
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePolymeric copper(II) and dimeric oxovanadium(V) complexes of amide–imine conjugate: bilirubin recognition and green catalysis†
Jayanta Dasa,
Sabyasachi Ta a,
Noor Salamab,
Sudipta Dasc,
Subhasis Ghosha and
Debasis Das*a
a,
Noor Salamab,
Sudipta Dasc,
Subhasis Ghosha and
Debasis Das*a
aDepartment of Chemistry, The University of Burdwan, Burdwan 713104, WB, India. E-mail: ddas100in@yahoo.com; Fax: +91-342-2530452; Tel: +91-342-2533913, ext. 424
bDepartment of Chemistry, Surendranath College, 24/2 MG Road, Kolkata 700009, WB, India
cRaina Swami Bholananda Vidyayatan, Burdwan 713421, WB, India
First published on 28th April 2023
Abstract
An exceptionally simple amide–imine conjugate, (E)-N′-(4-(diethylamino)-2-hydroxybenzylidene)-4-methylbenzohydrazide (L), derived by the condensation of 4-methyl-benzoic acid hydrazide (PTA) with 4-(diethylamino)-2-hydroxybenzaldehyde was utilized to prepare a dimeric oxo-vanadium (V1) and a one-dimensional (1D) copper(II) coordination polymer (C1). The structures of L, V1 and C1 were confirmed by single crystal X-ray diffraction analysis. The experimental results indicate that V1 is a promising green catalyst for the oxidation of sulfide, whereas C1 has potential for a C–S cross-coupling reaction in a greener way. Most importantly, C1 is an efficient ‘turn-on’ fluorescence sensor for bilirubin that functions via a ligand displacement approach. The displacement equilibrium constant is 7.78 × 105 M−1. The detection limit for bilirubin is 1.15 nM in aqueous chloroform (chloroform/water, 1/4, v/v, PBS buffer, and pH 8.0).
Introduction
The significance of amide functionality has attracted special attention in pharmaceutical chemistry1,2 as reflected by the fact that ∼25% of drug molecules contain amide functionalities.3 Moreover, conformational diversities allow amide moieties to be useful in synthetic chemistry.4–10 However, metal complexes having variable oxidation states are promising catalysts for organic transformation and the synthesis of bio-active compounds and functional materials.In this context, oxo-vanadium(V) complexes play a vibrant role in several biochemical processes, such as insulin mimicking activities,11–13 anti-cancer activities,14–17 nitrogen fixation,18 halo peroxidation,19 phosphorylation20 and sulphide oxidation.21 Sulphide oxidation plays a crucial role in the activation of enzymes and useful synthetic intermediates in several chemical and bio-active molecules.22–24 Traditional synthesis generates a significant amount of waste25–27 and causes over-oxidation, leading to sulfones.28 To overcome the issues regarding selectivity and yield, metal complexes may play a significant role as catalysts.29–35
However, Cu(II) complexes have occasionally been employed as catalyst, antiviral, antibacterial and anti-tumor agents.36–43 Several Cu(II) complexes have also been used for trace level detection and estimation of amino acids.37–40 Most notably, copper-catalyzed Ullmann-type C–S cross coupling reaction is very useful.44 Aryl sulphide component is commonly encountered in several drugs for the treatment of diseases, such as Alzheimer's, Parkinsons, cancer, HIV, diabetes and inflammation.45 The transition metal complex catalyzed C–S cross-coupling reactions have been relatively less explored compared to other carbon–heteroatom bonds, viz. C–N, C–O and C–P, probably owing to the associated S–S coupling and poisoning of the catalyst, particularly thiophilic Pd-based catalysts.46 To overcome catalyst poisoning, different systems, such as palladium compounds,47–50 nickel compounds,51,52 CoI2(dppe)/Zn,53 In(OTf)3,54 FeCl3 (ref. 55 and 56) and copper compounds,57–63 have been employed. However, it requires unfavourable reaction conditions, such as toxic polar solvent, high temperature (over 200 °C) and high catalyst loading.64 Hence, there is ample scope to contribute to the development of environment-/reaction-friendly and efficient catalysts for Ullmann-type C–S cross coupling reactions.
In addition, the literature suggests that metal complexes are potential candidates for the recognition of bio-molecules65–68 and bio-relevant analytes.69–73 Bilirubin (C33H36N4O6), a natural yellow colored linear tetrapyrrole compound, is catabolically produced by the degradation of red blood cells (RBCs) in the body.74,75 After its formation in the tissue, heme is transported to the liver in an unconjugated form using serum albumin as a carrier protein. In the liver, bilirubin predominantly transforms into bilirubin diglucuronide via an enzymatic conjugation reaction. This conjugated bilirubin, unlike unconjugated bilirubin, is fairly water soluble and thus excreted via the bile.76,77 The normal bilirubin concentration in the blood of a healthy and adult person lies in the range of 0.3–1.9 mg dL−1, including conjugated bilirubin (CB) (0.1–0.4 mg dL−1), unconjugated bilirubin (UCB or free bilirubin) (0.2–0.7 mg dL−1) and a trace amount of non-protein-bound unconjugated bilirubin.78,79 It is already known that bilirubin level in new born babies is almost ten times higher than that of adults.80 A low level of serum bilirubin concentration is associated with several clinical symptoms viz. iron deficiency and coronary artery disease, whereas its higher concentration (>2.5 mg dL−1) leads to hyper-bilirubinemia or jaundice. The elevated level of serum bilirubin in new-born babies often causes neonatal jaundice with symptoms of yellow skin and sclera, liver diseases (viz. cirrhosis and hepatitis), hearing loss, athetosis, brain damage related to lifelong neurologic sequelae (kernicterus) or even death.81,82
Although several methods of bilirubin determination, such as Jendrassik–Grofmethod, spectrophotometric method,83,84 HPLC,85,86 chemiluminescence combined with flow injection analysis,87,88 capillary micellar electrokinetic chromatography89,90 and bilirubin oxidase enzymatic analysis are available,91,92 fluorescence technique has more advantage over others owing to its operational simplicity, instantaneous response, high sensitivity, and non-destructive methodology.93–95 Hence, the development of fluorescence-based bilirubin sensors has attracted immense attention. For example, recognition of unconjugated, conjugated and total bilirubin has been performed using a D-glucuronic acid appended polymer (PF-Ph-GlcA),96 modified MOF (UIO-66-PSM),97 Zr-MOF: Eu3+ probe,98 HSA-AuNCs,99 L-cysteine modulated-CuNCs,100 BSA-CuNCs,101 Fe(III)-modulated BSA-CuNCs,102 2,2′-((1E,1′E)-(6-bromopyridine-2,3-diyl) bis-(azanylylidene)) bis-(methanylylidenediphenol),103 fiber optic fluorescent bio-sensors,80,104 Ru(bipy)32+,105 S,N-doped CDs106 and 4,4,4-trifluoro-1-(thiophen-2-yl)-butane-1,3-dione europium(III) complex (Eu(tta)3),107 pyrene based Schiff-base,108 polystyrene based probe,109 and N-doped graphene QDs/CoFe2O4-DOPA/H2O2 system.110
A careful review of the literature indicates that very few metal complex-based bilirubin sensors111,112 are available that function via a fluorescence quenching mechanism, a highly undesirable protocol. Herein, we explored a 1D copper(II) coordination polymer (C1) for the recognition of bilirubin via “turn-on fluorescence” without any significant interference from competing bio-molecules. We proposed that the recognition of bilirubin proceeds via the “ligand displacement approach”113,114 in which the amide–imine conjugate (L) from the C1 is displaced by bilirubin in solution, resulting in the fluorescence enhancement of the system. Moreover, it is found that the dimeric oxovanadium(V) (V1) and 1D copper(II) coordination polymer (C1) of the amide–imine conjugate efficiently catalyse the oxidation of sulphide and C–S cross-coupling reactions in a greener way, respectively.
Experimental
Synthesis
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) and 1271 ν(N–N).
O) and 1271 ν(N–N).![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ’ space group. The crystallographic refinement parameters are listed in Table S1, ESI.† Anal. calcd (%) for C19H23N3O2: C, 70.13; H, 7.12 and N, 12.91; found: C, 71.01; H, 7.27 and N, 12.87. 1H NMR (Fig. S5, ESI†) [400 MHz, CDCl3, TMS, J (Hz), δ(ppm)]: (a) 11.792 (1H, s, –CONH), (b) 11.506 (1H, s, –OH), (c) 8.410 (1H, s, –N–CH–), (d) 2.372 (3H, s, –CH3), (e) 3.369 (4H, m), (f) 1.09 (6H, t, J = 7.2), 7.815 (2H, d, J = 8), 7.326 (2H, d, J = 8), 7.178 (1H, d, J = 8.8), 6.116 (1H, d, J = 2.4) and 6.116 (1H, d, J = 2.4).13C NMR (Fig. S6, ESI†): 162.55, 160.17, 150.56, 150.24, 142.19, 132.09, 130.71, 129.50, 127.95, 106.91, 104.08, 97.94, 44.26, 21.50 and 13.00. ESI-MS (p+) (Fig. S7, ESI†) m/z calculated. For C19H23N3O2: 325.18, found: 326.48 (100%). FTIR (KBr, cm−1) (Fig. S8, ESI†), 3279 ν(N–H); 1595 ν(CN); 1632 ν(CO); 3029 ν(C–H); 1279 ν(N–N). The quantum yield is 0.78.’ space group. The crystallographic refinement parameters are listed in Table S1, ESI.† Anal. calcd (%) for C40H48V2N6O8: C, 57.01; H, 5.74 and N, 9.97; found: C, 57.37; H, 5.48 and N, 9.82. ESI-MS (p+) (Fig. S9, ESI†) m/z for one asymmetric unit C20H24VN3O4 calculated: 421.37, found: 422.14; selected FTIR bands (KBr, cm−1) (Fig. S10, ESI†): 3185 ν(O–H); 2970 ν(C–Har); 1244 ν(N–N); 1188 ν(C–O), stretching vibration; 963 ν(VO); 830 ν[V–(μ-O)–V] mode and 1586 ν(CN). The UV-vis spectrum in aqueous methanol (1
’ space group. The crystallographic refinement parameters are listed in Table S1, ESI.† Anal. calcd (%) for C19H23N3O2: C, 70.13; H, 7.12 and N, 12.91; found: C, 71.01; H, 7.27 and N, 12.87. 1H NMR (Fig. S5, ESI†) [400 MHz, CDCl3, TMS, J (Hz), δ(ppm)]: (a) 11.792 (1H, s, –CONH), (b) 11.506 (1H, s, –OH), (c) 8.410 (1H, s, –N–CH–), (d) 2.372 (3H, s, –CH3), (e) 3.369 (4H, m), (f) 1.09 (6H, t, J = 7.2), 7.815 (2H, d, J = 8), 7.326 (2H, d, J = 8), 7.178 (1H, d, J = 8.8), 6.116 (1H, d, J = 2.4) and 6.116 (1H, d, J = 2.4).13C NMR (Fig. S6, ESI†): 162.55, 160.17, 150.56, 150.24, 142.19, 132.09, 130.71, 129.50, 127.95, 106.91, 104.08, 97.94, 44.26, 21.50 and 13.00. ESI-MS (p+) (Fig. S7, ESI†) m/z calculated. For C19H23N3O2: 325.18, found: 326.48 (100%). FTIR (KBr, cm−1) (Fig. S8, ESI†), 3279 ν(N–H); 1595 ν(CN); 1632 ν(CO); 3029 ν(C–H); 1279 ν(N–N). The quantum yield is 0.78.’ space group. The crystallographic refinement parameters are listed in Table S1, ESI.† Anal. calcd (%) for C40H48V2N6O8: C, 57.01; H, 5.74 and N, 9.97; found: C, 57.37; H, 5.48 and N, 9.82. ESI-MS (p+) (Fig. S9, ESI†) m/z for one asymmetric unit C20H24VN3O4 calculated: 421.37, found: 422.14; selected FTIR bands (KBr, cm−1) (Fig. S10, ESI†): 3185 ν(O–H); 2970 ν(C–Har); 1244 ν(N–N); 1188 ν(C–O), stretching vibration; 963 ν(VO); 830 ν[V–(μ-O)–V] mode and 1586 ν(CN). The UV-vis spectrum in aqueous methanol (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v, 10 μM, Fig. S11, ESI†): λ, nm (ε, M−1 cm−1); 371 (37000).
:1, v/v, 10 μM, Fig. S11, ESI†): λ, nm (ε, M−1 cm−1); 371 (37000).
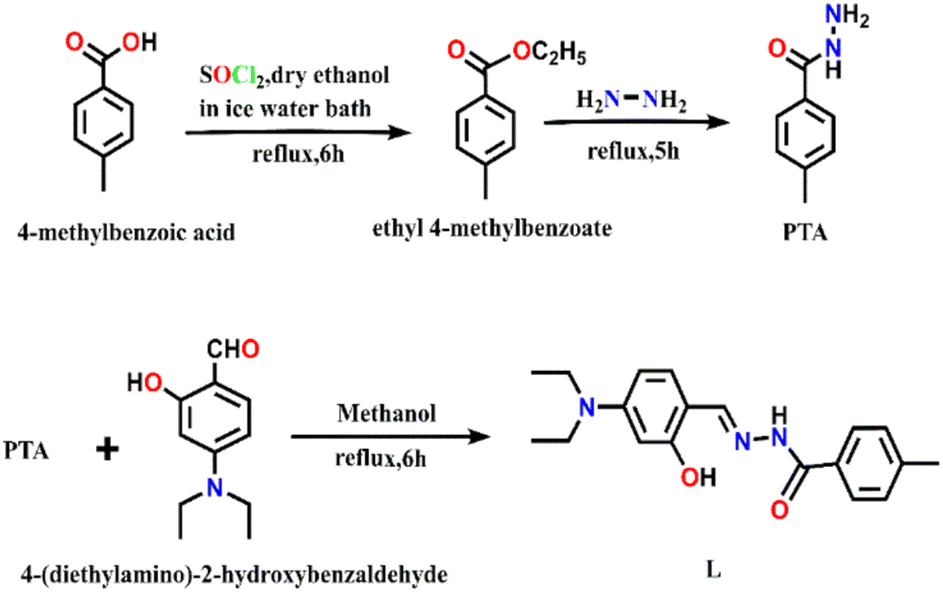 | ||
| Scheme 1 Synthesis of PTA and L. | ||
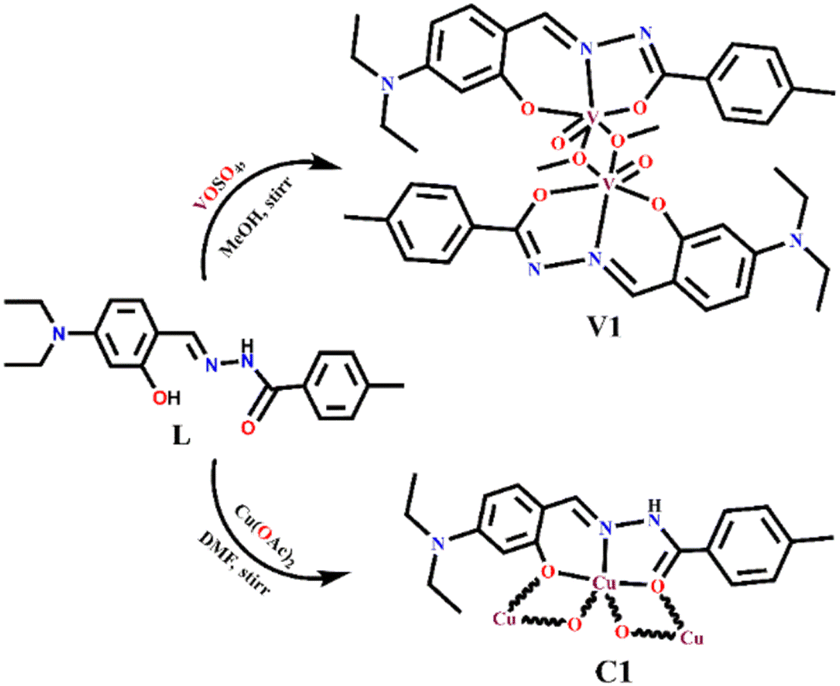 | ||
| Scheme 2 Synthesis of V1 and C1. | ||
’ space group. The crystallographic refinement parameters are listed in Table S1, ESI.†ESI-MS (p+) (Fig. S12, ESI†) m/z calcd for C19H22Cu3N3O4: 547.04, found: 547.99. FTIR (KBr, cm−1): 3085 ν(N–H); 3370 ν(OH); 2970 ν(C–Har); 1601 ν(CN) (Fig. S13, ESI†). UV-vis. spectrum in CHCl3 solution λmax/nm (εmax/M−1 cm−1): 395 (64000), 415 (53000) (Fig. S14, ESI†).
General procedure for catalytic studies
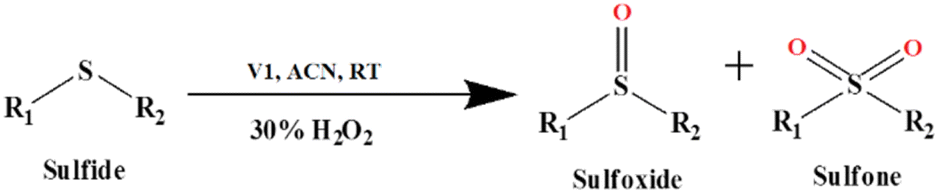 | ||
| Scheme 3 The V1 catalyzed oxidation of sulfide. | ||
Results and discussion
Single crystal X-ray structures of L, V1 and C1
The ORTEP view of V1 along with the atom labelling scheme is presented in Fig. 1a. A summary of the crystallographic data and refinement details for L, V1 and C1 is presented in Table S1 (ESI†). The V1 belongs to the orthogonal crystal system with space group P. Each unit in the symmetric dinuclear vanadium(V) complex is coordinated by one nitrogen (N2) and two oxygen (O1 and O2) donors of the ligand. One terminal oxo (O4) donor is also present along with a methoxy group (O3). The latter (O3) acts as a μ2-bridging unit to bind two identical parts of the complex together. Two square pyramidal asymmetric units form the complete structure of V1, where the central μ2-bridging methoxy group imposes a V⋯V separation of ∼3.337 Å, with a V–O–V bridging angle of ∼107.30(12)° and O–V–O angles of 72.70(12)° (Fig. 1b). The methoxido groups display two different V–O distances, 1.853(3) Å and 2.278(3) Å. The former is quite longer than other reported oxovanadium(V) complexes116 but similar to other bis-(μ-methoxy) bridged vanadium(V) dimer, 1.8207(13) Å.117 Other V–O bonds are V–Ooxide, 1.597(3) Å, V–Oketone, 1.955(3) Å and V–Ophenolate, 1.857(3) Å. The vanadium–oxygen bond lengths follow the order V–Ooxido < V–Oalkoxo < V–Ophenolate < V–Oketone. The V–Nimine bond length of 2.090(4) Å is comparable to the literature values.118 Thus, V1 is highly distorted, and vanadium centres deviate from the equatorial plane. The selected bond lengths and angles for V1 are presented in Table S2 (ESI†).
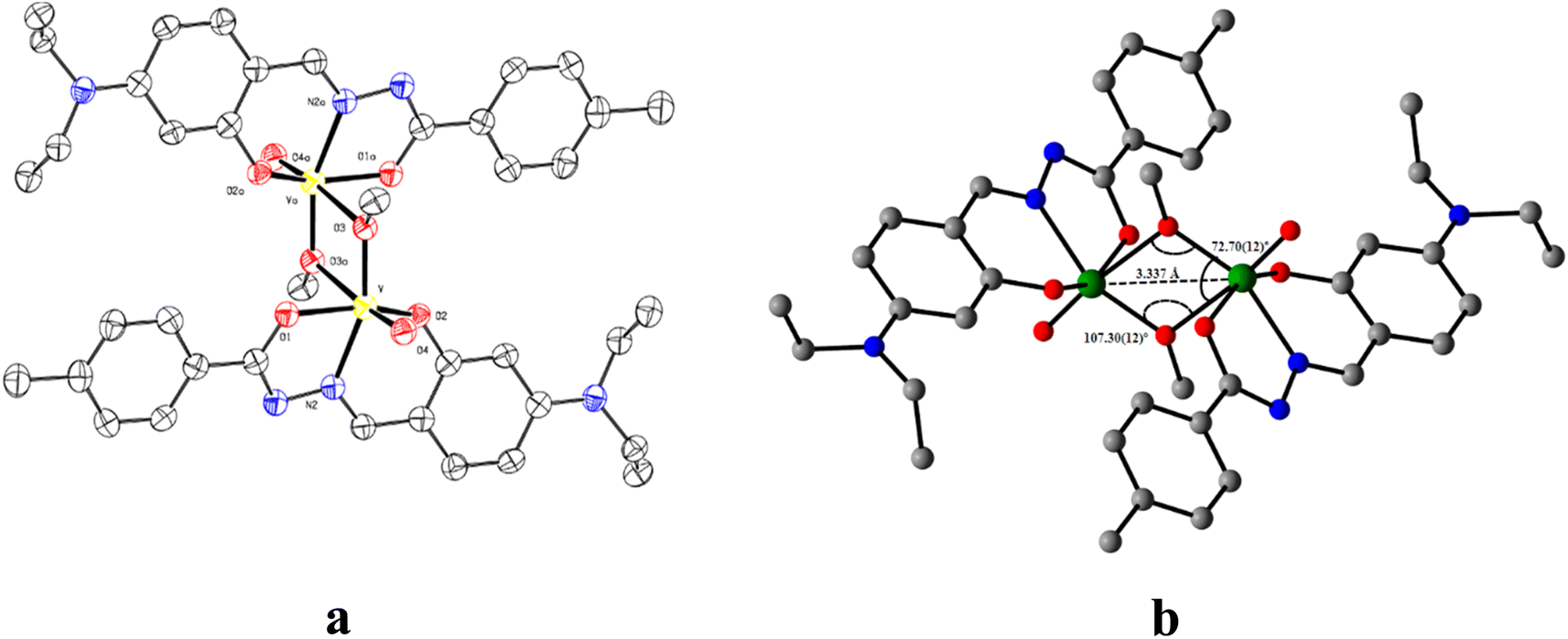 | ||
| Fig. 1 (a) ORTEP of V1 with 50% ellipsoid probability and (b) significant features around the bridging portion. H atoms are omitted for clarity. | ||
The ORTEP view of C1 is presented in Fig. 2a along with the atom labelling scheme. The C1 crystallizes in triclinic space group P, while the asymmetric unit comprises the independent molecule [C19H21CuN3O2] where the Cu(II) centre is attached to imine nitrogen (N2) and two oxygen (O1 and O2 from hydrazide and aldehyde moiety, respectively) atoms of the ligand. O1 and O2 act as end-on bridging groups and link [C19H21CuN3O2] moieties into a one-dimensional (1D) coordination polymer chain (Fig. 2b). The asymmetric unit [C19H21CuN3O2] grows in a 1D polymer chain by two μ-oxo bridges having a bond angle of 86.96° with the other plane, resulting in two different Cu⋯Cu separations of ∼3.589 Å and 3.080 Å. In one identical unit, the Cu–O1 distance is 1.982(5) Å, which differs from the other, Cu–O2, 2.012(5) Å, while the Cu–N2 distance is 1.896(5) Å, which varies in the 1D-polymer chain. In another plane, the distances are Cu–O1, 2.675 Å and Cu–O2, 1.972(4) Å. Significant distances around the central copper ion in one identical square plane are 3.203 Å, 2.807 Å, 2.529 Å and 2.518 Å (Fig. 2c). Important inter atomic distances and angles relevant to the copper(II) coordination sphere are presented in Table S3 (ESI†).
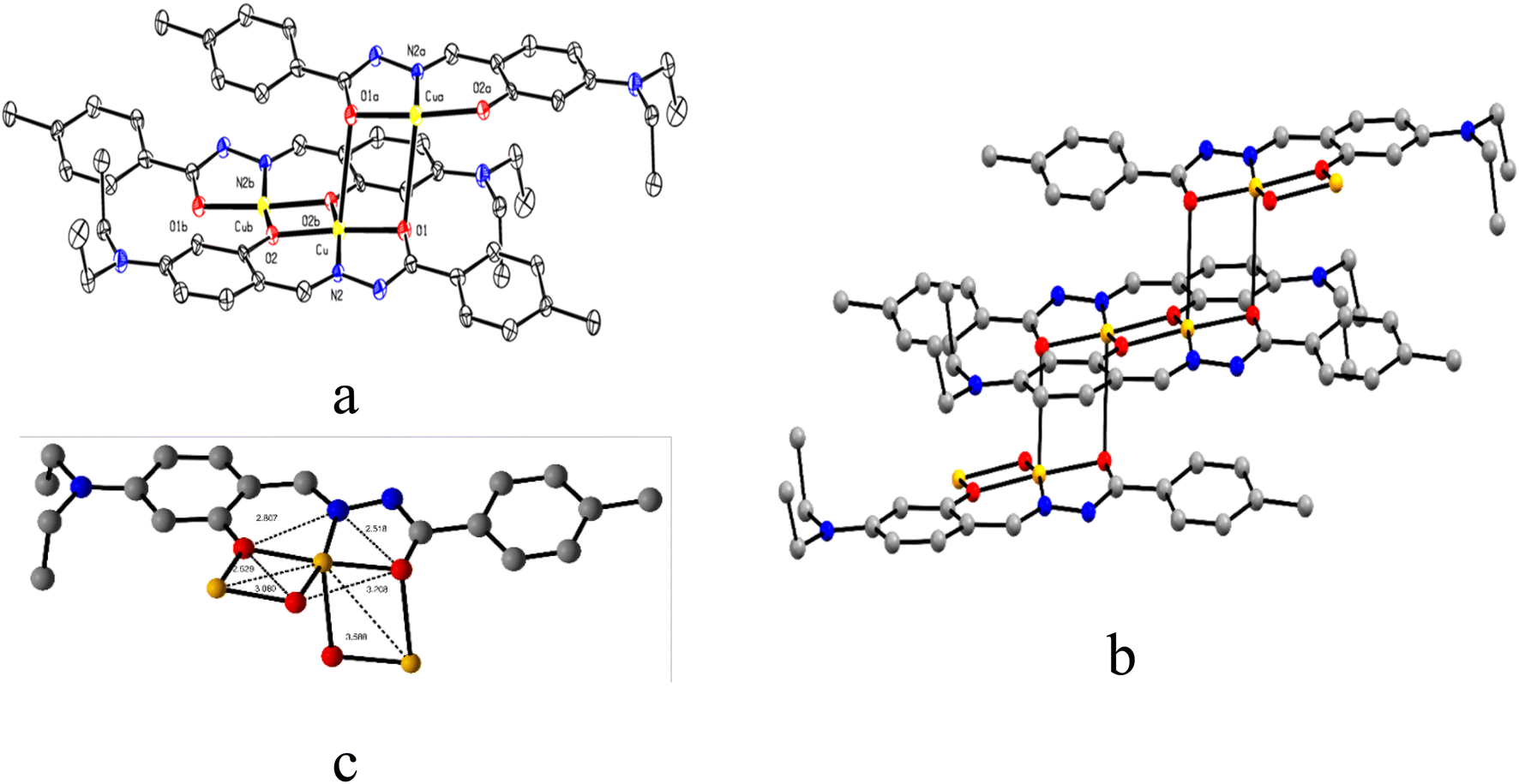 | ||
| Fig. 2 (a) ORTEP view of C1 with 50% ellipsoid probability, (b) 1D coordination polymer chain of C1, and (c) important distances around the central atom. H atoms are omitted for clarity. | ||
The ORTEP view along with the atom labelling scheme of L is presented in Fig. 3a. The L belongs to a triclinic crystal system with a P space group. The molecules of L are self-assembled via a hydrogen bond between amide groups (N⋯O = 2.935 Å and angle N–N⋯O = 105.96°), leading to the formation of a 1D chain (Fig. 3b). Important bond distances and angles are presented in Table S4 (ESI†).
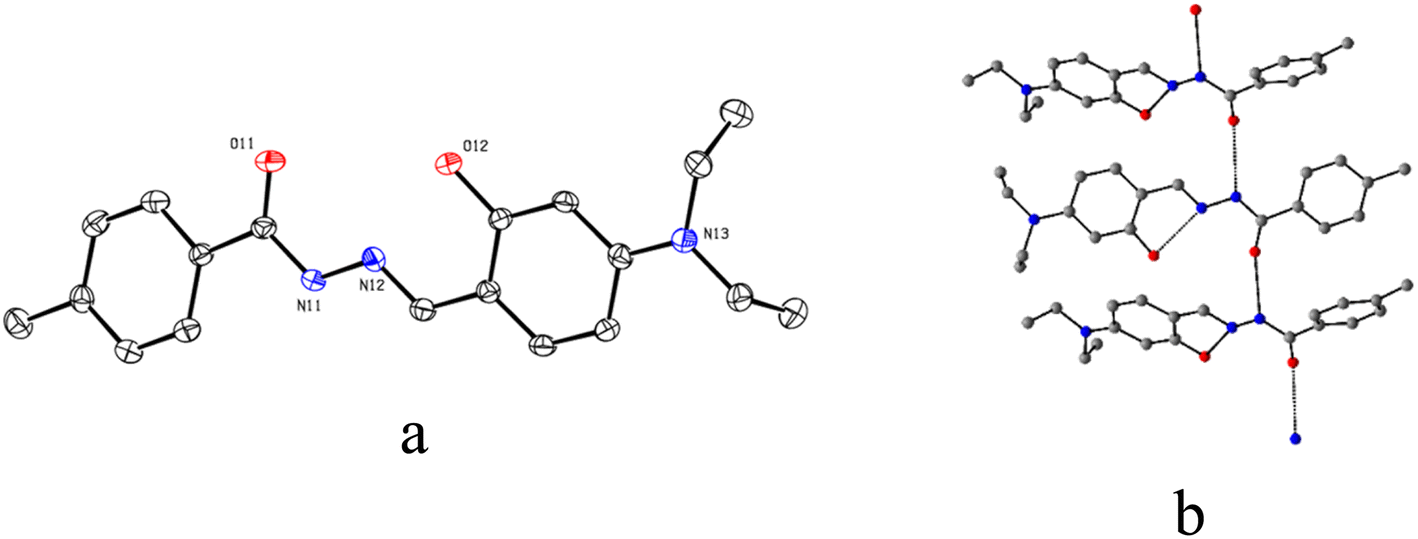 | ||
| Fig. 3 (a) ORTEP view of L at 50% ellipsoid probability and (b) centro-symmetric disposition of three units self-assembled through hydrogen bond (black dashes). H atoms are omitted for clarity. | ||
Fluorescence recognition of bilirubin using C1
To demonstrate the ability of C1 to act as a bilirubin sensor, first, the media and their pH were optimized. Bilirubin is practically insoluble in water but soluble in bio-compatible organic solvent chloroform.119 Therefore, to optimize solvent composition, several sets of solutions of C1 and bilirubin have been prepared in chloroform/water (v/v), viz. 5:1, 4:1, 3:1, 2:1,1:1, 1:2, 1:3, 1:4 and 1:5 in PBS buffer at pH 8.0. It is observed that the solvent composition 1:4 (chloroform/water, v/v, PBS buffer, pH 8.0) is the best to monitor their interaction using spectroscopy (emission and absorption). The C1 (20 μM) exhibits absorptions at 377 nm, 395 nm, 415 nm and 461 nm in the media, as stated earlier. Upon the gradual addition of bilirubin to C1, the shoulder at 461 nm further weakens markedly, whereas the absorbance at 395 nm increased gradually with an isobestic point at 434 nm (Fig. 4a), indicating their interaction.
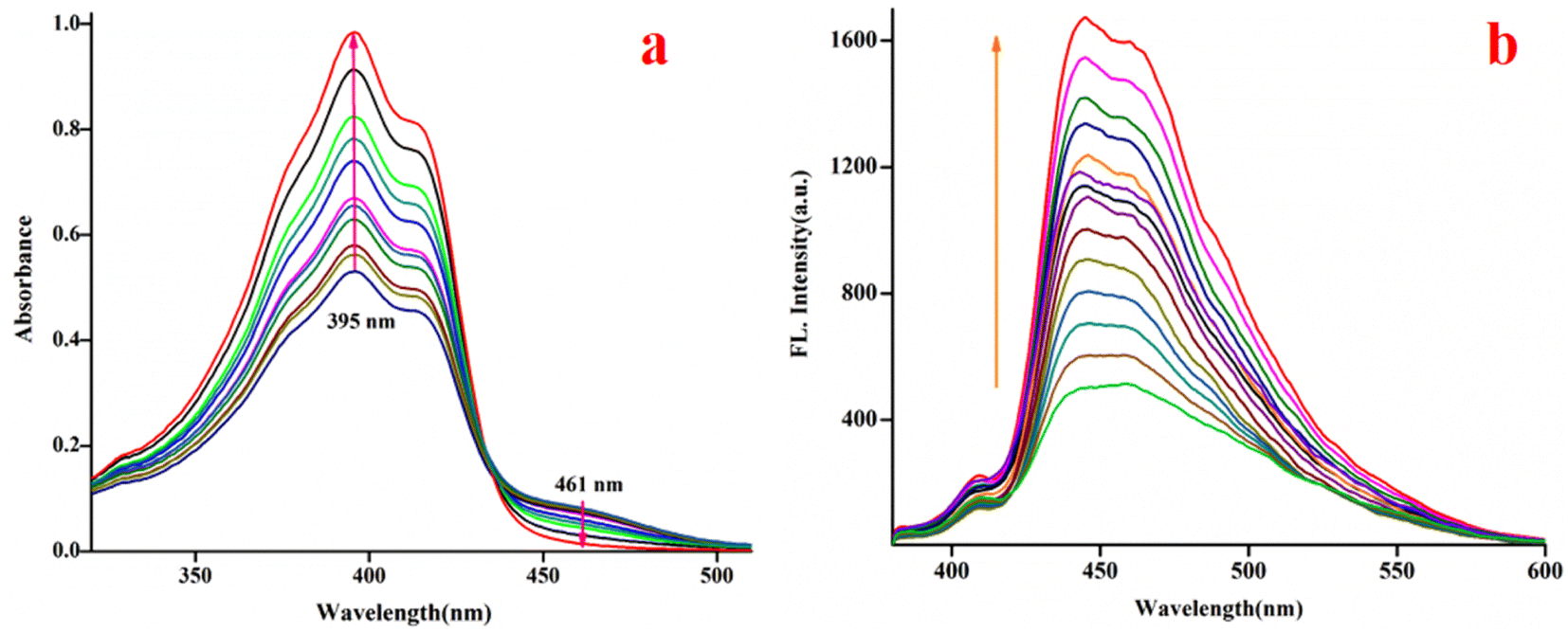 | ||
| Fig. 4 Changes in the spectra of C1 (20 μM, λex, 369 nm, λem, 444 nm) upon the gradual addition of bilirubin (0–2500 μM, CHCl3/water, 1/4, v/v, 0.1 M PBS buffer, pH 8.0), (a) absorption; (b) emission. | ||
The fluorescence experiments were performed in the same solvent system (chloroform/water, 1/4, v/v, PBS buffer, pH 8.0) at 25 °C. The C1 (λex, 369 nm) is weakly fluorescent owing to the quenching (via energy transfer and/or electron transfer from the excited state of fluorescence probe to a low-lying empty d-orbital of the Cu2+) nature of paramagnetic Cu2+ ion (d9-system).120–122 The C1 in solution alone (20 μM) exhibited weak emission at 444 nm and 460 nm with almost the same intensity. The incremental addition of bilirubin to C1 (20 μM) resulted in significant fluorescence enhancement at both wavelengths (Fig. 4b). However, the emission band centred at 444 nm increased to a higher extent than the emission band at 460 nm. The fluorescence enhancement of C1 in the presence of externally added bilirubin occurred because of the displacement of fluorescent amide-imine moiety (L, as an indicator) from the coordination sphere of C1 (via IDA process113,114), or in other words, Cu2+ is extracted from C1 by bilirubin to liberate free fluorescent L, resulting in fluorescence enhancement (Scheme 4).
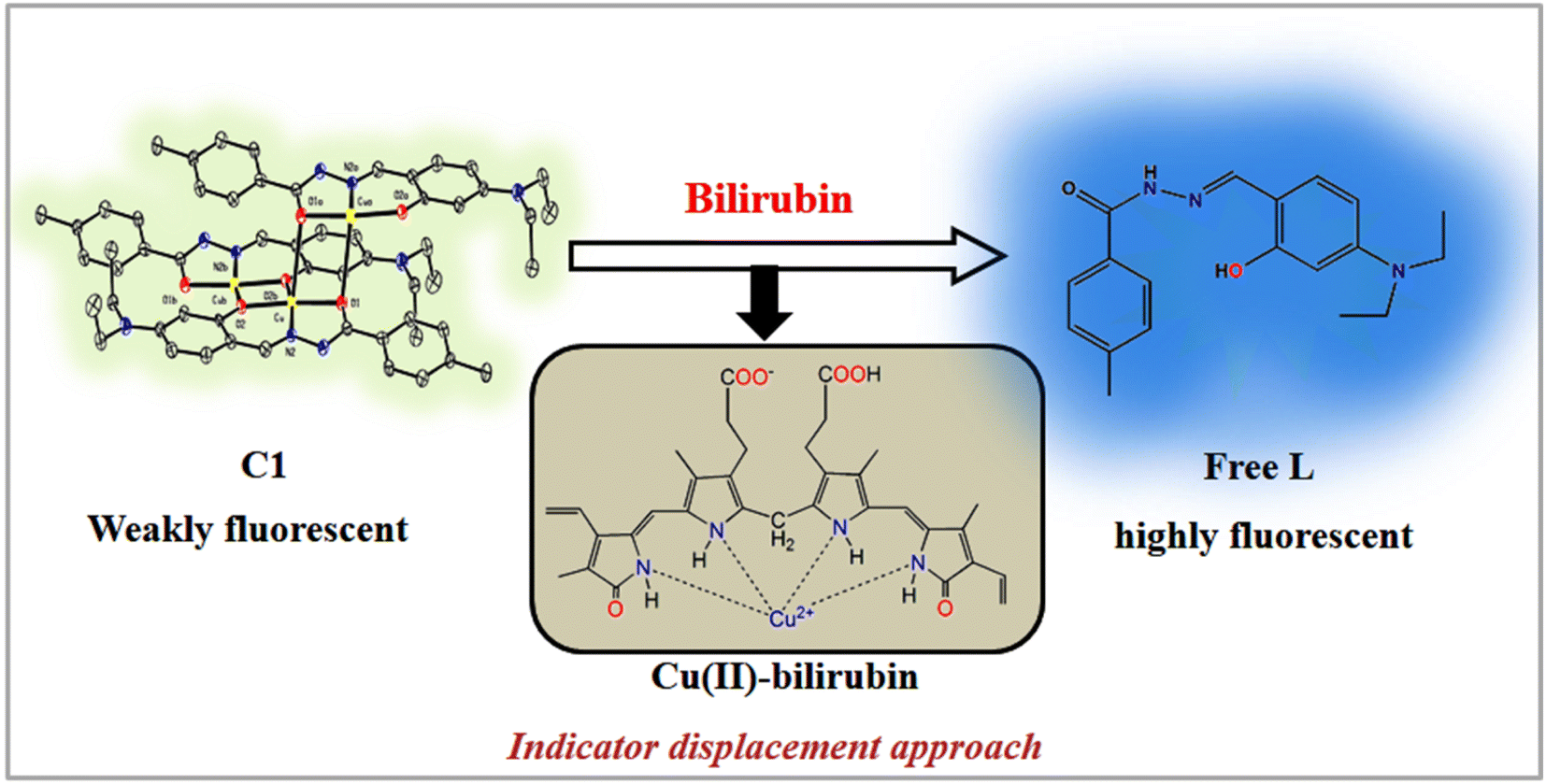 | ||
| Scheme 4 Proposed bilirubin sensing mechanism of C1. | ||
This displacement event is also corroborated by Job's plot (Fig. S15†), mass spectrum (Fig. S16†) and FTIR spectrum (Fig. S17 and S18†) of the C1-bilirubin molecular assembly. In the mass spectrum of the C1-bilirubin molecular assembly, we found peaks at m/z = 647.35, 675.47, and 697.52 for the C1 and m/z = 370.30 for the displaced L as indicators. Again, comparing the FTIR spectrum of the C1-bilirubin molecular assembly with the FTIR spectrum of pure bilirubin123 and C1, we found distinctively different band positions and shapes of those spectra that indicate the formation of new adducts (Fig. S17 and S18†). The characteristics of IR stretching frequencies for free bilirubin and its adduct with C1 are summarized in Table S5.†
However, fluorescence titration indicates that after the addition of a certain amount of bilirubin (2500 μM), the emission intensity remains almost unaltered (Fig. S19†). The displacement equilibrium constant is evaluated using Hill's equation:124,125
| log[Y/(1 − Y)] = nlog[G] + logK, |
Fig. 5 where Y, n, [G] and Kapp represent the fraction of ligand binding sites filled, Hill coefficient, and the concentration of guest (bilirubin) and displacement equilibrium constant, respectively. The value of the Hill coefficient describes the cooperativity of ligand binding in the following way: n > 1, positive cooperativity; n = 1, non-cooperativity and n < 1, negative cooperativity. Y is determined from the following equation: Y = (Fx − F0)/(Fmax − Fx), where F0, Fx, and Fmax are emission intensities of C1 in the absence of bilirubin, at intermediate bilirubin concentration, and at the concentration of complete interaction, respectively. The value of K is 7.78 × 105 M−1 (Fig. S20†). The detection limit for bilirubin, determined by 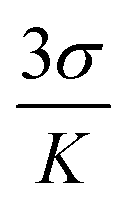 method,126 is 1.15 nM (Fig. S21†). We know that the development of sensors specific to any analyte in the complex background of potentially competing species is very challenging and requires extensive studies for any potential interferences. Hence, the effects of several bio-relevant ions viz. Na+, K+, Ca2+, Mg2+, α-amino acids viz. methionine, arginine, cysteine, proline, lysine, tryptophan, phenylalanine, glycine, aspartic acid and bio-molecules viz. cytochrome C, superoxide dismutase, bisphenol, insulin, creatinine, glutathione and uric acid are tested (Fig. 5a and b). No significant interferences are observed (Fig. S22†).
method,126 is 1.15 nM (Fig. S21†). We know that the development of sensors specific to any analyte in the complex background of potentially competing species is very challenging and requires extensive studies for any potential interferences. Hence, the effects of several bio-relevant ions viz. Na+, K+, Ca2+, Mg2+, α-amino acids viz. methionine, arginine, cysteine, proline, lysine, tryptophan, phenylalanine, glycine, aspartic acid and bio-molecules viz. cytochrome C, superoxide dismutase, bisphenol, insulin, creatinine, glutathione and uric acid are tested (Fig. 5a and b). No significant interferences are observed (Fig. S22†).
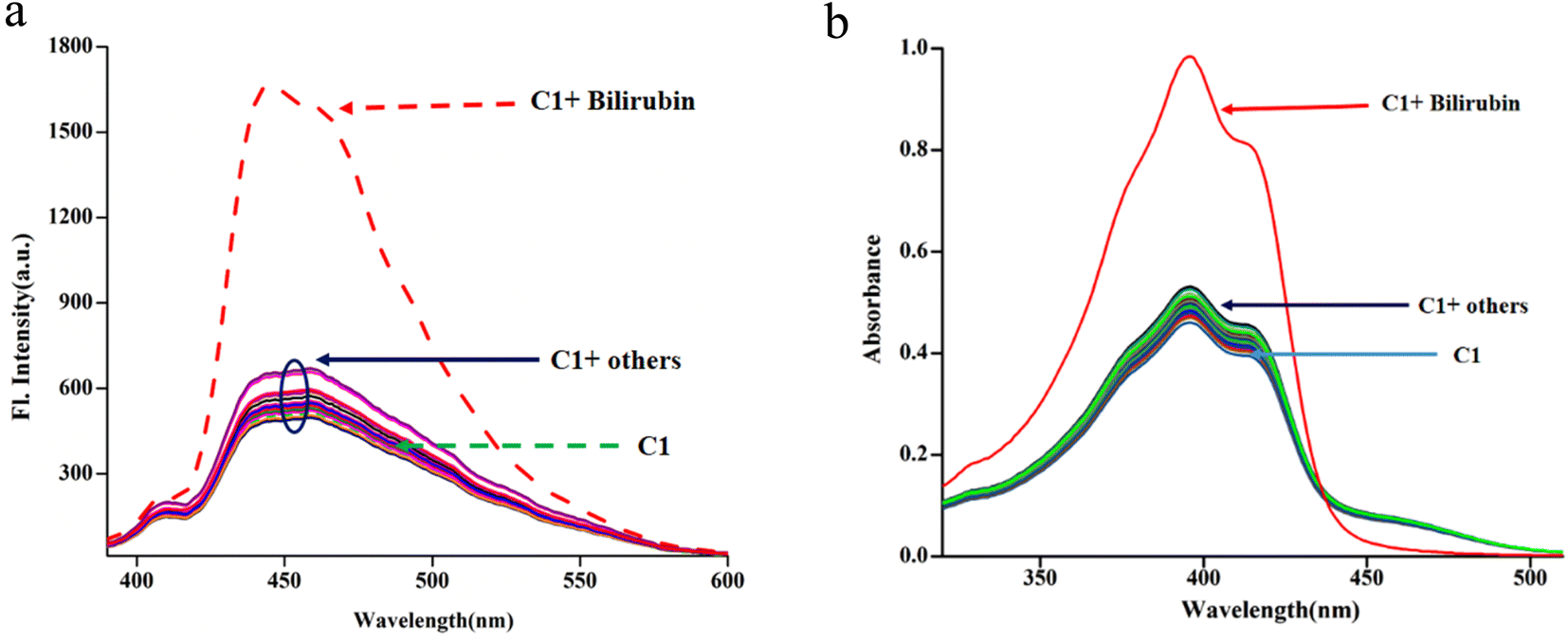 | ||
| Fig. 5 Spectra of C1 (20 μM, λex, 369 nm) in the presence of amino acids, bio-relevant ions and biomolecules viz. Met, Arg, Cys, Pro, Lys, Trp, Phe, Gly, Asp, Na+, K+, Ca2+, Mg2+ and Cyt C, SOD, bisphenol, insulin, creatinine, GSH, uric acid (100 μM; chloroform/water, 1/4, v/v, PBS buffer, pH, 8.0), (a): emission and (b): absorption. | ||
V1 catalyzed H2O2-assisted oxidation of sulphide
Oxidation of sulphide to sulfoxide and sulfone is important for the synthesis of several chemicals and pharmaceuticals. Hence, the catalytic activity of V1 for the oxidation of sulphide in diphenyl sulphide using hydrogen peroxide (Scheme 5) is investigated as a model reaction.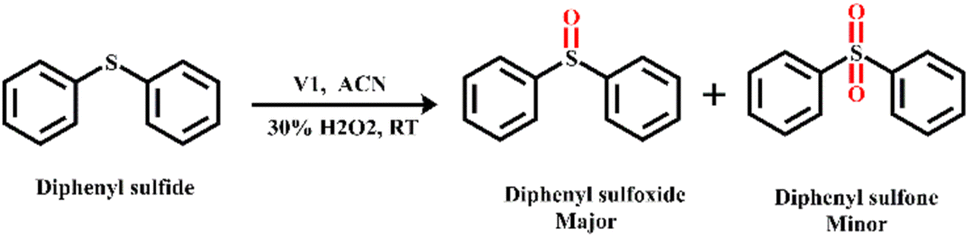 | ||
| Scheme 5 H2O2-assisted oxidation of diphenyl sulphide using V1 as a catalyst. | ||
To optimize reaction conditions, first, the effect of solvent (methanol, DMF, acetonitrile, CHCl3 and toluene) and amount of H2O2 were tested (Table S5, ESI†). The highest yield of the product is observed in acetonitrile (Table S5, entry 3, ESI†). Protic solvents favour yield, whereas less polar solvents are very ineffective (Table S5, entries 4, 5, ESI†). Solvents, such as ethyl cyanide and iso-propanol, give low product yield (Table S6, entries 6, 7, ESI†).
At a low concentration of H2O2 (5 mmol), the yield of the product is low and requires a longer reaction time. The increasing amount of H2O2 from 5 mmol (Table S5,† entry 8) to 10 mmol (run 3) increases the product yield. However, a further increase to 15 mmol of H2O2 (Table S6,† entry 9) did not improve the yield but led to a higher amount of sulfone over sulfoxide.
To expand the scope of the conversion, oxidation reactions with several other sulphides (aliphatic and aromatic sulphides) were investigated and found to have a significantly high conversion rate and selectivity for sulfoxide under mild conditions (Table 1). It is interesting to note that the reaction rate becomes slow with the increased chain length of the alkyl group of the sulphide moiety. This may be due to the orientation of the hydrophobic alkyl chain around the sulphur centre. In addition, the reactivity and the conversion rate depend on the nature of the substituents present in the aryl moiety of ArSMe. The electron donating substituents accelerate the rate, whereas electron withdrawing substituents retard the rate (Table 1, entries 7 and 8). Interestingly, the yield of diphenyl sulfoxide is lower than that of methyl phenyl sulfoxide probably due to the steric effect. The catalyst (V1) exhibited excellent chemo-selectivity towards the thio-centre over other oxidation prone functional groups, viz. CC and OH (Table 2, entries 9 and 10).
| Entry | Sulfide | Conversion (%)b (time, h) | Selectivity of sulfoxideb (%) |
|---|---|---|---|
| a Reaction conditions: sulphide (5 mmol); 30% aqueous H2O2 (10 mmol); CH3CN (10 mL), RT; 50 mg catalyst.b Conversion and selectivity were determined using GC.c Without catalyst. | |||
| 1 |  |
98 (3) | 96 |
| 2 |  |
96 (3) | 91 |
| 3 |  |
91 (5) | 90 |
| 4 |  |
83 (6) | 93 |
| 5 |  |
81 (4) | 81 |
| 6 | 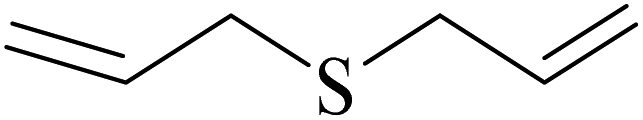 |
78 (4) | 79 |
| 7 | 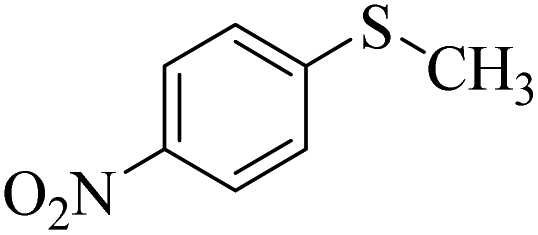 |
98 (3) | 94 |
| 8 | 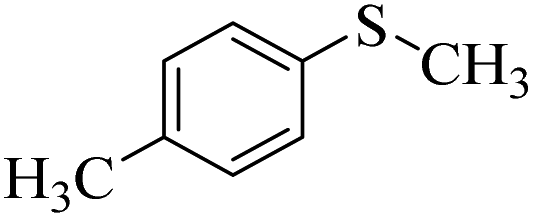 |
91 (3) | 93 |
| 9 | 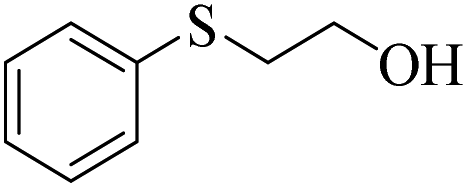 |
90 (5) | 81 |
| 10 | 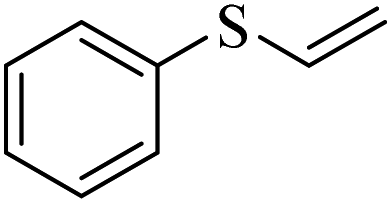 |
81 (5) | 87 |
| 11 | 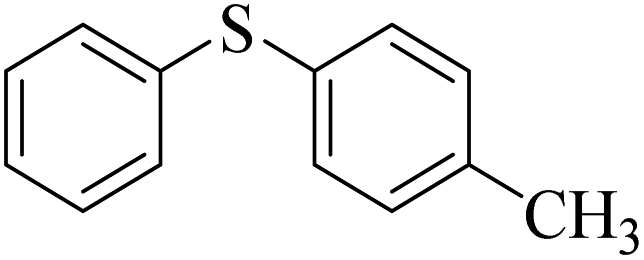 |
85 (5) | 94 |
| 12c | 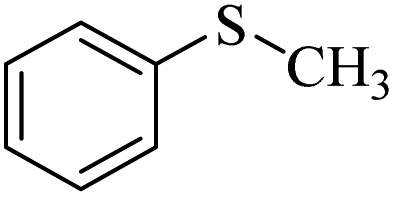 |
Trace | Trace |
| Entry | ArI | Ar–SH | Products | Yieldb (%) |
|---|---|---|---|---|
| a Reaction conditions: aryl iodides (1 mmol), thiophenol (1.1 mmol), water (3 mL), K2CO3 (2 mmol) and C1 (15 mg), temperature (90 °C), and time (12 h).b Yields were determined by GC analysis.c Without catalyst. | ||||
| 1 | 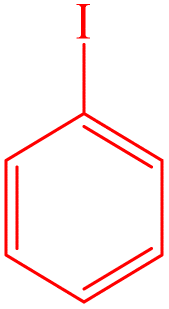 |
 |
 |
98 |
| 2 |  |
 |
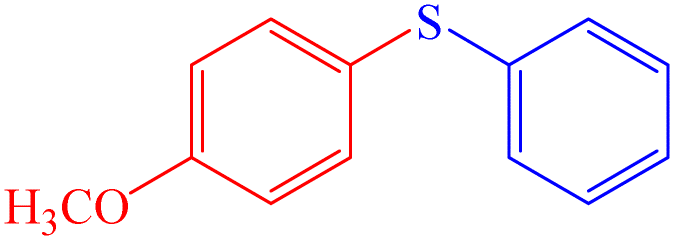 |
70 |
| 3 | 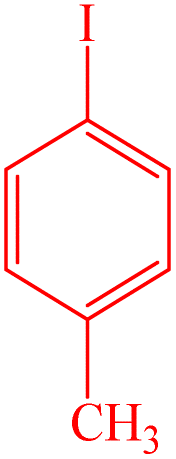 |
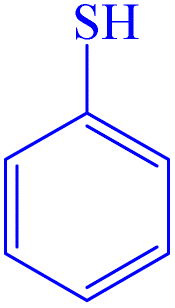 |
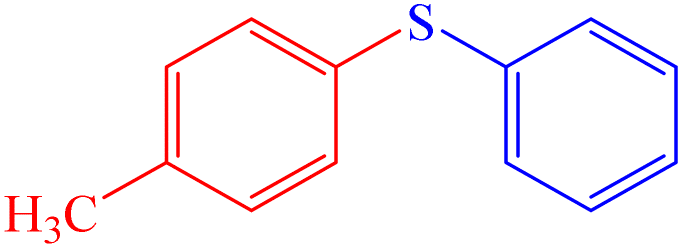 |
81 |
| 4 | 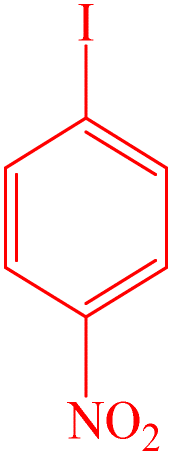 |
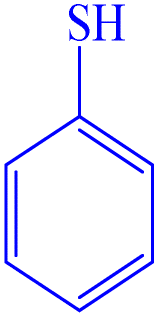 |
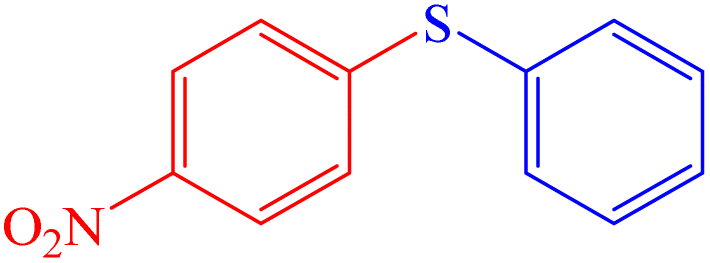 |
89 |
| 5 |  |
 |
 |
38 |
| 6 |  |
 |
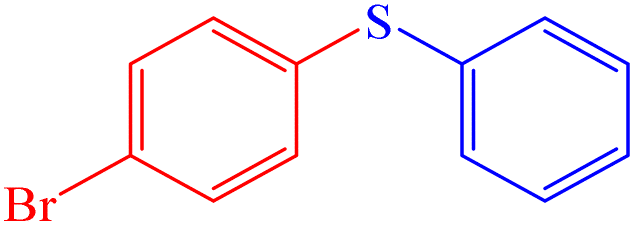 |
91 |
| 7 | 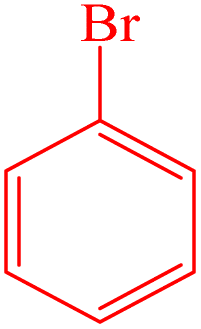 |
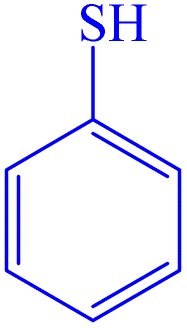 |
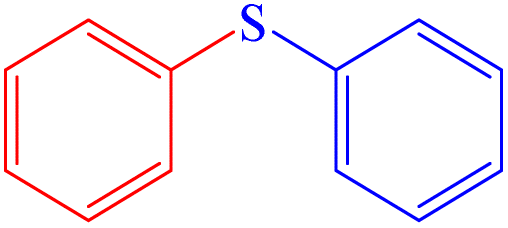 |
60 |
| 8 |  |
 |
 |
39 |
| 9 |  |
 |
 |
70 |
| 10c |  |
 |
No reaction | No reaction |
Aryl-sulphur coupling of thiophenol with aryl iodide using C1 as a catalyst
The catalytic efficiency of C1 for aryl-sulphur (C–S) cross-coupling in a model reaction involving thiophenol and aryl iodide to prepare diaryl sulphide is investigated (Scheme 6).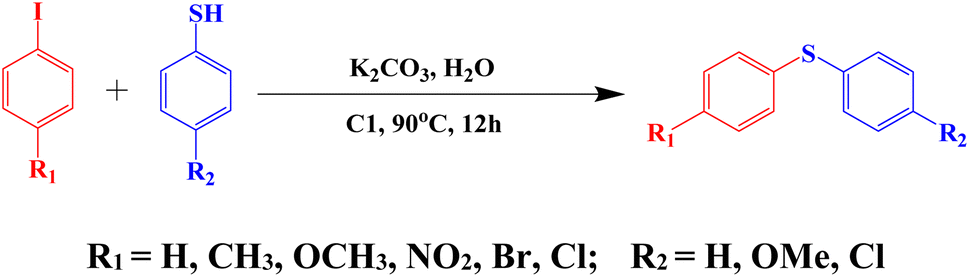 | ||
| Scheme 6 The C–S cross-coupling reaction of thiophenol with aryl iodide using C1 as a catalyst. | ||
The effects of base and solvent are summarized in Table S7 (ESI†). Among different solvents that have been examined for the S-arylation reaction, viz. DMSO, DMF and H2O, it is found that the yield is highest in water, ∼98% (Table S7,† entry 3). The effect of the base is also investigated. Among various inorganic and organic bases tested, such as K2CO3, Et3N, Cs2CO3 and KOH, it is found that the efficiency of KOH is poor, while that of Cs2CO3 is moderate and K2CO3 performs the best. Other bases are not effective at all (Table S7,† entry 3). It is interesting to note that the reaction does not proceed in the absence of any base (Table S7,† entry 6). Extreme moisture-sensitivity and cost of Cs2CO3 compelled us to choose more cheap and stable K2CO3 as the base for the C–S cross-coupling reaction.
To evaluate the scope and limitations and cover a broad range of aryl thiols and aryl iodides, the developed method is tested with substrates having different types of substituents. Under optimized conditions, as outlined in Table 2, the reactions are very clean and smooth with moderate yield. It is observed that aryl iodides with the electron withdrawing group were more reactive than those with the electron donating group. For example, aryl iodides containing methyl/methoxy group produced corresponding biphenyl derivatives in 70–81% yield (Table 2, entries 2 and 3). However, aryl iodides with nitro substituents produced the desired products with an 89% yield in water (Table 2, entry 4). The effect of halogen substituents on the C–S cross-coupling reactions involving thiophenol has the following reactivity order: iodobenzene > bromobenzene > chlorobenzene (Table 2).
Conclusion
Single crystal X-ray structurally characterized 1D copper(II) coordination polymer (C1) of an amide–imine conjugate (L), derived by condensation of 4-methyl-benzoic acid hydrazide (PTA) and 4-(diethylamino)-2-hydroxybenzaldehyde, efficiently recognizes bilirubin via ‘turn-on’ fluorescence through a ligand displacement approach. The displacement equilibrium constant is significantly high, 7.78 × 105 M−1; the detection limit for bilirubin is 1.15 nM. The present method is superior to the available methods in terms of mechanism that imparts high selectivity, specificity and the lowest level of detection. Almost all the reported probes are of unimpressive, undesirable “turn-off” kind. The only one of the “turn on” kind has LOD that is much higher than our present probe. According to the literature, the present probe has the lowest LOD for bilirubin (Table S8†). In addition, C1 acts as a potential green catalyst for the C–S cross-coupling reaction in water. A dimeric oxo–vanadium complex (V1) of the L functions as a promising catalyst for the oxidation of sulphide. The structures of both V1 and L were confirmed by SC-XRD analysis.Conflicts of interest
There are no disagreement to mention.Acknowledgements
We thank USIC, BU for extending SC-XRD facility.References
- G. Arthur, The Amide Linkage: Selected Structural Aspects in Chemistry, Biochemistry, and Materials Science, Wiley-Interscience, 2000 Search PubMed
.
- J. M. Humphrey and A. R. Chamberlin, Chem. Rev., 1997, 97, 2243–2266 CrossRef CAS PubMed
- X. Liu, L. Lin and X. Feng, Chem. Commun., 2009, 6145–6158 RSC
- V. R. Pattabiraman and J. W. Bode, Nature, 2011, 480, 471–479 CrossRef CAS PubMed
- R. C. Larock, Comprehensive Organic Transformation, VCH, New York, 1999, 102 Search PubMed
- S. Roy, S. Roy and G. W. Gribble, Tetrahedron, 2012, 68, 9867–9923 CrossRef CAS
- J. Lee, S. K. Muthaiah and S. H. Hong, Adv. Synth. Catal., 2014, 356, 2653–2660 CrossRef CAS
- S. C. Ghosh, J. S. Y. Ngiam, A. M. Seayad, D. T. Tuan, C. L. L. Chai and A. J. Chen, J. Org. Chem., 2012, 77, 8007–8015 CrossRef CAS PubMed
- R. Cadoni, A. Porcheddu, G. Giacomelli and D. L. Luca, Org. Lett., 2012, 14, 5014–5017 CrossRef CAS PubMed
- D. M. Flanigan, F. Romanov-Michailidis, N. A. White and T. Rovis, Chem. Rev., 2015, 115, 9307–9387 CrossRef CAS PubMed
- P. Noblia, M. Vieites, B. S. Parajo n-Costa, E. J. Baran, H. Cerecetto, P. Draper, M. Gonzalez, O. E. Piro, E. E. Castellano, A. Azqueta, D. L. Cerain A, A. Monge-Vega and D. Gambino, J. Inorg. Biochem., 2005, 99, 443–451 CrossRef CAS PubMed
- Y. Shechter, I. Goldwaser, M. Mironchik, M. Fridkin and D. Gefel, Coord. Chem. Rev., 2003, 237, 3–11 CrossRef CAS
- A. M. B. Bastos, J. G. da Silva, P. I. S. Maia, V. M. Deflon, A. A. Batista, A. V. M. Ferreira, L. M. Botion, E. Niquet and H. Beraldo, Polyhedron, 2008, 27, 1787–1794 CrossRef CAS
- S. Naskar, M. Corbella, A. J. Blake and S. K. Chattopadhyay, Dalton Trans., 2007, 11, 1150–1159 RSC
- L. Reytman, O. Braitbard, J. Hochman and E. Y. Tshuva, Inorg. Chem., 2016, 55, 610–618 CrossRef CAS PubMed
- K. K. Bedia, O. Elçin, U. Seda, K. Fatma, S. Nathaly, R. A. Sevim and A. Dimoglo, Eur. J. Med. Chem., 2006, 41, 1253–1261 CrossRef CAS PubMed
- P. Melnyk, V. Leroux, C. Sergheraert, P. Grellier and C. Sergheraert, Bioorg. Med. Chem. Lett., 2006, 16, 31–35 CrossRef CAS PubMed
- R. R. Eady, Coord. Chem. Rev., 2003, 237, 23–30 CrossRef CAS
- A. Butler and J. V. Walker, Chem. Rev., 1993, 93, 1937–1944 CrossRef CAS
- K. H. Thompson, J. H. McNeill and C. Orvig, Chem. Rev., 1999, 99, 2561–2572 CrossRef CAS PubMed
- M. Nikoorazm, A. Ghorbani-Choghamarani and N. Noori, Appl. Organometal. Chem., 2015, 29, 328–333 CrossRef CAS
- M. C. Carreno, Chem. Rev., 1995, 95, 1717–1760 CrossRef CAS
- X. F. Wu, Tetrahedron Lett., 2012, 53, 4328–4331 CrossRef CAS
- V. Conte, F. Di Furia and G. Licini, Appl. Catal., A, 1997, 157, 335–361 CrossRef CAS
- B. Li, A. H. Liu, L. N. He, Z. Z. Yang, J. Gao and K. H. Chen, Green Chem., 2012, 14, 130–135 RSC
- H. J. Kim and J. S. Kim, Tetrahedron Lett., 2006, 47, 7051–7055 CrossRef CAS
- M. Hirano, S. Yakabe, J. H. Clark and T. Morimoto, J. Chem. Soc., Perkin Trans. 1, 1996, 1, 2693–2698 RSC
- A. Rostami, B. Tahmasbi, F. Abedi and Z. Shokri, J. Mol. Catal. A: Chem., 2013, 378, 200–205 CrossRef CAS
- A. M. henkin, G. Leitus and R. Neumann, J. Am. Chem. Soc., 2010, 132, 11446–11448 CrossRef PubMed
- J. Gao, H. Guo, S. Liu and M. Wang, Tetrahedron Lett., 2007, 48, 8453–8455 CrossRef CAS
- R. Das and D. Chakraborty, Tetrahedron Lett., 2010, 51, 6255–6258 CrossRef CAS
- R. Das and D. Chakraborty, Synthesis, 2011, 2, 277–280 Search PubMed
- D. Chakraborty, P. Malik and V. K. Goda, Appl. Organomet. Chem., 2012, 26, 21–26 CrossRef CAS
- P. Malik and D. Chakraborty, Tetrahedron Lett., 2012, 53, 5652–5655 CrossRef CAS
- R. Fazaeli, H. Aliyan, S. P. Foroushani, Z. Mohagheghian and Z. Heidari, Iran J. Catal., 2013, 3, 129–137 CAS
- F. Lebon, N. Boggetto, M. Ledecq, F. Durant, Z. Benatallah, S. Sicsic and M. Reboud-Ravaux, Biochem. Pharmacol., 2002, 63, 1863 CrossRef CAS PubMed
- B. Kumari, S. Kundu, K. Ghosh, M. Banerjee, S. K. Pradhan, S. M. Islam, P. Brandao, V. Felix and D. Das, Dalton Trans., 2018, 47, 14008 RSC
- Y. Zhou, J. Won, J. Y. Lee and J. Yoon, Chem. Commun., 2011, 47, 1997 RSC
- Z. Li, X. Lou, Z. Li and J. A. Qin, ACS Appl. Mater. Interfaces, 2009, 2, 232 CrossRef PubMed
- Y. Zhou and J. Yoon, Chem. Soc. Rev., 2012, 41, 52 RSC
- I. P. Beletskaya and V. P. Ananikov, Chem. Rev., 2011, 111, 1596–1636 CrossRef CAS PubMed
- A. Gangjee, Y. Zeng, T. Talreja, J. J. McGuire, R. L. Kisliuk and S. F. Queener, J. Med. Chem., 2007, 50, 3046–3053 CrossRef CAS PubMed
- I. P. Beletskaya and A. V. Cheprakov, Copper in cross-coupling reactions: the post-Ullmann chemistry, Coord. Chem. Rev., 2004, 248, 2337–2364 CrossRef CAS
- S. Di Bella and I. Fragala, New J. Chem., 2002, 26, 285–290 RSC
- G. De Martino, M. C. Edler, G. La Regina, A. Cosuccia, M. C. Barbera, D. Barrow, R. I. Nicholson, G. Chiosis, A. Brancale, E. Hamel, M. Artico and R. Silvestri, J. Med. Chem., 2006, 49, 947–954 CrossRef CAS PubMed
- A. Jabbari, H. Mahdavi, M. Nikoorazm and A. Ghorbani-Choghamarani, J. Porous Mater., 2015, 22, 1111–1118 CrossRef CAS
- G. Y. Li, Angew. Chem., Int. Ed., 2001, 40, 1513–1516 CrossRef CAS PubMed
- M. Murata and S. L. Buchwald, Tetrahedron, 2004, 60, 7397–7403 CrossRef CAS
- M. A. Fernández-Rodríguez, Q. Shen and J. F. Hartwig, Chem.–Eur. J., 2006, 12, 7782–7796 CrossRef PubMed
- T. Itoh and T. Mase, Org. Lett., 2004, 6, 4587–4590 CrossRef CAS PubMed
- Y. Zhang, K. N. Ngeow and J. Y. Ying, Org. Lett., 2007, 9, 3495–3498 CrossRef CAS PubMed
- S. Jammi, P. Barua, L. Rout, P. Saha and T. Punniyamurthy, Tetrahedron Lett., 2008, 49, 1484–1487 CrossRef CAS
- Y. C. Wong, T. T. Jayanth and C. H. Cheng, Org. Lett., 2006, 8, 5613–5616 CrossRef CAS PubMed
- V. P. Reddy, K. Swapna, A. V. Kumar and K. R. Rao, J. Org. Chem., 2009, 74, 3189–3191 CrossRef CAS PubMed
- A. Correa, M. Carril and C. Bolm, Angew. Chem., Int. Ed., 2008, 47, 2880–2883 CrossRef CAS PubMed
- W. Y. Wu, J. C. Wang and F. Y. Tsai, Green Chem., 2009, 11, 326–329 RSC
- J. She, Z. Jiang and Y. G. Wang, Tetrahedron Lett., 2009, 50, 593–596 CrossRef CAS
- F. Y. Kwong and S. L. Buchwald, Org. Lett., 2002, 4, 3517–3520 CrossRef CAS PubMed
- C. G. Bates, P. Saejueng, M. Q. Doherty and D. Venkataraman, Org. Lett., 2004, 6, 5005–5008 CrossRef CAS PubMed
- Y. J. Chen and H. H. Chen, Org. Lett., 2006, 8, 5609–5612 CrossRef CAS PubMed
- M. S. Kabir, M. Lorenz, M. L. Van-Linn, O. A. Namjoshi, S. Ara and J. M. Cook, J. Org.Chem., 2010, 75, 3626–3643 CrossRef CAS PubMed
- R. Xu, J. Wan, H. Mao and Y. Pan, J. Am. Chem. Soc., 2010, 132, 15531–15533 CrossRef CAS PubMed
- F. Luo, C. Pan, L. Li, F. Chen and J. Cheng, Chem. Commun., 2011, 47, 5304–5306 RSC
- D. K. Dumbrea, P. R. Selvakannana, S. K. Patil, V. R. Choudhary and S. K. Bhargava, Appl. Catal., A, 2014, 476, 54–60 CrossRef
- S. Khanra, S. Ta, A. Paladhi, M. Ghosh, S. Ghosh, S. K. Hira, P. P. Manna, P. Brandão, V. Félix and D. Das, Chem. Commun., 2020, 56, 6563–6566 RSC
- D. L. Ma, V. P. Y. Ma, D. S. H. Chan, K. H. Leung, H. Z. He and C. H. Leung, Coord. Chem. Rev., 2012, 256, 3087–3113 CrossRef CAS
- L. You, D. Zha and E. V. Anslyn, Chem. Rev., 2015, 115(15), 7840–7892 CrossRef CAS PubMed
- D. L. Ma, W. L. Wong, W. H. Chung, F. Y. Chan, P. K. So, T. S. Lai, Z. Y. Zhou, Y. C. Leung and K. Y. Wong, Angew. Chem., Int. Ed., 2008, 47, 3735–3739 CrossRef CAS PubMed
- Y. Zhou and J. Yoon, Chem. Soc. Rev., 2012, 41, 52–67 RSC
- M. Bonizzoni, L. Fabbrizzi, G. Piovani and A. Taglietti, Tetrahedron, 2004, 60, 11159–11162 CrossRef CAS
- G. Klein and J. L. Reymond, Angew. Chem., 2001, 113, 1821–1823 CrossRef
- H. Aït-Haddou, S. L. Wiskur, V. M. Lynch and E. V. Anslyn, J. Am. Chem. Soc., 2001 Search PubMed
- S. Das, A. Banerjee, S. Lohar, B. Sarkar, S. K. Mukhopadhyay, J. S. Matalobos, A. Sahana and D. Das, New J. Chem., 2014, 38, 2744–2747 RSC
- P. A. Zunszain, J. Ghuman, A. F. McDonagh and S. Curry, J. Mol. Biol., 2008, 381, 394–406 CrossRef CAS PubMed
- K. Abha, J. Nebu, J. S. A. Devi, R. S. Aparna, R. R. Anjana, A. O. Aswathy and S. George, Sens. Actutors, B, 2019, 282, 300–308 CrossRef CAS
- P. Novotna, F. Kralik and M. Urbanova, Biophys. Chem., 2015, 205, 41–50 CrossRef CAS PubMed
- M. Martelanc, L. Ziberna, S. Passamonti and M. Franko, Talanta, 2016, 154, 92–98 CrossRef CAS PubMed
- B. Batra, S. Lata, J. S. Rana and C. Shekhar, Biosens. Bioelectron., 2013, 44, 64–69 CrossRef CAS PubMed
- L. Ngashangva, V. Bachu and P. Goswami, J. Pharm. Biomed. Anal., 2019, 162, 272–285 CrossRef CAS PubMed
- X. P. Li and Z. Rosenzweig, J. Anal. Chim. Acta, 1997, 353, 263–273 CrossRef CAS
- S. S. Tikmani, H. J. Warraich, F. Abbasi, A. G. Rizvi, L. Darmstadt and A. K. Zaidi, Trop. Med. Int. Health, 2010, 15, 502–507 CrossRef CAS PubMed
- S. Sherlock and J. Dooley, Diseases of the Liver and Biliary System, Blackwell Science Ltd, 2007, pp. 205–218 Search PubMed
- S. C. Kazmierczak, A. F. Robertson, P. G. Catrou, B. L. Briley and G. R. Gourley, Clin. Chem., 2002, 48, 1096–1097 CrossRef CAS
- A. Sevillanocabeza, M. C. Alberolagarbi and P. Campinsfalco, Microchem. J., 1994, 50, 14–27 CrossRef CAS
- J. Zelenka, M. Lenícek, L. Muchová, M. Jirsa, M. Kudla, P. Balaz, M. Zadinová, J. D. Ostrow, R. J. Wong and L. Vítek, J. Chromatogr. B, 2008, 867, 37–42 CrossRef CAS PubMed
- S. Itoh, K. Isobe and S. Onishi, J. Chromatogr. A, 1999, 848, 169–177 CrossRef CAS PubMed
- C. Lu, J. M. Lin and C. W. Huie, Talanta, 2004, 63, 333–337 CrossRef CAS PubMed
- H. S. Lee, M. M. Karim, S. M. Alam and S. H. Lee, Luminescence, 2007, 22, 331–337 CrossRef CAS PubMed
- J. W. Liu, X. M. Zhou and M. G. Zhang, Chin. J. Chromatogr., 1998, 16, 369 CAS
- Z. Nie and Y. S. Fung, Electrophoresis, 2008, 29, 1924–1931 CrossRef CAS PubMed
- K. Kurosaka, S. Senba, H. Tsubota and H. Kondo, Clin. Chim. Acta, 1998, 269, 125–136 CrossRef CAS PubMed
- A. Kosaka, C. Yamamoto, Y. Morishita and K. Nakane, Clin. Biochem., 1987, 20, 451–458 CrossRef CAS PubMed
- V. Amendola, L. Fabbrizzi, F. Forti, M. Licchelli, C. Mangano, P. Pallavicini, A. Poggi, D. Sacchi and A. Taglieti, Coord. Chem. Rev., 2006, 250, 273–299 CrossRef CAS
- K. Rurack and U. Resch-Genger, Chem. Soc. Rev., 2002, 31, 116–127 RSC
- S. Das, M. Dutta and D. Das, Anal. Methods, 2013, 5, 6262–6285 RSC
- T. Senthilkumar and S. K. Asha, Macromolecules, 2015, 48, 3449–3461 CrossRef CAS
- Y. Du, X. Li, X. Lv and Q. Jia, ACS Appl. Mater. Interfaces, 2017, 9, 30925–30932 CrossRef CAS PubMed
- C. Xia, Y. Xu, M. M. Cao, Y. P. Liu, J. F. Xia, D. Y. Jiang, G. H. Zhou, R. J. Xie, D. F. Zhang and H. L. Li, Talanta, 2020, 212, 120795 CrossRef CAS PubMed
- M. Santosh, S. R. Chinnadayyala, A. Kakoti and P. Goswami, Biosens. Bioelectron., 2014, 59, 370–376 CrossRef PubMed
- S. K. Anand, M. R. Mathew and K. G. Kumar, J. Fluoresc., 2019, 30, 63–70 CrossRef PubMed
- M. Jayasree, R. S. Aparna, R. R. Anjana, J. S. A. Devi, N. John, K. Abha, A. Manikandan and S. George, Anal. Chim. Acta, 2018, 1031, 152–160 CrossRef CAS PubMed
- S. Ellairaja, K. Shenbagavalli, S. Ponmariappan and V. S. Vasantha, Biosens. Bioelectron., 2017, 91, 82–88 CrossRef CAS PubMed
- S. M. Babin and R. M. Sova, Anal. Chem. Insights, 2014, 9, 59–64 CrossRef CAS PubMed
- S. M. Wabaidur, G. E. Eldesoky and Z. A. Alothman, Luminescence, 2018, 33, 625–629 CrossRef CAS PubMed
- R. R. Anjana, J. S. A. Devi and M. Jayasree, Microchim. Acta, 2018, 185, 11 CrossRef PubMed
- Y. Wei, F. X. Jin, H. Z. Guo and Y. J. Dan, Anal. Bioanal. Chem., 2018, 410, 6459–6468 CrossRef PubMed
- V. Srinivasan, M. A. Jhonsi, N. Dhenadhayalan, K. C. Lin, D. A. Ananth, T. Sivasudha, R. Narayanaswamy and A. Kathiravan, Spectrochim. Acta, Part A, 2019, 221, 117150 CrossRef CAS PubMed
- S. K. Makkad and S. K. Asha, ACS Appl. Polym. Mater., 2019, 1, 1230–1239 CrossRef CAS
- M. Zhang, L. Xu, Q. Ma, H. Yu, H. Fang, Z. Lin, Q. Zhang and Z. Chen, ACS Appl. Mater. Interfaces, 2018, 10, 42155–42166 CrossRef CAS PubMed
- W. Bian, N. Zhang and C. Jiang, Luminescence, 2011, 26, 54–58 CrossRef CAS PubMed
- W. Bian, N. Zhang and L. Wang, Anal. Sci., 2010, 26, 785–789 CrossRef CAS PubMed
- B. T. Nguyen and E. V. Anslyn, Coord. Chem. Rev., 2006, 250, 3118–3127 CrossRef CAS
- S. Das, S. Guha, A. Banerjee, S. Lohar, A. Sahana and D. Das, Org. Biomol. Chem., 2011, 9, 7097–7104 RSC
- S. Ta, M. Ghosh, K. Ghosh, P. Brandão, V. Félix, S. K. Hira, P. P. Manna and D. Das, ACS Appl. Bio Mater., 2019, 2, 2802–2811 CrossRef CAS PubMed
- B. Mondal, M. G. Drew and T. Ghosh, Inorg. Chim. Acta, 2009, 362, 3303–3308 CrossRef CAS
- E. Seena, N. Mathew, M. Kuriakose and M. P. Kurup, Polyhedron, 2008, 27, 1455–1462 CrossRef CAS
- H. H. Monfared, R. Bikas, P. M. Anarjan, A. J. Blake, V. Lippolis, N. B. Arslan and C. Kazak, Polyhedron, 2014, 69, 90–102 CrossRef
- R. Brodersen, J. Biol. Chem., 1979, 254, 2364–2369 CrossRef CAS PubMed
- Y. Q. Weng, F. Yue, Y. R. Zhong and B. H. Ye, Inorg. Chem., 2007, 46, 7749–7755 CrossRef CAS PubMed
- O. Sahin and M. Yilmaz, Tetrahedron, 2011, 67, 3501–3508 CrossRef CAS
- L. J. Tang and M. J. Cai, Sens. Actuators, B, 2012, 173, 862–867 CrossRef CAS
- M. Maity, S. Das and N. C. Maiti, Phys. Chem. Chem. Phys., 2014, 16, 20013–20022 RSC
- S. Shinkai, M. Ikeda, A. Sugasaki and M. Takeuchi, Acc. Chem. Res., 2001, 34, 494–503 CrossRef CAS PubMed
- M. Ghosh, S. Ta, J. S. Matalobos and D. Das, Dalton Trans., 2018, 47, 11084–11090 RSC
- A. Ghorai, J. Mondal, R. Chandra and G. K. Patra, Dalton Trans., 2015, 44, 13261–13271 RSC
- M. Ghosh, S. Ta, S. Lohar, S. Das, P. Brando, V. Felix and D. Das, J. Mol. Recognit., 2018, 32, 2771–2778 CrossRef PubMed
Footnote |
| † Electronic supplementary information (ESI) available: NMR, Mass, FTIR, UV-vis, fluorescence spectra. CCDC 1837654, 1837659 and 1837665. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ra00702b |
| This journal is © The Royal Society of Chemistry 2023 |