
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynchronized electrochemical detection of hydroquinone and catechol in real water samples using a Co@SnO2–polyaniline composite
Qasar Saleema,
Sammia Shahida,
Mohsin Javeda,
Shahid Iqbal *b,
Abdur Rahimc,
Sana Mansoora,
Ali Bahadur*d,
Nasser S. Awwade,
Hala A. Ibrahiumfg,
Rasmiah S. Almufarijh and
Eslam B. Elkaeedi
*b,
Abdur Rahimc,
Sana Mansoora,
Ali Bahadur*d,
Nasser S. Awwade,
Hala A. Ibrahiumfg,
Rasmiah S. Almufarijh and
Eslam B. Elkaeedi
aDepartment of Chemistry, School of Science, University of Management and Technology, Lahore 54770, Pakistan
bDepartment of Chemistry, School of Natural Sciences (SNS), National University of Science and Technology (NUST), H-12, Islamabad, 46000, Pakistan. E-mail: shahidgcs10@yahoo.com
cDepartment of Chemistry, COMSATS University Islamabad, Pakistan
dDepartment of Chemistry, College of Science and Technology, Wenzhou-Kean University, Wenzhou 325060, China. E-mail: abahadur@wku.edu.cn
eChemistry Department, Faculty of Science, King Khalid University, P. O. Box 9004, Abha 61413, Saudi Arabia
fBiology Department, Faculty of Science, King Khalid University, P. O. Box 9004, Abha 61413, Saudi Arabia
gDepartment of Semi Pilot Plant, Nuclear Materials Authority, P. O. Box 530, El Maadi, Egypt
hDepartment of Chemistry, College of Science, Princess Nourah bint Abdulrahman University, P. O. Box 84428, Riyadh 11671, Saudi Arabia
iDepartment of Pharmaceutical Sciences, College of Pharmacy, AlMaarefa University, Riyadh 13713, Saudi Arabia
First published on 29th March 2023
Abstract
The conductive composite Co@SnO2–PANI was successfully synthesized using hydrothermal/oxidative synthesis. Using differential pulse voltammetry, a glassy carbon electrode modified with a CoSnO2–PANI (polyaniline)-based electrochemical biosensor has been created for the quick detection of two phenolics, hydroquinone (Hq) and catechol (Cat). Differential pulse voltammetry (DPV) measurements revealed two well-resolved, strong peaks for GCE@Co–SnO2–PANI, which corresponded to the oxidation of Hq and Cat at 275.87 mV and +373.76 mV, respectively. The oxidation peaks of Hq and Cat mixtures were defined and separated at a pH of 8.5. High conductivity and remarkable selectivity reproducibility was tested by electrochemical impedance spectroscopy, chronoamperometry, and cyclic voltammetry techniques in standard solution and real water samples. The proposed biosensor displayed a low detection limit of 4.94 nM (Hq) and 1.5786 nM (Cat), as well as a large linear range stretching from 2 × 10−2 M to 2 × 10−1 M. The real-sample testing showed a good recovery for the immediate detection of Hq (96.4% recovery) and Cat (98.8% recovery) using the investigated sensing apparatus. The synthesized biosensor was characterized by XRD, FTIR, energy dispersive spectroscopy and scanning electron microscopy.
Introduction
Catechol (Cat) and hydroquinone (Hq), are two frequently coexisting isomers of di-hydroxy-benzene, which are widely used in industry but are incredibly poisonous and difficult to decompose in the natural environment.1 Ecosystems have a high level of toxicity of these chemicals.2 Catechol and its isomers are included as a category of hazardous water contaminants in both the EPA and EU databases.3–5 Due to the similarities in their chemical structures and properties, simultaneous detection of Hq and Cat is difficult. Numerous techniques, including liquid chromatography, chemiluminescence, colorimetric, and electrochemical approaches, have been used for their detection to date.6–8 Electrochemical detection technologies are advantageous for real-time, on-site detection. In recent years, extensive progress has been made in detecting Hq and Cat in freshwater; although there is a long way to go, detection can be achieved in seawater with complex conditions.9,10More than 70% of Earth's surface is covered by ocean, which provides us with a wealth of resources. However, owing to human activities in recent years, it has become more and more contaminated.11 Phenolic concentrations are quite high in coastal waters, mostly as a result of industrial pollution; stable algae in the intertidal zone is another source.12 Due to bioaccumulation or direct contact, water pollution with hydroquinone (Hq) and catechol (Cat) may be detrimental to human or aquatic health. Additionally, it encourages marine mussel adhesion and erosion to maritime infrastructure, which is detrimental to the development of marine resources.13–15 Pollutants like hydroquinone and its isomers must be detected in saltwater if the ocean economy is to grow sustainably. Detection pretreatment methods, such as decreasing pH and removing contaminant ions,8,16,17 are hindered by seawater's alkaline environment and ion concentration. It is therefore crucial to develop an electrode material capable of direct detection of Hq and Cat in marine settings.
Co@SnO2 is extensively used in the domains of heterogeneous catalysts,18 super-capacitors,19 and electrochemical biosensors.20 Co@SnO2 possesses the ideal empty electron orbit for Co(II) and Co(III) coexistence due to its many oxidation states and stability of redox charge transfer, making it simple to absorb electrons throughout the reaction phase and promoting the forward reaction.21 Co@SnO2 can primarily sustain its catalytic activity in an alkaline environment, allowing the detection of Hq and Cat in seawater. To improve the electrical conductivity of Co@SnO2, a conductive polyaniline (PANI) composite with Co@SnO2 was synthesized.22 Electron interaction between PANI and phenolic isomers may enhance the response signal. Furthermore, the diffusion of Hq and Cat during the detection process may be accelerated by the presence of a lot of mesoporous PANI networks.23 Modifying PANI with Co@SnO2 nanomaterial for the detection of Hq and Cat in saltwater is thus fascinating.24
This work fabricated a modified glassy carbon electrode GCE@Co@SnO2–PANI, for the electrochemical detection of Hq and Cat. Under mildly alkaline circumstances (pH 8.5), as the pH of seawater may vary between 7.4 and 8.5 due to the absorption of ambient carbon dioxide, simultaneous detection of Cat and Hq was accomplished with high sensitivity and remarkable stability. The sensor exhibited a large linear range from 2 × 10−2 to 2 × 10−1 M (Hq & Cat) and a low detection limit of 4.94 nM (Hq) and 1.5786 nM (Cat). Detecting Hq and Cat in real water using this method provided encouraging results. Cat was recovered 98.8% of the time in saltwater samples, whereas Hq was recovered 96.4% of the time. It successfully detects phenolic pollutants in real water samples, but when compared to the available online literature on this subject the results of synthesized biosensors were outshined.
Experimental
Chemicals
All of the compounds utilized in this study were of analytical grade and required no additional purification. Co(Ac)2·4H2O, ethanol, citrate (CA), NH3H2O (25–28 wt%), Na2HPO4·12H2O, catechol (Cat), di-ethyl tin dichloride [Sn(C2H5)2Cl2], polyvinylpyrrolidone-40 (PVP-40) as a capping agent, and hydroquinone (Hq) were all provided by Sinopharm Chemical Regent Co., Ltd. Chengdu Organic Chemicals Company Ltd. The solutions were prepared in double distilled water. A real water sample was prepared in a saline sample obtained from a pharmaceutical source.Synthesis of SnO2 and Co@SnO2 particles
4.802 g of di-ethyl tin dichloride (240.1 g mol−1) was added in a 20 mL mixture of ethanol and distilled water (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) in two beakers followed by 0.5 g of Co(Ac)2·4H2O in one of the beakers before beginning the sonication process. The mixture was allowed to stir for 30 minutes. 12.5 mL of NH3(aq) was added dropwise to a solution containing 4.0 g of PVP-40 followed by vigorous stirring. After two hours, homogeneous slurries in both beakers were generated and placed in 40 mL stainless steel Teflon-lined autoclaves. The autoclaves were sealed and heated at 150 °C for 180 min. Small amounts of ethanol and water were applied for washing while filtering the centrifuged contents at room temperature. The SnO2 and Co@SnO2 nanoparticles were dried at 60 °C for 48 hours in the oven.
:1) in two beakers followed by 0.5 g of Co(Ac)2·4H2O in one of the beakers before beginning the sonication process. The mixture was allowed to stir for 30 minutes. 12.5 mL of NH3(aq) was added dropwise to a solution containing 4.0 g of PVP-40 followed by vigorous stirring. After two hours, homogeneous slurries in both beakers were generated and placed in 40 mL stainless steel Teflon-lined autoclaves. The autoclaves were sealed and heated at 150 °C for 180 min. Small amounts of ethanol and water were applied for washing while filtering the centrifuged contents at room temperature. The SnO2 and Co@SnO2 nanoparticles were dried at 60 °C for 48 hours in the oven.
Synthesis of polyaniline (PANI) and Co@SnO2–PANI particles
Two 250 mL beakers were filled with a mixture of 100 mL of absolute ethanol and double deionized water (1:1), then put in an ice bath. 10 mL of 37% HCl and 2 mL of 98% aniline were added to each beaker, along with 0.01 mg of Co@SnO2 particles in one of the beakers. The solution was sonicated for almost 1 hour. 0.025 M solution of an oxidant (ammonium persulfate) was poured drop by drop into the beakers through two burettes after every ten seconds. After 24 hours, a clear filtrate and blue-black polymer (PANI) and Co@SnO2–PANI particles were produced. By centrifuging, the blue-black particles were separated from the solution. The particles were washed with DDIW and acetone. The synthesized particles were dried in an oven at 60 °C for 48 hours.
Instrumentation
The morphology and composition of synthesized particles, SnO2, polyaniline (PANI), and Co@SnO2–PANI were determined by scanning electron microscopy (Nova NanoSEM S-450). The compound's particle size and phases were identified using X-ray diffraction (XRD) patterns by an X-ray diffractometer (D-8 Bruker). Bonding within synthesized material was confirmed by the FTIR technique by using Thermo-Nicolet iS50 FTIR Spectrometer (USA). Electrochemical experiments were recorded using a Gamry-3000 model 43120 electrochemical workstations with traditional three-electrode systems in 0.1 M Na2HPO4 buffer solution (PBS) at pH 8.5. The working electrodes were the bare GCE and modified electrodes (GCE@SnO2, GCE@PANI, GCE@Co@SnO2, and GCE@Co@SnO2-PANI), while the counter and reference electrodes were a platinum (Pt) wire electrode and an Ag/AgCl electrode that had been saturated with 3 M KCl. The pH of the mixture was determined using PSH-3D.Electrode preparations
A Gamry polishing pad and aluminum slurry (3 μm) were used to polish the Gamry glassy carbon electrodes (GCE; 0.37714 cm2). After 2 hours of polishing, the electrodes were sonicated in a DDIW/ethanol solution for 5 minutes. The cleaned electrodes were warmed for 60 minutes at 60 °C to dry them out. SnO2, Co@SnO2, PANI, and Co@SnO2–PANI slurries were independently produced in 10 μL acetone and applied to four glassy carbon electrodes. With 10 μL pipettes, slurries were cast onto the active face of glassy carbon electrodes. For 10 minutes, electrodes were held under a 60 watt illuminated Philips bulb to achieve a dry coating. The electrodes that had been prepared were identified as bare GCE, GCE@SnO2, GCE@PANI, GCE@Co@SnO2, and GCE@Co@SnO2–PANI.Results and discussion
The phase structures of the SnO2, Co@SnO2, PANI, and Co@SnO2–PANI composite were determined with XRD data. As in Fig. 1(a-1), the PANI spectra show large diffraction peaks at 23, 26, and 28, which correspond to the planes (002) and (100). At values of, the diffraction peak is at 18, 31, 36, 38, 45, 55, 59, and 65 in Co@SnO2–PANI nanocomposite correlates to the distinct planes (110), (011), (020), (021), and (020). The spectrum of Co@SnO2 was matched with data bank JCPDS card no. 96-150-8984. The distinct peaks and strong intensity indicate that Co@SnO2–PANI has the superior crystallographic quality and SnO2 was successfully doped with cobalt particles. Fig. 1(a-3) and (a-4) indicate a shift from 27.79 to 29.43° which is evidence of the successful doping of Co with SnO2. Characteristic peaks in Fig. 1(a-4) compared to Fig. 1(a-3) show a decrease in peak intensities. No other contaminants were discovered, indicating that Co@SnO2–PANI nanocomposites were successfully synthesized. Phase simulation was conducted with the X'pert High Score Plus analysis tool and the cubic structure of SnO2 particles was confirmed (inset Fig. 1) by matching the data with the file (1000003) available at the COD data bank online.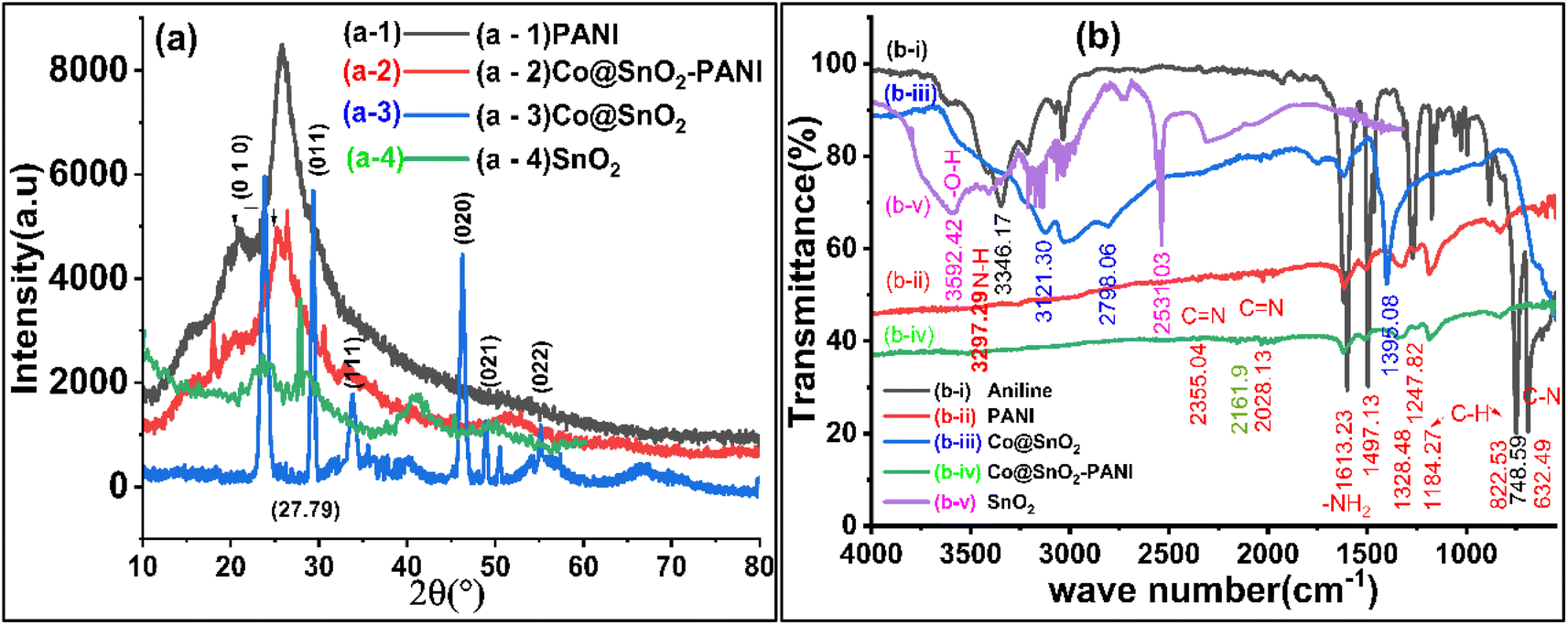 | ||
| Fig. 1 (a) XRD data plots of (a-4) SnO2, (a-1) PANI, (a-3) Co@SnO2, and (a-2) Co@SnO2–PANI (b) FTIR plots of (b-i) aniline, (b-ii) PANI, (v) SnO2, (b-iii) Co@SnO2, and (b-iv) Co@SnO2–PANI. | ||
The structural diagram of the Co@SnO2–PANI nanocomposite can be derived from the preceding results in Fig. 1a. The two main factors that led to the formation of this structure are the PANI's ability to engage with Co atoms in the third dimension and the NH2's ability to connect with Co atoms in the third dimension.25 In addition, the acid treatment of PANI has introduced an abundance of oxygen functional groups on its surface, allowing for the development of Co@SnO2–PANI particles. This arrangement not only increases the specific surface area of Co@SnO2–PANI but also speed up the electron transportation process26 during Hq and Cat detection.
FTIR spectroscopy
FTIR was used to evaluate bonding in the synthesized material, as shown in Fig. 1b, which shows strong SnO2 peaks at 3592, 3216, and 2539 cm−1 shifted. FTIR spectrum of PANI nanoparticles produced by oxidizing aniline in an acidic persulfate solution. Fig. 1b FTIR's spectra show peaks for C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bending at 632 cm−1, C–H bending at 748 cm−1, C–H bending at 822 cm−1, C–H bending at 872 cm−1, C–N stretching aromatic amine at 1247 cm−1, C–C stretching aromatic ring at 1497 cm−1, CC stretching conjugated at 1613 cm−1, and N–H stretching aliphatic primary The C–H in-plane bending has a band at 1184 cm−1 attributed to it. The composite of Co@SnO2 with PANI shown in Fig. 1(b-iv) had a negligible right shift, but there was a significant decrease in intensities of all peaks that were observed in PANI (Fig. 1(b-ii)). This shows that Co@SnO2 particles were uniformly dispersed in polyaniline and that there was no chemical bonding between the polymer (PANI) and Co@SnO2 particles. Physical interaction between Co@SnO2 and the polar ends of PANI was anticipated. SnO2 and Co@SnO2 particles were also analyzed, and the doping of SnO2 with Co was verified. Characteristic peaks of SnO2 at 3592 (–O–H) and 2531 cm−1 were significantly shifted, and new peaks at 3121 and 2598 cm−1 were observed. The results of FTIR peaks data showed that there were bonds or interactions between Co@SnO2 and PANI. This transformation increased the specificity and selectivity of the Co@SnO2–PANI composite for the detection of Cat and Hq.
C bending at 632 cm−1, C–H bending at 748 cm−1, C–H bending at 822 cm−1, C–H bending at 872 cm−1, C–N stretching aromatic amine at 1247 cm−1, C–C stretching aromatic ring at 1497 cm−1, CC stretching conjugated at 1613 cm−1, and N–H stretching aliphatic primary The C–H in-plane bending has a band at 1184 cm−1 attributed to it. The composite of Co@SnO2 with PANI shown in Fig. 1(b-iv) had a negligible right shift, but there was a significant decrease in intensities of all peaks that were observed in PANI (Fig. 1(b-ii)). This shows that Co@SnO2 particles were uniformly dispersed in polyaniline and that there was no chemical bonding between the polymer (PANI) and Co@SnO2 particles. Physical interaction between Co@SnO2 and the polar ends of PANI was anticipated. SnO2 and Co@SnO2 particles were also analyzed, and the doping of SnO2 with Co was verified. Characteristic peaks of SnO2 at 3592 (–O–H) and 2531 cm−1 were significantly shifted, and new peaks at 3121 and 2598 cm−1 were observed. The results of FTIR peaks data showed that there were bonds or interactions between Co@SnO2 and PANI. This transformation increased the specificity and selectivity of the Co@SnO2–PANI composite for the detection of Cat and Hq.
SEM analysis
The surface morphology of synthesized particles was evaluated by SEM results, as shown in Fig. 2. Cubic particles of Co@SnO2 were found in the 40 to 70 nm range. SEM results were evaluated with the analytical software ImageJ version 1.53. The average particle size of composite Co@SnO2–PANI was about 120 nm. The surface of the composite supported the distinction of two isomers (Cat and Hq).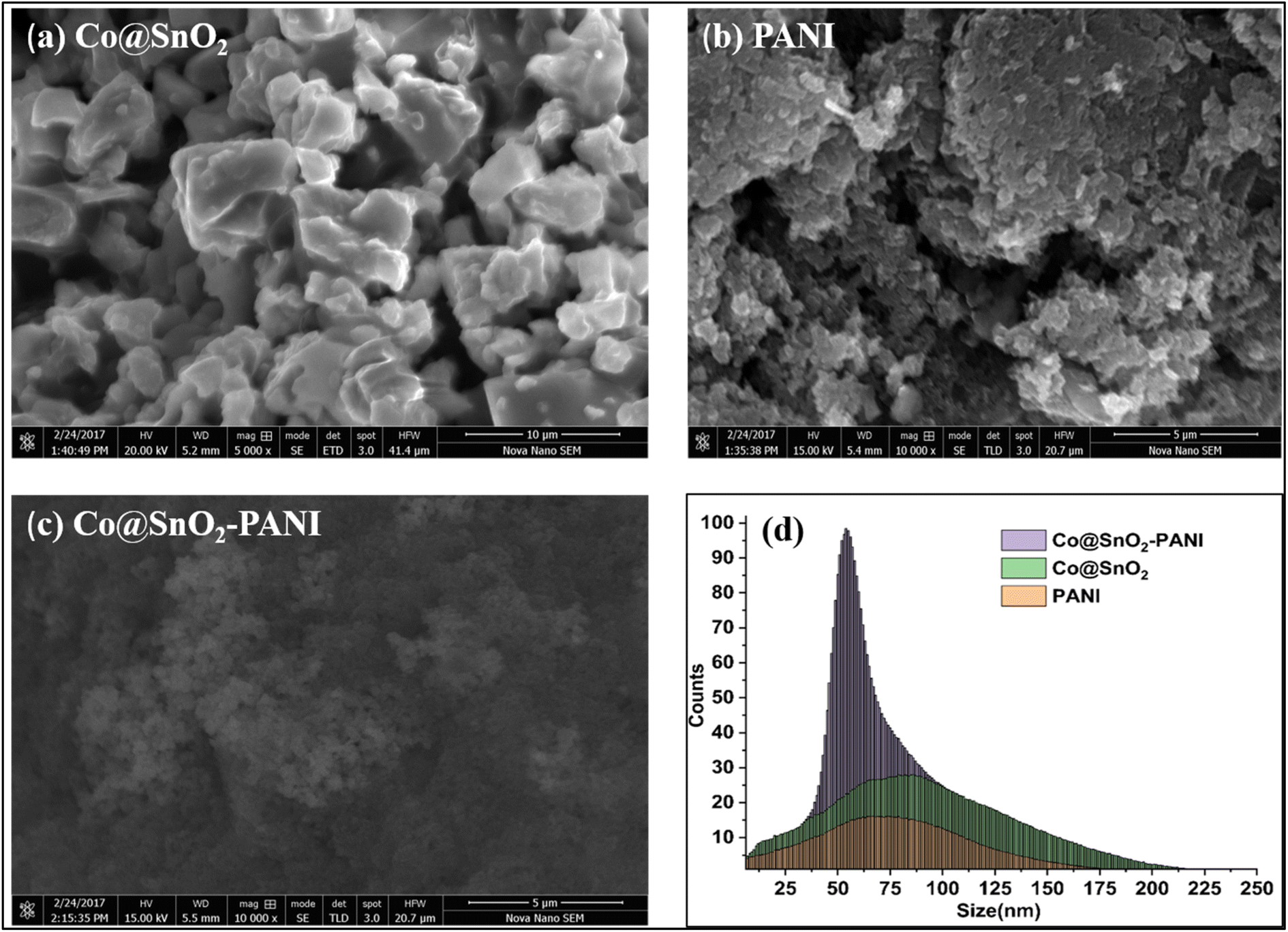 | ||
| Fig. 2 (a) SEM image of Co@SnO2 particles (b) SEM image of PANI (c) SEM image of composite Co@SnO2–PANI particles (d) Co@SnO2–PANI, Co@SnO2, PANI particle size distributions vs. comparative regression. | ||
EDS analysis
EDS analysis provides information about the elemental composition of fabricated composite. The EDS spectra of Co@SnO2, PANI, and Co@SnO2–PANI are shown in Fig. 3a–c respectively. The presence of Sn, Co, C, and O, shows the formation of the desired composite. The cps eV−1 value at 3.6 keV (Fig. 3a) dropped from 8 eV to 3.5 cps eV−1 at 3.6 keV (Fig. 3c), indicating that Co@SnO2 particles were successfully embedded in polyaniline. Co particles were intact with SnO2 and maintained their position in the composite. On the other hand, we have performed SEM analysis (Fig. 3d) to check the morphology of the synthesized sample after testing. We observed that there was no change in the morphology of the catalyst after the test. | ||
| Fig. 3 (a) EDS analysis of Co@SnO2 (b) EDS analysis of PANI (c) EDS analysis of Co@SnO2–PANI and (d) SEM image of Co@SnO2–PANI after test. | ||
Preliminary test of simultaneous Hq and Cat detection
In 25 mL of 0.1 M phosphate buffer solution (PBS) with 1.0 M KCl as the supporting electrolyte, the electrochemical responses of all modified working electrodes (GCE@SnO2, GCE@PANI, GCE@Co@SnO2, GCE@CoSnO2–PANI, and bare GCE) were assessed in the presence of 100 μL catechol/hydroquinone ([Cat + Hq] = 2.0 × 10−2 μM). Fig. 4a demonstrates the cyclic-voltammetry (CV) responses of modified electrodes with a scan rate of 50 mV s−1 and a potential range of −200 mV to 800 mV at pH 8.5. The electrochemical response of the modified working electrode GCE@Co@SnO2–PANI was superior to that of the other modified electrodes and the bare GCE. In the presence of 100 μL Cat/Hq in 0.1 M PBS/1.0 M KCl buffer solution, a well-defined oxidation peak (Ipa = 19.05 μA, Epa = 545 mV) and two reduction peaks (Ipc) at −7.36 μA and −6.9 μA were seen at Epc = 30 mV and 150 mV, respectively. The electrochemical biosensor for the simultaneous detection of catechol (Cat) and hydroquinone (Hq) in a real water sample used GCE@Co@SnO2–PANI as the working electrode and platinum wire as the counter electrode (CE). The selected modified electrode was tested by EIS, DPV, and chronoamperometry techniques. The electrochemical response of composite Co@SnO2–PANI is much superior to that of Co@SnO2 and PANI, as shown in Fig. 4a. The electrochemical study has proven that PANI has effectively enhanced the catalytic strength of Co@SnO2 particles. All the other modified electrodes have shown single oxidative and reductive peaks, but GCE@Co@SnO2–PANI has differentiated the reductive peaks of Cat and Hq at 275.87 mV and 373.27 mV, respectively. As both isomers have very close oxidative and reductive potentials, it was therefore difficult to identify them. But, the modified electrode GCE@Co@SnO2–PANI used in this study has successfully overcome this hurdle.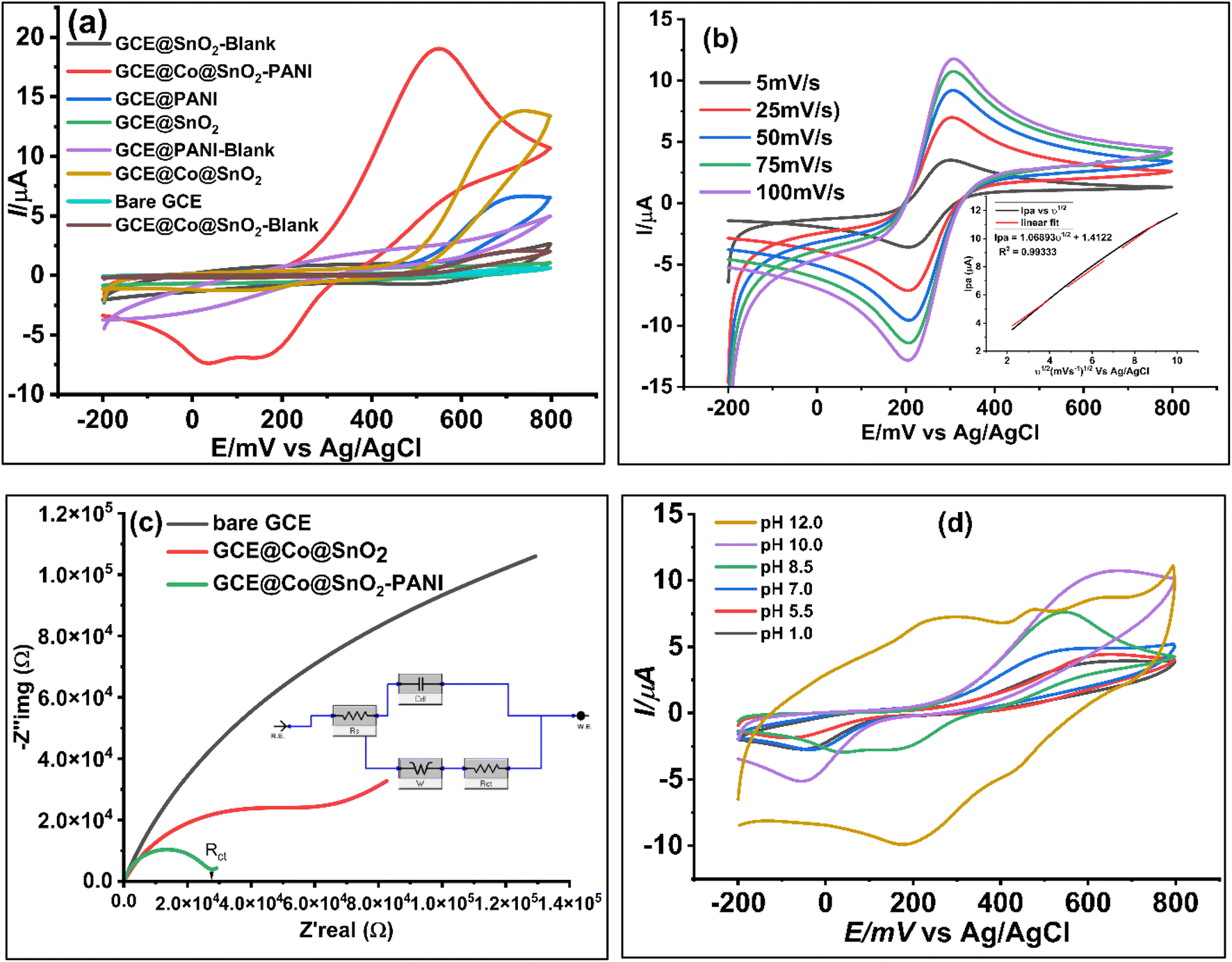 | ||
| Fig. 4 (a) Comparative CV plots of modified electrodes with bare GCE and blank GCE@Co@SnO2–PANI, blank GCE@SnO2, blank GCE@PANI, GCE@Co@SnO2 and in the presence of 100 μL (2 × 10−2 μM) in 0.1 M PBS/KCl Cat/Hq solution to validate the biosensor. (b) Scan rate voltammograms at 5 mV s−1 to 100 mV s−1 (inset) regression plot of the square root of scan rate vs. [Cat] (c) comparative Nyquist plots of bare GCE, modified electrodes GCE@Co@SnO2–PANI and GCE@Co@SnO2 (d) CV voltammograms at pH range 1 to 12 pH at potential range −200 mV to 800 mV at scan rate 50 mV s−1 vs. Ag/AgCl. | ||
Influence of scan rate
The behavior of catechol immobilized on GCE@Co@SnO2–PANI was studied by cyclic voltammograms, and responses at different scan rates (ν) are represented in Fig. 4b. With GCECo@SnO2–PANI as the working electrode, 25 mL PBS/KCl buffer with 100 μL ferro/ferri internal redox reference solution containing 100 μL Cat/Hq was scanned at several scan rates (5 mV s−1 to 100 mV s−1). This research was done to examine if diffusion is the sole mechanism that controls mass transfer throughout the redox process and to estimate the kinetics of the redox reaction between Cat/Hq immobilized on the surface of a GCE@Co@SnO2–PANI electrode. On cyclic voltammograms, the Cat's oxidation and reduction peaks were recorded at 305 mV and 202 mV, respectively. The applied scan rate potential causes levels of the oxidation and reduction peak signal (Ipc) to gradually rise. The direct proportionality of increasing scan rates and the linear correlation of this redox reaction point to a reduction in the diffusion layer on the electrode surface, which raises peak currents in cyclic voltammograms. The fact that this study gives the 50 mV s−1 acceptable scan rate for the pH inquiry adds to its relevance. The peak current at this scan rate is an average of all other scan rates used, which range from 5 to 100 mV s−1. The Fig. 4b inset illustrates how the peak currents produced at this range are proportional to the scan rate's square root, demonstrating that mass transfer is the GCE@Co@SnO2–PANI surface's defining step-by-step response. The reaction is freely diffusing at the electrode surface, as shown by the correlation equation, since the peak-to-peak separation was consistent and reproducible throughout a number of potential cycles. A quasi-reversible electrochemical electron transport process is the redox reaction.Randles–Sevcik equation:
| Ip = 0.4463nFAC(nFνD/RT)1/2 or Ip = 2.69 × 105n3/2ACD1/2ν1/2 | (1) |
| Ipa (μA) = 1.06893ν1/2 (mV s−1)1/2 + 1.4122, R2 = 0.99333 | (2) |
Slope of eqn (2) = 2.69 × 105n3/2ACD1/2 (from eqn (1)).
√D1/2 = √(1.06893/2.69 × 105n3/2AC), hence, D = 2.6 × 10−6 cm2 s−1.
Active surface area (A) = 0.29971218 cm2.
The data of the anodic peak current and logarithm of scan rate revealed a linear relation with a 0.59 slope. Theoretically, this number falls within the predicted range of 0.5 for a regulated current that is solely diffused. The finding demonstrates that the redox reaction is regulated by diffusion, since the electro-active species of Cat/Hq in the bulk solution diffuse to the electrode surface. This is also reflected in the equation:
|
Epa (mV) = 0.59logν (mV s−1) − 0.71, R2 = 0.989
| (3) |
Laviron's model predicts two straight lines, one with a slope of −2.3RT/αnF for the cathodic peak and another with a slope of 2.3RT/(1 − α)nF for the anodic peak, when the peak potential is plotted against the logarithm of the scan rate. The electron-transfer coefficient (α) for Hq was found to be 0.49, whereas that for Cat was found to be 0.59 using the aforementioned calculations. Using this equation, we were able to calculate the apparent heterogeneous electron transfer rate constant (ks).
|
Logks = α(1 − α) + (1 − α)α − logRT/nFα − (1 − α)αFΔEp/2.3RT
| (4) |
The effect of pH
In 0.1 M PBS/KCl, 2 × 10−2 μM [Cat/Hq] immobilized on the GCE@Co@SnO2–PANI was studied to see how pH affected peak potential and peak current. The study's shifting pH levels fall between the ranges of 1.0, 5.5, 7.0, 8.5, 10.0, and 12.0. The peak potential's change from 0.67 V to 0.39 V in Fig. 4d indicates that the investigation was substantially pH-dependent and that adsorption on the electrode surface took place. At pH 10.0, the Ipa is around its highest, and it starts to decline at pH 12.0. This drop in Ipa, along with a rise in pH, suggests that both adsorption and the thickness of the diffusion layer to the electrode surface have been reduced. The Ipa of the Cat/Hq decreases at a pH of 8.5. This is a result of the negatively charged ions' electrostatic repulsion effect, which limits the electrochemical reaction between the electrode surface and electrolyte solution. A pH change was unable to distinguish between the oxidative current peak (Ipa) and the reductive current peak (Ipc) of Cat and Hq. At pH 8.5, only the clear separation of the reductive peaks at 30 mV, −7.47 A of Cat and 147 mV, −7.9 A of Hq were seen, whilst the oxidative peaks at 546 mV, 19 A were combined. At pH 8.5, another electrochemical investigation was carried out.Electrochemical impedance spectroscopy study (EIS)
EIS was used to analyze the conductivity diffusion factor at the surface of Co@SnO2–PANI nanocomposite. EIS analysis framework was set as DC voltage 0.6 V vs. reference, AC voltage 5 V, initial frequency 0.1 Hz, maximum frequency 100000 Hz, delay range 0–100, estimated impedance value 100 Ω and open circuit voltage was 0.212285 V. An open circuit was compiled and tested as shown in Fig. 4c. In the presence of 100 μL (2 × 10−2 μM) of Cat/Hq solution in 5 mL of 1.0 mM ferro/ferri as internal redox reference in 25 mL of 0.1 M PBS/KCl buffer scanned with GCE@Co@SnO2–PANI, bare GCE and GCE@Co@SnO2. EIS study was conducted to determine electrochemical behavior of electrodes. Nyquist plots (Fig. 4c), the charge transfer resistance/polarization resistance (Rct) of GCE@Co@SnO2–PANI was less than that of GCE@Co@SnO2 and bare GCE, indicating that the PANI has effectively enhanced the conductivity of Co@SnO2 nanoparticles. The flow of electrons and diffusion process of the analyte on the surface of GCE@Co@SnO2–PANI were determined by the electrochemical impedance (EIS) technique with the potentiostat (Gamry-3000 model 43120). As represented in Fig. 4c, the imaginary impedance (−Z′′img), as well as real impedance (Z′real), were highest in bare GCE electrodes whereas it was least in GCE@CoSnO2–PANI. This shows the ability of GCE@CoSnO2–PANI to pass current was superior to the other competitor electrodes, GCE@Co@SnO2 and bare GCE. PANI significantly lowered the resistance and allows maximum current to pass through the electrode to oxidize Cat and Hq at the surface of the working electrode. An equivalent circuit model was compiled (Fig. 4c) and successfully tested for constant phase element data. Diffusion factor (Wd = 3.597 × 10−3 cm2 s1/2) line at 45° charge transfer resistance (Rct = 4.739 × 103 Ω), and solution resistance (Rs = 14.14 Ω) were calculated by using Gamry Echem analyst software for all working electrodes. Scan results with all electrodes were recorded in the presence of 5 mM ferro/ferri 100 μL internal reference solution and 25 mL PBS/KCl buffer. The line at 45° indicates the diffusion process at electrode surface whereas radius of circular arc represents the charge transfer resistance (Rct = 12.62 × 103 Ω), whereas the double layer capacitance (Cdl) was 6.464 × 10−3 μF. Admittance (Y0) value of GCE@Co@SnO2–PANI was found 5.159 × 10−6 S s−1. There are many ways to calculate the double layer thickness, in this study the following expression was used to find the thickness of double layer,
Thickness of double layer (σ) = double layer capacitance/scan rate (Cdl/(Ipa/ν1/2)) = 0.006047.
Cdl (6.464 × 10−3 μF) was calculated by using Gamry EIS analysis tool, whereas ν = 50 mV s−1, and Ipa/ν1/2 is the slope of regression equation of Ipa vs. ν1/2(1.05893).
The process for determining Hq and Cat simultaneously
Fig. 5a and b showed the possible electro-catalytic route to Hq and Cat at the Co@SnO2–PANI surface. The Hq molecule has a smaller electron cloud density than the Cat molecule, which increases its susceptibility to oxidation and lowers its oxidative potential. The two isomers may be detected simultaneously thanks to the difference in oxidation potential. The separation of the reduction/oxidation potentials, as well as sensitivity and selectivity, were all significantly improved by Co@SnO2–PANI. In a mildly alkaline environment (pH 8.5), Co@SnO2–PANI nanoparticles could retain a high degree of catalytic activity, and their tight association with PANI enabled all nanoparticles to take part in electrochemical detection.27 The catalysis of Hq and Cat at pH 8.5 might be improved by the well distributed Co@SnO2 nanoparticles on or inside PANI. These particles have a high specific surface area. By serving as an electron sink, the PANI may interact with di-hydroxybenzene through a conjugated electron system and improve the redox interaction between Hq and Cat on Co@SnO2–PANI nanoparticles. As a result, it was possible to determine Hq and Cat simultaneously in alkaline circumstances. The oxidative peaks of Hq and Cat are not well separated from one another, as illustrated in Fig. 5b, but reduction peaks are well segregated from one another. At a scan rate of 50 mV s−1, highly noticeable oxidative peaks at 275.87 mV and 373.27 mV (Fig. 5a) were observed because differential pulse voltammetry is more sensitive than cyclic voltammetry. A mixture of 2.0 × 10−2 μM of Cat and Hq was prepared as an analyte solution along 2.0 × 10−2 μM solutions of Cat and Hq separately. 100 μL (2.0 × 10−2 μM) of analyte solution was dropped in 25 mL KCl/PBS buffer against every scan. Under same conditions, Cat and Hq solutions were scanned with Co@SnO2–PANI working electrode against Ag/AgCl reference electrode as shown in inset of Fig. 5(a-i) and (a-ii). Broader oxidative peaks (Ipa) were formed at 331.4 mV for Cat and 271.35 mV of Hq were recorded respectively. Oxidation potential shift towards positive direction, recorded in case of scanning the mixture of Cat and Hq (Fig. 5a). In cyclic-voltammetry plots, shift in Epa was more significant, a shift of 249.32 mV was noticed. As gap between reductive peaks of Cat and Hq (ΔEpc = 170 mV, Fig. 5b) was very significant.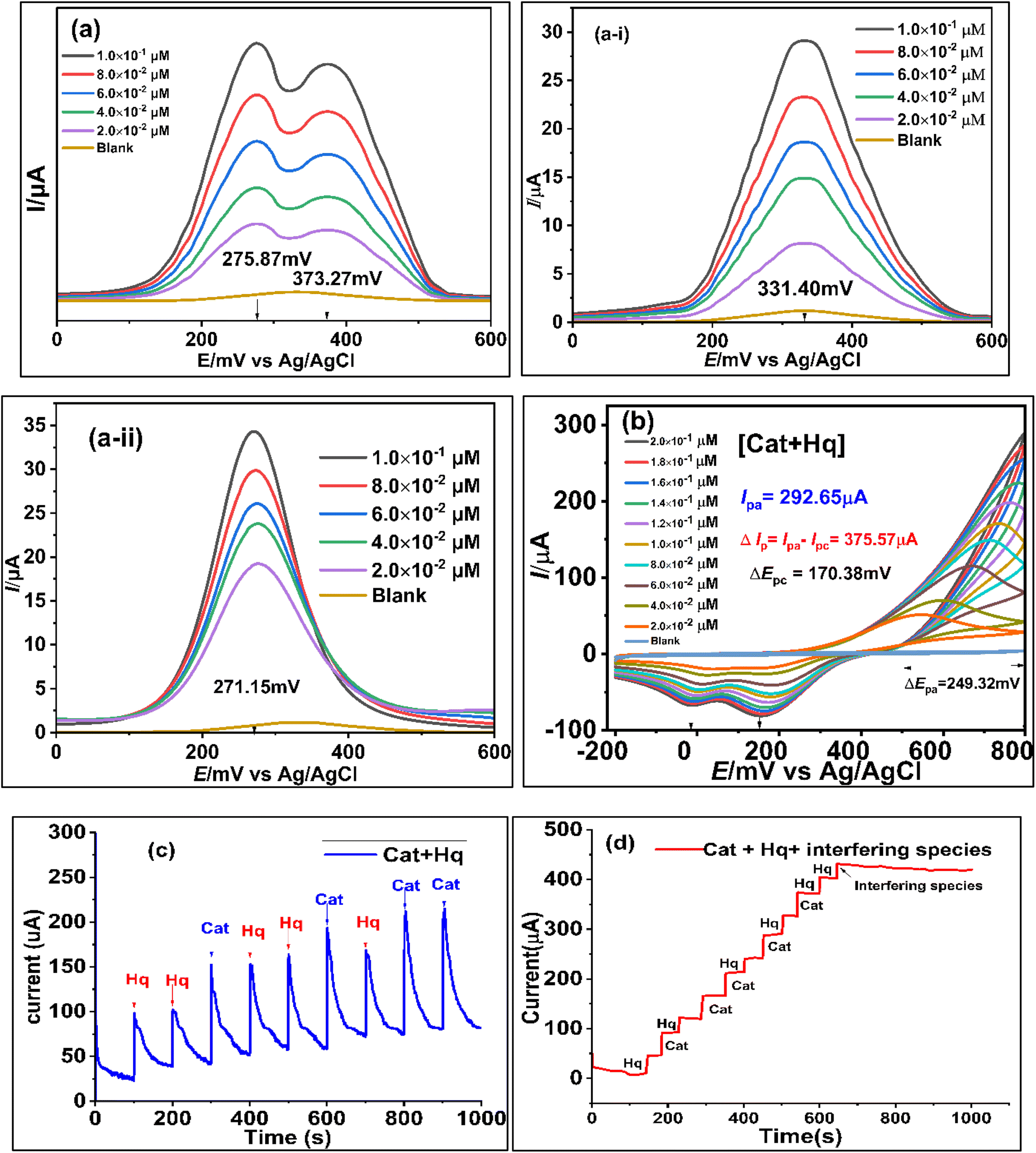 | ||
| Fig. 5 (a) DPV plots of [Hq + Cat] 2 × 10−2 to 2 × 10−1 μM at the surface of Co@SnO2–PANI, (a-i) DPV plot of [Cat] 2 × 10−2 to 2 × 10−1 μM, (a-ii) DPV plots of [Hq] 2 × 10−2 to 2 × 10−1 μM (b) CV plots of [Hq + Cat] 2 × 10−2 to 2 × 10−1 μM at the surface of Co@SnO2–PANI (c) chronoamperometry plot of [Hq] and [Cat] at constant alternatively injection rate (2 × 10−2 μM/100 s) (d) chronoamperometry plots of [Hq] and [Cat] in the presence of interfering species added at 600 s to onward till 1100 s in the real water sample. | ||
Ipa vs. concentrations of Cat and Hq were plotted and correlation equations for Cat and Hq were derived from the data as shown in eqn (5) and (6). High regression values of both Cat (R2 = 0.9909) and Hq (R2 = 0.9896) showed the effective response of catalyst GCECo@SnO2–PANI in selectivity and detection of Cat and Hq in real water sample. Epa values for Hq and Cat were 275.87 and 373.27 mV, a significant value of ΔEpa (97.83 mV) as shown in Scheme 1c is reflecting efficient catalysis at the surface of GCECo@SnO2–PANI.
| Ipa[Cat] = 4.7997[Cat] + 0.1422; R2 = 0.9909 | (5) |
| Ipa[Hq] = 3.6294[Hq] + 12.173; R2 = 0.9896 | (6) |
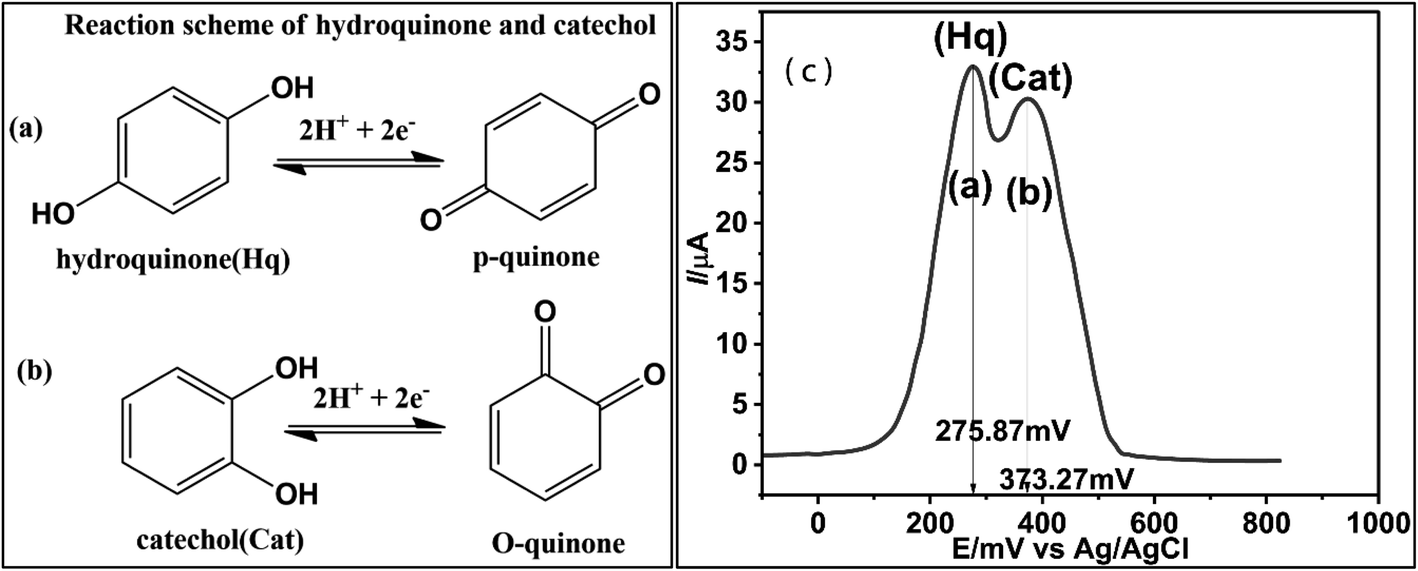 | ||
| Scheme 1 Reaction scheme of oxidation/reduction of Hq (a), Cat (b) and (c) DPV plot at the surface of GCE@Co@SnO2–PANI in the presence of 2 × 10−1 μM mixture of Hq and Cat. | ||
From regression eqn (5) and (6) it can be predicted the diffusion be predicted that oxidation and reduction of Hq and Cat was involved in transfer of two electrons as shown in Scheme 1(a) and (b).
Simultaneous estimation of Hq and Cat using DPV, CV and chronoamperometry
A more sensitive technique DPV was used to analyze the oxidation peak currents under the situation. Hq and Cat (5 mM) were mixed with 5 mM Cat solution in equal volumes. In three-electrode assembly Co@SnO2–PANI (WE), Ag/AgCl (RE) and Pt wire as CE were used. Three electrode cells were charged with 25 mL of 0.1 M PBS/1.0 M KCl. 100 μL of [Hq + Cat] was injected into the cell assembly to obtain each scan plot. Epa steadily moved to the right with each rise in analyte concentration, and two instances show that the presence of one analyte has no appreciable impact on the oxidation current (Ipa) and oxidation potential (Epa) of another. According to the previous two experiments, the concentrations of Hq and Cat were simultaneously increased from 2 × 10−2 to 2 × 10−1 μM for simultaneous determination. The oxidation maxima of Hq and Cat appeared at 275.87 mV and 373.27 mV, respectively, and their oxidation currents increased proportionally with the concentrations. Peak currents and Hq and Cat concentrations are related using linear regression, respectively. Least detection limit and LOQ values were calculated from the scan data by using 3.3 × S/N (signal-to-noise ratio) method as shown below.| Ipa[Hq] = 3.6294[Hq] + 12.173; R2 = 0.9896 | (7) |
| Ipa[Cat] = 4.7997[Cat] + 0.1422; R2 = 0.9909 | (8) |
LOD = 4.94 nM (Hq), and LOD for Cat 1.5786 nM.
LOQ (Hq) = 14.96 μM and LOQ for Cat 14.78 μM.
Sensitivity for Hq is 9.68 μA cm−2 and for Cat 12.80 μA cm−2.
By using the relation of signal-to-noise ratio (S/N), LOD = 3.3 × SD/slope and LOQ = 10 × SD/slope were calculated from calibration data. GCE@Co@SnO2–PANI at pH 8.5 was able to detect Hq and Cat simultaneously without interfering with each other, as demonstrated by this data. GCE@Co@SnO2–PANI not only detected Cat and Hq in a mild alkaline environment but also has a broader linear detection range when compared to other electrode materials (Table 1). Table 1 offers a comparison from the literature.
| Sensor material | Technique | LOD | Reference | |
|---|---|---|---|---|
| Hq | Cat | |||
| Cu-THHP (copper tetra kis(4-carboxyphenyl)porphayrine) | DPV | 3.4 × 10−3 μM | 2.3 × 10−3 μM | 28 |
| UiO-bpydc-Cu (2,2-bypyridine-5,5-dicarboxylic acid) | DPV | 0.361 μM | 0.245 μM | 29 |
| NCACNT/Ni (nickel@N-doped carbon core–shell nanoclusters in carbon aerogels) | DPV | 0.36 μM | 0.41 μM | 30 |
| NiCoFe-LDH/NF's (lactate dehydrogenase/nickel foam) | DPV | 0.15 μM | 0.12 μM | 31 |
| Fe-L-Zif-8 (zeolite imidazolate framework-8) | DPV | 0.17 μM | 0.50 μM | 32 |
| ITO/APTES (indium tin oxide/3-amino-propyltriethoxysilane) | DPV | 0.82 μM | 1.95 μM | 33 |
| TiO2–ZnO–rGO | DPV | 0.03 μM | 0.05 μM | 34 |
| Reduced graphene oxide–carboxylated carbon nanotubes/gold nanoparticles composite | DPV | 0.119 μM | 0.083 μM | 35 |
| Co@SnO2–PANI | DPV | 4.94 × 10−3 μM | 1.5786 × 10−3 μM | This work |
X. Li28 research work although shown competitive results for Cat and Hq detection but Co@SnO2–PANI used in this study has shown more sensitivity for both isomers.
Simultaneous detection by chronoamperometry
The sensor's validity was checked by using the chronoamperometry technique for the simultaneous detection of Hq and Cat. After every 100 seconds, 100 μL of the 5 mM (2 × 10−2 μM) Hq and Cat solutions were individually injected into 25 mL of 0.1 M PBS/1.0 M KCl buffer. Fig. 5c illustrates the significant increases in Ipa against both target analytes. The increase in current against [Hq] was greater than that against [Cat], and it is consistent with observations made using the DPV and CV approaches, as illustrated in the plots in Fig. 5a and b. 500 μL of HQ and 400 μL of Cat were injected. A typical current increase against a 900 μL analyte was 225.0 μA.Selectivity, stability, and interference of the GCE@Co@SnO2–PANI biosensor in the real water sample
Chronoamperometry was repeated to establish selectivity, stability and interference study of the proposed sensor GCE@Co@SnO2–PANI. 5 mM solutions of Hq and Cat were prepared in physiological water (4.0% NaCl) and one solution of interfering species (NaCl, KCl, KBr, KI, oxalic acid, Na2SO4, (NH4)2SO4, C3H3NaO3, CA, and NH4NO3, uric acid, nitrophenol, benzoic acid, gallic acid) of 25 mM. 25 mL of 0.1 M PBS/1.0 M KCl mixture was charged in Bob cell, GCE Co@SnO2–PANI (WE), Pt (CE) and an Ag/AgCl reference electrodes were fitted at a maximum limit of 5 V and 3 mA, Gamry potentiostat framework window. A chronoamperometry scan was run for 1100 seconds in a continuous mode without stirring; up to 600 seconds, 100 μL of Hq and Cat were alternatively injected every 50 seconds. The current rises linearly with every addition of 100 μL of Hq and Cat. After 600 seconds, 100 μL of interfering species solution was injected instead of Hq and Cat at the same rate for 500 s (600 to 1100 s) as depicted in Fig. 5d there was a kinky plot with declining gradient as shown in Fig. 5d.Nitrophenol, iodide, chloride and bromide ions had no discernible effect on detecting Hq and Cat (signal deviation less than 3%). This demonstrates that the portable Hq and Cat detection capability of the GCE@Co@SnO2–PANI biosensor was designed for use with actual water samples. Remarkable short-term repeated stability was shown by the relative standard deviations (RSD) for Hq and Cat, which were 1.98 percent and 4.2 percent, respectively. As shown in Fig. 5d, the sensitivity of Hq and Cat remained greater than 96.4% and 98.8% of the initial value after GCE@Co@SnO2–PANI was stored at 4 °C for 5 weeks. The results demonstrate the sensors exhibit high repeatability and consistency.
Practical real water sample determination
Using the conventional addition method, calibration samples were investigated in seawater to evaluate the performance of GCE@Co@SnO2–PANI in real-world applications. The recovery range for Hq in real water was 92.0 to 96.4 percent, whereas the recovery range for Cat was 95.0 to 98.8 percent. This whole activity was longed for five weeks. In addition, the RSD oscillated between 1.8% and 1.7%. All of the aforementioned results indicated that the novel biosensor might be used to assess Cat and Hq in marine environments. As seawater normally contain between 3% and 4% NaCl and organic compounds in the ppm range, the salt content is between 3% and 4%. Table 3 shows the GCE@Co@SnO2–PANI recovery data collected over a five week period for both analytes. An intra-day recovery procedure was also followed for five consecutive days in the first week. As 100 μL of 2 × 10−2 M samples of Hq and Cat were charged to get a CV scan every day the results of RSD are represented in Table 2.| Day | [Hq] μM | [Cat] μM | Recovery%, (n = 5) | |||
|---|---|---|---|---|---|---|
| Added | Found | Added | Found | Hq | Cat | |
| 1 | 100.0 | 99.5 | 100.0 | 99.8 | 99.5 | 99.8 |
| 2 | 100.0 | 99.2 | 100.0 | 99.6 | 99.2 | 99.6 |
| 3 | 100.0 | 99.1 | 100.0 | 99.3 | 99.1 | 99.3 |
| 4 | 100.0 | 98.7 | 100.0 | 98.8 | 98.7 | 98.8 |
| 5 | 100.0 | 98.2 | 100.0 | 98.3 | 98.2 | 98.3 |
| Sample | [Hq] μM | [Cat] μM | Recovery%, (n = 5) | |||
|---|---|---|---|---|---|---|
| Added | Found | Added | Found | Hq | Cat | |
| 1 | 1.0 | 0.92 | 1.0 | 0.955 | 92.0 | 95.0 |
| 2 | 3.0 | 2.91 | 3.0 | 2.87 | 97.0 | 95.6 |
| 3 | 6.0 | 5.78 | 6.0 | 5.93 | 96.3 | 98.8 |
| 4 | 8.0 | 7.39 | 8.0 | 7.64 | 92.4 | 95.5 |
| 5 | 10.0 | 9.64 | 10.0 | 9.88 | 96.4 | 98.8 |
For Hq and Cat, the average intra-day recovery was 98.94% and 99.14%, respectively. After one week, five weeks were used for the inter-day research to collect data on recovery.
Conclusion
An efficient electrochemical sensor based on a Co@SnO2–PANI modified glassy carbon electrode for simultaneous detection of Hq and Cat based has been developed. The sensor material was synthesized using a straightforward and practical process. The manufactured electrode GCE@Co@SnO2–PANI demonstrated outstanding sensitivity, selectivity, and catalytic character in determining Hq and Cat at a mildly alkaline pH of 8.5. The sensor's low detection limits for both isomeric compounds are convincing, such as 4.94 nM for Hq and 1.5786 nM for Cat. PANI has enhanced the catalytic activity of Co@SnO2 particles. The encouraging inter-day and intra-day recovery percentages (1.8% to 1.7%) in both analytes, 92.0 to 97.0 in Hq and 95.0 to 98.8% in Cat respectively, make the employment of composite in the effective detection of Hq and Cat in saline fluids alluring. The proposed catalyst has created a wide gap (ΔEpa) in the oxidation peak potentials of both isomers (ΔEpa = 97.83 mV), which favors its usage in detecting Hq and Cat in tap water or saline ocean water.Conflicts of interest
The authors declare that they have no known financial or intellectual conflicts of interest that could have affected this research.Acknowledgements
The authors extend their appreciation to the Deanship of Scientific Research at King Khalid University for supporting this work through research groups program under grant number RGP.2/273/44. This research was funded by Princess Nourah bint Abdulrahman University researchers supporting project number (PNURSP2023R316), Princess Nourah bint Abdulrahman University, Riyadh, Saudi Arabia. This research effort is supported by the Department of Chemistry, University of Management and Technology Lahore, Pakistan, and the Department of Chemistry and IRCBM (COMSAT) Lahore.References
- H. Qi and C. Zhang, Simultaneous Determination of Hydroquinone and Catechol at a Glassy Carbon Electrode Modified with Multiwall Carbon Nanotubes, Electroanalysis, 2005, 17(10), 832–838, DOI:10.1002/elan.200403150.
- N. Sultana, et al., Cobalt Oxide Nanorod-Modified GCE as Sensitive Electrodes for Simultaneous Detection of Hydroquinone and Catechol, Processes, 2022, 10(2), 390 CrossRef CAS , https://www.mdpi.com/2227-9717/10/2/390.
- Z.-C. Fan, et al., Longquan lignite-derived hierarchical porous carbon electrochemical sensor for simultaneous detection of hazardous catechol and hydroquinone in environmental water samples, Microchem. J., 2022, 182, 107880, DOI:10.1016/j.microc.2022.107880.
- T. S. S. K. Naik, et al., Low cost, trouble-free disposable pencil graphite electrode sensor for the simultaneous detection of hydroquinone and catechol, Mater. Chem. Phys., 2022, 278, 125663, DOI:10.1016/j.matchemphys.2021.125663.
- K. Harikrishnan, G. Singh, A. Kushwaha, V. P. Singh, U. K. Gaur and M. Sharma, 2D/2D heterojunction of graphitic carbon nitride and hexagonal boron nitride nanosheets mediated electrochemical detection of hazardous hydroquinone with high selectivity and sensitivity, J. Environ. Chem. Eng., 2022, 10(6), 108717, DOI:10.1016/j.jece.2022.108717.
- N. S. Prinith, J. G. Manjunatha, A. A. Al-Kahtani, A. M. Tighezza and M. Sillanpää, Highly Selective and Sensitive Voltammetric Method for the Detection of Catechol in Tea and Water Samples Using Poly(gibberellic acid)-Modified Carbon Paste Electrode, ACS Omega, 2022, 7(28), 24679–24687, DOI:10.1021/acsomega.2c02553.
- G. Singh, A. Kushwaha and M. Sharma, Electrochemistry of Gd2(MoO4)3-rGO nanocomposite for highly sensitive and selective detection of hazardous hydroquinone and chloramphenicol, J. Environ. Chem. Eng., 2021, 9(6), 106713, DOI:10.1016/j.jece.2021.106713.
- N. J. S. Arya, S. Saisree and K. Sandhya, Picomolar level electrochemical detection of hydroquinone, catechol and resorcinol simultaneously using a MoS 2 nano-flower decorated graphene, Analyst, 2022, 147(13), 2966–2979 RSC.
- K. Chetankumar, B. E. K. Swamy, S. C. Sharma and S. A. Hariprasad, An efficient electrochemical sensing of hazardous catechol and hydroquinone at direct green 6 decorated carbon paste electrode, Sci. Rep., 2021, 11(1), 15064, DOI:10.1038/s41598-021-93749-w.
- A. Padmanaban, et al., Electrochemical determination of harmful catechol and rapid decolorization of textile dyes using ceria and tin doped ZnO nanoparticles, J. Environ. Chem. Eng., 2021, 9(5), 105976, DOI:10.1016/j.jece.2021.105976.
- K. Harikrishnan, G. Singh, A. Kushwaha, V. P. Singh, U. K. Gaur and M. Sharma, 2D/2D heterojunction of graphitic carbon nitride and hexagonal boron nitride nanosheets mediated electrochemical detection of hazardous hydroquinone with high selectivity and sensitivity, J. Environ. Chem. Eng., 2022, 108717 CrossRef CAS.
- L.-W. Bai, et al., A Two-Dimensional NiMoO4 Nanowire Electrode for the Sensitive Determination of Hydroquinone in Four Types of Actual Water Samples, J. Anal. Test., 2022, 1–11 Search PubMed.
- S. A. B. Bukhari, et al., Supramolecular assemblies of carbon nanocoils and tetraphenylporphyrin derivatives for sensing of catechol and hydroquinone in aqueous solution, Sci. Rep., 2021, 11(1), 1–13 CrossRef PubMed.
- M. Kumar, et al., Electrochemical activation of copper oxide decorated graphene oxide modified carbon paste electrode surface for the simultaneous determination of hazardous di-hydroxybenzene isomers, Microchem. J., 2021, 168, 106503 CrossRef CAS.
- A. Chaudhary, M. Q. Khan, R. A. Khan, A. Alsalme, K. Ahmad and H. Kim, Fabrication of CeO2/GCE for Electrochemical Sensing of Hydroquinone, Biosensors, 2022, 12(10), 846 CrossRef CAS PubMed.
- S. Ganesan, et al., Novel delipidated chicken feather waste-derived carbon-based molybdenum oxide nanocomposite as efficient electrocatalyst for rapid detection of hydroquinone and catechol in environmental waters, Environ. Pollut., 2022, 293, 118556, DOI:10.1016/j.envpol.2021.118556.
- A. Al-Shekaili, W. Al-Shukaili and E. A. Khudaish, A surface network based on oxidative graphene oxide for the determination of hydroquinone and catechol in ground and wastewater samples, J. Electroanal. Chem., 2022, 919, 116509, DOI:10.1016/j.jelechem.2022.116509.
- C. C. Maciel, L. F. de Lima, A. L. Ferreira, W. R. de Araujo and M. Ferreira, Development of a flexible and disposable electrochemical sensor based on poly (butylene adipate-co-terephthalate) and graphite for hydroquinone sensing, Sensors and Actuators Reports, 2022, 4, 100091, DOI:10.1016/j.snr.2022.100091.
- A. Paul, S. Ghosh, H. Kolya, C.-W. Kang, N. C. Murmu and T. Kuila, High performance asymmetric supercapacitor device based on lanthanum doped nickel-tin oxide/reduced graphene oxide composite, J. Energy Storage, 2022, 55, 105526, DOI:10.1016/j.est.2022.105526.
- S. S. Soumya and T. S. Xavier, Effect of cobalt doping on the microstructural, optical and electrical properties of SnO2 thin films by sol-gel spin coating technique, Phys. B, 2022, 624, 413432, DOI:10.1016/j.physb.2021.413432.
- M. Zhang, Y. Wang, Y. Ma, X. Wang, B. Zhao and W. Ruan, Study of charge transfer effect in Surface-Enhanced Raman scattering (SERS) by using Antimony-doped tin oxide (ATO) nanoparticles as substrates with tunable optical band gaps and free charge carrier densities, Spectrochim. Acta, Part A, 2022, 264, 120288, DOI:10.1016/j.saa.2021.120288.
- N. Shanmugam, et al., Influence of Cerium and Nickel Co-Doping on ZnO Nanostructures for Electrochemical Behavior of H2O2 Sensing Applications, Sustainability, 2022, 14(10), 6353 CrossRef CAS , https://www.mdpi.com/2071-1050/14/10/6353.
- A. K. Rana, F. Scarpa and V. K. Thakur, Cellulose/polyaniline hybrid nanocomposites: design, fabrication, and emerging multidimensional applications, Ind. Crops Prod., 2022, 187, 115356, DOI:10.1016/j.indcrop.2022.115356.
- S. Cai, et al., Self-Assembled Deposition of Polyaniline/Cobalt Porphyrin Based on Flexible PET to Improve Sensing of Room-temperature NH3 Sensor, J. Alloys Compd., 2022, 167566, DOI:10.1016/j.jallcom.2022.167566.
- M. Kandasamy, et al., Experimental and theoretical investigations of a multiwalled carbon nanotubes/SnO2/polyaniline ternary nanohybrid electrode for energy storage, Surf. Interfaces, 2022, 30, 101978, DOI:10.1016/j.surfin.2022.101978.
- H. Wang, J. Lin and Z. X. Shen, Polyaniline (PANi) based electrode materials for energy storage and conversion, J. Sci.: Adv. Mater. Devices, 2016, 1(3), 225 CrossRef.
- T. P. Anandhu, R. R. Mohan, J. Cherusseri, R. Rohith and S. J. Varma, High areal capacitance and enhanced cycling stability of binder-free, pristine polyaniline supercapacitor using hydroquinone as a redox additive, Electrochim. Acta, 2022, 425, 140740, DOI:10.1016/j.electacta.2022.140740.
- L. Ji, Q. Wang, L. Peng, X. Li, X. Zhu and P. Hu, Cu-TCPP Nanosheets-Sensitized Electrode for Simultaneous Determination of Hydroquinone and Catechol, Materials, 2022, 15(13), 4625 CrossRef CAS PubMed , https://www.mdpi.com/1996-1944/15/13/4625.
- H.-j. Zhang, X. Zou, W.-y. Chen, Q. Sun and E.-q. Gao, A Cu-functionalized MOF and multi-walled carbon nanotube composite modified electrode for the simultaneous determination of hydroquinone and catechol, Anal. Methods, 2022, 14(40), 3961–3969, 10.1039/D2AY01230H.
- Y. Chen, et al., Carbon aerogels with nickel@N-doped carbon core-shell nanoclusters as electrochemical sensors for simultaneous determination of hydroquinone and catechol, Electrochim. Acta, 2022, 414, 140199, DOI:10.1016/j.electacta.2022.140199.
- H. He, S. Lv, Y. Kang, J. Yi, Y. Zhang and Y. Cong, In situ preparation of NiCoFe-LDH nanoflowers on carbon cloth toward simultaneous detecting hydroquinone and catechol, J. Electroanal. Chem., 2022, 919, 116540, DOI:10.1016/j.jelechem.2022.116540.
- Q. Wang, J. Zhang, N. Dou and J. Qu, Sensitive simultaneous determination of catechol and hydroquinone based on iron and nitrogen doped carbon nanonets derived from MOFs, J. Electroanal. Chem., 2022, 913, 116290, DOI:10.1016/j.jelechem.2022.116290.
- G. Wang, et al., Simultaneous detection of acetaminophen, catechol and hydroquinone using a graphene-assisted electrochemical sensor, RSC Adv., 2022, 12(37), 23762–23768, 10.1039/D2RA03900A.
- X.-Y. Lu, et al., Facile synthesis of TiO2-ZnO-rGO nanocomposites for highly sensitive simultaneous determination of hydroquinone and catechol, Microchem. J., 2021, 166, 106246, DOI:10.1016/j.microc.2021.106246.
- F. Wu, J. Zhao, D. Han, S. Zhao, R. Zhu and G. Cui, A three-electrode integrated electrochemical platform based on nanoporous gold for the simultaneous determination of hydroquinone and catechol with high selectivity, Analyst, 2021, 146(1), 232–243, 10.1039/D0AN01746A.
| This journal is © The Royal Society of Chemistry 2023 |