DOI:
10.1039/D3RA00211J
(Review Article)
RSC Adv., 2023,
13, 6246-6263
Ruthenium-catalyzed hydroarylation reactions as the strategy towards the synthesis of alkylated arenes and substituted alkenes
Received
11th January 2023
, Accepted 13th February 2023
First published on 21st February 2023
Abstract
Metal-catalyzed hydroarylation reactions are always powerful tools in organic synthesis since they can form C–C or C-heteroatom bonds in an atom and step economic manner. Medicinally and biologically relevant scaffolds can be easily and efficiently synthesized using this strategy. By tuning the directing groups that are present on the arenes, regioselectivity can be induced to the C–H activation. Metals like cobalt, rhodium and ruthenium are well known as catalysts in this type of reaction. But due to their easy availability and efficiency, Ru catalysts are found to be more preferable for hydroarylation purposes. In this review, the Ru-catalyzed hydroarylation of alkenes and alkynes, intramolecular Ru-catalyzed hydroarylation of olefin tethered arenes, modifications in the catalytic system to improve the catalytic efficiency, and carboxylate-assisted Ru-catalyzed hydroarylation reactions are discussed in detail, covering literature from 2016 to 2022.
 Salam Thowfik | Salam Thowfik was born in Kerala, India, in 2001. Currently he is pursuing his M.Sc degree at Indian Institute of Technology (I. I. T), Madras. He obtained his interdisciplinary BSc degree from Institute for Integrated Programmes and Research in Basic Sciences (IIRBS), Mahatma Gandhi University, Kerala in 2022. He has qualified for the CSIR-UGC National Eligibility Test 2022 with a research fellowship and GATE 2022 examinations. His research interest includes enantioselective and metal mediated organic synthesis. |
 C. M. A. Afsina | C. M. A. Afsina was born in Kerala, India, in 1993. She obtained her BSc degree from Kannur University (Government Brennen College, Thalassery) in 2014 and her M.Sc degree from the School of Chemical Sciences, Mahatma Gandhi University, Kottayam, Kerala, in 2016. She has qualified for the CSIR-UGC National Eligibility Test 2019 with a research fellowship and is currently pursuing her doctoral research under the guidance of Dr G. Anilkumar in the School of Chemical Sciences, Mahatma Gandhi University, Kottayam. Her research interests are in the areas of transition metal catalysis and synthetic methodology. |
 Gopinathan Anilkumar | Gopinathan Anilkumar was born in Kerala, India and took his PhD in 1996 from Regional Research Laboratory (renamed as National Institute for Interdisciplinary Science and Technology NIIST-CSIR), Trivandrum with Dr Vijay Nair. He did postdoctoral studies at University of Nijmegen, The Netherlands (with Professor Binne Zwanenburg), Osaka University, Japan (with Professor Yasuyuki Kita), Temple University, USA (with Professor Franklin A. Davis), Leibniz-Institüt für Organische Katalyse (IfOK), Rostock, Germany (with Professor Matthias Beller) and Leibniz-Institüt für Katalyse (LIKAT), Rostock, Germany (with Professor Matthias Beller). He was a senior scientist at AstraZeneca (India). Currently he is Professor in Organic Chemistry at the School of Chemical Sciences, Mahatma Gandhi University in Kerala, India. His research interests are in the areas of organic synthesis, medicinal chemistry, heterocycles and catalysis. He has published more than 190 papers in peer-reviewed journals, 7 patents, 7 book chapters and edited two books entitled “Copper Catalysis in Organic Synthesis” (Wiley-VCH, 2020) and “Green Organic Reactions” (Springer, 2021). He has received Dr S Vasudev Award from Govt of Kerala, India for best research (2016) and Evonik research proposal competition award (second prize 2016). |
1 Introduction
The reactions that involve the addition of an aromatic species and a hydrogen to an alkene or alkyne are basically defined as “hydroarylation reactions”. Hydroarylation of an aryl group with an alkene will yield a Csp2–Csp3 bond, and with an alkyne, a Csp2–Csp2 bond. It can also be considered as an example for Csp2–H activation reaction. Since transition metal catalyzed hydroarylation reactions1 enable the synthesis of biologically and pharmaceutically relevant compounds, they have received a lot of attention in recent years. The hydroarylation reactions of olefins and alkynes with aromatic compounds enable the synthesis of alkylated arenes and trisubstituted olefinic compounds respectively. An aromatic compound with directing groups allows the site selective C–H activation reaction or site selective construction of C–C (ref. 2) or C–X (X= N, O, S etc.) bonds.3 Moreover, transition metal catalyzed C–H bond activation is an environmentally-benign, atom-economic, and step economic method of C–C or C-heteroatom bond construction. For the hydroarylation reactions, catalysts derived from metals such as Rh, Co, and Pd were also used. But the easier and cheaper availability of Ru makes its use more popular. Studies on Ru(II)-catalyzed hydroarylations of olefins were pioneered by Murai,4 Chatani,5 and Ackermann.6
The previous review in this field reported by Jeganmohan et al.7 in 2015, dealt mainly with the hydroarylation of alkynes with arenes. In this review, we have included the discussions on different kinds of Ru-catalyzed hydroarylation reactions in a more detailed manner by covering all the literature from 2016 to 2022. In this review, the recent advancements in the field of Ru-catalyzed hydroarylation reactions are grouped together into six different categories for the sake of better understanding. The categories include1 Ru-catalyzed hydroarylation reactions of olefins for the synthesis of alkylated arenes,2 intramolecular hydroarylation reactions in alkene tethered arenes,3 Ru(II)-catalyzed hydroarylation reaction of alkynes: synthesis of trisubstituted alkenes carboxylate directed C–H activation using Ru(II) catalyst,4 traceless-carboxylate directing group mediated Ru(II)-catalyzed hydroarylation reaction,5 attempts to improve the efficiency of Ru catalyst, and6 the effect of reaction parameters on the catalyst decomposition of ethylene hydrophenylation.
2 Ru-catalyzed hydroarylation-classification
2.1 Ru-catalyzed hydroarylation reaction of olefins: synthesis of alkylated arenes
7-Oxa and 7-azabenzonorbornene derivatives are synthetically important since it can act as a precursor in the synthesis of partially hydrogenated naphthalene scaffolds, which are famous for their role in medicinal chemistry.8 Bolm et al. disclosed a novel strategy in 2015 for the synthesis of the aforementioned bicyclic compounds via Ru(II)-catalyzed hydroarylation reactions.9 The reaction does not require any additives or metal salts, which are mandatory for other Ru-catalyzed reactions for the catalyst activation, and the dioxygen plays an inextricable role here in the catalytic cycle which is still undiscovered.
The reaction was carried out between arenes or (hetero)arenes with oxa or azabicyclic alkenes using [RuCl2(p-cymene)]2 (1 mol%) catalyst in toluene and O2. Interestingly, when optimizing the reaction conditions, they noted that the replacement of the oxidant air with dioxygen and the absence of any of the additives or metal salts drove the reaction to a good yield. On adjusting the temperature to 120 °C and the catalyst concentration to 1 mol%, the yield was found to be increasing again, reaching 92%. Studies on substrate scope revealed that the arene (2-phenyl pyridine) with both electron-donating and electron-withdrawing substituents underwent the reaction with good yields ranging from 65% to 94% (Scheme 1). Moreover, pyridine with aromatic substituents such as thiophenyl, pyrrolyl, indolyl etc. also afforded a good yield. When the alkene starting material was varied with a range of oxa and azabicyclic alkenes, all of them proceeded with high efficiency with yields ranging from 61% to 92%.
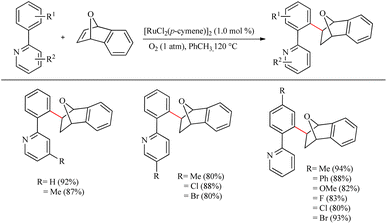 |
| | Scheme 1 Scope of (hetero)arenes in Ru(II)-catalyzed hydroarylation reaction of aza-and oxabicyclic alkene via Ru(II) catalyst. | |
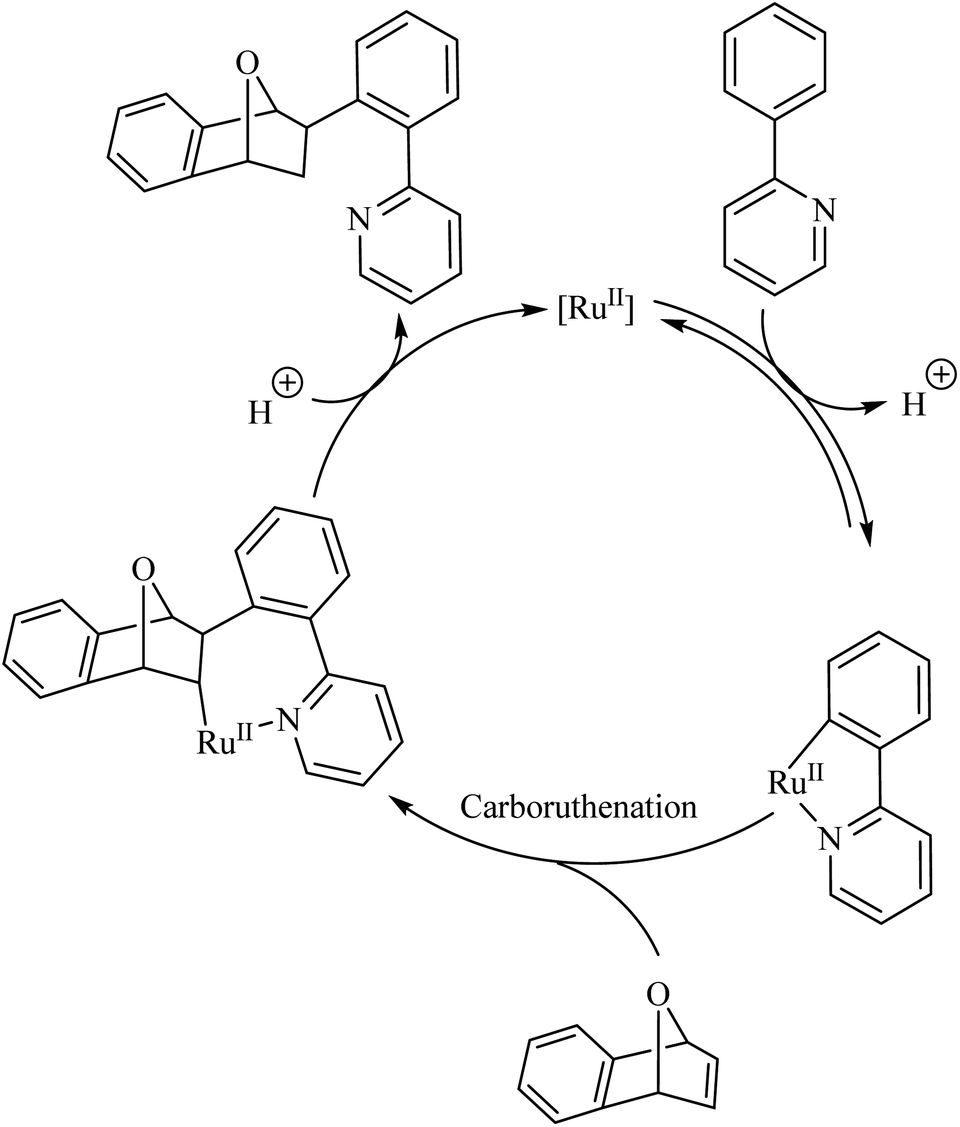
Deuterium labelling studies showed that the first step of the catalytic cycle involves a deprotonation followed by the formation of a ruthenacycle through the insertion of catalyst into the C–H bond (Scheme 2). The reaction of the ruthenacycle thus formed with the alkene (carboruthenation) forms a new Ru compound in the next step. Protonation of this compound leads to product formation and catalyst regeneration.
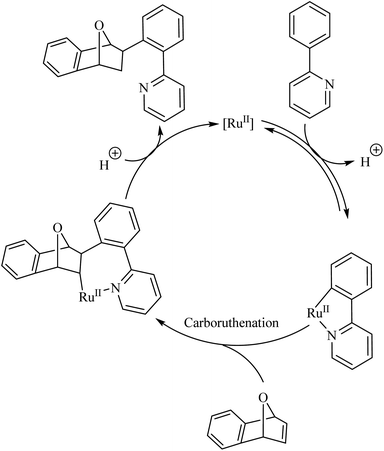 |
| | Scheme 2 Proposed catalytic cycle of Ru(II)-catalyzed hydroarylation reaction of aza-and oxabicyclic alkene via Ru(II) catalyst. Reproduced with the permission of ref. 9. | |
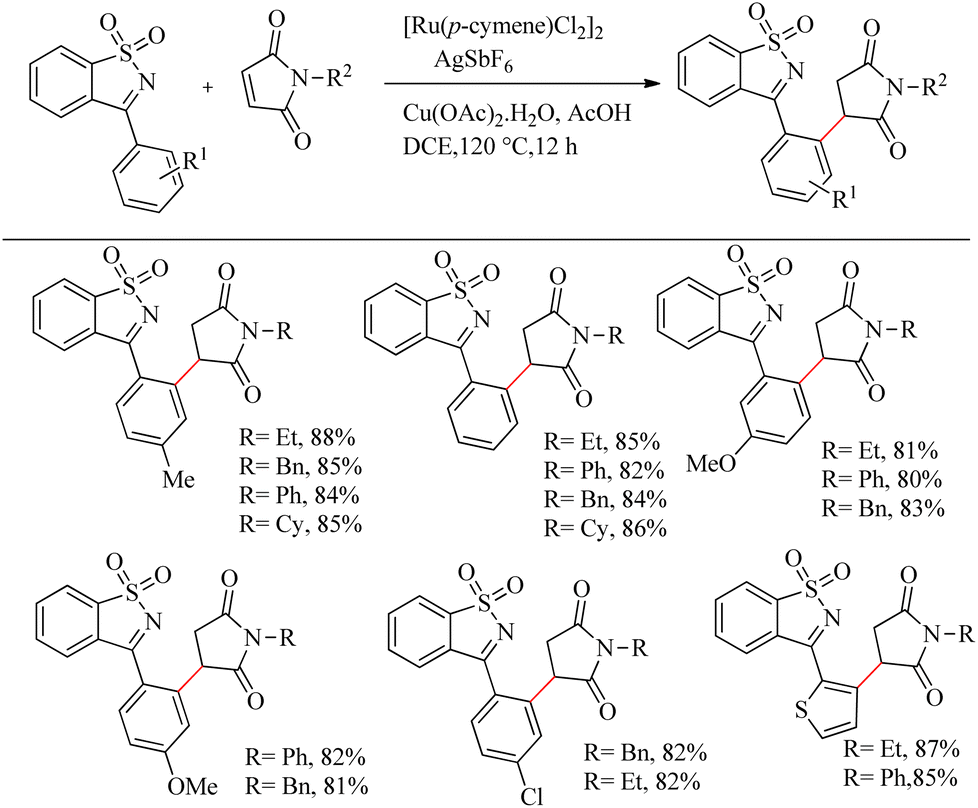
Reddy et al. recently reported a novel strategy for the synthesis of biologically relevant 3-[(isothiazol)-3-yl-phenyl] succinimide systems through the ortho-C–H bond activated Ru(II)-catalyzed hydroarylation reaction of maleimides with cyclic N-sulfonyl ketimines.10 The proposed strategy was simple, pharmaceutically relevant, atom-economical as well as a step economical method. Reaction conditions were optimized with 0.2 mmol of 3-aryl cyclic N-sulfonyl ketimine, 0.2 mmol of N-phenyl maleimide, 5 mol% [Ru(p-cymene)Cl2]2 as catalyst and Cu(OAc)2·H2O/AgSbF6/AcOH as additive in DCE solvent at 120 °C. The optimized conditions were extended to a variety of ketimines and N-protected maleimides which afforded the expected products smoothly. Ketimines bearing electron-donating groups such as chloro, methoxy and methyl groups afforded excellent yields. 2-Thienyl substituted cyclic N-sulfonyl ketimines also underwent the reaction efficiently. In the case of meta-substituted aryl rings, substitution proceeded at the less hindered side. When the reaction was performed by varying the substituents on nitrogen, reaction went very smoothly in all cases except for the N-t-butyl, N-allyl and N-Boc substituted ones. The reaction was also found to be efficient with substituted thiazole ring (Scheme 3).
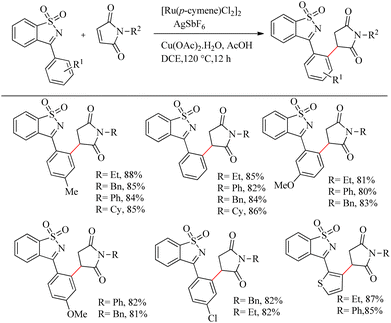 |
| | Scheme 3 Synthesis of 3-[(isothiazol)-3-yl-phenyl] succinimide via Ru(II)-catalyzed hydroarylation reaction of maleimides with cyclic N-sulfonyl ketimines. | |
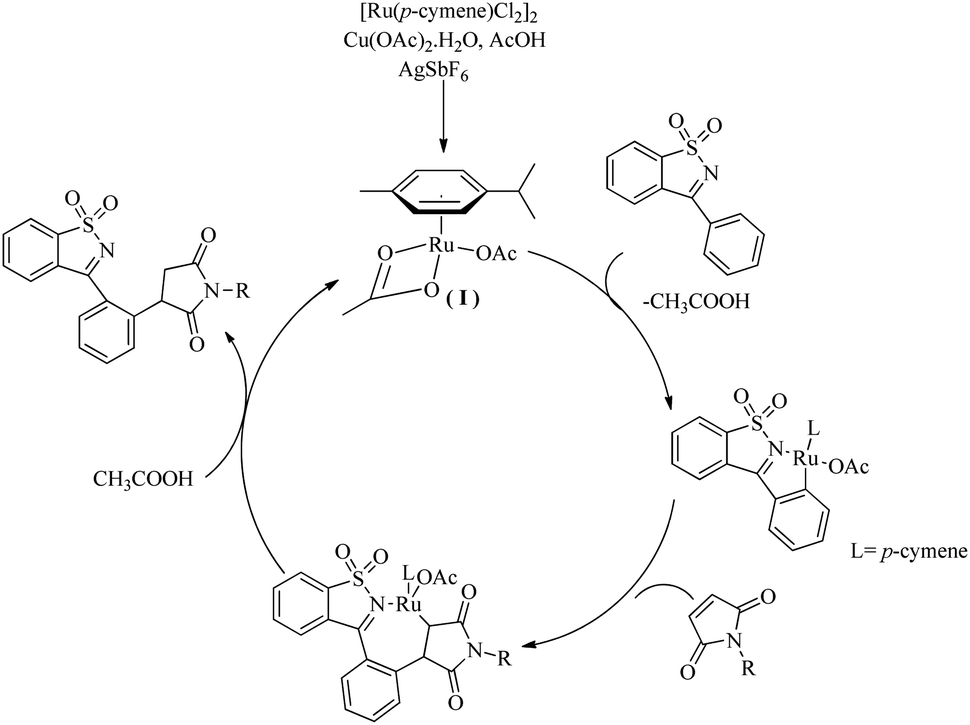
Based on a series of mechanistic investigations, they proposed a plausible mechanism in which the first step of the catalytic cycle is the formation of a metallocycle(I) through the C–H activation at the aromatic ring of ketimine (Scheme 4). Protodemetallation of the species thus formed via the hydroarylation of maleimide affords the desired product.
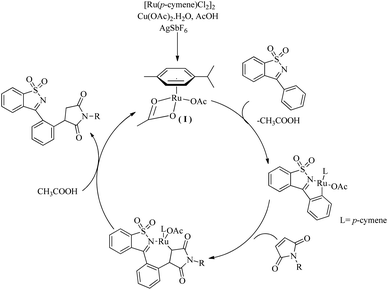 |
| | Scheme 4 Plausible reaction pathway of Ru(II)-catalyzed hydroarylation reaction of maleimides with cyclic N-sulfonyl ketimines via ortho C–H bond activation. Reproduced with the permission of ref. 10. | |
Recently Baidya et al. reported an elegant method for the synthesis of pharmaceutically and agrochemically relevant benzoannulated azaheterocycles via a Ru(II)-catalyzed hydroarylation and annulation reaction cascade.11 They proved the efficiency of this synthetic route relative to other modes via the reaction of o-arylaniline derivatives and diversely substituted maleimides. Experiments for optimizing the reaction conditions resulted in the selection of a mixture of 5 mol% [Ru(p-cymene)Cl2] as the catalyst, 20 mol% AgSbF6 as halide scavenger, acetic acid (7.5 equiv.) and 50 mol% of 1-AdCO2H additive in THF solvent (0.1 M) as the catalytic system. The reaction was found to be more fruitful at 80 °C. Both electron-donating and electron-withdrawing substituents at either of the two aryl rings afforded excellent yields of the product (Scheme 5). Substrate scope studies of substituted maleimides revealed that all types of substituents on the nitrogen atom drive the reaction to a good yield, independent of the electronic nature of them. Various maleimides bearing a variety of amino acid residues on the nitrogen atom underwent the reaction very smoothly (Scheme 6). Mechanistic studies strongly supported the involvement of a carboxylate directed concerted metallation deprotonation (CMD) process and a reversible C–H ruthenation step in the reaction pathway. Further studies with the radical scavengers such as TEMPO, BHT etc. refute the possibility of radical intermediates.
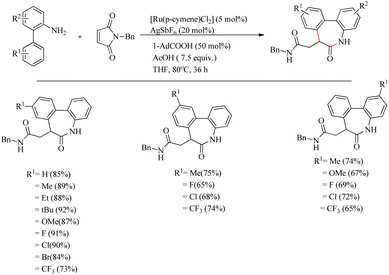 |
| | Scheme 5 Synthesis of benzoannulated azaheterocycles via Ru(II)-catalyzed hydroarylation. | |
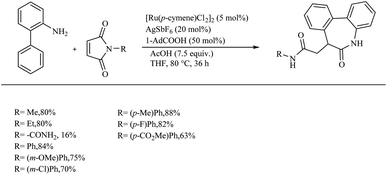 |
| | Scheme 6 Ru(II)-catalyzed hydroarylation for the synthesis of benzoannulated azaheterocycles. | |
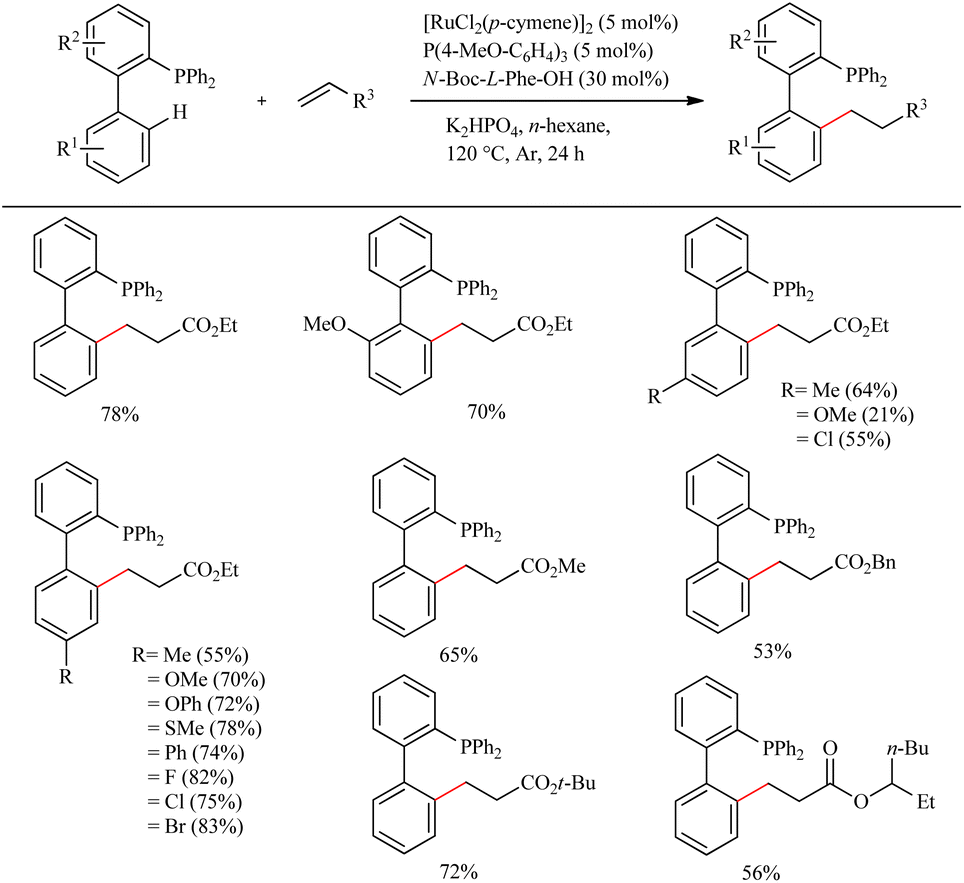
Due to their efficient electronic and steric compatibility, tertiary phosphines are widely used in transitional metal catalysis. But in most of the cases, the phosphorous atom should be in the P(V) oxidation state to avoid the coordination poisoning of them with metals. Zeng et al. pioneered the ligand promoted Ru(II)-catalyzed hydroarylation reaction of biaryl phosphines with olefins in which phosphorous atom is in (III) oxidation state.12 P(III) instead of P(V) eradicate the additional protection, deprotection step. This study paved the way for the direct synthesis of a group of Buchwald type phosphines with varying alkyl groups with high atom economy. On optimizing the reaction conditions they found that less hindered and electronically more denser (4-MeO-C6H4)3P (5 mol%) in presence of [RuCl2(p-cymene)]2 (5 mol%) as the catalyst and N-Boc-L-Phe-OH (30 mol%) as the ligand afforded the product with a maximum yield of 80% when using K2HPO4 as base in hexane for 24 h at 120 °C under Ar atmosphere (Scheme 7). Amino acid plays a critical role here since the reaction without amino acid was found to be passive. A variety of substituents on both the aryl ring exhibit high efficiency irrespective of their electronic effects or their relative position. Aromatic moieties such as naphthalene, pyridine, furan etc. are found to afford good yields. The substrate scope studies on the olefinic partner show that activated acrylates of any type resulted in a good yield of the alkylated product. Instead of acrylates, acrylamides are also found to be viable in the reaction.
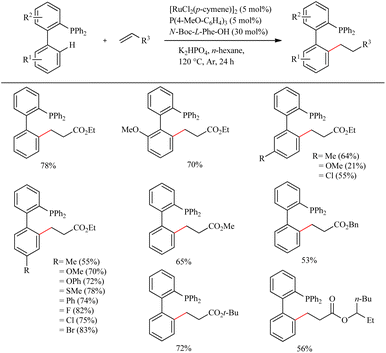 |
| | Scheme 7 Ru(II)-catalyzed hydroarylation reaction of biaryl phosphines with olefins. | |
The first step in the catalytic cycle, according to mechanistic studies is the formation of an active Ru(II) complex in presence of K2HPO4 and N-Boc-L-Phe-OH (Scheme 8). The orthoruthenation of the active Ru(II) complex in the presence of base results in the formation of a six-membered cycloruthenated complex in the second step. Then an intermediate Ru complex (Int A) is formed in the next step via the coordination of alkene with the cycloruthenated complex followed by it′s insertion into the Ru–C bond. Proto-demetallation of this intermediate complex leads to the regeneration of the catalyst and the formation of the linear alkylation product.
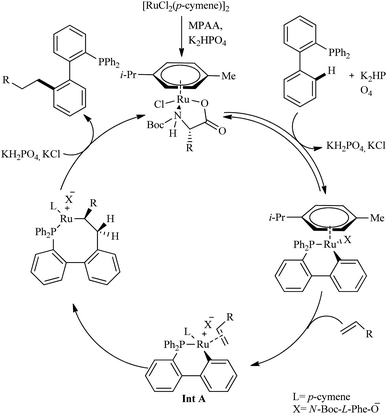 |
| | Scheme 8 Plausible mechanistic pathway for Ru(II)-catalyzed hydroarylation reaction of biaryl phosphines with olefins. Reproduced with the permission of ref. 12. | |
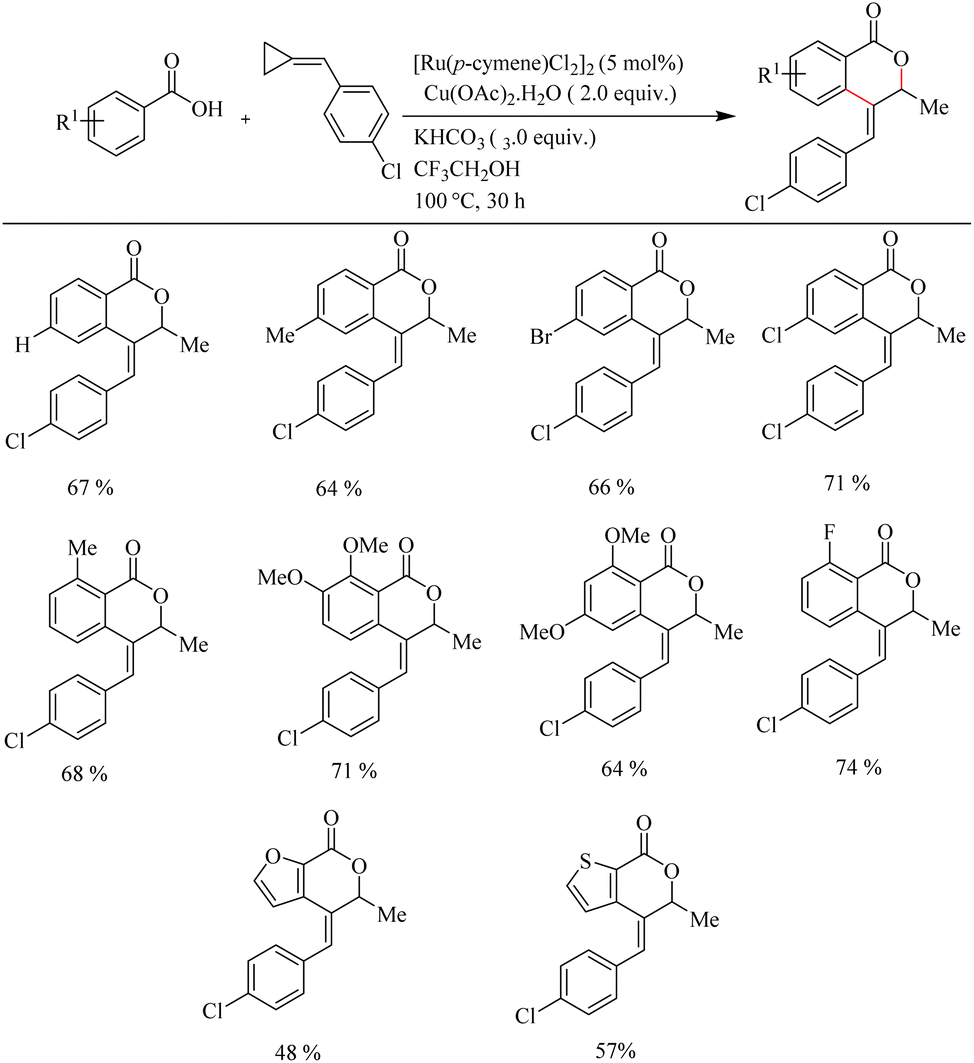
Recently Jeganmohan et al.13 reported a stereoselective synthesis of isochromanones – a privileged oxygen containing heterocycle system via Ru(II)-catalyzed hydroarylation of aromatic acids with alkylidenecyclopropanes. This method is an efficient way to synthesize (E)-stereoselective 4-aryl isochroman-(E)-stereoselective-1-one since its synthesis is very challenging and not yet developed. The optimum reaction conditions were found to be the use of [Ru(p-cymene)Cl2]2 (5.0 mol%) as the catalyst, Cu(OAc)2·H2O (2.0 equiv.) as the additive, 3.0 equiv. of KHCO3 as the base in trifluroethanol (3.0 equiv.) as the solvent at 100 °C for 3 h. When the reaction was performed with electronically and sterically diverse benzoic acids under optimized conditions, it is found that both electron-donating and electron-withdrawing substituents gave their corresponding products in relatively good yields. Also aryl heterocycles such as 2-furoic acids and 2-thiophene carboxylic acids drive the reaction in a moderate yield to afford a mixture of products (Scheme 9). The reaction was also performed by varying the alkyl substitution in alkylidine cyclopropanes, and all of them gave rise to their products independent of their electronic property (Scheme 10).
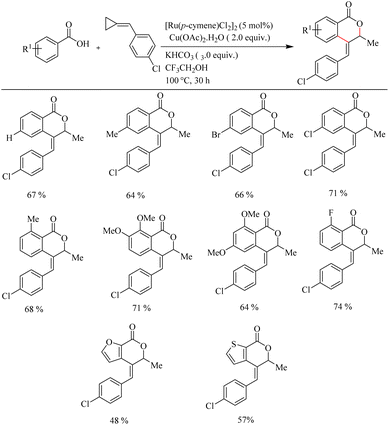 |
| | Scheme 9 Scope of substituted benzoic acids in Ru(II)-catalyzed hydroarylation with alkylidenecyclopropanes. | |
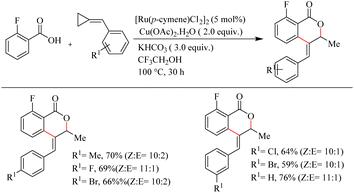 |
| | Scheme 10 Scope of substituted alkylidenecyclopropanes in Ru(II)-catalyzed hydroarylation with aromatic acids. | |
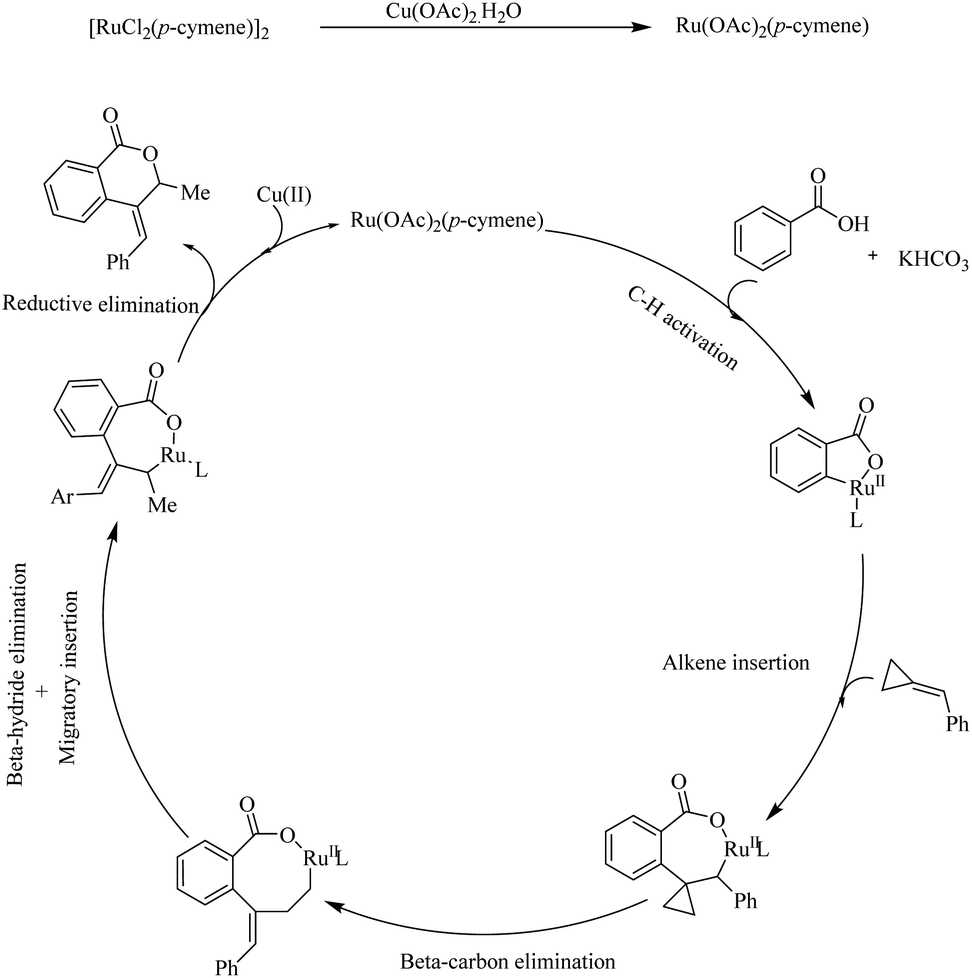
Mechanistic investigations revealed that the first step in the catalytic cycle is the reaction of [RuCl2(p-cymene)2]2 with a Cu based additive to form the activated species [Ru(OAc)2(p-cymene)]. Then the aryl carboxylic acids react with the activated species, followed by cyclometallation in the presence of KHCO3 base which leads to the formation of a five membered metallacycle. Then the alkene undergoes coordinative insertion with the five membered metallacycle in presence of AcOH. β-Carbon elimination of the formed species leads to an intermediate which undergoes β-hydride elimination to provide the arylated butadiene intermediate. Then the RuH bond is inserted into the less substituted double bond of this intermediate to form another cyclometallated species. Finally the reductive elimination of this species gives rise to the product and the regenerates the catalyst (Scheme 11).
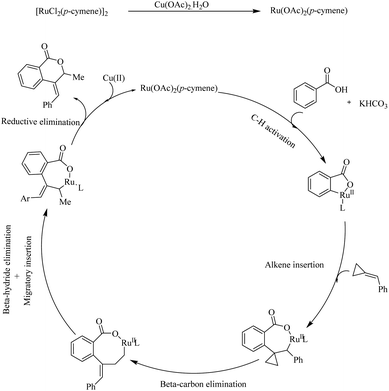 |
| | Scheme 11 Proposed mechanism of Ru(II) catalyzed hydroarylation of aromatic acids with alkylidenecyclopropanes. Reproduced with the permission of ref. 13. | |
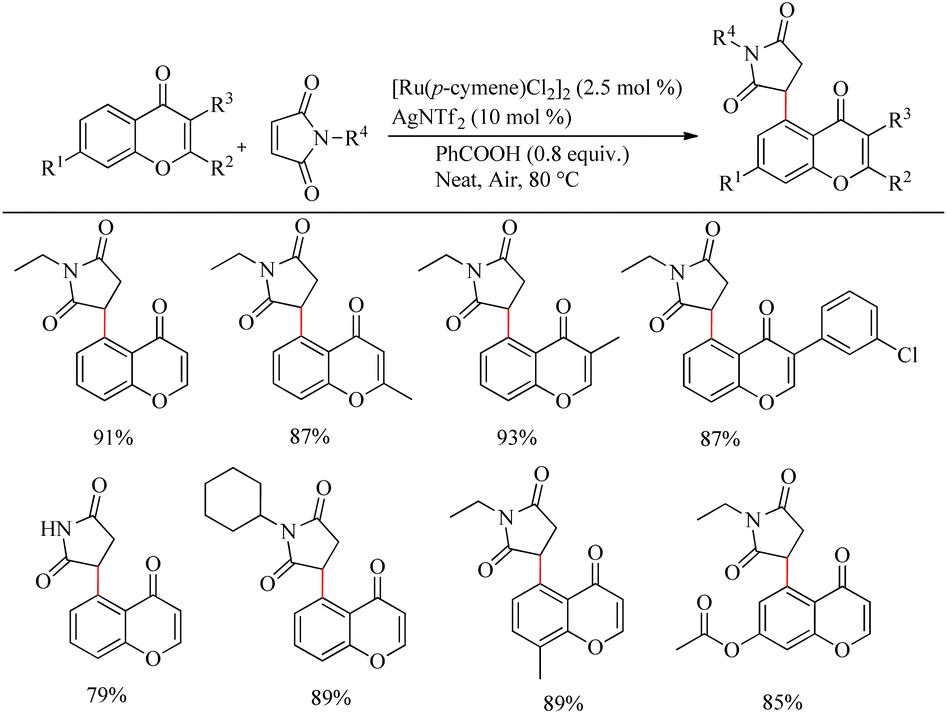
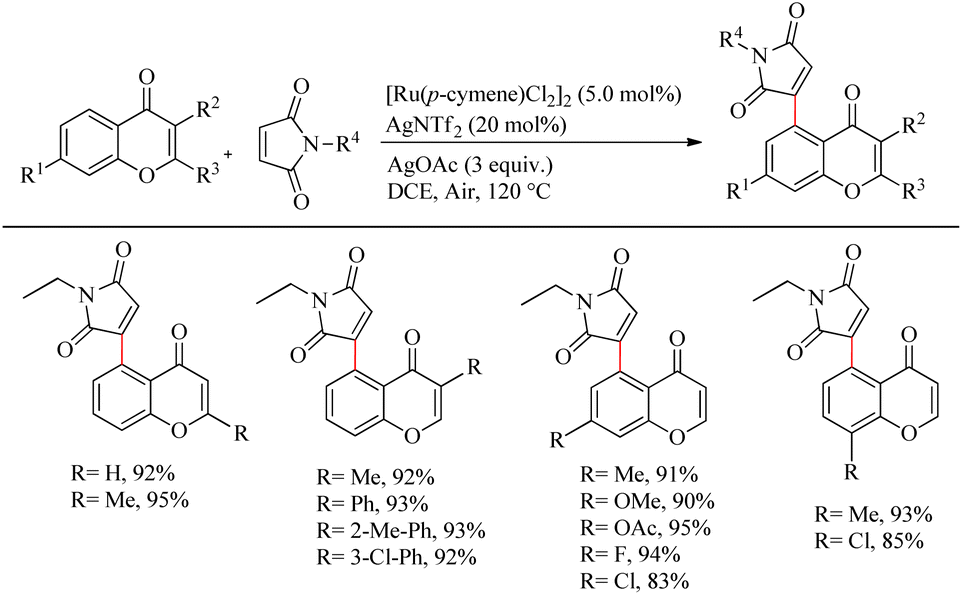
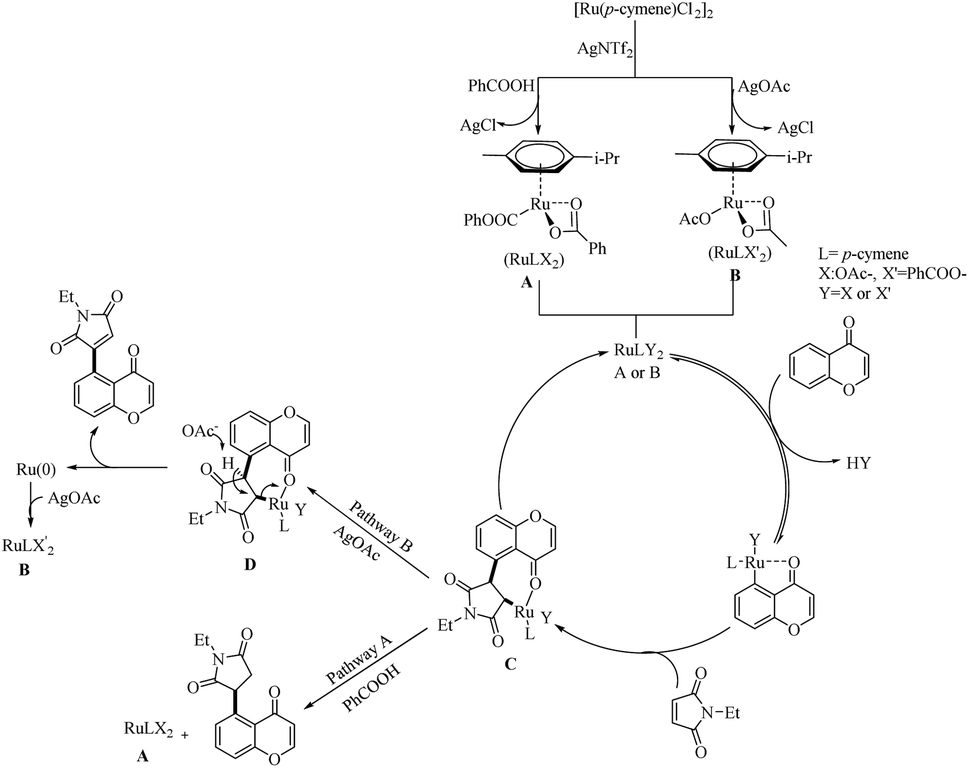
Zhou et al. reported an efficient way to synthesize various succinimide/maleimide containing chromone scaffolds via selective Ru(II)-catalyzed hydroarylation of maleimides with chromones at their C5-position.14 Interestingly it was noted that by varying the additives, both 1,4 addition products and oxidative Heck type coupling products could be obtained. Under solvent free conditions, using benzoic acid as the additive afforded 1, 4 addition products, whereas under the application of solvent with silver acetate as the additive resulted in oxidative Heck type coupling. An excellent feature of this proposed scheme was it’s short reaction time. For 1, 4 addition, the optimized reaction conditions involve the use of N-ethyl maleimide (2.5 equiv.), [Ru(p-cymene)Cl2]2 (2.5 mol%) as the catalyst, AgNTf2 (10 mol%), benzoic acid (0.8 equiv.) under neat conditions at 80 °C in air (Scheme 12). The optimized condition for Heck type coupling was N-ethyl maleimide (2.5 equiv.), [Ru(p-cymene)Cl2]2 as the catalyst (1.5 mol%), AgOAc (3 equiv.), AgNTf2 (20 mol%) in 2 mL DCE at 120 °C in air (Scheme 13). Substrate scope studies showed that, independent of the electronic effects, a wide variety of the substituted maleimides and chromones underwent the 1, 4 addition very smoothly. The substituents on the nitrogen also did not show a vital role in the rate of the reaction. Substituents like bromo, ethyl, acetyl and nitro were tolerated in this reaction and the protocol was also found to be feasible with acrylates and phenyl vinyl sulfone.
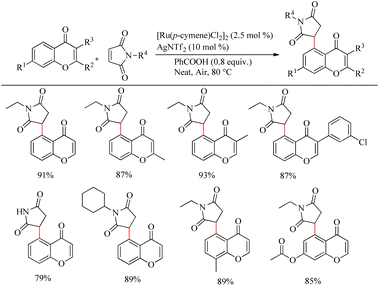 |
| | Scheme 12 Investigation of chromones and maleimides towards the Ru-catalyzed hydroarylation reaction. | |
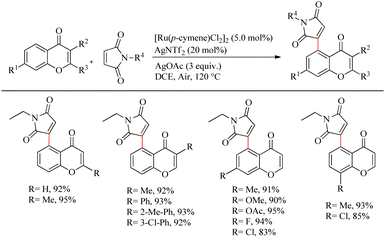 |
| | Scheme 13 Ru-catalyzed hydroarylation of chromones and maleimides towards the Heck-type coupling. | |
Mechanistic investigations showed that, the first step of catalytic cycle involves the generation of an active catalyst (A or B) via the reaction of Ru(II)-catalyst with AgNTf2 and benzoic acid or AgOAc. Consequent C–H bond activation and the coordination of ketone oxygen to the Ru centre yielded a metallocycle. This follows the formation of a bicyclic intermediate (C) through the insertion of maleimide into the C–Ru bond of the previously formed metallocycle. The nature of the additives used influences the subsequent reaction of C. If the additive is benzoic acid, 1,4-addition occurs and the catalyst A regenerates through a protodemetallation step. AgOAc as the additive promotes the Heck-type coupling and the regeneration of Ru(0) species through the deprotonation of β-hydrogen of C by –OAc. Ru(II) catalyst will then be regenerated from the Ru(0) species easily through its reaction with the AgOAc (Scheme 14).
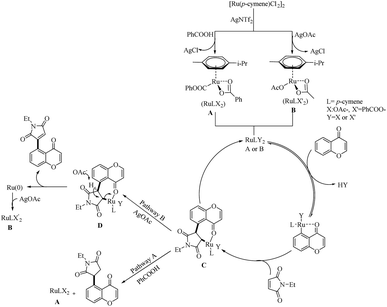 |
| | Scheme 14 Plausible reaction pathway of Ru(II)-catalyzed hydroarylation and Heck-type coupling of chromone moieties with maleimides. Reproduced with the permission of ref. 14. | |
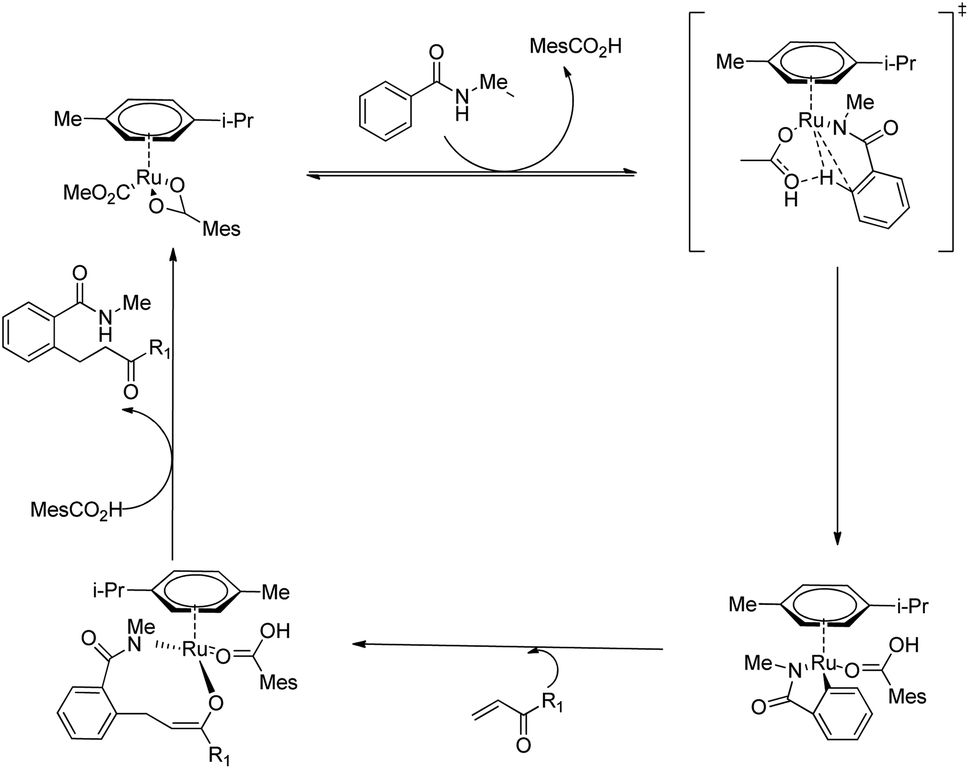
Ackermann and co-workers recently reported a very efficient and chemoselective method for the preparation of β-arylketones through the hydroarylation reaction of electron-deficient ketone with monodentate amides via the assistance of carboxylate group.15 In addition to this, the oxidative cascade annulation of electron-deficient ketone was found to yield quinolone scaffolds. Reaction conditions were optimized for the Ru(II)-catalyzed (5.0 mol%) hydroarylation reaction of benzamide (0.5 mmol) with methyl vinyl ketone (1.0 mmol). The reaction showed significant improvement only during the supply of co-catalytic amounts of KOAc and stoichiometric amounts of HOAc as additives (Scheme 15). Modification of the reaction with stoichiometric amount of MesCO2H along with MesCO2K (30 mol%) under nitrogen atmosphere for 20 h at 120 °C afforded the desired result.
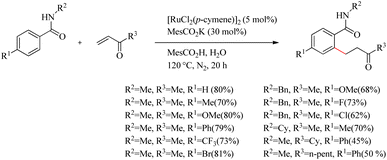 |
| | Scheme 15 Synthesis of β-arylketones through the hydroarylation reaction of ketone with monodentate amides via the assistance of carboxylate group. | |
Both electron-rich and electron-deficient para-substituted aryl amides afforded good results. The substitution on amide nitrogen with electronically diverse substituents also showed excellent yields. Steric hindrance in the ortho-position reduced the product yield. They extended the same methodology towards indole derivatives too. The catalyst [Ru(MesCO2)2(p-cymene)] afforded the major product without the use of any additives which implies the importance of carboxylate assistance. Based on the experimental studies, they proved that the catalytic cycle is consisting of carboxylate assisted reversible C–H bond activation, migratory insertion and reductive elimination respectively (Scheme 16).
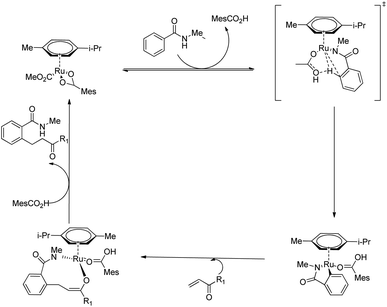 |
| | Scheme 16 Proposed catalytic cycle of Ru(II)-catalyzed hydroarylation of monodentate amides with conjugated alkenes via carboxylate assistance. | |
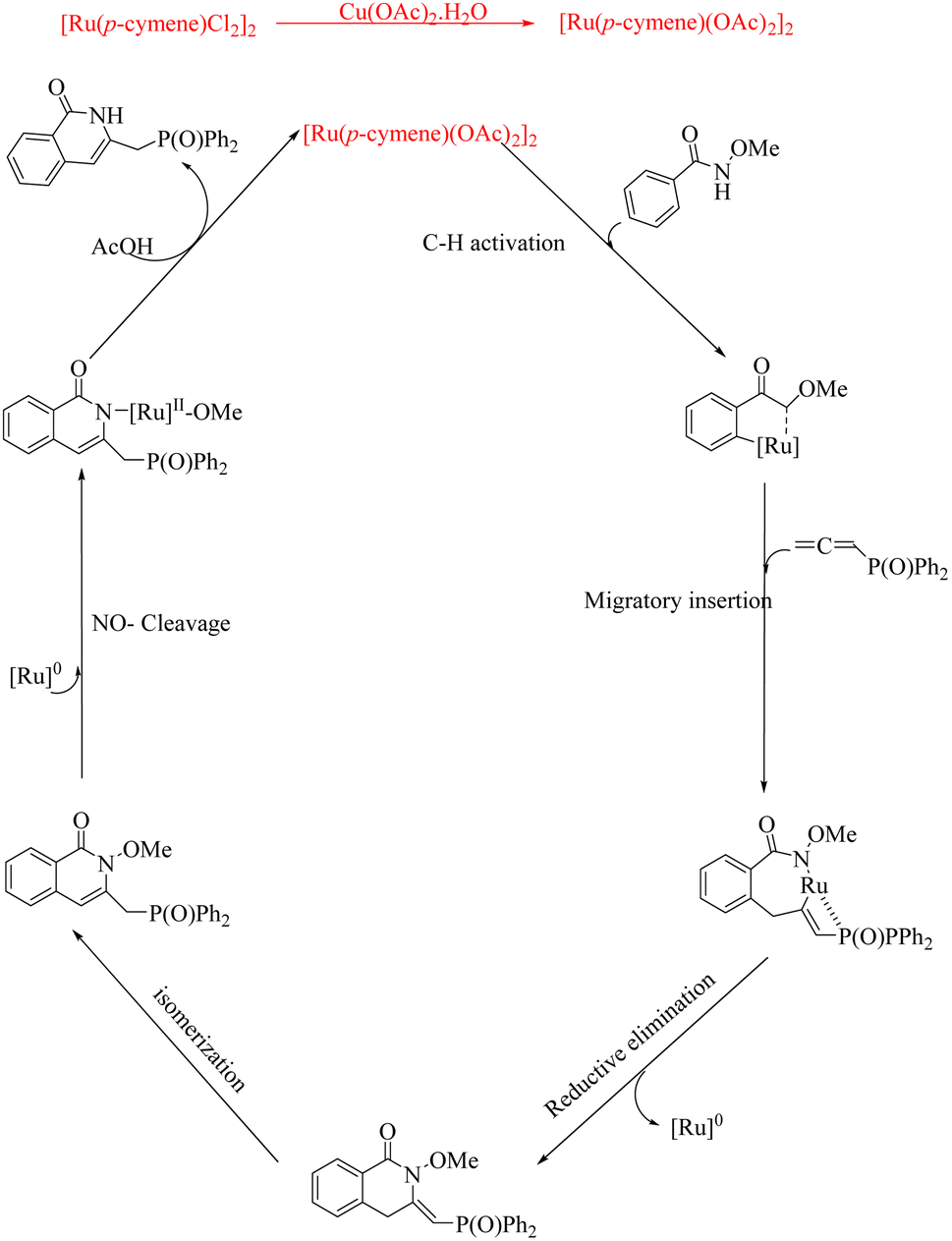
Recently Baidya et al.16 reported a strategic approach to synthesize pharmaceutically relevant isoquinolinones via Ru catalyzed hydroarylation of arylamides with diversely substituted allenes. This fascinating Ru(II)-catalyzed (4 + 2) annulation of aromatic amides with allenes is the first report in this field to deal with Ru catalysis where the terminal steps of the CH activation are β-hydride elimination and protonation. Attempts to optimize the reaction conditions resulted in the identification of desired reaction parameters as 5 mol% of [Ru(p-cymene)cl2]2 as the catalyst, Cu(OAc)2·H2O (1 equiv.) as the base and hexafluroisopropanol (HFIP) as the solvent (at 60 °C for 12 h). Substrate scope studies revealed that the rate of this transformation was independent of the electronic properties of benzamides (Scheme 17). But the N–OMe group on the amide was found to be specific in the reaction since the reaction is unproductive with the N–Me group. Also, the Substrate scope studies on various allenes show that both the electron-donating and electron-withdrawing substituents on the allenes can drive the reaction to relatively good yields (Scheme 18). The allenes without the phosphine oxide moiety are found to be resistant towards the given reaction conditions.
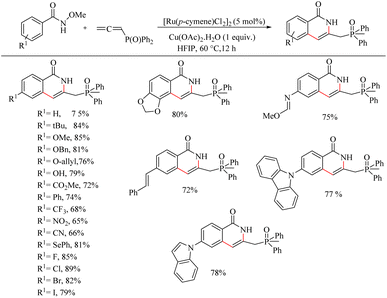 |
| | Scheme 17 Scope of substituted benzamide systems in Ru(II) catalyzed hydroarylation with allenes. | |
 |
| | Scheme 18 Scope of diversely substituted allenes in Ru(II) catalyzed hydroarylation with aryl amides. | |
Experiments on mechanistic investigations revealed that the first step in the catalytic cycle is a C–H activation which results in the formation of a ruthenacycle (A). Then this undergoes migratory insertion with allene species to form Ru-sigma alkenyl intermediate (B). Subsequent sequential reductive elimination and isomerization result in the formation of another intermediate (C). The species C on protonation yields the isoquinolinone product along with the regenerated Ru catalyst (Scheme 19).
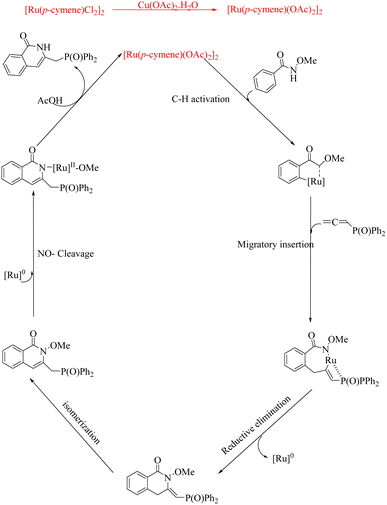 |
| | Scheme 19 Plausible mechanism of Ru-catalyzed hydroarylation of arylamides with diversely substituted allenes to form isoquinolinones. Reproduced with the permission of ref. 16. | |
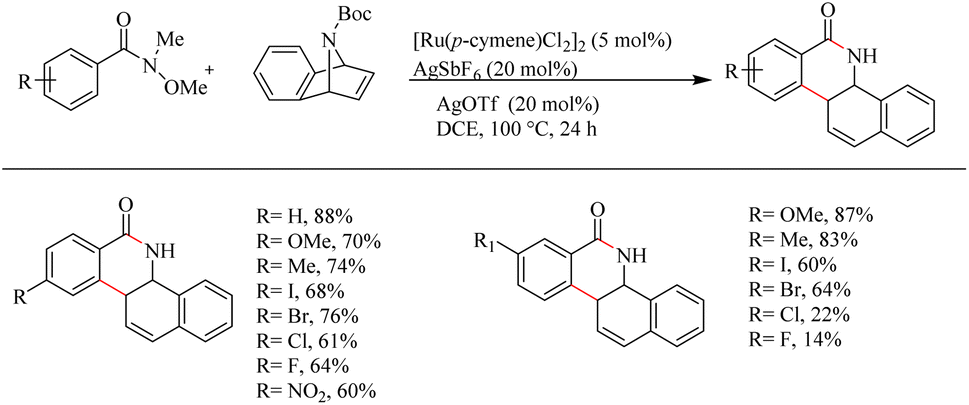
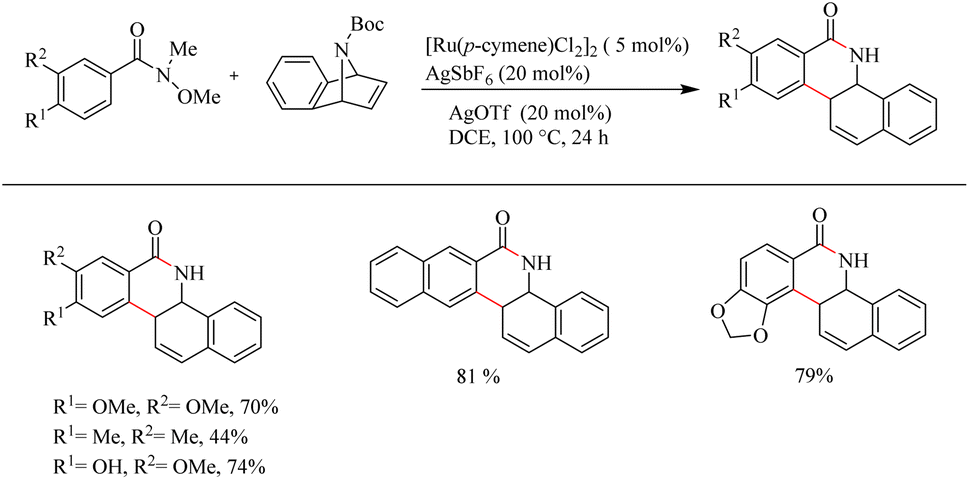
Jeganmohan et al.17 recently reported an intriguing method to synthesize pharmaceutically relevant dihydrobenzo[c]phenanthridinones through Ru-catalyzed hydroarylation of arylamides and 7-azabenzonorbornadienes. The amide group in the substrate can effectively act as both the leaving group and the directing group. Optimization studies revealed that the use of [Ru(p-cymene)Cl2]2 (5 mol%) as the catalyst, AgOTf (20 mol%) as the additive in DCE solvent at 100 °C under N2 atmosphere can be considered as the desired reaction parameters. Substrate scope studies on substituted aryl amides under the optimized reaction conditions have shown that both electron-donating and electron-withdrawing groups at the aryl part can afford the products with relatively good yields, except in the case of meta-fluro/chloro substituted aryl species (Scheme 20). The experiments with a variety of amides lead to the conclusion that the Weinreb amide is the most suitable leaving group/directing group in this reaction. When the monosubstituted aryl moiety was replaced with unsymmetrical disubstituted aryl moieties, the reaction was found to proceed at the less hindered site except in the case of piperonylic amide. With symmetrical disubstituted aryl moieties, it was found that C–H activation is proceeding at the ortho site as usual (Scheme 21). Both electron-donating and electron-withdrawing group substitutions at the N-Boc-azabenzonorbornadiene drive the reaction in a moderate to good yield (Scheme 22).
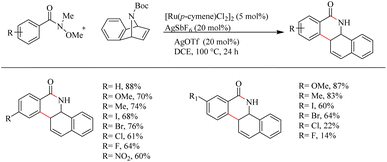 |
| | Scheme 20 Scope of monosubstituted aryl amides in Ru(II)-catalyzed hydroarylation with 7-azabenzonorbornadienes. | |
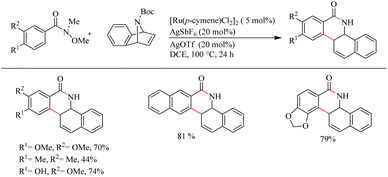 |
| | Scheme 21 Scope of disubstituted aryl amides in Ru(II)-catalyzed hydroarylation with 7-azabenzonorbornadienes. | |
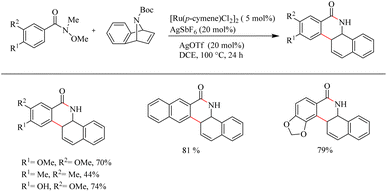 |
| | Scheme 22 Scope of diversely substituted azabenzonorbornadienes in Ru(II)-catalyzed hydroarylation with aryl amides. | |
2.2 Ru(II)-catalyzed intramolecular hydroarylation reactions in akene tethered arenes
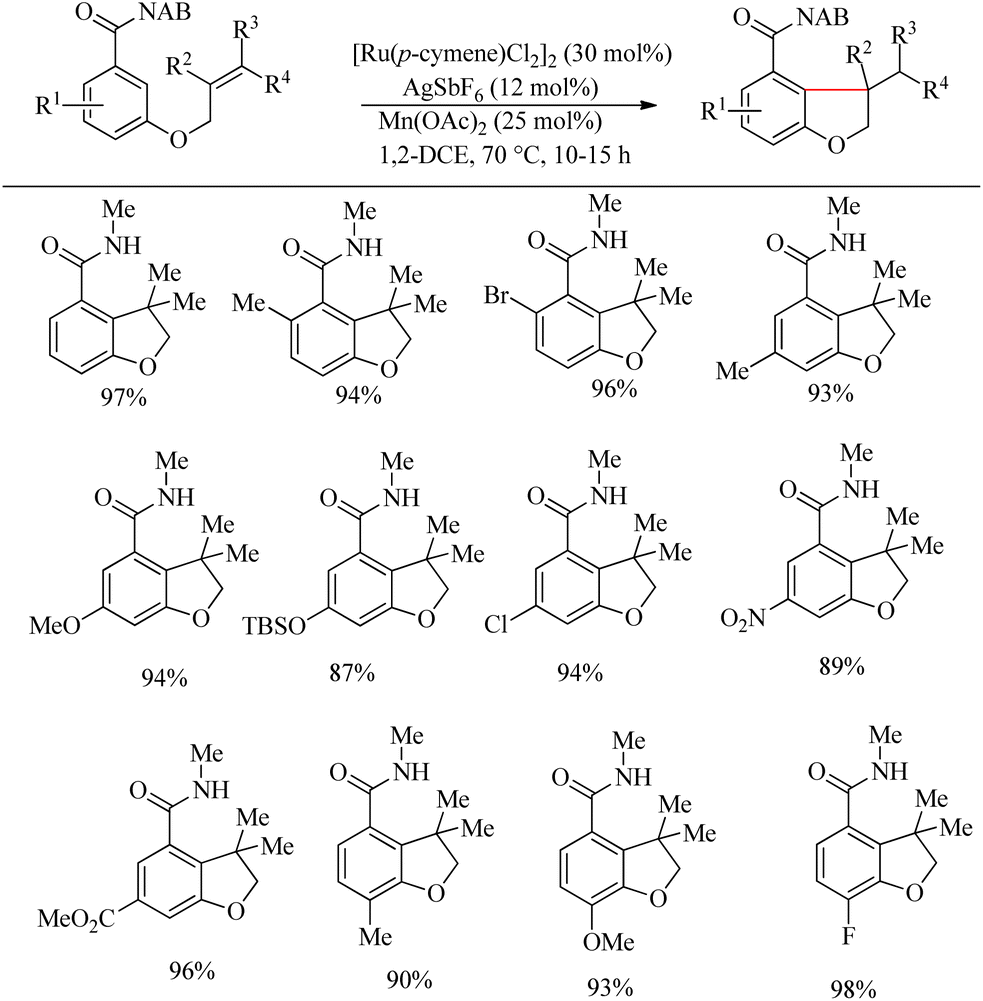
Inspired from the pioneering work of Murai4 on directing group aided Ru-catalyzed hydroarylation reaction, Sahoo et al. reported an atom-economic and step-economic18 for the synthesis of various heterocycles via amide directed Ru-catalyzed intramolecular hydroarylation on alkene tethered benzamide derivatives. The proposed methodology was found to show wide substrate scope, good functional group tolerance and labile protecting units. Attempts to optimize the reaction conditions resulted in [Ru(p-cymene)Cl2]2 (30 mol%) as catalyst, AgSbF6 (12 mol%) as additive in 1-2 DEC solvent and catalytic amount of Mn(OAc)2 (25 mol%) as the base. In the absence of base, the reaction was found to be passive. Various alkene tethered benzamides were subjected to the optimized reaction conditions to analyse the substrate scope. Both electron-donating and electron-withdrawing substituents afforded good yields (Scheme 23). Labile groups such as OTBS were found to be resistant to the applied conditions. The directing group ability of the ester moiety remained ineffective under the applied conditions. Independent of the electronic parameters, a range of substituents on nitrogen afforded excellent yields.
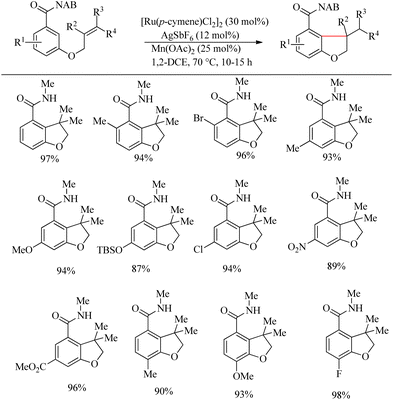 |
| | Scheme 23 Synthesis of O-heterocycles through amide directed Ru-catalyzed intramolecular hydroarylation on alkene tethered benzamide derivatives. | |
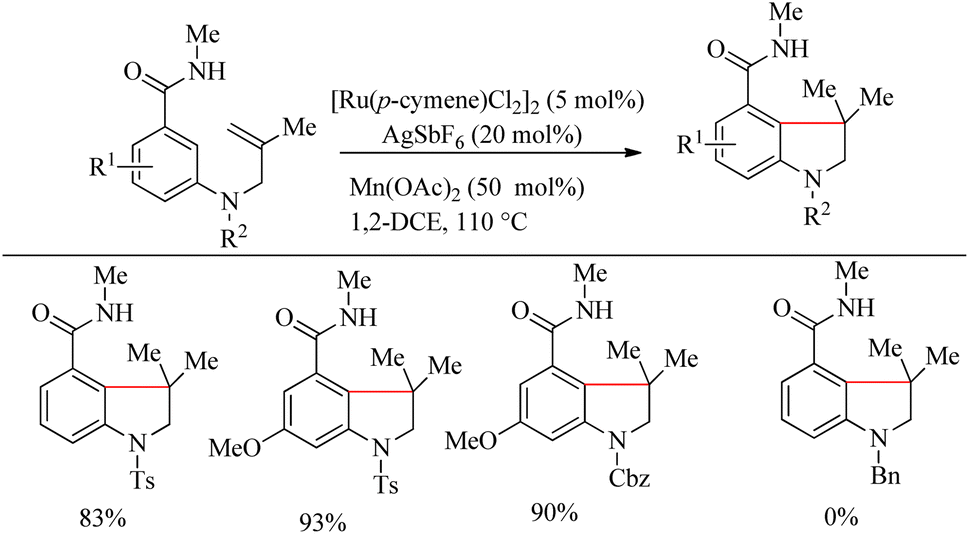
The optimized reaction condition was extended to the N-tethered olefin substrate in presence of Mn(OAc)2 (50 mol%) at 110 °C to construct the indoline scaffold in excellent yields of 83–93% (Scheme 24). N-Bn protected benzamide did not afford any product due the coordination of its nitrogen lone pair with the Ru species. To synthesize the chroman scaffold, which is an important pharmacophore, the optimized reaction conditions were applied to 3-O-homoallyl tethered benzamides, and relatively good yields were reported. This is the first known Ru-catalyzed amide group directed hydroarylation for the synthesis of chroman derivative.
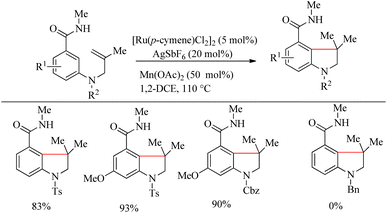 |
| | Scheme 24 Synthesis of N-heterocycles through amide directed Ru-catalyzed intramolecular hydroarylation on alkene tethered benzamide derivatives. | |
Based on a series of mechanistic investigations, they proposed a plausible mechanism for this amide directed intramolecular hydroarylation reaction. The first step is the formation of a cycloruthenated complex (A) through the ortho-C–H bond activation and the coordination of the active catalyst to the amide group. The next step involves the activation of the sterically hindered C–H bond via the additional interaction of oxygen tethered olefin with the metal centre. Finally, the consecutive migratory insertion and proto-demetallation yield the product and the regenerated catalyst (Scheme 25).
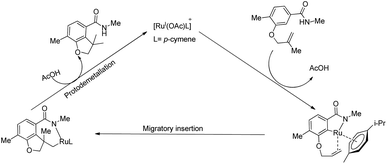 |
| | Scheme 25 Proposed mechanism for the amide directed intramolecular hydroarylation reaction. Reproduced with the permission of ref. 18. | |
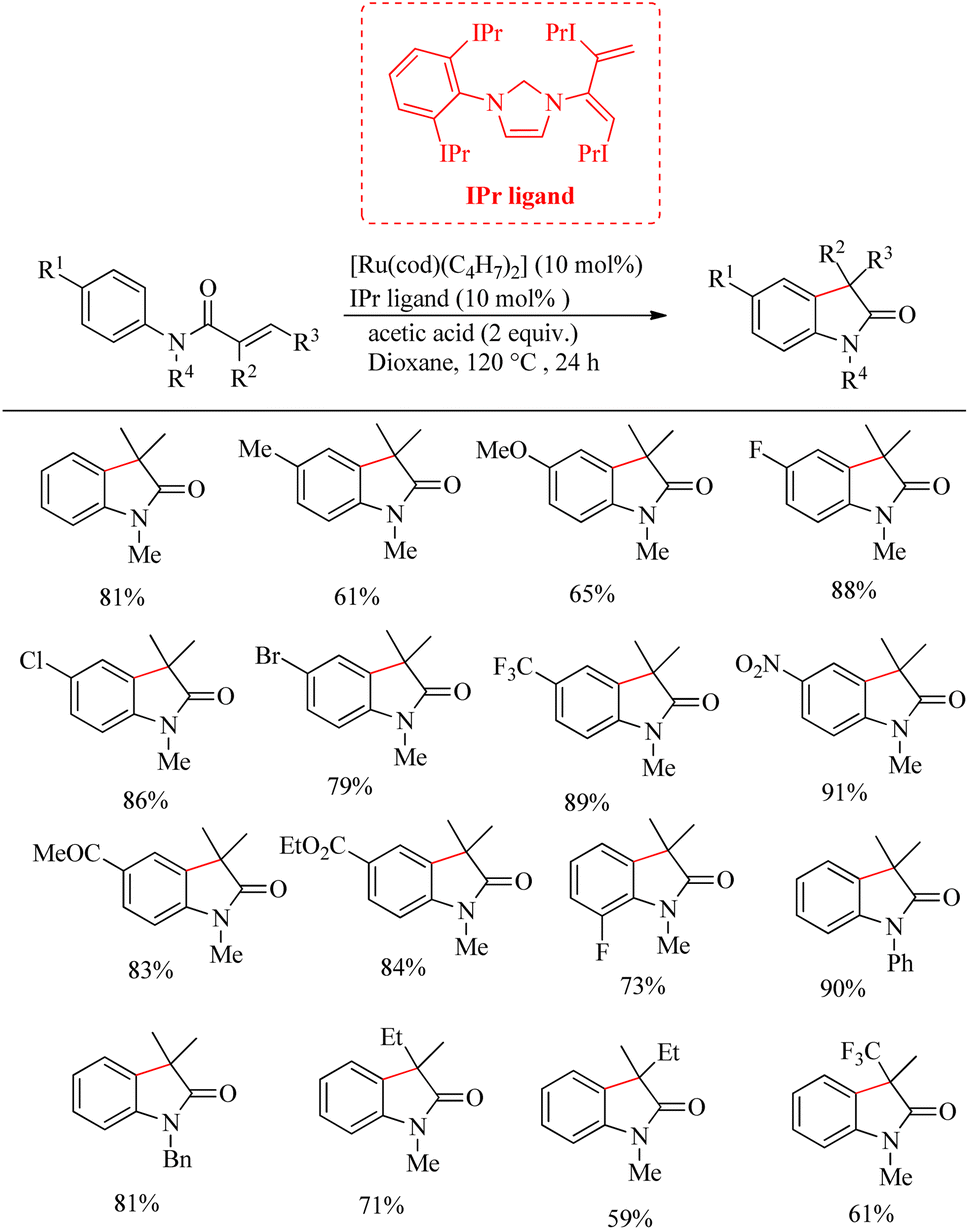
Zhao et al. successfully derived an atom-economic strategy for the preparation of biologically important oxindole scaffolds via the Ru-catalyzed intramolecular alkene hydroarylation of N-aryl alkene tethered amide moiety.19 The major challenge with all the previous reports in this field was unsatisfactory substrate scope. They further hypothesized that the insertion of the appropriate directing group into the alkene moiety of the system would lead to the elimination of additional directing groups and wider substrate scope for the reaction. Inspired from their previous work20 on Ru/NHC catalysed [3 + 2] annulation between ketimines and alkynes, they proposed a new catalyst system for this reaction. They optimized reaction condition as 10 mol% [Ru(cod)(η3-C4H7)2]IPr and 2 equiv. of acetic acid at 120 °C in dioxane solvent over 24 h (Scheme 26). Interestingly no other byproducts were detected. Studies on substrate scope revealed that para-electron-withdrawing groups with the exception of iodine and cyano groups provided good yields. In the case of p-tolyl substrates the acetic acid additive was replaced with trifluoro acetic acid. For running the reaction with ortho-fluro substituent, harsher reaction condition was needed. Replacement of methyl groups on nitrogen with phenyl or benzyl groups also afforded satisfactory yields. They also synthesized high value spirooxindoles by using 1-cyclopentene carboxylic amide as the substrate.
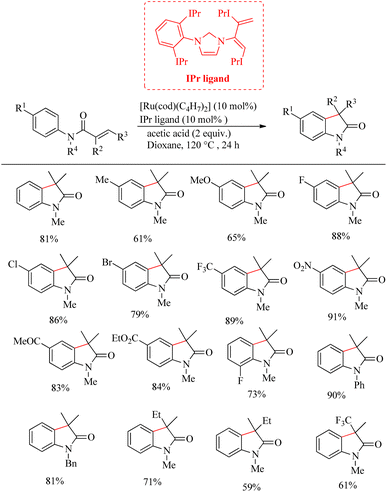 |
| | Scheme 26 Ru-catalyzed intramolecular alkene hydroarylation for oxindole synthesis. | |
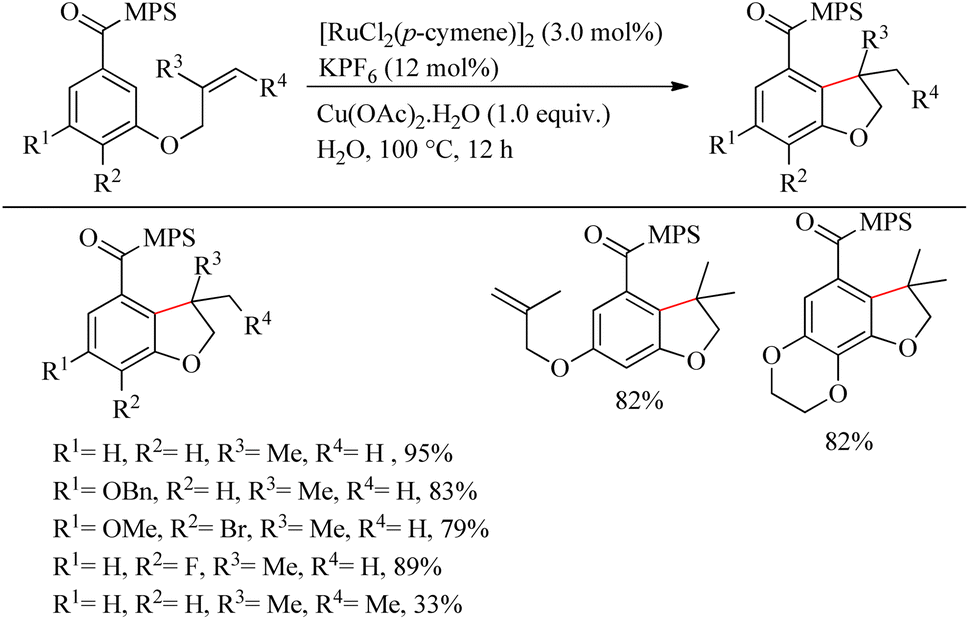
Sahoo et al. reported green synthesis of a biologically and pharmaceutically relevant dihydrobenzofuran motif via the sulfoximine or amide directed intramolecular hydroarylation reaction of olefin tethered arenes.21 Through this revolutionary approach, they overcame many challenges such as the immiscibility of organic substrates in water medium, the incompatibility of the reactive catalytic species in water medium, etc. When attempts to optimize the reaction conditions were performed with [3-(2-methyl allyloxy)benzoyl] methyl phenyl sulfoximine, the desired results were obtained at 100 °C, for 12 h of reaction with [RuCl2(p-cymene)]2 (3.0 mol%) as the catalyst, KPF6 (12 mol%) and Cu(OAc)2·H2O (1.0 equiv.) as additives (Scheme 27). The methyl phenyl sulfoximine groups drive the electron-rich arenes to undergo the reaction smoothly. The fluoro and the bromo groups on the aromatic ring were found to survive throughout the reaction. m,m′-di-o-allyl methyl phenyl sulfoximine [MPS] containing carboxylates yielded monohydroarylation product exclusively under the optimized conditions. Hydroarylation with trisubstituted olefinic part resulted in the formation of 3,3-methyl ethyl bearing dihydro benzofuran in 33% yield. The Me, OMe and NO2 substitution at the para-position of the aromatic ring decreased the yield of the reaction to zero due to the immiscibility of those in water even at higher temperatures. But when the sulfoximine group is replaced with an amide group, those aryl systems have undergone the reaction smoothly and yielded their corresponding products in presence of AgSbF6. The probable reason is attributed to the salt formation of amide moiety in presence of the acetate base and thereby drives the desired reaction at an elevated temperature.
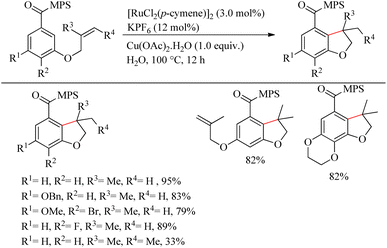 |
| | Scheme 27 Ru-catalyzed synthesis of dihydrobenzofuran motifs via sulfoximine or amide directed intramolecular hydroarylation reaction of olefin tethered arenes. | |
Experimentally supported mechanistic studies (Scheme 28) showed that the initial step of catalytic cycle involves the formation of an active Ru catalyst as a result of the reaction of [RuCl2(p-cymene)]2 with KPF6 and Cu(OAc)2·H2O. The next step involves the generation of a cyclometallated complex A via the ortho-C–H bond activation which is promoted by the coordination of Ru centre to the MPS directing group. Consequent migratory insertion of A yields B and the final step is the protodemetallation of B to afford the desired product regenerating the catalyst.
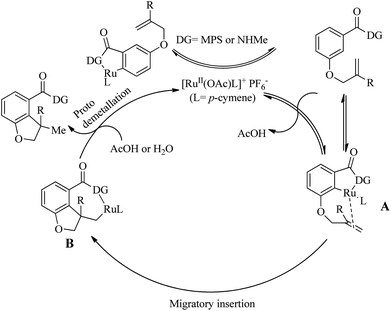 |
| | Scheme 28 Proposed catalytic cycle for the synthesis of dihydrobenzofuran catalyzed by Ru(II). Reproduced with the permission of ref. 21. | |
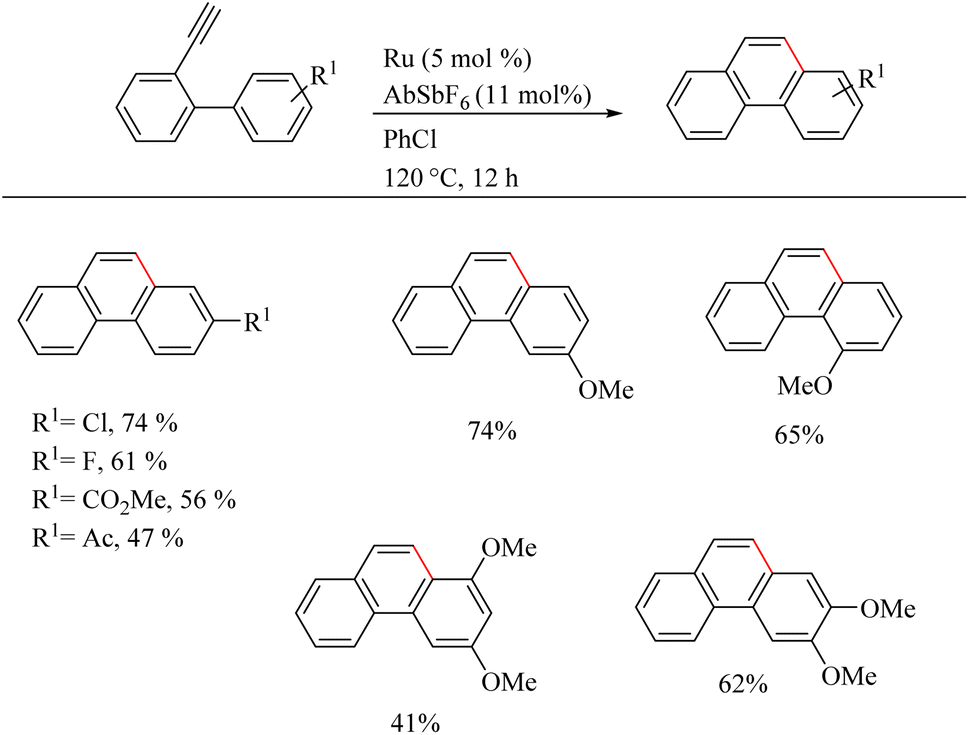
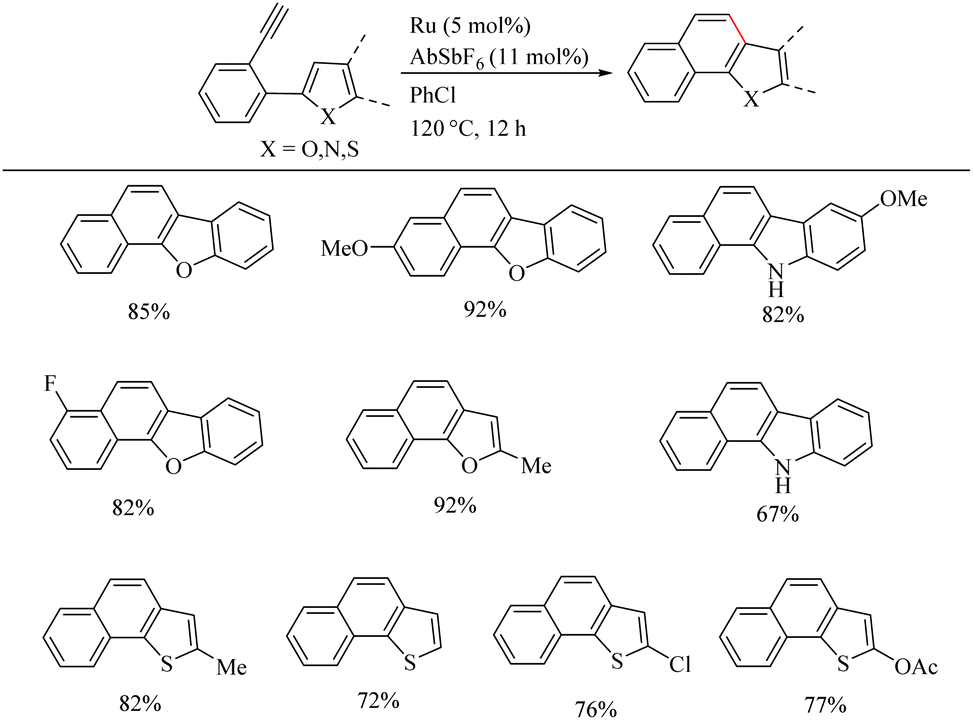
Yamamoto et al.22 reported an atom-economic synthesis of phenanthrene and its derivative scaffolds via Ru catalyzed intramolecular hydroarylation of ethyl and phenyl moieties of 2-ethynyl biphenyl systems. They developed a new catalytic system themselves for this purpose. After a series of investigations, they chose [(p-cymene)RuCl2(PPh3)] (5 mol%) as the desired catalyst and 11 mol% AgSbF6 (additive), PhCl (solvent) at 120 °C for 12 h as the optimized reaction conditions. Substrate scope studies show that the electron-withdrawing groups on the phenyl ring that undergoes the C–H activation reaction lower the yield of the reaction and electron-donating groups increase the rate of the reaction. When the substitution pattern on the phenyl ring was studied, the methoxy group in the 3′ position was found to afford a mixture of products and at 2′ position afford the corresponding product with a 65% yield due to steric factors. When the ethynyl group substituted phenyl ring contains electron-donating groups, it seems to have negative effect in the overall reaction rate. But the electron-withdrawing fluorine group gave relatively good yield of around 63% (Scheme 29). The substrate scope studies on the intramolecular heteroarene C–H hydroarylation reaction showed that numerous such scaffolds underwent the reaction smoothly with both electron-donating and electron-withdrawing groups on the ethyne substituted phenyl ring (Scheme 30).
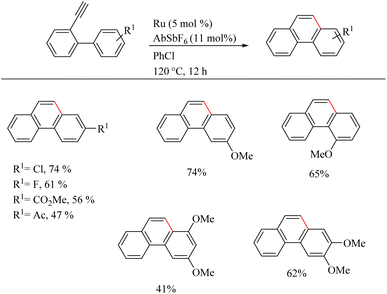 |
| | Scheme 29 Substrate scope of various 2-ethynyl biphenyl systems in Ru(II) catalyzed intramolecular hydroarylation. | |
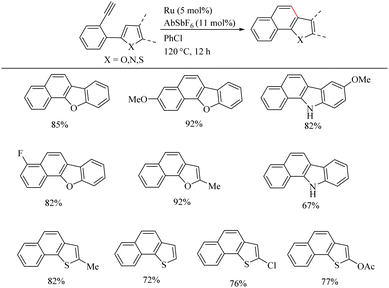 |
| | Scheme 30 The substrate scope of heteroarenes in Ru(II) catalyzed intramolecular hydroarylation. | |
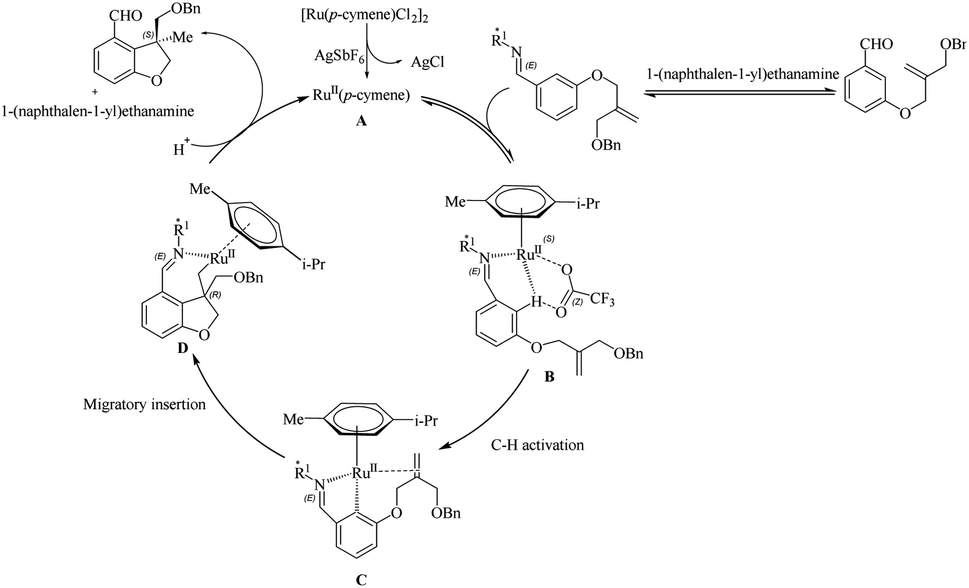
Wang et al.23 proposed a novel strategy for the synthesis of biologically important 2,3-dihydro benzofuran scaffolds24 via the relatively inexpensive Ru(II) catalyst through the chiral transient group directing intramolecular asymmetric hydroarylation reactions. Surprisingly, the Ru(II) catalyst produced products with high yield and enantioselectivity. This study built on previous work by the same group25 on the same synthetic route catalyzed by a structurally similar Rh(III) catalyst. They used various amino acids with different stereochemical configurations as the chiral reagents in this transformation. The optimization of reaction conditions shows that 1-(naphthalen-1-yl)ethanamine (20 mol%) is the most efficient amino acid and the L-tert-leucine amminoacid is observed to give no result. Acid is playing an inextricable role here in the catalytic cycle since without it the reaction does not proceed, and TFA is found to be the more efficient among various acids. The reaction for a duration of 60 h at 60 °C with 20 mol% of TFA and the aforementioned amino acid with AgSbF6 as the additive (20 mol%) under nitrogen atmosphere in DCE solvent (0.3 mL) yielded excellent results. Both electron-donating and electron-withdrawing substituents on the arene yielded the corresponding products with good yield and enantioselectivity (Scheme 31). Moreover, the extension of this methodology to the synthesis of indoline derivatives was found less successful. The strategy was successfully employed towards the synthesis of the CB2 receptor agonist MDA7.
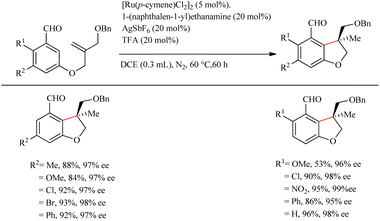 |
| | Scheme 31 Chiral transient group directed asymmetric intramolecular hydroarylation reaction via the Ru(II) catalyst. | |
The first step of the proposed catalytic cycle involves the generation of an active species A through the dechlorination of Ru(II) catalyst by AgSbF6 (Scheme 32). The next step is the coordination of the imine, which is formed by the reaction of the substrate with the chiral amino acid, to the Ru centre in the active species to form a new intermediate B. This intermediate undergoes a concerted metalation-deprotonation step (CMD) assisted by trifluroacetate to form a ruthenacycle C. Further step involves the formation of a new intermediate D which is formed by the coordination of tethered olefin followed by migratory insertion. The protonation of the C–Ru bond in D and the subsequent hydrolysis of imine gives the product regenerating the catalyst.
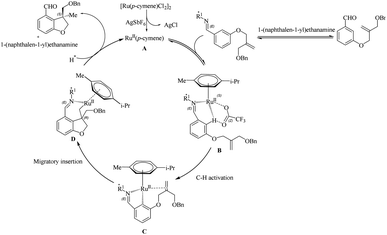 |
| | Scheme 32 Proposed catalytic cycle involving chiral transient group directed asymmetric intramolecular hydroarylation reaction via the Ru(II) catalyst. Reproduced with the permission of ref. 23. | |
2.3 Ru(II)-catalyzed hydroarylation reaction of alkynes: synthesis of trisubstituted alkenes
Urriolabeitia et al. reported a novel method for the synthesis of pharmaceutically relevant sulfur containing moieties via the sulfur group directed Ru(II)-catalyzed hydroarylation reaction of benzyl thioethers with alkynes.26 The reaction was found to be regioselective, atom-economic and step economic with a wide variety of electron-rich alkynes. This work pioneered the use of cheaper Ru complexes for the sulfur directed C–H functionalization reactions. Interestingly, for a variety of thioethers and electron-rich alkynes, the reaction yielded ortho-alkenylated products in a very short reaction time of 30 min under microwave irradiation conditions. The reaction conditions were optimized with benzyl phenyl thioether and Hex-3-yne. The yields were maximum at 100 °C for 24 h in MeOH and in presence of Cu(OAc)2 as additive. Hexafluroisopropanol was found to be the best choice of solvent. Under microwave irradiation, the reaction time was drastically reduced. Other –OAc sources such as AgOAc, NaOAc etc. were not efficient. In order to eliminate the possibility of bis-hydroarylation and the consequent formation of the mixture of products, one of the ortho-positions of the benzyl fragment was blocked with a variety of substituents. Better yield were noted with the Me substituent. Excellent results were obtained for the coupling of electron-rich alkynes such as 3-hexyne, 2-butyne and 2-hexyne. The addition of alkynes to arenes was found to be syn selective resulting in a trisubstituted vinylic fragment that was E in stereochemistry. Good regioselectivity was observed with the alkyne 1-phenylpropyne since the molar ratio of the regioisomers thus formed was 9.1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, but not with 2-hexyne where equimolar mixture of the products was formed. When the starting alkyne was introduced with two aryl rings, the yield of the reaction rapidly declined. The presence of electron-withdrawing groups at the ring where C–H activation is proceeding proved to have no effect on the rate of the reaction. When the electron density at the sulfur atom was varied, notable changes in the reaction yield were observed. In the presence of the phenyl group that containing sulphur (–SPh), electron-donating groups such as 4-OMe or 4-tBu produced a high yield of alkenylated products in a short reaction time of 30 minutes. But due to steric effects, the substitution at the 2′ position leads to a drop in the yield. The presence of electron-withdrawing groups at the SPh moiety decreased the rate of the reaction. The presence of a single electron-withdrawing group resulted in the requirement of a relatively higher time of irradiation of microwaves (30 min. to 2 h). The introduction of alkyl moieties such as tert-butyl, cyclohexyl or ethyl groups to the sulfur of thioether drives the reaction to excellent yields. But with the SMe moiety the reactions were not proceeding (Scheme 33).
:1, but not with 2-hexyne where equimolar mixture of the products was formed. When the starting alkyne was introduced with two aryl rings, the yield of the reaction rapidly declined. The presence of electron-withdrawing groups at the ring where C–H activation is proceeding proved to have no effect on the rate of the reaction. When the electron density at the sulfur atom was varied, notable changes in the reaction yield were observed. In the presence of the phenyl group that containing sulphur (–SPh), electron-donating groups such as 4-OMe or 4-tBu produced a high yield of alkenylated products in a short reaction time of 30 minutes. But due to steric effects, the substitution at the 2′ position leads to a drop in the yield. The presence of electron-withdrawing groups at the SPh moiety decreased the rate of the reaction. The presence of a single electron-withdrawing group resulted in the requirement of a relatively higher time of irradiation of microwaves (30 min. to 2 h). The introduction of alkyl moieties such as tert-butyl, cyclohexyl or ethyl groups to the sulfur of thioether drives the reaction to excellent yields. But with the SMe moiety the reactions were not proceeding (Scheme 33).
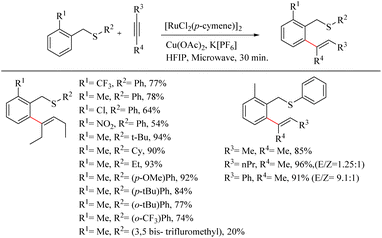 |
| | Scheme 33 Synthesis of sulfur containing moieties through Ru(II)-catalyzed hydroarylation reaction of benzyl thioethers with alkynes. | |
According to the proposed reaction pathway (Scheme 34), the first step of the catalytic cycle involves the S-bonding from the thioether to Cu(OAc)2. In the next step this S-bonded intermediate (A) transfers one-OAc and the thioether species to the Ru centre of the catalyst. This ruthenated species (B) then interacts with the alkynic part via migratory insertion to form (C). The last step of the catalytic cycle involves the protodemetellation of (C) and generation of the ortho-vinyl thioether.
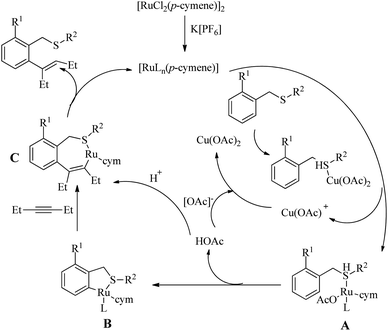 |
| | Scheme 34 Plausible reaction pathway of Ru-catalyzed hydroarylation reaction of alkynes with benzyl thioethers via the directing effect of sulfur. Reproduced with the permission of ref. 26. | |
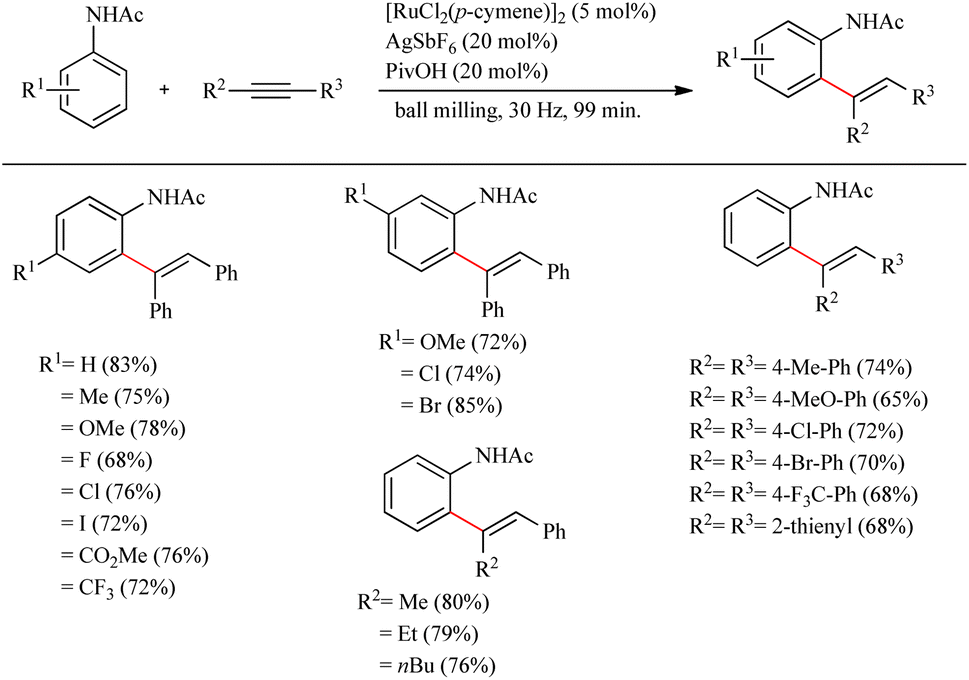
Recently Bolm et al. reported a revolutionary approach in hydroarylation reactions by adopting the mechanochemical approach of ball-milling.27 By employing the less expensive [RuCl2(p-cymene)]2 they performed the hydroarylation of various alkynes with acetanilide and its derivatives via ball milling process. They found that ball milling is very effective in hydroarylation reaction and proved that it is faster than the its solution based counterpart even in the absence of external heating. For obtaining the optimization conditions, a series of screening was carried out which revealed that unlike in the case of solution based method, if the amount of pivalic acid additive was reduced to 20 mol%, in presence of 5 mol% [RuCl2(p-cymene)]2 as the catalyst and 20 mol% AgSbF6 as the silver salt, the yield of the product was increased up to 82% (milling time = 198 min). By employing the same pivalic acid additive, but at a much shorter milling time of 99 min, surprisingly 83% yield of the product was obtained. Moreover they observed that the para-substituted acetanilide derivatives with both electron-donating and electron-withdrawing substituents exhibited high efficiency with yields ranging from 68% to 33% (Scheme 35). Also various alkynes were applied with the acetanilide and found that p-substituted diaryl alkynes with electron-donating or electron-withdrawing substituents afforded good yields. The di-substituted alkyne was found to show high regioselectivity in the reaction as the addition was proceeding exclusively at the alkyl substituted carbon.
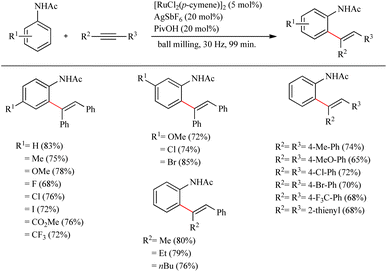 |
| | Scheme 35 Ru-catalyzed hydroarylation of alkynes with acetanilide. | |
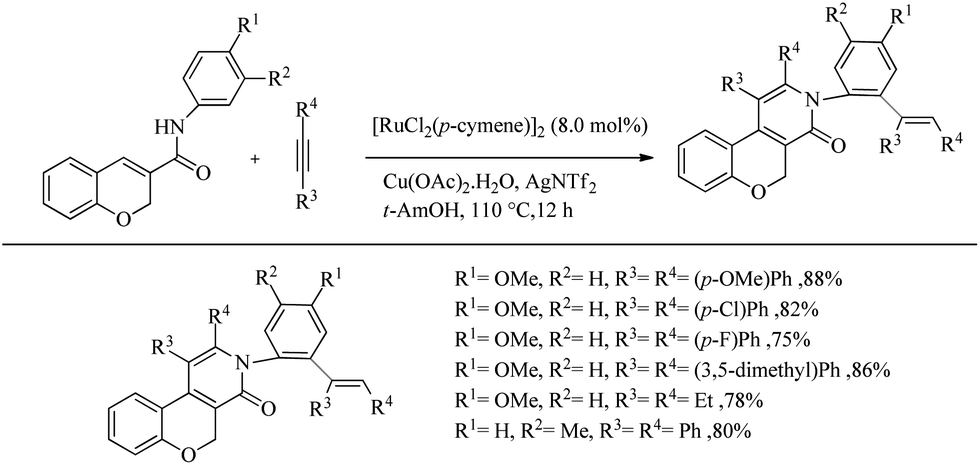
Swamy et al. reported an amide directed Ru-catalyzed oxidative annulation of 2H-chromene-3-carboxamides which lead to the formation of biologically active benzopyran fused pyridine scaffolds.28 Both symmetrical and unsymmetrical alkynes underwent the reaction smoothly. With unsymmetrical alkynes, the extend of regioselectivity was high and by supplying an excess of alkyne they also reported regioselective and stereoselective one-pot double C–H activation where both oxidative annulation and hydroarylation were proceeding via the dual role of catalyst. Oxidative annulation was found to proceed with the involvement of Ru–N covalent bond formation while double C–H activation was involved with the Ru–O coordinate bond formation. The optimized conditions for the oxidative annulation reaction was proved as [RuCl2(p-cymene)2]2 (5.0 mol%) as the catalyst, AgNTf2 (30 mol%) as the additive and Cu(OAc)2·H2O (1.0 mmol) as the oxidant with 1 equiv. of alkyne at 100 °C for 14 h in tert-AmOH solvent. When the scope of various alkynes for the oxidative annulation was examined with N-(4-methoxy phenyl)-2H-chromene-3-caraboxamide as the partner, symmetric diaryl acetylenes underwent the reaction very smoothly independent of the electronic nature of the substituents present in them. Both heteroaryl alkynes and dialkyl acetylenes furnished the 2-pyridone derivatives smoothly. 1-Phenyl-1-propyne yielded one regioisomer exclusively whereas 1-methoxy-4-(phenylethynyl) benzene afforded equimolar mixture of 2 regioisomers. They also studied the effect of substituents on the nitrogen of amide with diphenyl acetylene as the partner, which showed that both N-aryl and N-alkyl substituents furnished the reaction smoothly.
The optimized condition for the double C–H activation reaction was proved to be 2 equiv. of alkyne with 8.0 mol% of [RuCl2(p-cymene)2]2 as the catalyst, Cu(OAc)2·H2O (2.0 equiv.) as the oxidant and AgNTf2 (30 mol%) as the additive in t-AmOH solvent at 110 °C For 12 h. The substrate scope studies showed that a wide variety of symmetrical and unsymmetrical alkynes with electron-donating and electron-withdrawing substituents are compatible with the optimized conditions. Dialkyl acetylenes also underwent the reaction smoothly. When the 3-methyl substituted chromene-3-carboxamide was subjected to the reaction with diphenyl acetylene, reaction was found to proceed exclusively at the less hindered side (Scheme 36).
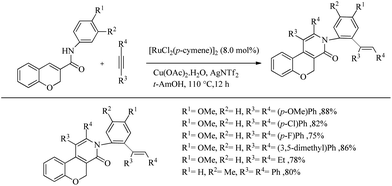 |
| | Scheme 36 Substrate scope studies on Ru-catalyzed double C–H activation reaction using 2H-chromene-3-carboxamide. | |
The vinyl trifluromethylated residues are of much importance in organic synthesis since it is found in several important categories of compounds like trifluridine, panomifene etc. But the direct introduction of vinyl trifluromethylated residues to an aryl ring via transition metal catalyzed C–H activation is practically difficult due to the low boiling point of coupling partners such as 3,3,3-trifluropropene and 3,3,3-trifluropropyne. Besset et al. recently reported an important strategy for synthesizing this scaffolds by the in situ generation of 3,3,3-trifluro-1-propyne from inexpensive non-toxic 2-bromo-3,3,3-trifluropropene (BTP) and the Ru(II)-catalyzed hydroarylation of this alkyne with the aryl species of our interest.29 The reaction conditions were found to be optimum with 5 mol% [RuCl2(p-cymene)]2 as the catalyst, 2.0 equiv. tBuONa as the base and DMF as the solvent at 90 °C for 24 h with 3-methyl-2-phenyl pyridine as the model substrate. Substrate scope studies on various arenes under the optimized reaction conditions showed that, compound bearing halogens, electron-withdrawing substituents as well as electron-donating substituents (except NHBoc) at the para position afford the desired mono-functionalized product in good yields (Scheme 37). Functionalization was found to proceed exclusively at the less hindered site. When arenes get substituted at the meta position with various electronically and sterically divergent groups, all of them have undergone the mono-funtionalization at the less hindered side. All those transformations are functional group tolerant towards various functional groups such as cyano, ketone, nitro and carabamate. When the phenyl group was replaced with heteroaromatic rings such as furan or thiophene, both were found to afford the products in relatively good yields.
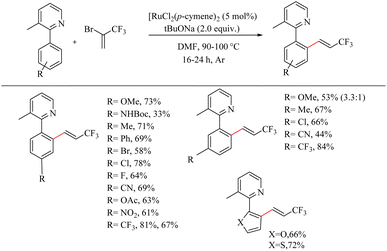 |
| | Scheme 37 Scope of substituted arenes in Ru(II) catalyzed hydoarylation with in situ generated 3,3,3-trifluro-1-propyne. | |
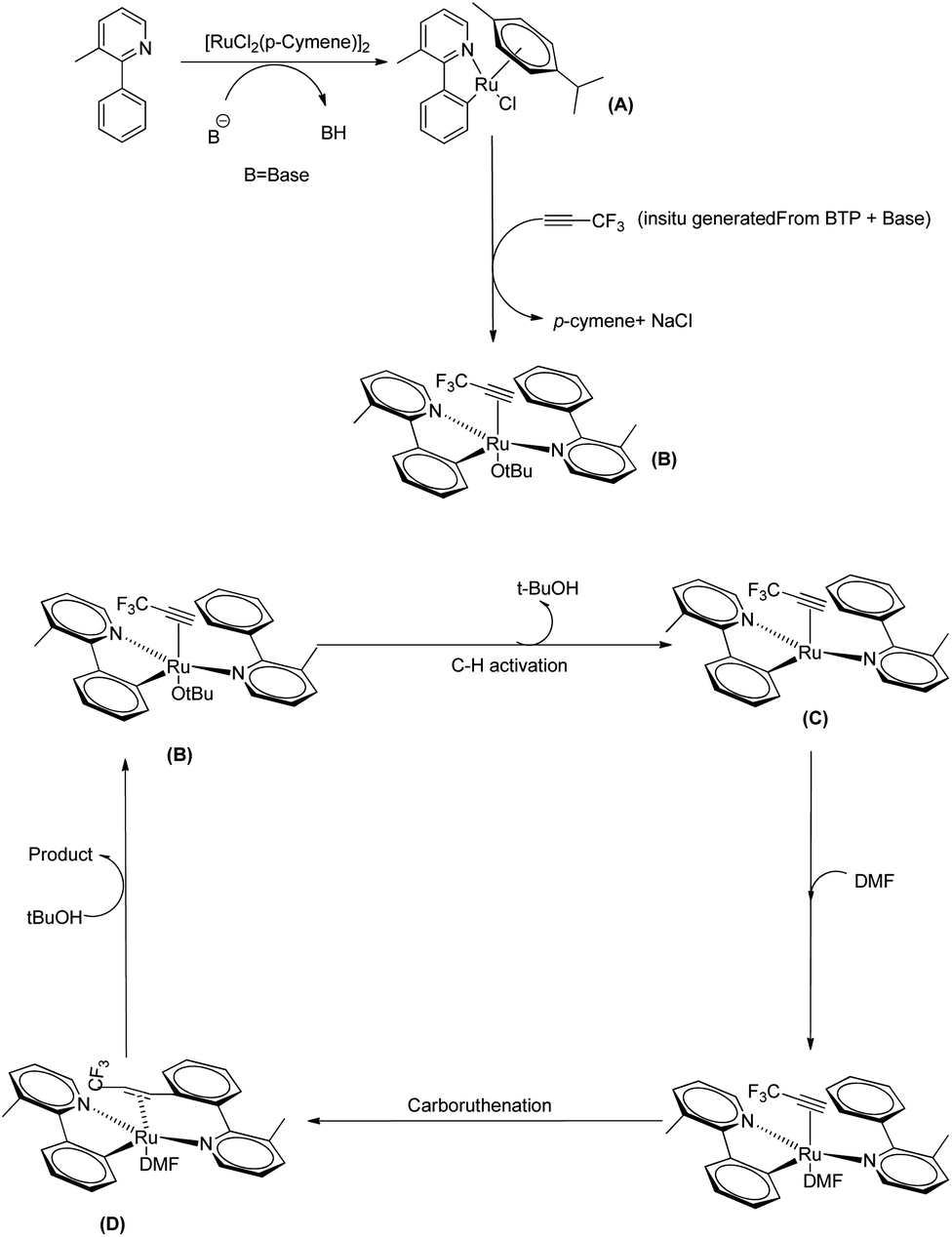
They proposed a plausible mechanistic pathway mainly based on DFT studies in which the first step involves a C–H bond activation which results in the formation of a monocylometallated Ru species (A) (Scheme 38). Then as a result of the de-coordination of p-cymene, an arene-free ruthenacycle (B) is generated in presence of in situ generated alkyne from BTP and tBuONa in DMF solvent. The next step involves the formation of a bis-cyclometallated Ru(II) complex (C). Then the DMF gets coordinated to the metal centre and a subsequent carboruthenation gives rise to the formation of (D). After a ligand exchange and protonation step it selectively provides the expected beta-trifluromethyl styrene and the regenerated active form of the catalyst.
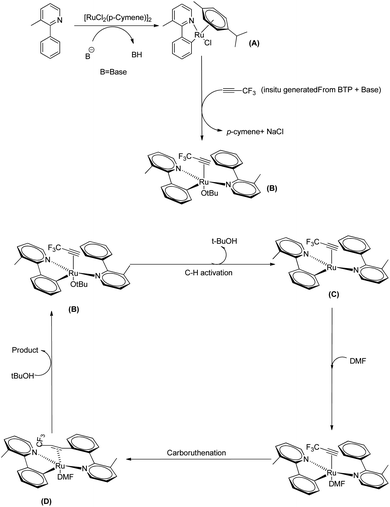 |
| | Scheme 38 Plausible mechanism of Ru(II) catalyzed hydoarylation of 3-methyl-2-phenyl pyridine and in situ generated 3,3,3-trifluro-1-propyne. Reproduced with the permission of ref. 29. | |
2.4 Traceless carboxylate directing group mediated C–H activation using Ru(II) catalyst
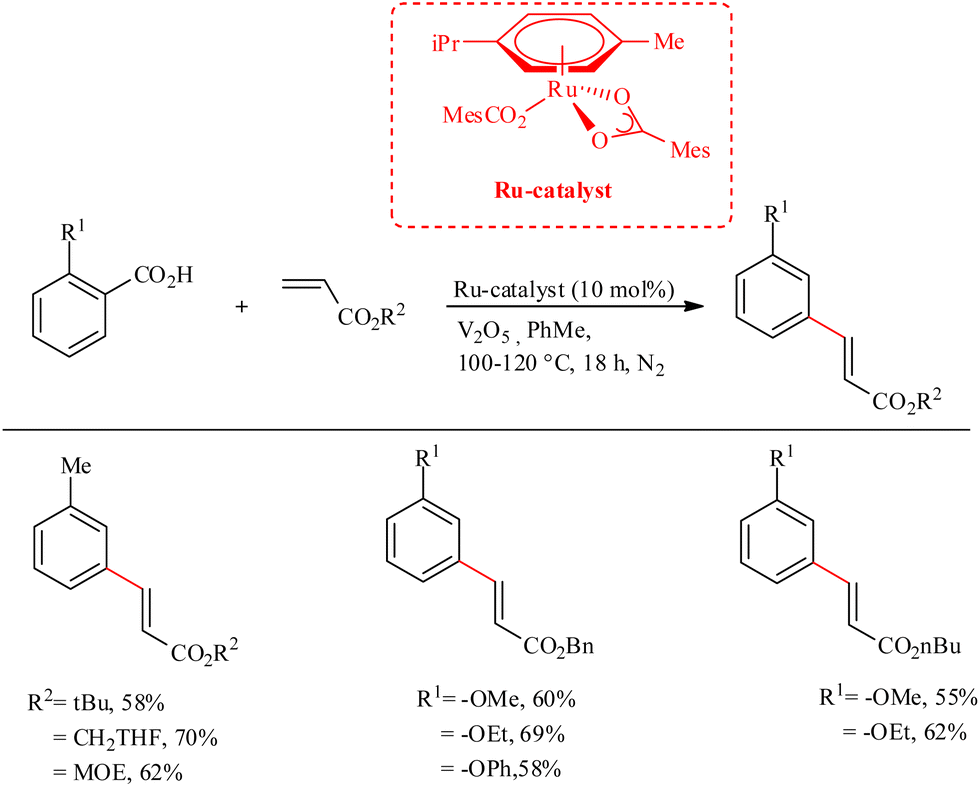
Ackermann et al. recently reported a strategic methodology for the decarboxylative C–H activation on substituted aryl carboxylic acids which finally led to the formation of meta-alkenylated arenes.30 Under ambient aerobic conditions, decarboxylation did not proceed, and such pathways resulted in isocoumarin and phthalide derived scaffolds via sequential C–H/C–H and C–H/O–H cleavage in the dehydrogenative C–H functionalization sequence. The use of Ru(II) catalyst eliminated the need for Ag(I) or Cu(II) salts, which were previously required in the corresponding synthetic routes using Pd or Rh-containing catalyst. Furthermore, Ru(II) bis (carboxylate)s necessitate a relatively low reaction temperature (100–120 °C). The catalyst can also set the stage for the neutral redox hydroarylation of alkynes. An inert atmosphere of argon or nitrogen proved to possess beneficial effects on the reaction. The use of additives such as V2O5 with 10 mol% of the aforementioned Ru(II) catalyst for 18 h gave the optimal result in toluene as the solvent. The catalyst has excellent functional group tolerance towards ester, chloro, bromo groups, as well as towards the cholesteryl moiety. During the catalytic cycle, the catalyst is found to show high chemo, regio and positional selectivity, and showed a wide range of substrate scope (Scheme 39).
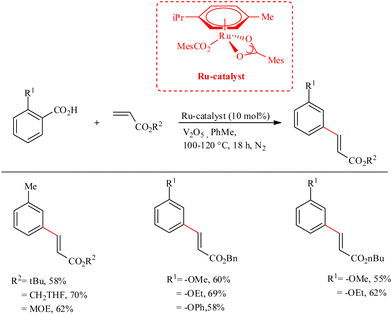 |
| | Scheme 39 Synthesis of meta-alkenylated arenes through Ru-catalyzed decarboxylative C–H activation on substituted aryl carboxylic acids. | |
Zhao et al. disclosed an efficient catalytic system for the preparation of biologically active alkenyl arenes via the decarboxylation incorporated and carboxy group assisted Ru(II)-catalyzed regio and stereo selective hydroarylation reactions of aromatic carboxylic acids with alkynes.31 Inspired by Murai and co-worker's study,32 various works were reported on chelation assisted Ru-catalyzed hydroarylation reaction. But in all cases, the removal of ortho-directing groups which are used for the regioselective hydroarylation reaction required complex additional chemical transformations. Since the alkenyl group has both the activating and ortho-directing property on metal mediated decarboxylation, Zhao et al. successfully overcame the difficulties by the development of ortho-substituted alkenyl arenes and pioneered strategies for the development of meta- or para-substituted alkenyl arenes through the hydroarylation pathway. Under the reported conditions, unlike other metal mediated decarboxylation, the reaction was found to proceed smoothly under mild and redox-neutral conditions. This characteristic property allowed the reaction of unprotected, oxidation sensitive functional groups such as anilines and phenols. The compatibility of the catalytic system was optimized with the reaction between benzoic acid and diphenyl acetylene. The optimized catalytic conditions were reported as Ru(p-cymene)(OAc)2 as the catalyst at 80 °C over 48 h in a mixture of solvent comprising of dioxane, mesitylene and heptane in 2:2:1 ratio. All of the hydroarylation reactions took place with syn stereoselectivity under these conditions. Para-substituted benzoic acids afforded the meta-substituted alkenyl arenes. Substrate scope studies showed that para-substituents with electron-donating properties such as methyl, methoxy, and dimethyl aminogroups etc. afforded the corresponding products with the maximum yield. The application of weekly electron-donating group such as the para-methyl thio group required a raise in the reaction temperature. Furthermore, even at high reaction temperatures, para-substitution with deactivating groups such as acetyl, carboxamide, CF3, and halogen results in a rapid decrease in product yield. Ortho-substituted benzoic acids also yielded meta-substituted alkyl arenes similar to the para-substituted benzoic acids or their derivatives (Scheme 40).
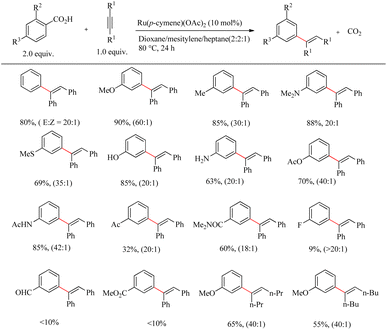 |
| | Scheme 40 Ru(II)-catalyzed regio and stereoselective hydroarylation reaction of aromatic carboxylic acids with alkynes. | |
Reaction with meta-substituted benzoic acids suggested that, depending upon the electronic and steric properties of the substituents, substitution can happen at both the ortho-sites. When the substitution in the meta-position is tried with sterically demanding groups, para-alkenylated product is found to form exclusively. A possible reason for this is attributed to the dominating power of electronic effects over steric effects. When the scope of various alkynes was investigated with 4-methoxy benzoic acid as the reaction partner, terminal alkynes such as phenyl acetylene were found to be non-reactive, whereas unsymmetrical aryl-alkynes reacted stereoselectively to form 1-alkyl-1-meta-anisyl alkene products (Scheme 41). The substitution of phenyl acetylenes with methyl, ethyl and n-butyl moieties afforded the corresponding products with a relatively low yield and required 20 mol% of Cu(OAc)2 as the additive. Surprisingly, methoxymethyl substituted aryl acetylenes reacted relatively quickly with no need for Cu(OAc)2 as an additive. In the case of diaryl acetylenes, ortho-substituted aryl groups produced higher yields than with meta- or para-substituted aryl groups.
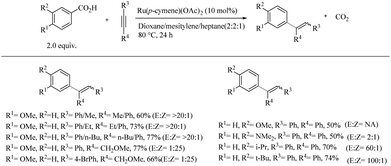 |
| | Scheme 41 Regio and stereoselective hydroarylation reaction of aromatic carboxylic acids with alkynes catalyzed by Ru(II). | |
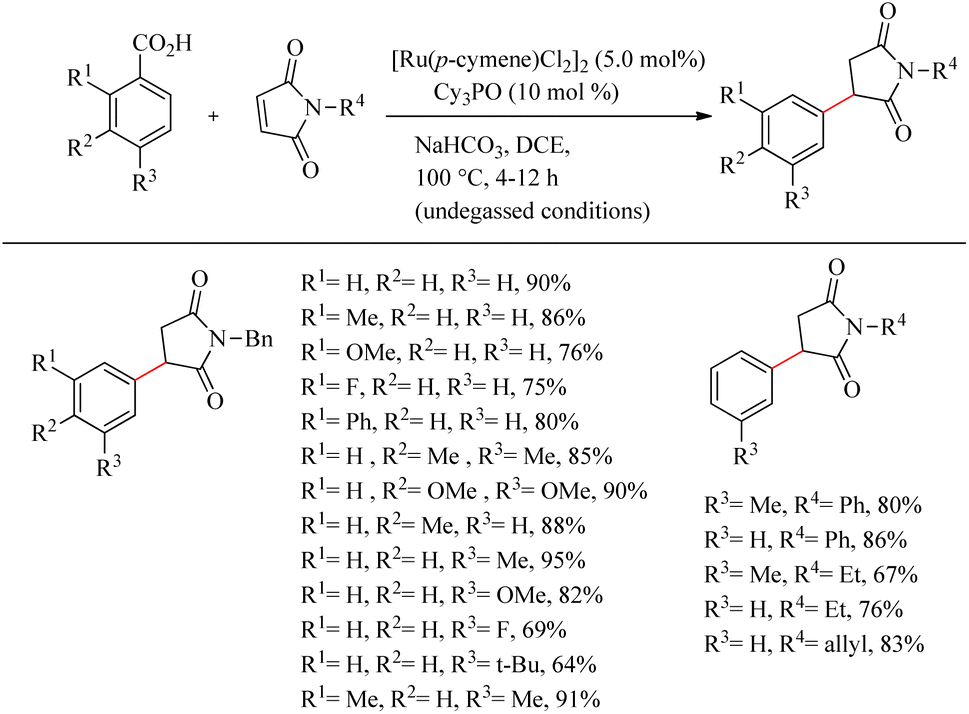
Baidya et al. reported a novel strategy for the atom-economic and step-economic synthesis of biologically important 3-aryl succinimide scaffolds via the Ru(II)-catalyzed hydroarylation reaction of maleimides by employing the directing effect of traceless carboxylate group.33 The synthetic scheme possesses wider substrate scope and operational simplicity. Moreover it avoids the use of expensive metal additives or oxidants. Optimization of reaction conditions commenced by studying carboxylate directed ortho-C–H alkylation reaction of p-toluic acid (0.2 mmol) with maleimide (0.3 mmol) (Scheme 42). Commercially available [Ru(p-cymene)Cl2]2 (5.0 mol%) was the preferred catalyst in DCE solvent. The reactivity of the catalyst was found to improve in the presence of phosphine oxide Cy3PO (10 mol%). The reaction was ineffective in the absence or replacement of the above mentioned Ru(II) catalyst and the base NaHCO3 (1.0 equiv.). The reaction was also smooth for sterically hindered di- or trisubstituted benzoic acids. But generally, the reaction proceeds faster with electron-donating substituents. Ortho- and para-substituted benzoic acids afforded the meta-substituted product, and meta-substituted benzoic acids resulted in the ortho-, para-substituted products. Both electron-donating and electron-withdrawing substituents in maleimide nitrogen effortlessly led to the desired product.
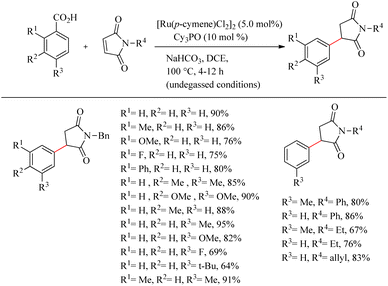 |
| | Scheme 42 Synthesis of 3-aryl succinimides through Ru(II)-catalyzed hydroarylation reaction of maleimides with aryl carboxylic acids. | |
2.5 Attempts to improve the efficiency of Ru catalyst
Schoztak et al. reported a novel strategy for the hydroarylation of industrially relevant alkynes using weakly coordinating ketones via the catalytic activity of in situ generated Ru(0) catalyst.34 The use of Ru(0) catalyst in addition to the conventional Ru(II) catalyst has got numerous advantages which include high atom economy of hydroarylation reactions under reductive conditions which eradicate the use of oxidants – a mandatory requirement on Ru(II) catalytic cycle, Ru(0) catalysts are economically more feasible, moreover the field of Ru(II)-catalyzed C–H activation has got notable progress in the recent times. RuH2(PR3)2 was prepared in situ from the commercially available [Ru(p-cymene)Cl2]2 by reacting it with sodium formate in presence of various phosphines.
For optimizing the reaction conditions, reactions were performed on a series of α-tetralone derivatives (1.0 equiv.) with diphenyl acetylene (2.0 equiv.) under various reaction conditions. The studies revealed that P(p-OMe-Ph)3 (15 mol%) is the most favourable ligand in 2.5 mol% of [Ru(p-cymene)Cl2]2 as the catalyst and toluene as solvent (1.0 M) (Scheme 43). The reaction was found to be more fruitful at 160 °C. Both electron-neutral and electron-deficient ligands showed considerably higher yields. The reaction was found to afford relatively good yields with various sterically and electronically different starting materials. The reaction conditions were also proved to be valid for the β-vinylic C–H activation of acyclic amide substrates. RuCl3 was found to be an efficient pre-catalyst for the in situ generation of Ru(0). Attempts to experimentally elucidate the mechanism of the reaction revealed certain useful information such as electron-deficient substrates are more favourable with the Ru(0) catalytic cycle, reactivity decreases with increase in the steric bulkiness of ketone etc. (via the intermolecular competition experiments).
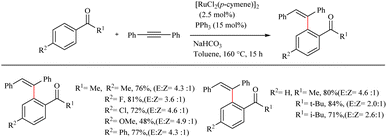 |
| | Scheme 43 Hydroarylation of ketones with alkynes using Ru-catalyst. | |
Miura et al. reported an elegant and environmentally-benign synthesis of CeO2 supported Ru species, which along with PPh3 and formaldehyde in 2-methoxy alcohol was found to generate a Ru(IV)-oxospecies on the surface of CeO2.35 This Ru(IV)-oxo species can effectively transform into a variety of low-valent Ru-species, which are found to be very useful for a wide range of organic transformations. Ru catalyst which is supported on other media like SeO2, Al2O3, TiO2 etc. were found to be passive for the hydoarylation purpose. The generated active form of the catalyst showed higher efficiency and reproducibility with a wide substrate scope. The Ru/CeO2 catalyst on treatment with 2-methoxy ethanol in presence of PPh3, formaldehyde (36% aqueous solution at 140 °C for 30 min) and adequate removal of solvent under reduced pressure yields the catalyst of interest (HCHO + 4PPH3)–Ru/CeO2(Scheme 44). To prove the catalytic activity and efficiency, the catalyst at the optimized reaction condition (0.023 mmol of Ru, toluene as the solvent (1.0 mL), and 140 °C) was applied to a range of alkenes, which afforded excellent yields. The catalyst also showed high activity with a variety of vinyl silanes. The substrate scope of various aromatic ketones with the catalytic system was also examined and all of them underwent the reaction smoothly. With the applied catalytic conditions, the ester and cyano groups are found to show poor directing effect.
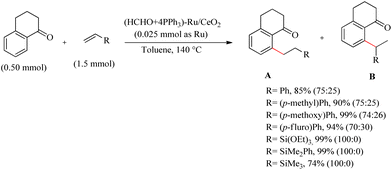 |
| | Scheme 44 Investigation of Ru-catalyzed hydroarylation of ketones with alkenes. | |
2.6 Effect of reaction parameters on the catalyst decomposition of Ru catalyzed ethylene hydrophenylation
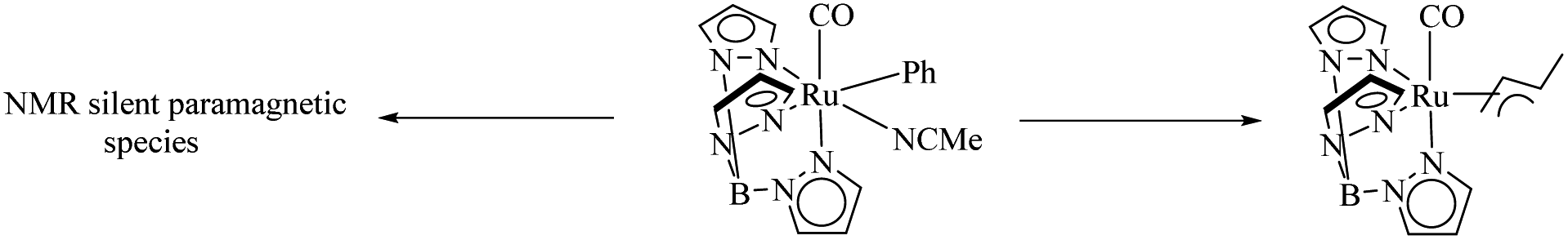
TpRu(CO)(NCMe)Ph is an efficient catalyst for the preparation of ethyl benzenes from benzene and ethylene via hydroarylation reaction.36 Gunnoe et al. studied in detail about the effect of concentration of the above catalyst and the ethylene pressure on the decomposition pathways of the catalyst.37 Studies show that there are two competing catalyst deactivation pathways in Ru catalyzed ethylene hydrophenylation and the dominant or recessive effect of these two pathways are dependent on the reaction conditions. Among the decomposition pathways one is a bimolecular decomposition mode which forms a paramagnetic species which is still uncharacterised and the second mode is an η3 allyl complex of catalyst formation via ethylene C–H activation (Scheme 45). Higher concentration of catalyst complex and lower concentration of ethylene species drive the catalytic decomposition via the uncharacterised paramagnetic species and at higher concentration of ethylene and lower concentration of catalyst complex, the decomposition pathway proceeds via the allyl complex of TpRu(CO)(η3-C4H7) formation. The maximum turnover is obtained at lower catalyst loading and at lower ethylene concentration.
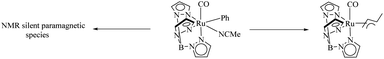 |
| | Scheme 45 Competing deactivation pathways. | |
3 Conclusion
The Ru-catalyzed hydroarylation reaction is an effective method for forming C–C and C-(heteroatom) bonds in organic synthesis. It is more interesting to explore the scope of this reaction due to the importance of hydroarylated products in pharmaceuticals and biologically active natural products. In this review, we have discussed the Ru-catalyzed hydroarylation of alkenes as an important tool for the synthesis of alkylated arenes, hydroarylation reactions of alkynes as a synthetic tool for trisubstituted alkenes, intramolecular Ru-catalyzed hydroarylation of olefin tethered arenes, modifications in the catalytic system to improve its efficiency, and carboxylate assisted Ru-catalyzed hydroarylation reactions in detail. We have also discussed the effect of directing groups that are present on the aryl systems on driving the reaction in a regioselective and stereoselective manner. Available mechanistic details of all kinds of hydroarylation reactions are explained which are also supported by strong experimental evidences such as deuterium labelling and kinetic studies etc.
The major challenge associated with most of the Ru-catalyzed hydroarylation reactions is the requirement of high temperature for the C–H activation. But in the future, probably we can overcome this by modifying the catalytic systems or proposing the appropriate reaction conditions with the already known catalysts. In our investigations, studies on the modification of catalytic system for their improved efficiency were found to be relatively few. As we discussed in detail, hydroarylation reactions are inextricable since they can act as a powerfull strategy for the synthesis of numerous biologically and pharmaceutically relevant compounds. So we believe that these drawbacks could be easily overcome in the future. In brief, numerous methods have been developed during the last decades, that can be used further to integrate better, more atom-economic, greener, and more powerful methods for the Ru-catalyzed hydroarylation of various compounds in synthetic organic chemistry.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
CMA thanks the Council of Scientific and Industrial Research (CSIR), New Delhi for the award of a junior research fellowship.
References
- Recent reviews:
(a) J. C. Lewis, R. G. Bergman and J. A. Ellman, Acc. Chem. Res., 2008, 41, 1013–1025 CrossRef CAS PubMed;
(b) L. Ackermann, R. Vicente and A. R. Kapdi, Angew. Chem., Int. Ed., 2009, 48, 9792–9826 CrossRef CAS PubMed;
(c) A. E. Wendlandt, A. M. Suess and S. S. Stahl, Angew. Chem., Int. Ed., 2011, 50, 11062–11087 CrossRef CAS PubMed;
(d) K. M. Engle, T. S. Mei, M. Wasa and J. Q. Yu, Acc. Chem. Res., 2012, 45, 788–802 CrossRef CAS PubMed;
(e) G. Song, F. Wang and X. Li, Chem. Soc. Rev., 2012, 41, 3651–3678 RSC;
(f) C. Zhu, R. Wang and J. R. Falck, Chem.–Asian J., 2012, 7, 1502–1514 CrossRef CAS PubMed;
(g) B. J. Li and Z. J. Shi, Chem. Soc. Rev., 2012, 41, 5588–5598 RSC.
- G. Caillot, J. Dufour, M. C. Belhomme, T. Poisson, L. Grimaud, X. Pannecoucke and I. Gillaizeau, Chem. Commun., 2014, 50, 5887–5890 RSC.
-
(a) C–N bond formation: J. Peng, Z. Xie, M. Chen, J. Wang and Q. Zhu, Org. Lett, 2014, 16, 4702–4705 CrossRef CAS PubMed;
(b) C–O bond formation: F. Yang, K. Rauch, K. Kettelhoit and L. Ackermann, Angew. Chem., Int. Ed., 2014, 53, 11285–11288 CrossRef CAS PubMed;
(c) C–S bond formation: C. Xu and Q. Shen, Org. Lett., 2014, 16, 2046–2049 CrossRef CAS PubMed.
- S. Murai, F. Kakiuchi, S. Sekine, Y. Tanaka, A. Kamatani, M. Sonoda and N. Chatani, Nature, 1993, 366, 529–531 CrossRef CAS.
- N. Chatani, T. Asaumi, S. Yorimitsu, T. Ikeda, F. Kakiuchi and S. Murai, J. Am. Chem. Soc., 2001, 123, 10935–10941 CrossRef CAS PubMed.
- S. I. Kozhushkov, D. S. Yufit and L. Ackermann, Org. Lett., 2008, 10, 3409–3412 CrossRef CAS PubMed.
- R. Manikandan and M. Jeganmohan, Org. Biomol. Chem., 2015, 13, 10420–10436 RSC.
- S. H. Watterson, T. G. M. Dhar, S. K. Ballentine, Z. Shen, J. C. Barrish, D. Cheney, C. A. Fleener, K. A. Rouleau, R. Townsend, D. L. Hollenbaugh and E. J. Iwanowicz, Bioorg. Med. Chem. Lett., 2003, 13, 1273–1276 CrossRef CAS PubMed.
- H. Cheng, W. Dong, C. A. Dannenberg, S. Dong, Q. Guo and C. Bolm, ACS Catal., 2015, 5, 2770–2773 CrossRef CAS.
- K. N. Reddy, M. V. Krishna Rao, B. Sridhar and B. V. Subba Reddy, ChemistrySelect, 2018, 3, 5062–5065 CrossRef CAS.
- D. Chowdhury, S. Dana, S. Maity and M. Baidya, Org. Lett., 2020, 22, 6760–6764 CrossRef CAS PubMed.
- J. W. Li, L. N. Wang, M. Li, P. T. Tang, N. J. Zhang, T. Li, X. P. Luo, M. Kurmoo, Y. J. Liu and M. H. Zeng, Org. Lett., 2020, 22, 1331–1335 CrossRef CAS PubMed.
- B. Ramesh and M. Jeganmohan, J. Org. Chem., 2022, 87, 5668–5681 CrossRef CAS PubMed.
- Y. Zhou, H. Liangong, Y. Sheng, S. Wang, Y. Gao, L. Zhan, Z. Zheng, M. Yang, G. Liang, J. Zhou, J. Deng and Z. Song, J. Org. Chem., 2020, 85, 9230–9243 CrossRef CAS PubMed.
- J. Li and L. Ackermann, Org. Chem. Front., 2015, 2, 1035–1039 RSC.
- D. Chowdhury, M. Koner, S. Ghosh and M. Baidya, Org. Lett., 2022, 24, 3604–3608 CrossRef CAS PubMed.
- N. Aravindan, V. Vinayagan and M. Jeganmohan, Org. Lett., 2022, 24, 5260–5265 CrossRef CAS PubMed.
- R. K. Rit, K. Ghosh, R. Mandal and A. K. Sahoo, J. Org. Chem., 2016, 81, 8552–8560 CrossRef CAS PubMed.
- P. Kilaru, S. P. Acharya and P. Zhao, Chem. Commun., 2018, 54, 924–927 RSC.
- J. Zhang, A. Ugrinov and P. Zhao, Angew. Chem., Int. Ed., 2013, 52, 6681–6684 CrossRef CAS PubMed.
- K. Mukherjee, E. Ramesh, K. Ghosh and A. K. Sahoo, Asian J. Org. Chem., 2018, 7, 1380–1384 CrossRef CAS.
- Y. Yamamoto, K. Matsui and M. Shibuya, Eur. J. Chem., 2015, 21, 7245–7255 CrossRef CAS PubMed.
- G. Li, Q. Liu, L. Vasamsetty, W. Guo and J. Wang, Angew. Chem., Int. Ed., 2020, 59, 3475–3479 CrossRef CAS PubMed.
- K. C. Nicolaou, S. A. Snyder, N. Giuseppone, X. Huang, M. Bella, M. V. Reddy, P. B. Rao, A. E. Koumbis, A. O'Brate and P. Giannakakou, J. Am. Chem. Soc., 2004, 126, 10174 CrossRef CAS PubMed.
- G. Li, J. Jiang, H. Xie and J. Wang, Eur. J. Chem., 2019, 25, 4688 CrossRef CAS PubMed.
- P. Villuendas and E. P. Urriolabeitia, Org. Lett., 2015, 17, 3178–3181 CrossRef CAS PubMed.
- H. Cheng, J. G. Hernández and C. Bolm, Org. Lett., 2017, 19, 6284–6287 CrossRef CAS PubMed.
- R. N. P. Tulichala, M. Shankar and K. C. K. Swamy, J. Org. Chem., 2017, 82, 5068–5079 CrossRef CAS PubMed.
- M. Vuagnat, V. Tognetti, P. Jubault and T. Besset, Eur. J. Chem., 2022, 28, e202201928 CAS.
- N. Y. P. Kumar, A. Bechtoldt, K. Raghuvanshi and L. Ackermann, Angew. Chem., Int. Ed., 2016, 55, 6929–6932 CrossRef CAS PubMed.
- J. Zhang, R. Shrestha, J. F. Hartwig and P. Zhao, Nat. Chem., 2016, 8, 1144–1151 CrossRef CAS PubMed.
- F. Kakiuchi, Y. Yamamoto, N. Chatani and S. Murai, Chem. Lett., 1995, 681–682 CrossRef CAS.
- A. Mandal, H. Sahoo, S. Dana and M. Baidya, Org. Lett., 2017, 19, 4138–4141 CrossRef CAS PubMed.
- F. Hu and M. Schoztak, Chem. Commun., 2016, 52, 9715–9718 RSC.
- H. Miura, M. Nagao, S. Hosokawa, T. Shishido, M. Inoue and K. Wada, Bull. Chem. Soc. Jpn., 2018, 91, 1397–1401 CrossRef CAS.
- N. A. Foley, J. P. Lee, Z. F. Ke, T. B. Gunnoe and T. R. Cundari, Acc. Chem. Res., 2009, 42, 585–597 CrossRef CAS PubMed.
- E. E. Joslin, B. A. McKeown, T. R. Cundari and T. B. Gunnoe, J. Organomet. Chem., 2017, 847, 289–293 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *b
*b