewater.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was financially supported by National Natural Science Foundation of China (No. 51978648) and Natural Science Foundation of Hunan Province (No. 2022JJ50156; 2021JJ50012).
References
- S. Li, Y. N. Wu, H. S. Zheng, H. B. Li, Y. J. Zheng, J. Nan, J. Ma, D. Nagarajand and J. S. Chang, Antibiotics degradation by advanced oxidation process (AOPs): recent advances in ecotoxicity and antibiotic-resistance genes induction of degradation products, Chemosphere, 2023, 311(2), 136977 CrossRef PubMed.
- A. Thiam, I. Sirés and E. Brillas, Treatment of a mixture of food color additives (E122, E124 and E129) in different water matrices by UVA and solar photoelectro-Fenton, Water Res., 2015, 81, 178–187 CrossRef CAS PubMed.
- Y. T. Yang, J. Chen, Z. Chen, Z. Yu, J. C. Xue, T. G. Luan, S. S. Chen and S. G. Zhou, Mechanisms of polystyrene microplastic degradation by the microbially driven Fenton reaction, Water Res., 2022, 223, 118979 CrossRef CAS PubMed.
- Z. Yang, C. Shan, B. Pan and J. J. Pignatello, The Fenton reaction in water assisted by picolinic acid: accelerated iron cycling and co-generation of a selective Fe-based oxidant, Environ. Sci. Technol., 2021, 55(12), 8299–8308 CrossRef CAS PubMed.
- T. Zhang, Y. Chen and T. O. Leiknes, Oxidation of refractory benzothiazoles with PMS/CuFe2O4: kinetics and transformation intermediates, Environ. Sci. Technol., 2016, 50(11), 5864–5873 CrossRef CAS PubMed.
- H. Li, C. Shan and B. Pan, Fe(III)-doped g-C3N4 mediated peroxymonosulfate activation for selective degradation of phenolic compounds via high-valent iron-oxo species, Environ. Sci. Technol., 2018, 52(4), 2197–2205 CrossRef CAS PubMed.
- Y. Gao, Y. Zhu, L. Tong, Z. Chen, Q. Jiang, Z. Zhao, X. Liang and C. Hu, Unraveling the high-activity origin of single-atom iron catalysts for organic pollutant oxidation via peroxymonosulfate activation, Environ. Sci. Technol., 2021, 55(12), 8318–8328 CrossRef CAS PubMed.
- H.-L. So, K.-Y. Lin, W. Chu and H. Gong, Degradation of triclosan by recyclable MnFe2O4-activated PMS: process modification for reduced toxicity and enhanced performance, Ind. Eng. Chem. Res., 2020, 59(10), 4257–4264 CrossRef CAS.
- S. Zuo, D. Li, Z. Guan, Y. Fan, H. Xu, D. Xia and J. Wan, Tailored d-band facilitating in Fe gradient doping CuO boosts peroxymonosulfate activation for high efficiency generation and release of singlet oxygen, ACS Appl. Mater. Interfaces, 2021, 13(42), 49982–49992 CrossRef CAS PubMed.
- L. Wu, Z. Sun, Y. Zhen, S. Zhu, C. Yang, J. Lu, Y. Tian, D. Zhong and J. Ma, Oxygen vacancy-Induced nonradical degradation of organics: critical trigger of oxygen (O2) in the Fe–Co LDH/peroxymonosulfate system, Environ. Sci. Technol., 2021, 55(22), 15400–15411 CrossRef CAS PubMed.
- C. Li, S. Yang, R. Bian, Y. Tan, X. Dong, N. Zhu, X. He, S. Zheng and Z. Sun, Clinoptilolite mediated activation of peroxymonosulfate through spherical dispersion and oriented array of NiFe2O4: upgrading synergy and performance, J. Hazard. Mater., 2021, 407, 124736 CrossRef CAS PubMed.
- Y. Zhao, H. An, F. Jing, Y. Ren and J. Ma, Impact of crystal types of AgFeO2 nanoparticles on the peroxymonosulfate activation in the water, Environ. Sci. Technol., 2019, 53(8), 4500–4510 CrossRef CAS PubMed.
- Q. Yi, W. Liu, J. Tan, B. Yang, M. Xing and J. Zhang, Mo0 and Mo4+ bimetallic reactive sites accelerating Fe2+/Fe3+ cycling for the activation of peroxymonosulfate with significantly improved remediation of aromatic pollutants, Chemosphere, 2020, 244, 125539 CrossRef CAS PubMed.
- Y. Zhang, J. Niu and J. Xu, Fe(II)-promoted activation of peroxymonosulfate by molybdenum disulfide for effective degradation of acetaminophen, Chem. Eng. J., 2020, 381, 122718 CrossRef CAS.
- B. Sheng, F. Yang, Y. Wang, Z. Wang, L. Qian, Y. Guo, X. Lou and J. Liu, Pivotal roles of MoS2 in boosting catalytic degradation of aqueous organic pollutants by Fe(II)/PMS, Chem. Eng. J., 2019, 375, 121989 CrossRef CAS.
- S. Wang, W. Xu, J. Wu, Q. Gong and P. Xie, Improved sulfamethoxazole degradation by the addition of MoS2 into the Fe2+/peroxymonosulfate process, Sep. Purif. Technol., 2020, 235, 116170 CrossRef CAS.
- D. He, Y. Cheng, Y. Zeng, H. Luo, K. Luo, J. Li, X. Pan, D. Barceló and J. C. Crittenden, Synergistic activation of peroxymonosulfate and persulfate by ferrous ion and molybdenum disulfide for pollutant degradation: theoretical and experimental studies, Chemosphere, 2020, 240, 124979 CrossRef CAS PubMed.
- L. Yu, Y. Feng, B. Yang, Z. Yang and K. Shih, Activation of peroxymonosulfate by molybdenum disulfide-mediated traces of Fe(III) for sulfadiazine degradation, Chemosphere, 2021, 283, 131212 CrossRef PubMed.
- X. Li, Y. Guo, L. Yan, T. Yan, S. Wen, R. Feng and Y. Zhao, Enhanced activation of peroxymonosulfate by ball-milled MoS2 for degradation of tetracycline: boosting molybdenum activity by sulfur vacancies, Chem. Eng. J., 2022, 429, 132234 CrossRef CAS.
- J. Lu, Y. Zhou and Y. Zhou, Efficiently activate peroxymonosulfate by Fe3O4@MoS2 for rapid degradation of sulfonamides, Chem. Eng. J., 2021, 422, 130126 CrossRef CAS.
- C. Yi, Z. He, Y. Hu, D. Liang, Y. Zhang and Y. Chen, FeOOH@MoS2 as a highly effective and stable activator of peroxymonosulfate-based advanced oxidation processes for pollutant degradation, Surf. Int., 2021, 27, 101465 CAS.
- J. Zhou, X. Guo, X. Zhou, J. Yang, S. Yu, X. Niu, Q. Chen, F. Li and Y. Liu, Boosting the efficiency of Fe-MoS2/peroxymonosulfate catalytic systems for organic pollutants remediation: insights into edge-site atomic coordination, Chem. Eng. J., 2022, 433, 134511 CrossRef CAS.
- Y. Sun, R. Li, C. Song, H. Zhang, Y. Cheng, A. Nie, H. Li, D. D. Dionysiou, J. Qian and B. Pan, Origin of the improved reactivity of MoS2 single crystal by confining lattice Fe atom in peroxymonosulfate-based Fenton-like reaction, Appl. Catal., B, 2021, 298, 120537 CrossRef CAS.
- M. Karroua, J. Ladrière, H. Matralis, P. Grange and B. Delmon, Characterisation of unsupported FeMoS catalysts: stability during reaction and effect of the sulfiding temperature, J. Catal., 1992, 138(2), 640–658 CrossRef CAS.
- X. Zhao, M. Xiao, Q. Lu, Q. Li, C. Han, Z. Xing and X. Yang, FeS2-doped MoS2 nanoflower with the dominant 1T-MoS2 phase as an excellent electrocatalyst for high-performance hydrogen evolution, Electrochim. Acta, 2017, 249, 72–78 CrossRef CAS.
- Y. Zhou, X. L. Wang, C. Y. Zhu, D. D. Dionysiou, G. C. Zhao, G. D. Fang and D. M. Zhou, New insight into the mechanism of peroxymonosulfate activation by sulfur containing minerals: role of sulfur conversion in sulfate radical generation, Water Res., 2018, 142, 208–216 CrossRef CAS PubMed.
- X. W. Liu, H. Li, S. Gao, Z. Bai and J. Tian, Peroxymonosulfate activation by different iron sulfides for bisphenol-A degradation: performance and mechanism, Sep. Purif. Technol., 2022, 289, 120751 CrossRef CAS.
- M. Huang, X. Wang, C. Liu, G. Fang, J. Gao, Y. Wang and D. Zhou, Mechanism of metal sulfides accelerating Fe(II)/Fe(III) redox cycling to enhance pollutant degradation by persulfate: metallic active sites vs. reducing sulfur species, J. Hazard. Mater., 2021, 404, 124175 CrossRef CAS PubMed.
- L. Wu, P. Guo, X. Wang, H. Li, A. Li and K. Chen, Mechanism study of CoS2/Fe(III)/peroxymonosulfate catalysis system: the vital role of sulfur vacancies, Chemosphere, 2022, 288, 132646 CrossRef CAS PubMed.
- C. Liang, C.-F. Huang, N. Mohanty and R. M. Kurakalva, A rapid spectrophotometric determination of persulfate anion in ISCO, Chemosphere, 2008, 73, 1540–1543 CrossRef CAS PubMed.
- L. Chang, H. Yi, B. Yang, F. Jia and S. Song, Activation of Fenton reaction by controllable oxygen incorporation in MoS2-Fe under visible light irradiation, Appl. Surf. Sci., 2021, 566, 150674 CrossRef.
- L. E. Aseman-Bashiz and H. Sayyaf, Synthesis of nano-FeS2 and its application as an effective activator of ozone and peroxydisulfate in the electrochemical process for ofloxacin degradation: a comparative study, Chemosphere, 2021, 274, 129772 CrossRef PubMed.
- X. Ma, Y. Cheng, J. Deng, A. Cai, L. Xiao, J. Li and X. Li, Elucidating the role of Fe(IV) and radical species for CBZ degradation in FeS2/PS system, Sep. Purif. Technol., 2021, 274, 118982 CrossRef CAS.
- H. Zhou, L. Lai, Y. Wan, Y. He, G. Yao and B. Lai, Molybdenum disulfide (MoS2): a versatile activator of both peroxymonosulfate and persulfate for the degradation of carbamazepine, Chem. Eng. J., 2022, 384, 123264 CrossRef.
- J.-C. E. Yang, M.-P. Zhu, X. Duan, S. Wang, B. Yuan and M.-L. Fu, The mechanistic difference of 1T–2H MoS2 homojunctions in persulfates activation: structure dependent oxidation pathways, Appl. Catal., B, 2021, 297, 120460 CrossRef CAS.
- M. Du, Q. Yi, J. Ji, Q. Zhu, H. Duan, M. Xing and J. Zhang, Sustainable activation of peroxymonosulfate by the Mo(IV) in MoS2 for the remediation of aromatic organic pollutants, Chin. Chem. Lett., 2020, 31(10), 2803–2808 CrossRef CAS.
- J. da Silveira Salla, K. da Boit Martinello, G. L. Dotto, E. García-Díaz, H. Javed, P. J. J. Alvarez and E. Luiz Foletto, Synthesis of citrate-modified CuFeS2 catalyst with significant effect on the photo-Fenton degradation efficiency of bisphenol A under visible light and near neutral pH, Colloids Surf., A, 2020, 595, 124679 CrossRef.
- S. H. Tian, Y. T. Tu, D. S. Chen, X. Chen and Y. Xiong, Degradation of acid orange II at neutral pH using Fe2(MoO4)3 as a heterogeneous Fenton-like catalyst, Chem. Eng. J., 2011, 169, 31–37 CrossRef CAS.
- Y. C. Huang, L. Lai, W. Huang, H. Zhou, J. Li, C. Liu, B. Lai and N. Li, Effective peroxymonosulfate activation by natural molybdenite for enhanced atrazine degradation: role of sulfur vacancy, degradation pathways and mechanism, J. Hazard. Mater., 2022, 435, 128899 CrossRef CAS PubMed.
- H. Kuang, Z. He, L. Mu, R. Huang, Y. Zhang, X. Xu, L. Wang, Y. Chen and S. Zhao, Enhancing co-catalysis of MoS2 for persulfate activation in Fe3+-based advanced oxidation processes via defect engineering, Chem. Eng. J., 2021, 417, 127987 CrossRef CAS.
- H. Luo, Y. Cheng, Y. Zeng, K. Luo, D. He and X. Pan, Rapid removal of organic micropollutants by heterogeneous peroxymonosulfate catalysis over a wide pH range: performance, mechanism and economic analysis, Sep. Purif. Technol., 2020, 248, 117023 CrossRef CAS.
- A. Mehdi and F. Ghanbari, Organic dye degradation through peroxymonosulfate catalyzed by reusable graphite felt/ferriferrous oxide: mechanism and identification of intermediates, Mater. Res. Bull., 2019, 111, 43–52 CrossRef.
- J. Nematollah, F. Ghanbari and Z. Amir, Coupling electro-oxidation and oxone for degradation of 2,4-dichlorophenoxyacetic acid (2,4-D) from aqueous solutions, J. Water Proc. Eng., 2018, 22, 203–209 CrossRef.
- X. Song, J. Ni, D. Liu, W. Shi, Y. Yuan, F. Cui, J. Tian and W. Wang, Molybdenum disulfide as excellent co-catalyst boosting catalytic degradation of sulfamethoxazole by nZVI/PDS process, Sep. Purif. Technol., 2022, 285, 120398 CrossRef CAS.
- M. M. Rashad, A. A. Ibrahim, D. A. Rayan, M. M. S. Sanad and I. M. Helmy, Photo-Fenton-like degradation of rhodamine B dye from waste water using iron molybdate catalyst under visible light irradiation, Environ. Nanotechnol., Monit. Manage., 2017, 8, 175–186 Search PubMed.
- S.-Y. Oh, S.-G. Kang, D.-W. Kim and P. C. Chiu, Degradation of 2,4-dinitrotoluene by persulfate activated with iron sulfides, Chem. Eng. J., 2011, 172(2–3), 641–646 CrossRef CAS.
- X. Zheng, X. Niu, D. Zhang, X. Ye, J. Ma, M. Lv and L. Zhang, Removal of Microcystis aeruginosa by natural pyrite-activated persulfate: performance and the significance of iron species, Chem. Eng. J., 2022, 428, 132565 CrossRef CAS.
- H. Peng, J. Zhu, Y. Chen, F. Chen, J. Zhu, M. Liu, K. Zhang and M. Gan, Pyrite-activated persulfate for simultaneous 2,4-DCP oxidation and Cr(VI) reduction, Chem. Eng. J., 2021, 406, 126758 CrossRef.
- Z.-H. Diao, J.-J. Liu, Y.-X. Hu, L.-J. Kong, D. Jiang and X.-R. Xu, Comparative study of rhodamine B degradation by the systems pyrite/H2O2 and pyrite/persulfate: reactivity, stability, products and mechanism, Sep. Purif. Technol., 2017, 184, 374–383 CrossRef CAS.
- S. Sühnholz, F.-D. Kopinke and K. Mackenzie, Reagent or catalyst? FeS as activator for persulfate in water, Chem. Eng. J., 2020, 387, 123804 CrossRef.
- X. B. Wang, Y. Wang, N. Chen, Y. Shi and L. Zhang, Pyrite enables persulfate activation for efficient atrazine degradation, Chemosphere, 2020, 244, 125568 CrossRef CAS PubMed.
- L. Zhou, W. Song, Z. Q. Chen and G. C. Yin, Degradation of organic pollutants in wastewater by bicarbonate activated hydrogen peroxide with a supported cobalt catalyst, Environ. Sci. Technol., 2013, 47, 3833–3839 CrossRef CAS PubMed.
- E. G. Heckert, S. Seal and W. T. Self, Fenton-like reaction catalyzed by the rare earth inner transition metal cerium, Environ. Sci. Technol., 2008, 42(13), 5014–5019 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a