
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePreparation of a single and reusable biopolymer-based film for the extraction and preconcentration of anti-inflammatory drugs from environmental water samples†
Maite V. Aguinaga Martínez,
Claudia E. Domini* and
Carolina C. Acebal *
*
INQUISUR, Departamento de Química, Universidad Nacional del Sur (UNS)-CONICET, Av. Alem 1253, Bahía Blanca, 8000, Argentina. E-mail: cdomini@criba.edu.ar; cacebal@uns.edu.ar
First published on 20th March 2023
Abstract
One of the main goals of green chemistry is to reduce the use of toxic materials and the generation of hazardous waste, both during method development and in the synthesis of the materials used. Thus, a biodegradable, single and reusable material composed of agarose and multi-walled carbon nanotubes was proposed. The film preparation was carefully optimized in order to obtain a one-piece sorbent, with high extraction efficiency and the possibility of reuse. The film was tested in the simultaneous extraction and preconcentration of three non-steroidal anti-inflammatory drugs (ketorolac, ketoprofen and piroxicam) from environmental water samples. The optimal extraction parameters were as follows: isopropyl alcohol as the activation solvent, a sample pH value of 3.0, extraction time of 30 min, 2.00 mL of acetonitrile as the eluent, an elution time of 5 minutes, and a sample volume of 250.00 mL. Under these conditions, the film was reusable 50 times without losing its extraction capacity significantly. HPLC with a photodiode array detector was used for the separation and determination. The method presented a linear range between 0.10 and 1.2 μg L−1, good sensitivity with limits of detection between 0.0075 and 0.0089 μg L−1, and quantification between 0.025 and 0.030 μg L−1. In addition, low RSD values (0.46–3.13%) were obtained demonstrating satisfactory precision. Stream water samples were analyzed, and recoveries between 82.0 and 109.0% were obtained.
1. Introduction
Solid-phase extraction (SPE) is probably the most frequently adopted technique used for the extraction and preconcentration of pollutants in environmental samples. In comparison with the conventional liquid–liquid extraction (LLE) technique, it is simpler and consumes lower amounts of organic solvents.1 In addition, a wide variety of sorbents of different selectivity allows its application for the extraction of several organic compounds.2 Sorbent materials have an important function in obtaining high extraction yields and high enrichment factors of analytes.3 In recent years, the development of new and better materials with higher extraction capacity and improved mechanical or physicochemical stability, compared to traditional sorbents, has drawn attention.4 On the other hand, there is a significant tendency to improve the sustainability of the sample preparation procedure.5 Particularly, biopolymers that are obtained from biological macromolecules have been used to replace chemically synthesized sorbents. These materials present the advantage of being highly renewable, biodegradable and non-toxic.6Agarose is a natural polymer obtained from seaweed formed by repeated units of 1,3-β-D-galactose and 3,6-anhydro-α-L-galactose.7,8 It is considered as a green material because of its biodegradable nature9 and its gelling capacity that facilitates the fabrication of film-based layers. In addition, the gel structure of agarose can be conveniently modified by adding different materials while the gelation process occurs, obtaining interesting hybrid materials that are suitable for extraction processes.6 In this sense, agarose was mainly combined with well-established sorbents as multi-walled carbon nanotubes (MWCNTs) and C18 to perform the extraction of different pollutants from water samples and some beverages.9–12
Non-steroidal anti-inflammatory drugs (NSAIDs) belong to the group of the most widely used pharmaceuticals, showing analgesic and anti-inflammatory effects.13 Although the Food and Drug Administration (FDA) has a guidance on the safe disposal of pharmaceuticals,14 this information is unknown for the majority of the population. Thus, NSAIDs are discarded and can enter into the environment through domestic, industrial and hospital wastewaters since the existing water treatment plants are not designed to entirely remove them.15 They present high solubility in water, relatively low environmental stability and can be biodegradated.16 However, this is offset by its continued introduction into surface waters.17 Different studies have demonstrated that NSAIDs can produce toxic effects on living organism in the aquatic environment, such as fish and invertebrates, even at low concentration levels.18 Thus, its quantification in different water sources is of great interest. Particularly, some authors were able to determine NSAIDs of massive consumption like ibuprofen and diclofenac found in urbanized areas of Argentina, specially from cities located in the province of Buenos Aires, Córdoba and La Pampa.19,20
Several materials have been exploited as sorbents to perform the SPE of NSAIDs from environmental water samples. In most methods, commercial sorbents, such as C18,21 Strata X cartridges22,23 and Oasis HLB24,25 have been used. Among new materials, metal–organic frameworks (MOFs) and MWCNTs have also been proposed to extract NSAIDs in these matrixes.26–29 In addition, core–shell polyaniline/polyacrylonitrile nanofibers3 and polyethyleneimine-600 decorated magnetic microporous organic network nanosphere30 have been synthetized for extracting some of these analytes with good results. Concerning renewable materials, Ibrahim et al. proposed a MWCNTs-agarose-chitosan composite to perform the solid-phase microextraction of naproxen, diclofenac and mefenamic acid by using non-reusable pieces of the film inserted in a hypodermic needle.31
In this work, a film composed of agarose and MWCNTs was prepared for the simultaneous extraction and determination of ketorolac, ketoprofen and piroxicam. The new film is utilized as a single piece and it can be reused several times, resulting in a high reproducibility, less reagent use (and waste generation) and lower analysis costs. In addition, this hybrid material is potentially biodegradable since both agarose and MWCNTs are materials that can be degraded by bacteria and enzymes.32,33 The film was successfully used in the extraction of the selected NSAIDs from environmental water samples of Bahia Blanca's region.
2. Experimental part
2.1 Reagents and materials
The appropriate amount of ketorolac (KEC), ketoprofen (KEN) and piroxicam (PIR) (Sigma-Aldrich, Germany) were diluted in methanol, MeOH (≥99%, Sigma-Aldrich, Germany), to prepare 200 mg L−1 stock solutions that were kept in dark containers (4 °C). Working solutions were prepared daily by diluting a suitable volume of the stock solutions with an acetic acid (Sintorgan, Argentina) solution (pH 3.0). Agarose (BioReagent, for molecular biology, low EEO) was bought from Sigma-Aldrich (Germany). Acetonitrile, ACN (≥99%, Sigma-Aldrich, Germany) and formic acid (Sintorgan, Argentina) were used for preparation of the mobile phase. Isopropyl alcohol, IPA (≥99.8%) was purchased from Dorwil (Argentina). The MWCNTs (13–16 nm, purity >95%) were supplied by Bayer®.2.2 Instrumentation
The HPLC separation was carried out on a HPLC Waters 600 (Waters, USA) combined with a photodiode array detector (PDA). The analytes were separated using a C18 Gemini column (150 mm × 4.6 mm, 5 μm, Phenomenex) under isocratic mode. The mobile phase was composed of ACN and water acidified with 0.07% of formic acid in a 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 v/v proportion. The column temperature, the injection volume and the flow rate were 25 °C, 10 μL and 1.0 mL min−1, respectively. The separated analytes were detected at 254 nm (KEN), 312 nm (KEC) and 326 nm (PIR).
:50 v/v proportion. The column temperature, the injection volume and the flow rate were 25 °C, 10 μL and 1.0 mL min−1, respectively. The separated analytes were detected at 254 nm (KEN), 312 nm (KEC) and 326 nm (PIR).
The MWCNTs solution was dispersed in a Cole-Parmer EW-08895-16 ultrasonic bath. The MWCNTs-agarose film was dried in a laboratory oven (Model SL30S, San Jor, Argentina). A vacuum evaporator system (Speed Vac SPD11V, Thermo Fisher Scientific, Argentina) was used to evaporate the solvent.
The characterization of the agarose and the MWCNTs-agarose films, before and after drying, was performed with a Scanning Electron Microscope, SEM (LEO, EVO 40 XVP) with a secondary electron (SE) detector.
2.3 Preparation of the MWCNTs-agarose film
The preparation of the MWCNTs-agarose film was based on the procedure developed by Hong Loh et al.9 with modifications (Fig. 1). Briefly, 0.1 g of agarose and 5.0 mL of water were mixed in a beaker and submerged into a water bath at 80 °C. The solution was magnetically stirred until the agarose was completely dissolved. In another glass beaker, 80 mg of MWCNTs were mixed with 5.0 mL of water and sonicated in an ultrasonic bath for 15 minutes. Then, the dispersed MWCNTs were added to the agarose solution. The final mixture was heated with magnetic stirring at 80 °C for 30 minutes in order to disperse the MWCNTs in the agarose and to achieve a final volume of 5.0 mL. The mixture was placed over a circular glass (5.0 cm diameter) and cooled at 25 °C. The obtained film was dried at 40 °C for 24 h. The images of the obtained MWCNTs-agarose film, before and after drying, are presented in the ESI (Fig. S1†). | ||
| Fig. 1 Schematic representation of the preparation of the MWCNTs-agarose film procedure. | ||
2.4 SPE procedure
The MWCNTs-agarose film was first conditioned by submerging it in IPA for 5 minutes, and then washed with distilled water. Next, the film was placed inside 250.00 mL of a standard solution or sample containing the mixture of target analytes. The pH of the standard solution or samples was 3.0. The solution was magnetically stirred and the extraction was performed for 30 minutes. Then, the film was separated from the solution and washed with water. After that, it was placed inside a glass beaker containing 2.00 mL of ACN and sonicated in an ultrasonic bath for 5 minutes for elution. The film was removed from the eluate that was evaporated to dryness using a vacuum evaporator system at 30 °C. Then, the extract was reconstituted in 600 μL of the mobile phase. The extraction blank was performed using distilled water or the sample applying the same procedure.2.5 Sampling and preparation of samples
The samples were taken from various sampling points of the Naposta stream. Two of them (1: P1 38° 40′ 51.822′′S 62° 14′ 13.707′′W; 2: P2 38°45′09.0′′S 62°14′10.2′′W) were situated in the city of Bahía Blanca and the other one (3: P3 38°32′00.1′′S 61°52′26.4′′W) was located in the village of Cabildo (Fig. S2, ESI†). The samples were acidified and kept in the fridge (4 °C) until their analysis. Before the extraction procedure, the samples were left at 25 °C, homogenized, and filtered with an ashless filter paper (4–12 μm pore, Microclar, USA). After that, 250.00 mL aliquots were taken, acidified until pH 3.0 and extracted following the procedure mentioned in Section 2.4.To assess the accuracy of the proposed method, a recovery study was performed. The samples were spiked with KEC, KEN and PIR at three concentration levels within the calibration range in triplicate at the beginning of the analytical process.
3. Results and discussion
3.1 Optimization of the MWCNTs-agarose film preparation procedure
Most of the SPE approaches using biosorbents that can be found in the literature9–12,31 involved the use of several films or multiple pieces of the same film. This fact can cause difficulties during the extraction procedure, especially during the washing and elution steps. Thus, the main objective of this work was to obtain a single piece sorbent with high extraction efficiency that can be reused as many times as possible. For that purpose, the optimization of the preparation process was performed. The concentration of agarose was the same in all the experiments.At first, the MWCNTs were dispersed directly in the agarose solution. The accomplishment of a homogeneous dispersion was time consuming since the MWCNTs tended to aggregate. Thus, in order to ensure the homogeneity of the final mixture the dispersion of the MWCNTs was performed in water, assisted by ultrasound, before mixing them with the agarose solution. It was observed that the total dispersion of the MWCNTs required 15 minutes. At lower times, the dispersion was incomplete, resulting in longer times to reach a homogeneous MWCNTs-agarose mixture.
To achieve a satisfactory extraction capacity of the MWCNTs-agarose film, a concentration of MWCNTs higher than that found in the literature was tested.3,8,11 Therefore, the amount of MWCNTs was evaluated in the range of 30 to 120 mg. The highest recovery values were obtained when the film was prepared with 80 mg of MWCNTs. When the amount of MWCNTs was higher than 80 mg, the film broke in the drying step.
The temperature of the agarose solution was kept in the interval between 80 °C and 85 °C to ensure the complete dissolution of the agarose. It was noticed that when the temperature dropped below 75 °C during the preparation of the film, it broke during drying. This was attributed to a more fragile structure, owing to an incomplete dissolution of the agarose, which hindered the gelation and drying processes.
Also, the MWCNTs film was tested with and without a drying step. When the film was not dry, the extraction of the analytes was not possible. Thus, the temperature and time of the drying step were evaluated. The temperature was assessed between 40 °C and 60 °C, and no difference in the film appearance and recovery values was observed. Therefore, 40 °C was selected as the drying temperature. The drying time was tested between 24 and 48 hours. Satisfactory recovery values were obtained with 24 hours of drying. At lower times, the film was not completely dry, leading to lower recovery values.
A comparison of the different procedures and formats for agarose-based sorbents are shown in Table 1 of the ESI.†
| Analytes | Linear range (μg L−1) | R2 | LODa (μg L−1) | LOQb (μg L−1) | RSD% intra-dayc | RSD% inter-dayd |
|---|---|---|---|---|---|---|
| a LOD calculated as 3 s A−1 and.b LOQ calculated as 10 s A−1, where, s: standard deviation, A: slope of the calibration curve.c n = 5, 0.6 μg L−1.d 0.6 μg L−1, measure by triplicate over 3 days. | ||||||
| KEC | 0.10–1.20 | 0.997 | 0.0089 | 0.030 | 0.46 | 3.13 |
| KEN | 0.10–1.20 | 0.998 | 0.0075 | 0.025 | 1.20 | 3.08 |
| PIR | 0.10–1.20 | 0.998 | 0.0082 | 0.027 | 0.99 | 2.90 |
3.2 Characterization of the agarose-MWCNTs films
The composite was examined by SEM to study the morphology of the MWCNTs-agarose film. The surface homogeneity of the MWCNTs in the agarose matrix was observed. Fig. 2 shows the surface of an agarose film after its drying (a), and the morphology of the MWCNTs-agarose film before (b) and after (c) its drying. As it can be seen, while the surface of the agarose film is smooth, the addition of MWCNTs generates the formation of a rough surface which facilitates the interaction between the active sites of the sorbent and the analytes and improves the mass transfer process and its adsorption capacity. Moreover, it can be observed that the surface of the film before drying is rougher than the surface of the film after drying. This fact can be confirmed by observing Fig. 2d and e that corresponds to the cross section of both films. In addition, Fig. 2e shows an adequate blend of the MWCNTs in the agarose matrix, suggesting good dispersion and adhesion.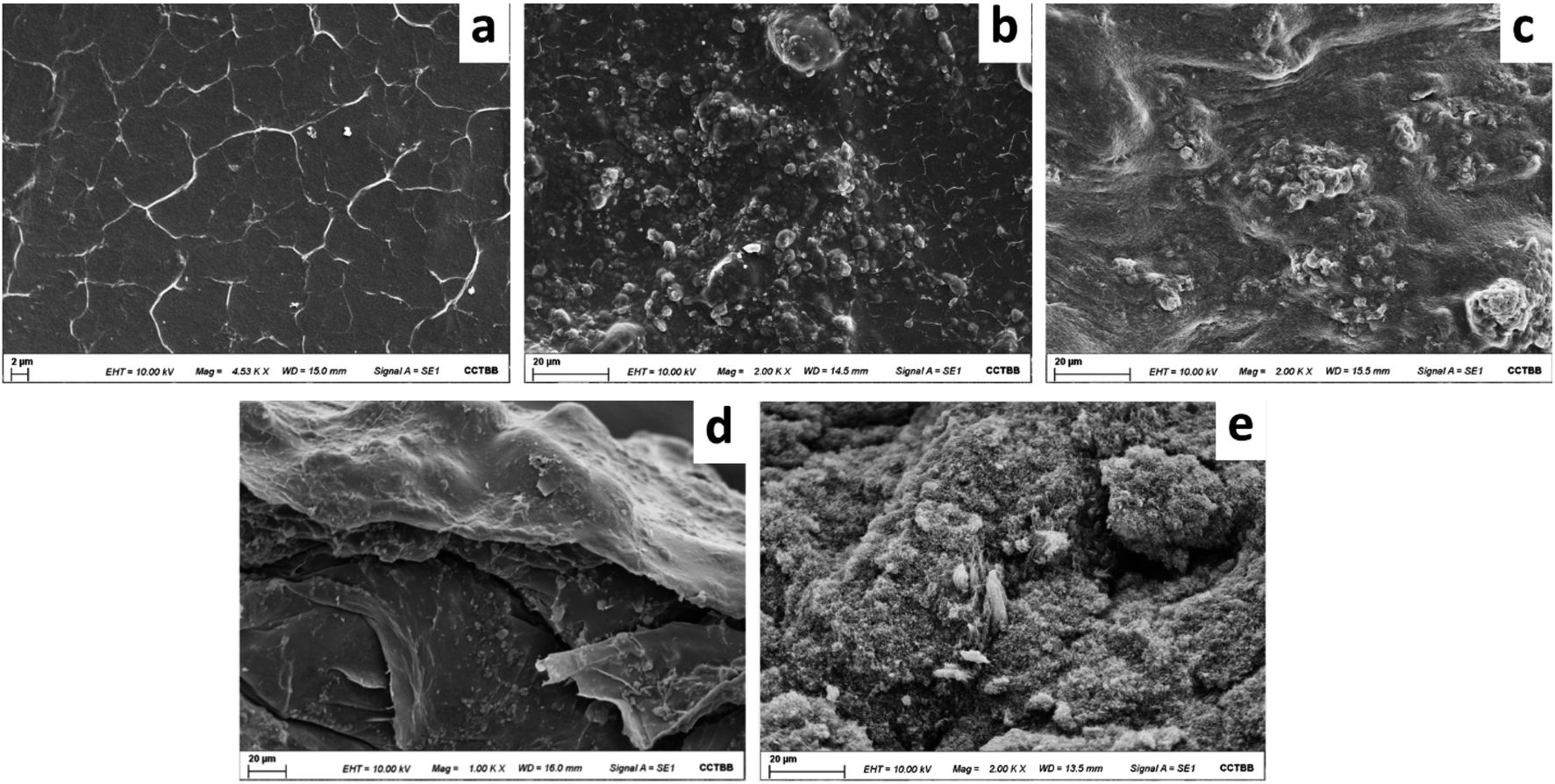 | ||
| Fig. 2 (a) SEM surface morphology of the agarose film after its drying, (b) SEM surface morphology of the MWCNTs-agarose film before its drying, (c) SEM surface morphology of the MWCNTs-agarose film after its drying, (d) SEM of the cross section of the MWCNTs-agarose film before drying (e) SEM of the cross section of the MWCNTs-agarose film after drying. | ||
3.3 Optimization of the extraction procedure
The extraction mechanism of the film is mainly due to the interactions between MWCNTs and the analytes through hydrophobic and π–π interactions.34 Particularly, the π–π interactions are favored by the structure of the analytes that possess aromatic rings and cyclic molecules with conjugated double carbon–carbon bonds. To confirm that the agarose did not intervene in the extraction, the extraction procedure was performed with a film made only of this material. The results can be seen in Fig. S30 (ESI†).Thus, in order to achieve a high extraction efficiency, different factors that can affect the interactions between the MWCNTs and the analytes were optimized, including activation solvent, extraction time, sample pH value, eluent and eluent volume, sample volume and elution time. All variables were tested for the three analytes and experiments were performed in triplicate using 50.00 mL of the standard solution with a concentration of 0.10 mg L−1 for the three compounds.
On the other hand, the extraction time was studied in the interval between 10 and 40 minutes (Fig. 3a). The highest recoveries were obtained after 30 minutes of extraction. At longer extraction times, recoveries remained constant.
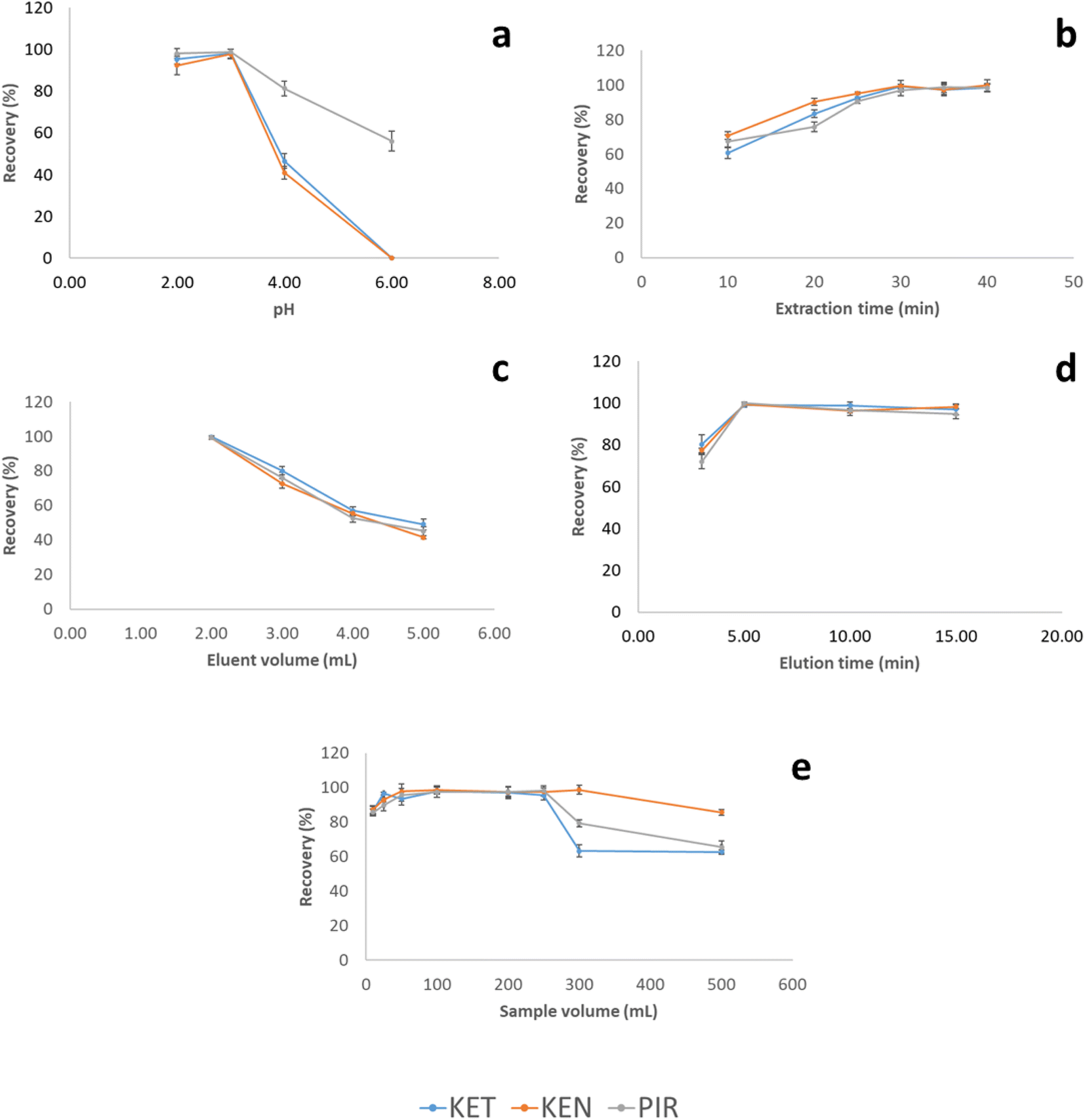 | ||
| Fig. 3 Effect of the sample pH (a), extraction time (b), eluent volume (c), elution time (d) and sample volume (e) on the NSAIDs recovery. | ||
The volume of ACN needed to elute quantitatively the analytes was studied in the range of 2.00 to 5.00 mL. 2.00 mL of ACN was the minimum volume needed to completely cover the MWCNTs-agarose film. Within this range, the volume of eluent to obtain the highest recovery value was 2.00 mL (Fig. 3b). The decrease in the recovery values above this volume can be attributed to a dilution effect. Due to the low elution volume, the elution step was assisted by ultrasound since the stirring of the solution led to the breaking of the film. The elution time in the ultrasonic bath was tested in the range of 2 to 15 minutes. The highest recovery was achieved at 5 minutes of elution, and there was no significant improvement in the recoveries for all NSAIDs at higher times.
3.4 Reusability of the MWCNTs-agarose film
The reuse and stability of the sorbent are important factors to assess its performance. To evaluate this, the MWCNTs-agarose film was subjected to consecutive cycles of adsorption/elution under the optimized conditions. At the end of the working day, the film was washed repeatedly with ACN and distilled water, and then it was dried until reuse. The obtained recovery percentage values were satisfactory and remained approximately constant up to 50 times, demonstrating that the film was stable and reusable, without losing the extraction capacity for the target analytes.The use of a biopolymers as supports for the sorbents is important not only because the material obtained is potentially non-toxic and biodegradable but also to avoid other types of immobilization that require organic reagents that can contaminate the environment. However, in the works that can be found in the literature, the proposed sorbents are not reusable or used few times and discarded after each sample extraction (Table S1, ESI†). This represents a great disadvantage since, although they are biodegradable materials, the preparation of different films for each extraction reduces the reproducibility of the method, increases the quantity of reagents needed and it is time-consuming.
3.5 Analytical performance
Table 1 shows the analytical figures of merit that were calculated to evaluate the proposed method. Calibration plots for the target compounds were built in the range of 0.10 to 1.20 μg L−1 by performing the extraction of a mixture of the analytes’ standard solutions. Correlation coefficients higher than 0.99 were obtained for all analytes. LOD values were 0.0089, 0.0075 and 0.0082 μg L−1 for KEC, KEN and PIR, respectively, and LOQ values were 0.030, 0.025 and 0.027 μg L−1 for KEC, KEN and PIR, respectively. The intra-day precision was assessed by calculating the relative standard deviation (RSD%) with the values obtained when the analysis was performed with a 0.6 μg L−1 standard solution of each compound five times within a day. In order to study the inter-day precision, the solutions with the same concentration were measured by triplicate on three consecutive days. The obtained RSD values demonstrated a high precision of the extraction method.3.6 Application to environmental water samples
The applicability of the MWCNTs-agarose film in the extraction of the analytes from surface water samples was evaluated. The samples were taken from different points of the Napostá stream (sampling points 1 and 2), which provides the city of Bahía Blanca with drinking water. The sampling point 3 was collected from the Napostá chico stream. Sampling sites were taken at strategic locations in and around the city. Point 1 and point 2 were selected before entering the city and before the mouth of the stream enters the estuary, respectively. These points were chosen due to the fact that there may be clandestine sewage discharges that arrive through freshwater courses'.39 On the other hand, the sampling point of the Naposta Chico stream was selected after the last town (Cabildo) that crosses the stream.Even though the sampling points were selected taking into account a potential contamination of the water, residues of the target compounds were not found at concentrations above the quantitation limit of the method.
To evaluate the accuracy of the proposed method, a recovery study was performed. For that, the samples were spiked at the beginning of the analytical process and treated as described in Section 2.4. Table 2 shows the added concentrations (0.30, 0.60 and 1.10 μg L−1) and the recoveries obtained for KEN, KEC and PIR, which ranged from 82.0% to 109.0%, with most of the results between 90.0 and 103.3%.
| Samplesa | Added concentration (μg L−1) | KEN | Recovery (%) KEC | PIR |
|---|---|---|---|---|
| a The recoveries were calculated in triplicate.b (1) and (2) were different sampling points. | ||||
| Napostá river (1)b | 0.30 | 90.0 ± 3.3 | 106.7 ± 6.7 | 97.0 ± 6.7 |
| 0.60 | 85.0 ± 6.7 | 108.3 ± 6.7 | 98.3 ± 1.7 | |
| 1.10 | 82.7 ± 1.8 | 109.0 ± 1.8 | 99.1 ± 3.6 | |
| Napostá river (2)b | 0.30 | 100.0 ± 13.3 | 106.7 ± 6.7 | 90.0 ± 16.7 |
| 0.60 | 91.7 ± 8.3 | 100.0 ± 3.3 | 96.0 ± 8.3 | |
| 1.10 | 96.4 ± 5.4 | 99.1 ± 4.5 | 99.1 ± 2.7 | |
| Napostá Chico stream | 0.30 | 103.3 ± 3.3 | 100.0 ± 3.3 | 100.0 ± 6.7 |
| 0.60 | 83.3 ± 1.7 | 99.1 ± 3.3 | 82.0 ± 15.0 | |
| 1.10 | 102.7 ± 2.7 | 99.7 ± 3.6 | 99.1 ± 4.5 | |
Fig. 4 shows the chromatograms of the 0.30 μg L−1 standard solutions of the analytes and spiked samples at the same concentration level for comparison.
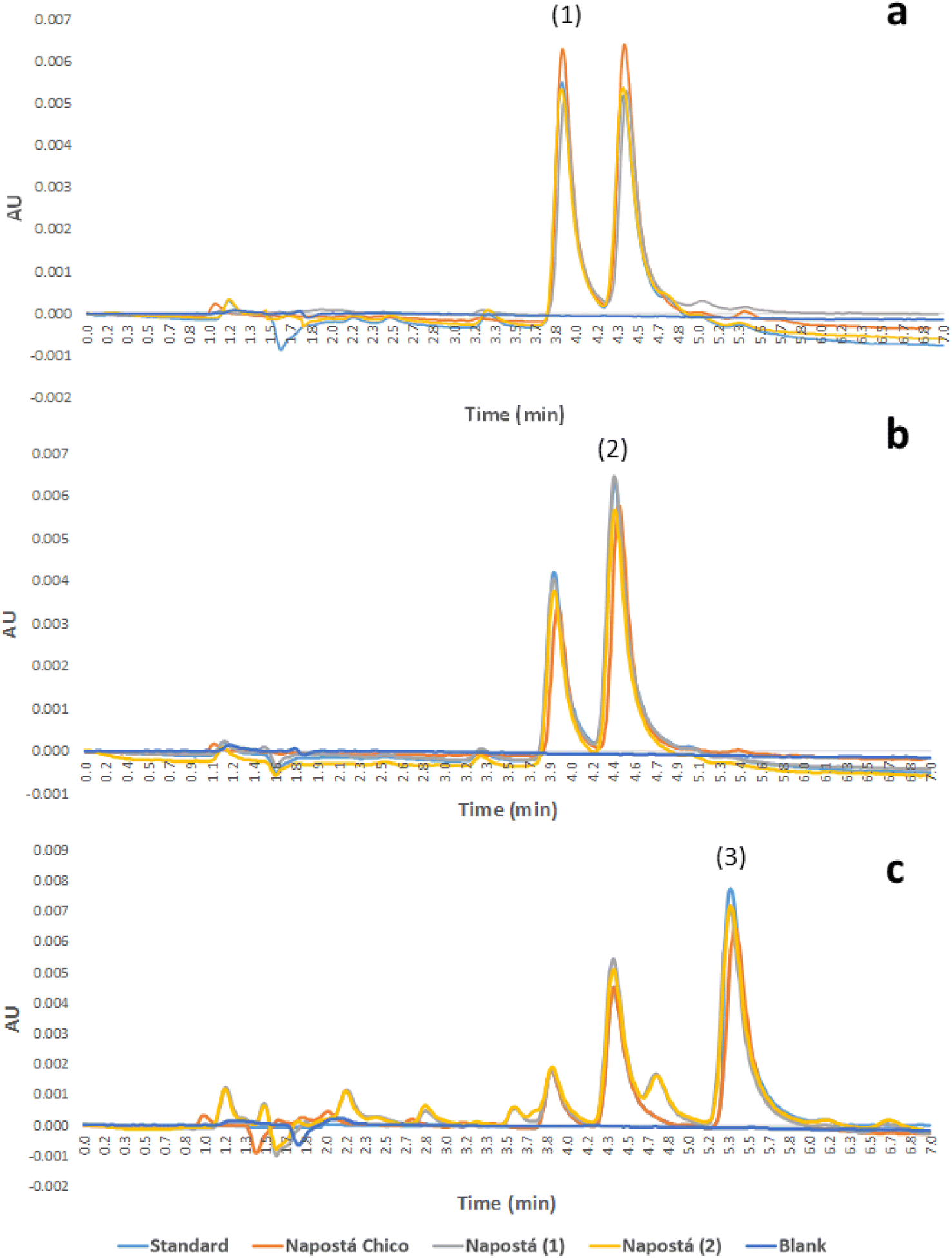 | ||
| Fig. 4 Chromatograms corresponding to the standard solution and spiked water samples measured at λ = 310 nm for KEC (a), λ = 326 nm for PIR (b) and λ = 254 nm for KEN (c). Peak identification: (1) KEC, (2) PIR and (3) KEN. Extraction conditions: concentration of NSAIDs, 0.30 μg L−1; sample volume, 250.00 mL; mass of MWCNTs, 80 mg; volume of desorption solvent, 2.00 mL; sample pH, 3.0. Chromatographic conditions: 150 mm × 4.6 mm, 5 μm particle size C18 Gemini Phenomenex column; mobile phase: ACN-water 0.07% formic acid (50:50 v/v); injection volume: 10 μL; flow rate: 1.0 mL min−1. | ||
The slope of the calibration plot and the slope of the calibration plot built using a sample spiked at the same concentration levels were compared to evaluate the matrix effect.40 At the calculated probability level (p > 0.1), there was no significant difference between the slopes. Thus, it can be concluded that no matrix effect was present.
Different SPE methods that have been found in the literature for the extraction of the studied analytes are shown in Table 3. They have been compared in terms of SPE-based extraction technique, type of sorbent, SPE parameters, reusability of the sorbent, detection technique, LOD and RSD values. The use of less amount of sorbent and lower volume of organic solvent were the main advantages, together with the reusability of the MWCNTs-agarose film several times. As it can be observed, it was possible to obtain preconcentration factors comparable with most of the methods that were found in the literature. In addition, the obtained LOD value was significantly lower than the LODs of the methods that use the same detection technique (molecular absorption spectrometry). Furthermore, it was comparable with some of the methods that use mass spectrometry as detection technique. Concerning RSD, the obtained values were similar or even lower than the published methods.
| Analytes | SPE and SPE-based extraction technique | SPE sorbent | Amount of sorbent (mg) | Eluent volume (mL) | Sample volume (mL) | Sorbent reused | Detection technique | LOD (μg L−1) | RSD (%) | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| a MSPE: magnetic solid phase extraction; SB-SPE: stir bar solid phase extraction.b Calculated from the samples.c Calculated from the samples at different concentration levels.d Calculated at different concentration levels. | ||||||||||
| Tolfenamic acid, naproxen, clofibric acid, diclofenac, indomethacin, ibuprofen, aspirin, ketoprofen | SPE | PANI/Pan NFsM | 3 | 0.5 | 10 | Yes (20 times) | UPLC-MS/MS | 0.2–5.0 ng L−1 | 0.4–11.1c | 3 |
| Ketorolac | SPE | C18 | 1000 | 5.0 | 250 | n. i. | HPLC-UV | 2.83 | 1.8 | 21 |
| Ketoprofen, naproxen, piroxicam, diclofenac, indometacin, carprofen, meclofenamic acid | SPE | Strata-X | 200 | 6.0 | 200 | n. i. | HPLC-Q-Orbitrap-HRMS | 3.7–11.7 ng L−1 | 6.2–32.7b | 22 |
| Ketoprofen, naproxen, diclofenac, ibuprofen | SPE | Strata-X | 200 | 6.0 | 200 | n. i. | HPLC-Q-Orbitrap-HRMS | n. i. | 4.2–56c | 23 |
| Ketoprofen | SPE | Oasis®HLB | n. i. | n. i. | 2.5 | n. i. | HPLC-MS/MS | n. i. | 2–20 | 24 |
| Ketoprofen, naproxen, flurbiprofen, diclofenac sodium and ibuprofen | SPE | CF@UiO-66-NH2 | 50 | 0.5 | 20 | Yes (5 times) | UPLC-PDA | 0.12–3.50 | 2.36–8.49 | 27 |
| Tolfenamic acid, diclofenac, flunixin, ibuprofen, carprofen, ketoprofen, mefenamic acid, meloxicam, naproxen, niflumic acid, vedaprofen, flufenamic acid | SPE | CNTs | 20 | 10.0 | 100.0 | n. i. | LC-QqQ-MS/MS | 0.01–1.3 ng L−1 | 2–14d | 28 |
| 1-Naphthylacetic acid, naproxen, ibuprofen, ketoprofen | MSPE | Fe3O4@MON-PEI600 | 4.0 | 0.4 | 10.0 | Yes (8 times) | HPLC-UV | 0.042–0.149 | 0.3–4.5d | 30 |
| Naproxen, diclofenac sodium salt, mefenamic acid | SPME | Agarose-chitosan-MWCNTs composite film | 0.6 (four discs per extraction) | 0.1 | 10 | No | HPLC-UV | 0.89–8.05 | 1.03–4.59c | 31 |
| Carprofen, mefenamic acid naproxen, diclofenac, ketoprofen | SB-SPE | MIL-101(Cr)/PVA | 60 mg mL−1 | 1.0 | 10.0 | Yes | HPLC-MS/MS | 0.011–0.093 | 1.67–9.52 | 41 |
| Ketorolac, piroxicam, ketoprofen | Agarose-MWCNTs composite film | 80 | 2.0 | 250.0 | Yes (50 times) | HPLC-PDA | 0.0075–0.0089 | 2.90–3.13 | Our work | |
4. Conclusions
Agarose, which is a non-toxic, renewable and biodegradable material, was used for the immobilization of MWCNTs. The MWCNTs-agarose film presented excellent sorbent properties and allowed the extraction and preconcentration of KEC, KEN and PIR, obtaining satisfactory analytical parameters. In addition, the film was reusable up to 50 times without losing its extraction capacity. Furthermore, it was possible to perform the extraction with a single film, which notably simplified the experimental procedure. The proposed method was successfully applied for the quantification of the selected analytes in natural water samples of Bahia Blanca's region with satisfactory results. As far as we know, it is the first time that these compounds were determined in this region, which is of great importance to obtain the state of the art of their presence/absence. In this case, the analytes were not found in the selected sampling points at the detection limits that were achieved by the proposed method.Conflicts of interest
The authors declare no competing interests.Acknowledgements
Authors gratefully acknowledge to Universidad Nacional del Sur (PGI 24/Q119, PGI 24/Q099) and Comisión Nacional de Investigaciones Científicas y Técnicas (CONICET), PIP 11220200102603CO. Also, the authors would like to thank to Agencia Nacional de Promoción Científica y Tecnológica (PICT-2019-04458).References
- R. E. Majors, in Handbook of Sample Preparation, ed. Pawliszyn J. and Lord H. L., Wiley & Sons, Inc, 2010, pp. 53–79 Search PubMed
.
- A. Azzouz, S. K. Kailasa, S. S. Lee, A. J. Rascón, E. Ballesteros, M. Zhang and K. H. Kim, Trends Anal. Chem., 2018, 108, 347–369 CrossRef
- N. Jian, L. Qian, C. Wang, R. Li, Q. Xu and J. Li, J. Hazard. Mater., 2019, 363, 81–89 CrossRef CAS PubMed
- J. Płotka-Wasylka, N. Szczepańska and J. Namieśnik, Trends Anal. Chem., 2016, 77, 23–43 CrossRef
- I. Pacheco-Fernández, D. W. Allgaier-Díaz, G. Mastellone, C. Cagliero, D. D. Díaz and V. Pino, Trends Anal. Chem., 2020, 125, 115839 CrossRef
- H. Tabani, M. Alexovič, J. Sabo and M. Ramos Payán, Talanta, 2021, 224, 121892 CrossRef CAS PubMed
- M. N. H. Rozaini, N. F. Semail, B. Saad, S. Kamaruzaman, W. N. Abdullah, N. A. Rahim and N. Yahaya, Talanta, 2019, 199, 522–531 CrossRef CAS PubMed
- M. K. Yazdi, A. Taghizadeh, M. Taghizadeh, F. J. Stadler, M. Farokhi, F. Mottaghitalab, P. Zarrintaj, J. D. Ramsey, F. Seidi, M. R. Saeb and M. Mozafari, J. Controlled Release, 2020, 326, 523–543 CrossRef PubMed
- S. Hong Loh, M. Marsin Sanagi, W. Aini Wan Ibrahim and M. Noor Hasan, Talanta, 2013, 106, 200–205 CrossRef PubMed
- S. Hong Loh, P. Ee Neoh, C. Tung Tai and S. Kamaruzaman, Malaysian J. Anal. Sci., 2018, 22, 1–7 Search PubMed
- M. Marsin Sanagi, M. Nazirul Muzakkir Mat Jais, S. Kamaruzaman, W. Aini Wan Ibrahim and U. Baig, Anal. Methods, 2015, 7, 2862–2868 RSC
- N. T. Ng, M. M. Sanagi, W. N. W. Ibrahim and W. A. W. Ibrahim, Food Chem., 2017, 222, 28–34 CrossRef CAS PubMed
- Y. Zhou, J. Xu, N. Lu, X. Wu and X. Hou, Talanta, 2021, 225, 121846 CrossRef CAS PubMed
- Food and Drug Administration, Information for Consumers and Patients, Safe disposal of medicines, https://www.fda.gov/drugs/ensuring-safe-use-medicine/safe-disposal-medicines, Accessed March 7, 2023 Search PubMed.
- A. Gogoi, P. Mazumder, V. K. Tyagi, G. T. Chaminda, A. K. An and M. Kumar, Groundw. Sustain. Dev., 2018, 6, 169–180 CrossRef
- K. Świacka, J. Maculewicz, D. Kowalska, M. Caban, K. Smolarz and J. Świeżak, J. Hazard. Mater., 2022, 424, 127350 CrossRef PubMed
- K. Świacka, A. Michnowska, J. Maculewicz, M. Caban and K. Smolarz, Environ. Pollut., 2021, 273, 115891 CrossRef PubMed
- H. Wang, H. Xi, L. Xu, M. Jin, W. Zhao and H. Liu, Sci. Total Environ., 2021, 788, 147819 CrossRef CAS PubMed
- Y. Elorriaga, D. J. Marino, P. Carriquiriborde and A. E. Ronco, Bull. Environ. Contam. Toxicol., 2013, 90, 397–400 CrossRef CAS PubMed
- Y. Elorriaga, D. J. Marino, P. Carriquiriborde and A. E. Ronco, Int. J. Environ. Health, 2013, 6(4), 330–339 CrossRef CAS
- H. H. Ortega Soto, J. J. Ramírez García, P. Gamboa Suárez and A. M. Dávila Estrada, Int. J. Photoenergy, 2017, 6781310 Search PubMed
- C. L. Chitescu, G. Kaklamanos, A. I. Nicolau and A. A. M. L. Stolker, Sci. Total Environ., 2015, 532, 501–511 CrossRef CAS PubMed
- I. Pugajeva, J. Rusko, I. Perkons and V. Bartkevics, J. Pharm. Biomed. Anal., 2017, 133, 64–74 CrossRef CAS PubMed
- J. Camilleria, R. Baudota, L. Wiesta, E. Vullieta, C. Cren-Olivéa and G. Daniele, Int. J. Environ. Anal. Chem., 2015, 95, 67–81 CrossRef
- A. Kot-Wasik, A. Jakimska and M. Śliwka-Kaszyńska, Environ. Monit. Assess., 2016, 188, 661 CrossRef CAS PubMed
- A. Amiri and F. Ghaemi, J. Chromatogr. A, 2021, 1648, 462168 CrossRef CAS PubMed
- Y. Gao, S. Wang, N. Zhang, X. Xu and T. Bao, J. Chromatogr. A, 2021, 1652, 462349 CrossRef CAS PubMed
- I. Reinholds, I. Pugajeva, Dz. Zacs, E. Lundanes, J. Rusko, I. Perkons and V. Bartkevics, Environ. Monit. Assess., 2017, 189, 568 CrossRef CAS PubMed
- T. Wang, S. Liu, G. Gao, P. Zhao, N. Lu, X. Lun and X. Hou, Microchim. Acta, 2017, 184, 2981–2990 CrossRef CAS
- X. Q. He, Y. Y. Cui and C. X. Yang, Talanta, 2021, 233, 122471 CrossRef CAS PubMed
- W. N. Wan Ibrahim, M. Marsin Sanagi, N. S. Mohamad Hanapi, S. Kamaruzaman, N. Yahaya and W. A. Wan Ibrahim, J. Sep. Sci., 2018, 41, 2942–2951 CrossRef CAS PubMed
- M. Chen, X. Qin and G. Zeng, Trends Biotechnol., 2017, 35(9), 836–846 CrossRef CAS PubMed
- C. Jiang, Z. Liu, D. Cheng and X. Mao, Biotechnol. Adv., 2020, 45, 107641 CrossRef CAS PubMed
- J. Płotka-Wasylka, N. Szczepańska, M. de La Guardia and J. Namieśnik, TrAC Trend. Anal. Chem., 2016, 77, 23–43 CrossRef
- National Center for Biotechnology Information, PubChem Compound Summary for CID 3826, Ketorolac, 2022, https://pubchem.ncbi.nlm.nih.gov/compound/Ketorolac, Accessed March 7, 2023 Search PubMed.
- National Center for Biotechnology Information, PubChem Compound Summary for CID 3825, Ketoprofen, https://pubchem.ncbi.nlm.nih.gov/compound/Ketoprofen, 2022, Accessed March 7, 2023 Search PubMed.
- National Center for Biotechnology Information, PubChem Compound Summary for CID 54676228, Piroxicam, https://pubchem.ncbi.nlm.nih.gov/compound/Piroxicam, 2022, Accessed March 7, 2023 Search PubMed.
- M. Razali, C. Didaskalou, J. F. Kim, M. Babaei, E. Drioli, Y. M. Lee and G. Szekely, ACS Appl. Mater. Interfaces, 2017, 9(12), 11279–11289 CrossRef CAS PubMed
- M. E. Streitenberger and M. D. Baldini, Rev. Int. Contam. Ambiental, 2016, 32(2), 243–248 CrossRef
- D. L. Massart, Handbook of Chemometrics and Qualimetrics, 1st edn, Elsevier Science, 1997 Search PubMed
- Y. Wang, M. Ji, X. Wu, T. Wang, J. Wang and X. Hou, Microchem. J., 2019, 146, 214–219 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra00166k |
| This journal is © The Royal Society of Chemistry 2023 |