
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and screening of cyclic diketone indanedione derivatives as future scaffolds for neutrophil elastase inhibition
Meena S. *a,
Jubie S.b,
Pramila C.a,
Manal T. N. A.a and
Gigi S.a
*a,
Jubie S.b,
Pramila C.a,
Manal T. N. A.a and
Gigi S.a
aDepartment of Pharmaceutical Sciences, College of Pharmacy, Shaqra University, Al Dawadmi, Kingdom of Saudi Arabia. E-mail: smchemsm@gmail.com
bDepartment of Pharmaceutical Chemistry, JSS College of Pharmacy, JSS Academy of Higher Education and Research, Ooty, Tamilnadu, India
First published on 17th April 2023
Abstract
Human neutrophil elastase (HNE) and proteinase 3 (Pr3) released from neutrophils at inflammatory sites are the major causes of pathogens in chronic obstructive pulmonary disease (COPD) and various lung tissue derangements, among which cystic fibrosis and blockade of airway passages are chronic. These proteolytic mediatory agents combined with induced oxidative reactions sustain pathogenicity. Cyclic diketone indane-1,3-dione derivatives were designed, and toxicity evaluation predictions were performed in silico. Benzimidazole and hydrazide derivatives of indanedione were synthesized and characterized. Synthesized compounds were run using neutrophil elastase inhibition assay protocols. The compounds exhibit considerable inhibition of neutrophil elastase enzymes.
1. Introduction
Human neutrophil elastase (HNE) (EC: 3.4.21.37) and proteinase 3 (Pr3) released from neutrophils at inflammatory sites are the major causes of pathogens in chronic obstructive pulmonary disease (COPD) and various lung tissue derangements, such as cystic fibrosis. These proteolytic mediatory agents combined with induced oxidative reactions sustain pathogenicity. The accumulation of mucus in the pulmonary air passages is a direct consequence of HNE and Pr3 stimulatory effects on goblet cells and is a key diagnostic feature in COPD and other chronic lung inflammatory disorders.1–3 The World Health Organization (WHO) has alerted the medical fraternity worldwide about this issue, more specifically the US-based NIH. This led to the collaborative programme, the global initiative for chronic obstructive lung disease (GOLD).Since the Covid infection threatened the human population, along with anti-viral drugs, the need to manage lung inflammation has attracted the attention of healthcare professionals. Covid-related lung lesions are a cause of the direct consequences of elastase hyper secretion. Histamine and elastase release are believed to be complementary to tissue damage. This necessity has directed our focus to targeting human neutrophil elastase (HNE).4
Cyclic diketone indane-1,3-dione derivatives were synthesized and screened for potential neutrophil elastase inhibition. Indane-1,3-dione derivatives constitute a distinctive property due to their 1,3-dicarbonyl nature, having specific physiochemical property, which offers wide scope for studies in the problems of theoretical organic chemistry, particularly based on tautomerism and dual reactivity. Specifically, β-cyclic diketones (the two carbonyl groups separated by a single C atom) exhibit differing reactivity for the two ketonic groups. The enolisation effect significantly affects the structural relationship activities of these carbonyl derivatives.5–7 Cyclic diketone derivatives are known for their broader synthetic applications and selective pharmacological responses. Studies on the medicinal chemistry of indane-1,3-dione have been very sporadic. Initially, these compounds were used as anti-coagulants, insecticidal and rodenticide agents.
Indanedione scaffolds were screened and reported for anti-viral and anti-inflammatory activities.8,9 Some scaffolds comprising anti-fungal, anti-bacterial and anti-parasitic activities have been published.10–12 Anti-proliferative activities have been reported for a series of fused indane-1,3-dione derivatives. Several tricyclic compounds have been synthesized using 2-alkene-1,3-indandione derivatives as precursors and have also been tested for anti-proliferative activity.13
Several reports are available on the efficacy of cyclic ketones as serine protease inhibitors. Pearson et al. in 2002 investigated an eight-member ring ketone.14 Later, Fengtian in 2006 reported the inhibition of plasmin by a series of cyclic diketones.15 This concept is further supported by a review article on the inhibition of serine and cysteine proteases by Powers et al. 2002.16 The underlying concepts in the aforementioned literature laid the foundation for the present study. To generate a stable tetrahedral complex, nucleophilic serine attacks the electron-deficient ketone. HNE's proteolytic action is mediated by a catalytic triad composed of Ser195–Asp102–His57, as shown in Fig. 1a and b, in which the strong nucleophile Ser195 OH group assaults the peptide bond's carbonyl carbon.17–19
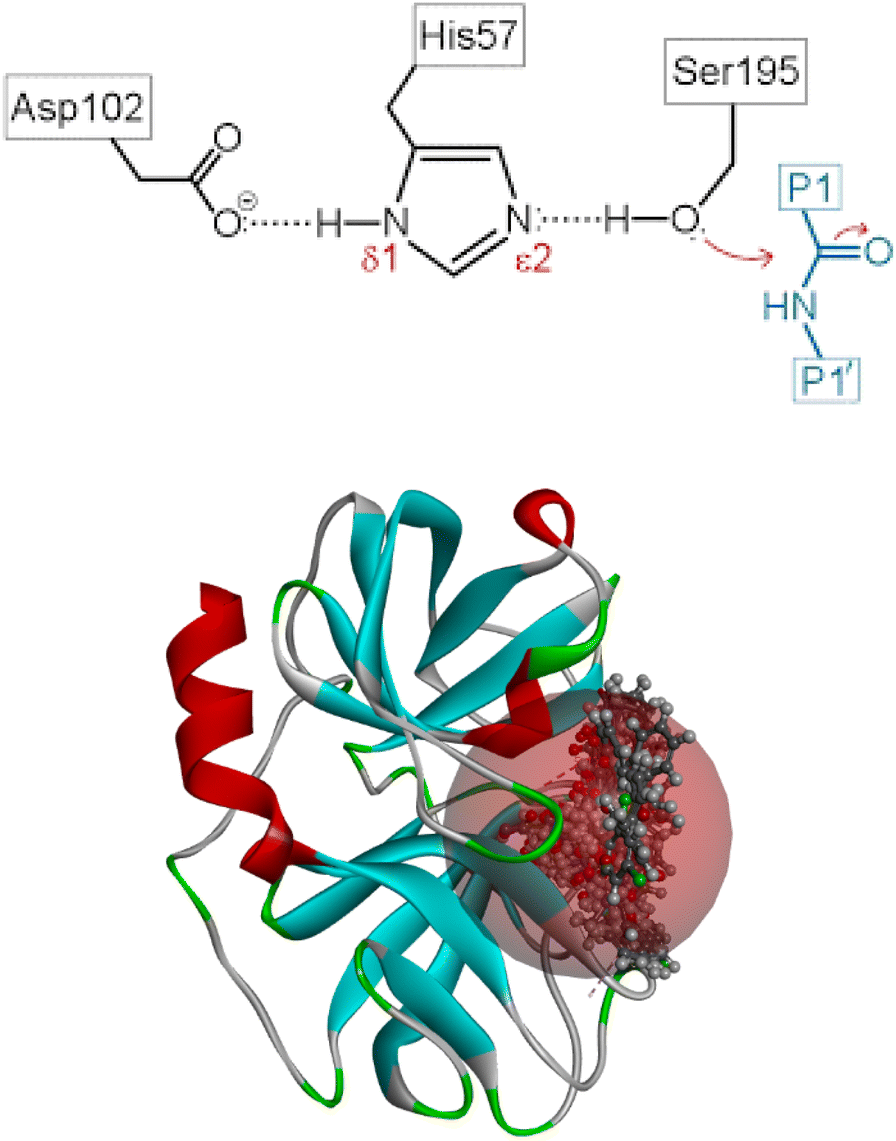 | ||
| Fig. 1 (a) Active site residues of the HNE. (b) Binding site of the designed compounds on HNE. | ||
The present Covid pandemic situation demands the need for potential HNE inhibitors, as lung lesions are the prominent hypersecretion of elastase. This study designs and identifies the candidate molecule in silico and synthesizes the required moiety using two schemes, one to prepare indanedione benzimidazoles and another for indanedione hydrazides, followed by its characterization and evaluation of the HNE inhibition capacity.
2. Materials and methods
Sigma Aldrich, SD Fine Chemicals and Carbanio supplied all the chemicals used, and they were of analytical grade. The solvents were purified and dried according to industry standards. A microwave synthesizer (Catalyst Systems) was used in the microwave-assisted synthesis. For analytical thin-layer chromatography, the plates used were MERCK aluminum back pre-coated silica gel 60-F 254 (0.5 mm) plates. An iodine chamber or UV light was used to visually identify the spots on TLC plates. The melting points of the synthesized compounds were measured in open capillaries using the Veego VMP-1 apparatus, expressed in degree Celsius (°C) and uncorrected. The IR spectra of the compounds were obtained using the KBr pellet technique on a Shimadzu FT-IR spectrometer and represented in cm−1. A Bruker 400 MHZ spectrometer was used to evaluate the 1H and 13C NMR data. Tetramethyl silane (TMS) was used as an internal standard, and the solvent was DMSO-d6. Chemical changes in parts per million were detected downfield from TMS (ppm). The Shimadzu instrument recorded the mass spectra. Elemental analysis was carried out using a LECO Truspec Micro Analyser (Micro CHNS module and oxygen module).A neutrophil elastase enzyme inhibitor activity kit was purchased from Abcam (118971). It contained ready to use assay buffer, substrate, neutrophil elastase and control. Microplates for fluorescent-based assays and 96-well (M33089) were purchased from Thermo Fischer. SpectraMax i3 Microplate Reader, Spincotech.
A molecular docking study of the generated compounds against the HNE receptor was carried out using Discovery Studio 4.1. Docking experiments, ADMET research, and TOPKAT investigations were conducted and assessed for the designed compounds.
2.1 Molecular docking studies
In silico investigations were conducted on the interaction of Human Neutrophil Elastase (HNE) with the inhibitor dihydropyrimidone (PDB id: 5A8Z). The chemical structures of indane 1,3-dione analogues (ligands) were developed and optimized using chem sketch software selected 5A8Z for docking studies because of the crystal structure of human neutrophil elastase in complex with a dihydropyrimidone inhibitor. The organism of 5A8Z is homo sapiens. The resolution of our protein is 2 Å, and the dihydropyrimidone is similar to our novel-designed indanedione compounds. The docking of all tautomers and isomers of the ligands with the lowest energy was accepted. The protein was produced by removing unnecessary water molecules, hetero atoms, tiny ions, and alternate conformations from the crystal structure of HNE using the inhibitor dihydropyrimidone, which was downloaded from the RCSB PDB. The missing loops were modelled to complete the framework by standardizing atom names, and the missing atoms were inserted. The prediction of pKa was used to protonate the titratable residues. Before and after protein reduction, the potential energy, van der Waals energy, electrostatic energy, and RMS gradient of the complex were measured, and the pH used was 7.4. Finally, the hydrogen receptors were merged into the target receptor molecule using the Discovery Studio 4.1 client.20 Prepared protein was validated through a Ramachandran plot (Fig. 2).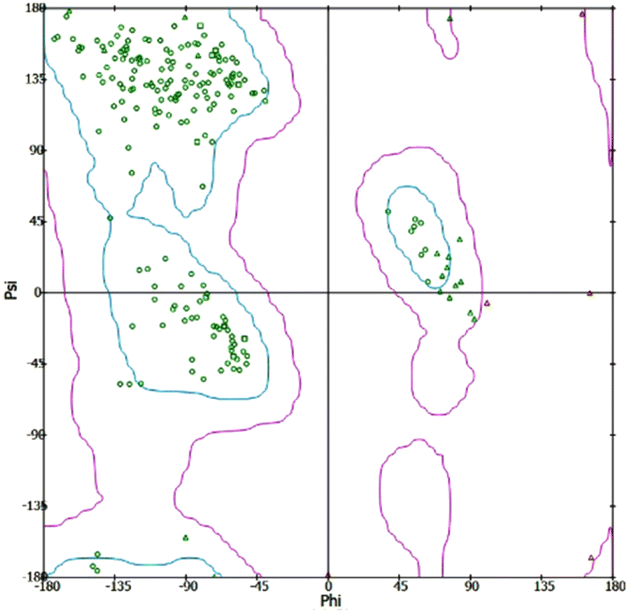 | ||
| Fig. 2 Ramachandran plot of the HNE. | ||
2.2 ADMET studies
The solubility of the drugs in water at 25 °C, human intestinal adsorption level after oral administration, metabolism of the administered compounds by the metabolizing enzyme cytochrome P450 (CYP2D6) using 2D input, hepatotoxicity of the compounds, plasma protein binding extent, 95 percent and 99 percent confidence ellipses in the ADMET PSA 2D, and ADMET AlogP98 programs were calculated using Discovery Studio (DS) 4.1.222.3 TOPKAT studies
TOPKAT is a commonly used system for determining the carcinogenicity of manufactured compounds (Oxford Molecular, Beaverton, OR). Predictions of carcinogenicity based on one- and two-atom fragment electro topological state descriptors TOPKAT were created to predict chemical carcinogenicity in four rodent models: male rat, female rat, male mouse, and female mouse.232.4 Molecular dynamic simulations
The protein–ligand complexes that are involved in the potent blocking of any receptor must be highly stable in their conformation. To gain better insight into the dynamics that occur between the ligand and receptor, our team conducted a molecular dynamics simulation study.24,25 To carry out the MD simulations, the Desmond module of the Schrodinger suite 2021-4 was used with the OPLS3e force field. The docked complexes served as the starting point for the MD simulations. TIP4P water molecules were added to an orthorhombic box to provide a 10 Å buffer zone between atoms or amino acid residues and box sides, thus enabling the explicit solution of the complexes.26 The minimization of the systems was achieved using the OPLS3e force field and the application of 200 steepest descent step algorithms, which were then followed by 1000 steps of the conjugate gradient method until a gradient threshold of 25 kcal mol−1 was reached. This study has greatly improved our understanding of the interactions that occur between the ligand and receptor during the potent blocking of any receptor.27 A smooth particle mesh Ewald approach was employed to account for both short- and long-range coulombic and electrostatic interactions. The cutoff radius for the short-range coulombic forces was set to 9.0. A tolerance of 1 × 10−9 was used for long-range electrostatic forces. To maintain a temperature of 300 k and pressure of 1 bar during the simulation, a Martyna–Tobias–Klein barostat and a Nose–Hoover thermostat were used. The system was simulated in an isothermal–isobaric ensemble (quantity of substance, pressure, and temperature [NPT]) for a duration of one hundred nanoseconds with a 2 femtosecond time step. A reversible system propagator algorithm (RESPA) with various time steps was used for bonded, far nonbonded, and near nonbonded interactions, and frames were generated every 100 picoseconds. The postures and 3D structures of the trajectory were inspected using the Maestro graphical user interface.262.5 Synthesis of indanedione compounds
Step 1: synthesis of indane 1,3-dione. Diethyl phthalate (0.53 mol, 125 g, and 106.5 ml) and sodium wire (1.09 mol, 25 g) were placed in a dry two-necked flask with a dropping funnel and reflux condenser, each protected by a calcium chloride guard tube. The mixture was refluxed; then, dry ethyl acetate (1.39 mol, 122.5 g, 136 ml) and absolute ethanol (2.5 ml) were added for 90 minutes. Refluxing was continued for about 6 h after the addition of all the contents at 80 °C. The mixture was then cooled, and 50 ml of ether was added. This solution was filtered, washed in ethyl acetate and dissolved in hot water (1400 ml). The solution was chilled and acidified (100 ml) with H2SO4 (H2SO4
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O; 3:1). Finally, the mixture was cooled to 15 °C, and the obtained indane-1,3-dione product was filtered and dried. With the addition of light petroleum, the chemical was recrystallized from a dioxane–benzene (1:1) mixture to obtain the pure product.10
:H2O; 3:1). Finally, the mixture was cooled to 15 °C, and the obtained indane-1,3-dione product was filtered and dried. With the addition of light petroleum, the chemical was recrystallized from a dioxane–benzene (1:1) mixture to obtain the pure product.10
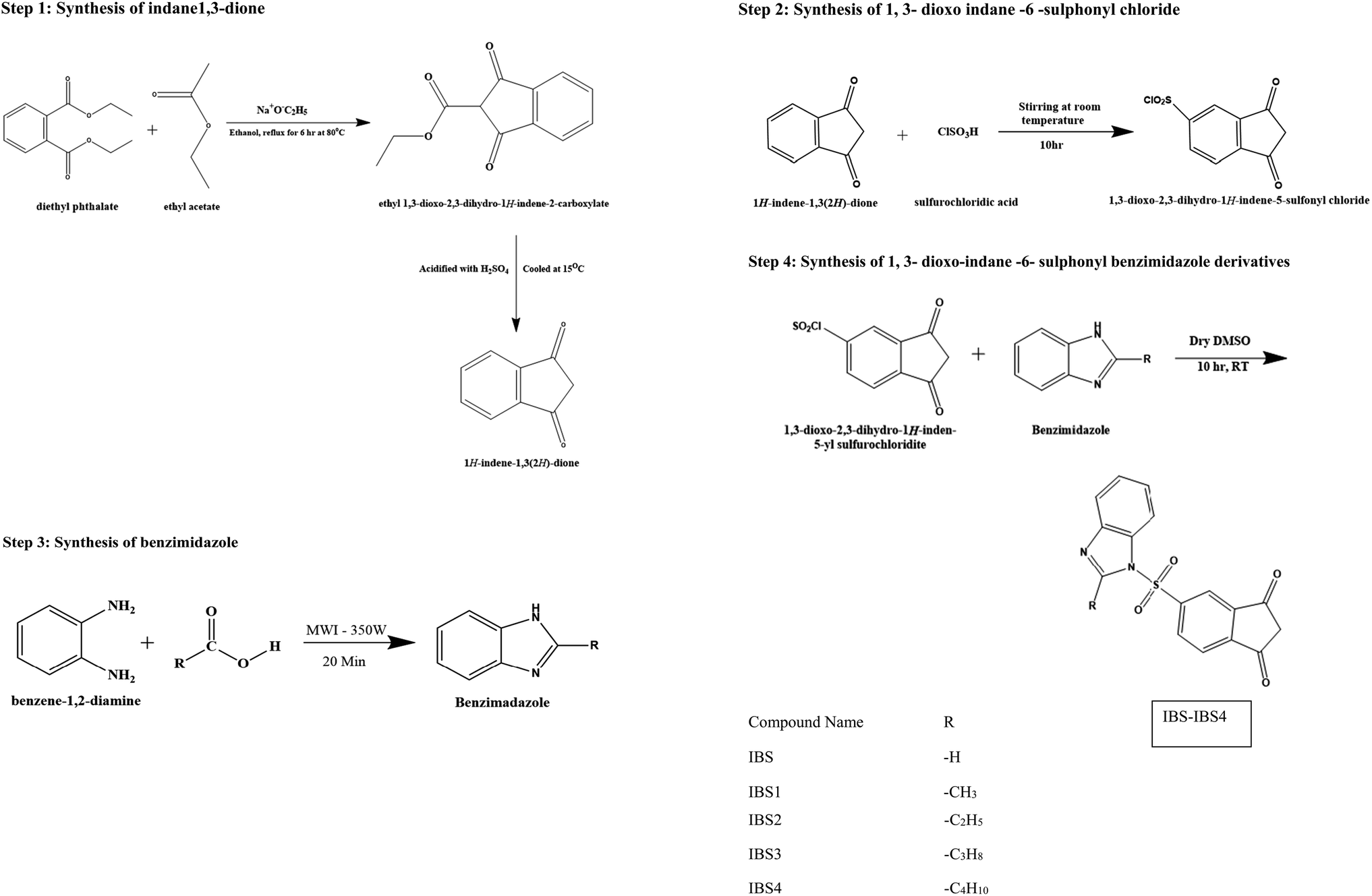 | ||
| Scheme 1 Indanedione benzimidazoles. | ||
Step 2: synthesis of 1,3-dioxo indane-6-sulphonyl chloride. Indane-1,3-dione (0.03 mol and 4.86 g) was added aliquot wise to chlorosulphonic acid (0.27 mol, 17.98 ml) at 0 °C. The reaction solution was added to ice-water and filtered after being agitated at room temperature for 10 hours. The solid was then washed with water to produce white crystals, which were then recrystallized from a dry dioxane–benzene combination.
Step 3: synthesis of benzimidazole. The microwave oven method was employed to synthesize benzimidazole. Then, o-phenylenediamine (0.0457 mol) was added to the monocarboxylic acid (0.0457 mol). The mixture was irradiated in a microwave synthesizer (MWI) at an emitted power of 350 W for 20 min. The resulting mixture was allowed to cool and be basified with 10% NaOH using litmus paper. The precipitate was filtered, washed with water and recrystallized from hot water.
Step 4: synthesis of 1,3-dioxo-indane-6-sulphonyl benzimidazole derivatives. The 1,3-dioxo-indane-6-sulphonyl chloride (0.01 mol, 2.4 g) was dissolved in dry dimethyl sulphoxide (20 ml); then, benzimidazole derivatives (0.02 mol) were added, and the mixture was stirred at room temperature for 10 hours. The resultant mixture was then put into 50 ml of hot water to produce a frothy precipitate, which was then recrystallized.
Step 1: synthesis of indane-1,3-dione. Diethyl phthalate (0.53 mol, 125 g, 106.5 ml) and sodium wire (1.09 mol, 25 g) were placed in a dry two-necked flask with a dropping funnel and reflux condenser, each protected by a calcium chloride guard tube. The mixture was refluxed; then, dry ethyl acetate (1.39 mol, 122.5 g, 136 ml) and absolute ethanol (2.5 ml) were added for 90 minutes. Refluxing was continued for about 6 h after the addition of all the contents at 80 °C. The mixture was then cooled, and 50 ml of ether was added. This solution was filtered, washed in ethyl acetate and dissolved in hot water (1400 ml). The solution was chilled and acidified (100 ml) with H2SO4 (H2SO4
:H2O; 3:1). Finally, the mixture was cooled to 15 °C, and the obtained indane-1,3-dione product was filtered and dried. With the addition of light petroleum, the chemical was recrystallized from a dioxane–benzene (1:1) mixture to obtain the pure product.10
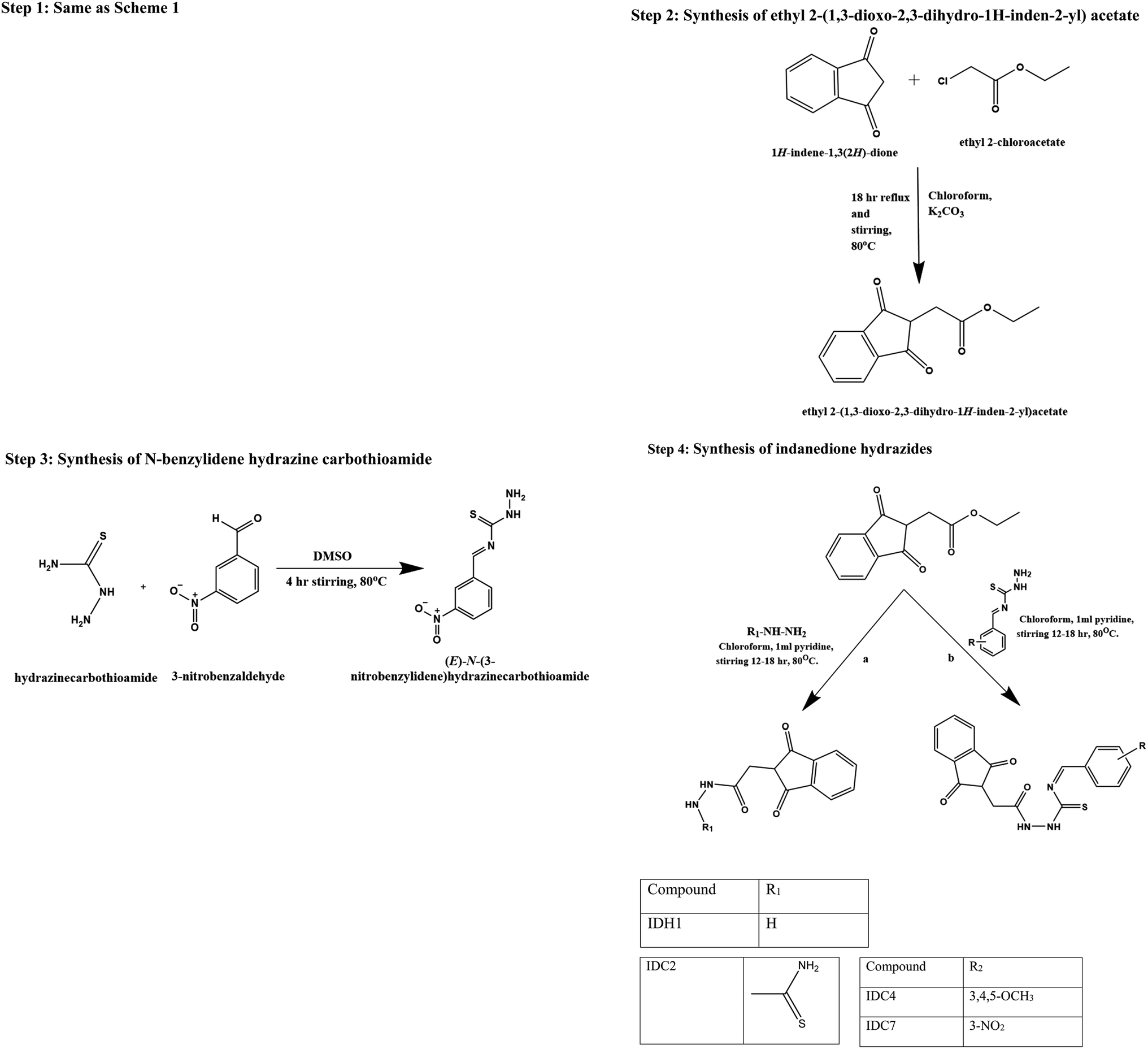 | ||
| Scheme 2 Indanedione hydrazides. | ||
Step 2: synthesis of ethyl 2-(1,3-dioxo-2,3-dihydro-1H-inden-2-yl)acetate. Indane-1,3-dione (0.01 mol, 3 g) was added aliquot wise to ethyl chloroacetate (0.02 mol, 2.5 ml) in RBF5 containing 15 ml of chloroform. To this, potassium carbonate (2 g) was added with continuous stirring and refluxed for 18 h at 80 °C. The reaction solution was incorporated into ice-water and filtered. The obtained product was washed with water; the precipitate was recrystallized from a dry dioxane–benzene mixture.
Step 3: synthesis of N-benzylidene hydrazine carbothioamide. Thiosemicarbazide (0.01 M, 0.91 g) was added to different substituted benzaldehydes (0.01 M) dissolved in DMSO (50 ml) and stirred for 4 hours at 60 °C. Then, the reaction mixture was transferred to crushed ice. A solid precipitate was formed and dried, and the melting point was determined.
Step 4: synthesis of indanedione hydrazides. Ethyl 2-(1,3-dioxo-2,3-dihydro-1H-inden-2-yl) acetate (0.01 mol, 300 mg) was added aliquot wise to the respective hydrazine derivatives (0.02 mol) in 15 ml of chloroform. To this solution, 1 ml of pyridine was added with continuous stirring and refluxed for 12–18 h at 80 °C. The reaction solution was incorporated into ice-water and filtered. The resulting solid was washed with water, resulting in a precipitated product, and it was recrystallized from a dry dioxane–benzene mixture.
2.6 Neutrophil elastase inhibitor activity
The synthesized test compounds (IBS, IBS1, IBS 2, IBS 3, IBS 4, IDH, IDC2, IDC 4 and IDC 7) were diluted in serial dilution in DMSO to the final desired test concentration (100 μl, 50 μl, 10 μl, 1 μl and 0.1 μl). Neutrophil elastase enzyme and substrates were reconstituted with assay buffer as per the manual instructions. In each well, 50 μl of diluted enzyme (1) and 25 μl of the test compound in various concentrations were added. After it was kept for 5 min at 37 °C, substrate (25 μl) was added and incubated for 30 min. Finally, the fluorescence intensity was measured (excitation at 400 nm and emission at 505 nm) in a fluorimeter. For the eight compounds, assays were performed in five concentrations in triplicate.3. Results and discussion
3.1 Cdocker results
An in silico docking study hypothesized the spatial organization of the chemicals in the HNE active site. To identify the probable binding mechanisms of the synthesized compounds, the crystalline structure of HNE (PDB ID: 5A8Z) was employed. The ligands were docked into the active site of a human elastase inhibitor that was previously described. The results are presented in detail in Table 1 and Fig. 3a–h.| S. no. | Compounds | Binding energy (kcal mol−1) |
|---|---|---|
| 1 | IBS4 | −81.13 |
| 2 | IBS2 | −76.76 |
| 3 | IBS3 | −40.63 |
| 4 | IDC2 | −68.62 |
| 5 | IDH1 | −66.32 |
| 6 | 5A8Z (co-crystal) | −65.92 |
| 7 | IBS | −65.34 |
| 8 | IDC7 | −59.51 |
| 9 | IDC4 | −55.30 |
| 10 | IBS1 | −54.48 |
| 11 | Sivelestat | −36.17 |
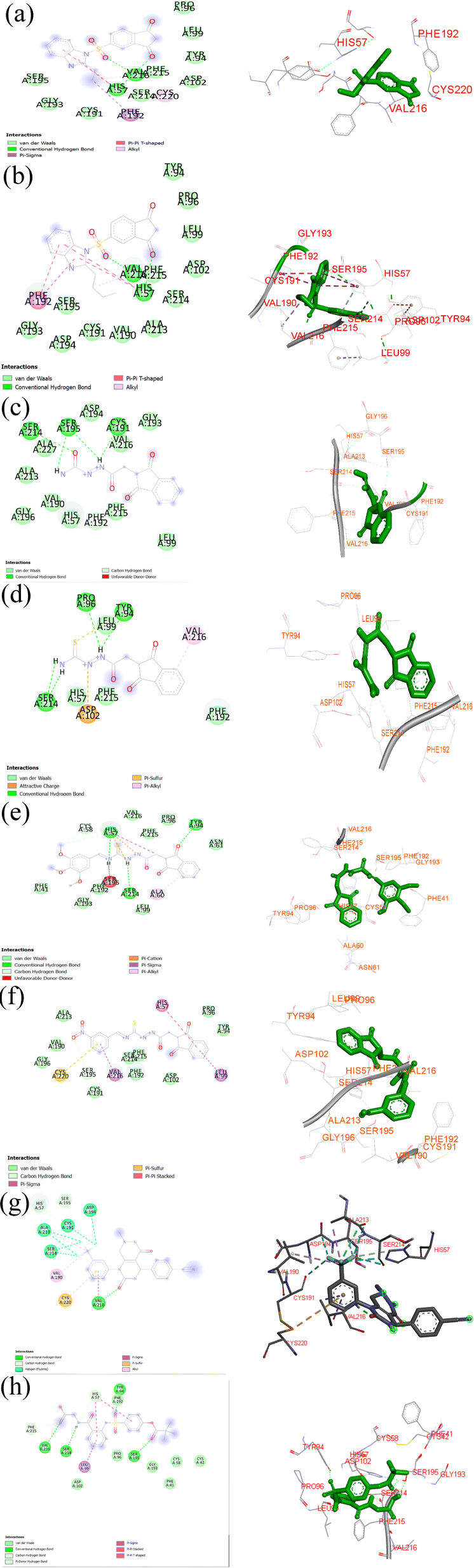 | ||
| Fig. 3 (a) 2D ligand–protein interactions for compound IBS-2. (b) 2D ligand–protein interactions for compound IBS-4. (c) 2D ligand–protein interactions for compound IDH-1. (d) 2D ligand–protein interactions for compound compound IDC-2. (e) 2D ligand–protein interactions for compound IDC-4. (f) 2D ligand–protein interactions for compound compound IDC-7. (g) 2D ligand–protein interactions for compound co-crystal. (h) 2D ligand–protein interactions for compound sivelestat. | ||
To test the proposed hypothesis (in silico level), whether the designed indanedione analogues inhibit the HNE or not, the binding affinities of designed compounds and HNE complexes were studied through in silico docking studies. To study the binding affinities, the binding energies of the ligand–receptor complexes were calculated. In addition, the extent of modulation of indanedione analogues with the natural ligand bound in the LBD of HNEs and the calculated binding energies were compared. The ligand molecules with the least binding energy were considered compounds with the highest binding affinity. This binding affinity indicated a focused interaction between the compounds and the HNEs. The findings of evaluating the binding affinities of synthesized compounds with the human elastase inhibitor receptor are shown in Table 1. The binding interaction energies of the chemicals were calculated. The co-crystal ligand dihydropyrimidone and standard sivelestat were used to compare the in silico results. When compared to the co-crystal (−65.92), the compounds IBS4 (−81.13) and IBS2 (−76.76) were discovered to be the highest scorers among the library of indane-1,3-dione designed. We obtained a preliminary conclusion that the designed analogues appreciably inhibit HNE.
We investigated the binding mechanisms of all the proposed compounds in the LBD of HNEs. Furthermore, the kind of core structure of the ligands significantly affected the binding affinity of the agonists and antagonists. Although the chemical signatures of the sivelestat and native ligand dihydropyrimidone entirely differed from those of the designed compounds, the designed indane dione compounds were similarly bound as the native ligand did. In fact, compounds IBS4 and IBS2 exhibited improved binding affinity compared to the standard sivelestat (−36.17).
The proposed hybrids developed in this study exhibited remarkable binding affinities with the HNE receptor compared to the standard silvestat control, which was further validated by studying the non-covalent interactions between the receptor and ligand molecules. The developed hybrids demonstrated a significantly higher number of non-covalent interactions, including conventional hydrogen bond interactions, hydrophobic interactions, and electrostatic interactions. For instance, IBS4 showed two conventional hydrogen bonding interactions with His57 and Val216 and multiple van der Waals interactions, such as Tyr 94, Pro 96, Leu 99, Asp 102, Phe 215, Ser 214, Cys 191, Gly 193, Ser 195, one pi–sigma interaction with Phe 192, and one alkyl interaction with Cys 220. The co-crystal structure revealed one conventional hydrogen bond interaction with Val 216, two van der Waals interactions with His 57 and Ser 195, and the standard silvestat showed four conventional hydrogen bonds with Ser 214, Val 216, Tyr 94, Ser 195, and multiple van der Waals interactions, such as His 57, Cys 42, Pro 96, Asp 102, Phe 215, Phe 192, and Gly 193 and one pi–sigma interaction with Leu 99. Overall, this study provides evidence to support the hypothesis that the developed hybrids exhibit higher binding affinities than the standard silvestat control, which is further demonstrated by the detailed analysis of the non-covalent interactions between the receptor and ligand molecules. The findings of this study could have numerous implications for the development of HNE-targeted drugs and potential therapeutic products. The proposed compounds interacted with the HNE (PDB ID: 5A8Z) receptor via hydrogen bonds, hydrophobic bonds, electrostatic bonds, and pi–pi interactions; the residues are tabulated in Table 2.
| Name | Distance (bond length in angstrom) | Category of the interaction | Type of the interaction | From (residue number) | From chemistry (atom in the compound) |
|---|---|---|---|---|---|
| IBS4 | |||||
| A:HIS57:HE1 – IDSB4:O10 | 2.41269 | Hydrogen bond | Carbon hydrogen bond | A:HIS57:HE1 | H-donor |
| A:CYS191:HA – IBS4:O11 | 2.43425 | Hydrogen bond | Carbon hydrogen bond | A:CYS191:HA | H-donor |
| A:SER195:HA – IBS4:O10 | 2.37374 | Hydrogen bond | Carbon hydrogen bond | A:SER195:HA | H-donor |
| IBS4:C26 – A:LEU99 | 4.62443 | Hydrophobic | Alkyl | IDSB4:C26 | Alkyl |
| IBS4 – A:VAL216 | 4.89974 | Hydrophobic | Pi–alkyl | IDSB4 | Pi-orbitals |
|
|||||
| IBS2 | |||||
| A:HIS57:HE1 – IBS2:O10 | 2.44463 | Hydrogen bond | Carbon hydrogen bond | A:HIS57:HE1 | H-donor |
| A:CYS191:HA – IBS2:O11 | 2.43105 | Hydrogen bond | Carbon hydrogen bond | A:CYS191:HA | H-donor |
| A:SER195:HA – IDSB2:O10 | 2.42215 | Hydrogen bond | Carbon hydrogen bond | A:SER195:HA | H-donor |
| A:PHE215 – IBS2 | 5.44236 | Hydrophobic | Pi–pi stacked | A:PHE215 | Pi-orbitals |
| IB2 – A:VAL216 | 4.99021 | Hydrophobic | Pi–alkyl | IDSB2 | Pi-orbitals |
|
|||||
| Sivelestat | |||||
| A:TYR94:HH – sivelestat:O8 | 1.91717 | Hydrogen bond | Conventional hydrogen bond | A:TYR94:HH | H-donor |
| Sivelestat:H40 – sivelestat:O6 | 2.11594 | Hydrogen bond | Conventional hydrogen bond | Sivelestat:H40 | H-donor |
| Sivelestat:H52 – A:ASP102:OD2 | 2.42706 | Hydrogen bond | Conventional hydrogen bond | Sivelestat:H52 | H-donor |
| A:PRO96:HA – sivelestat:O8 | 2.45981 | Hydrogen bond | Carbon hydrogen bond | A:PRO96:HA | H-donor |
| A:PHE192:HA – sivelestat:O3 | 2.61768 | Hydrogen bond | Carbon hydrogen bond | A:PHE192:HA | H-donor |
| Sivelestat:H50 – A:SER214:O | 3.01208 | Hydrogen bond | Carbon hydrogen bond | Sivelestat:H50 | H-donor |
| Sivelestat:C12 – A:VAL216 | 3.69712 | Hydrophobic | Alkyl | Sivelestat:C12 | Alkyl |
| Sivelestat:C14 – A:VAL190 | 5.31574 | Hydrophobic | Alkyl | Sivelestat:C14 | Alkyl |
| Sivelestat:C14 – A:VAL216 | 4.15712 | Hydrophobic | Alkyl | Sivelestat:C14 | Alkyl |
|
|||||
| IDH1 | |||||
| A:TYR94:HH – N:UNK0:O | 2.74255 | Hydrogen bond | Conventional hydrogen bond | A:TYR94:HH | H-donor |
| N:UNK0:H – A:SER214:O | 2.6267 | Hydrogen bond | Conventional hydrogen bond | N:UNK0:H | H-donor |
| N:UNK0:HN – A:HIS57:NE2 | 2.42746 | Hydrogen bond | Conventional hydrogen bond | N:UNK0:HN | H-donor |
| N:UNK0:C – A:HIS57:O | 3.39514 | Hydrogen bond | Carbon hydrogen bond | N:UNK0:C | H-donor |
| N:UNK0:C – A:CYS58:O | 3.49711 | Hydrogen bond | Carbon hydrogen bond | N:UNK0:C | H-donor |
| N:UNK0:H – A:HIS57 | 2.44198 | Hydrogen bond; electrostatic | Pi–cation; pi–donor hydrogen bond | N:UNK0:H | Positive; H-donor |
| N:UNK0:N – A:HIS57 | 4.45041 | Electrostatic | Pi–cation | N:UNK0:N | Positive |
| N:UNK0:C – A:HIS57 | 3.96704 | Hydrophobic | Pi–sigma | N:UNK0:C | C–H |
| N:UNK0 – A:ALA60 | 4.70655 | Hydrophobic | Pi–alkyl | N:UNK0 | Pi-orbitals |
|
|||||
| IDC2 | |||||
| N:UNK0:N – A:ASP102:OD2 | 4.45233 | Electrostatic | Attractive charge | N:UNK0:N | Positive |
| N:UNK0:H – A:TYR94:OH | 3.03165 | Hydrogen bond | Conventional hydrogen bond | N:UNK0:H | H-donor |
| N:UNK0:H – A:PRO96:O | 2.82547 | Hydrogen bond | Conventional hydrogen bond | N:UNK0:H | H-donor |
| N:UNK0:H – N:UNK0:O | 2.86157 | Hydrogen bond | Conventional hydrogen bond | N:UNK0:H | H-donor |
| N:UNK0:H – A:SER214:OG | 2.02313 | Hydrogen bond | Conventional hydrogen bond | N:UNK0:H | H-donor |
| N:UNK0:H – A:SER214:O | 2.34912 | Hydrogen bond | Conventional hydrogen bond | N:UNK0:H | H-donor |
| N:UNK0:H – N:UNK0:O | 2.11788 | Hydrogen bond | Conventional hydrogen bond | N:UNK0:H | H-donor |
| N:UNK0:S – A:TYR94 | 4.92128 | Other | Pi–sulfur | N:UNK0:S | Sulfur |
| N:UNK0 – A:VAL216 | 5.32015 | Hydrophobic | Pi–alkyl | N:UNK0 | Pi-orbitals |
|
|||||
| IDC4 | |||||
| A:SER195:CA – N:UNK0:O | 3.13474 | Hydrogen bond | Carbon hydrogen bond | A:SER195:CA | H-donor |
| A:LEU99:CD2 – N:UNK0 | 3.64879 | Hydrophobic | Pi–sigma | A:LEU99:CD2 | C–H |
| A:VAL216:CG2 – N:UNK0 | 3.89959 | Hydrophobic | Pi–sigma | A:VAL216:CG2 | C–H |
| A:CYS220:SG – N:UNK0 | 5.66345 | Other | Pi–sulfur | A:CYS220:SG | Sulfur |
| A:HIS57 – N:UNK0 | 5.20768 | Hydrophobic | Pi–pi stacked | A:HIS57 | Pi-orbitals |
|
|||||
| Co-crystal | |||||
| A:SER195:HG – 5A8Z:F19 | 2.64763 | Hydrogen bond; halogen | Conventional hydrogen bond; halogen (fluorine) | A:SER195:HG | H-donor; halogen acceptor |
| A:VAL216:HN – 5A8Z:O21 | 2.09957 | Hydrogen bond | Conventional hydrogen bond | A:VAL216:HN | H-donor |
| A:HIS57:HE1 – 5A8Z:F19 | 2.87142 | Hydrogen bond | Carbon hydrogen bond | A:HIS57:HE1 | H-donor |
| A:LEU99:HA – 5A8Z:N23 | 2.46794 | Hydrogen bond | Carbon hydrogen bond | A:LEU99:HA | H-donor |
| A:SER195:HA – 5A8Z:F19 | 2.35713 | Hydrogen bond; halogen | Carbon hydrogen bond; halogen (fluorine) | A:SER195:HA | H-donor; halogen acceptor |
| A:PHE215:HA – 5A8Z:O21 | 2.98793 | Hydrogen bond | Carbon hydrogen bond | A:PHE215:HA | H-donor |
| A:CYS191:O – 5A8Z:F20 | 3.63714 | Halogen | Halogen (fluorine) | A:CYS191:O | Halogen acceptor |
| A:ASP194:C – 5A8Z:F20 | 3.49529 | Halogen | Halogen (fluorine) | A:ASP194:C | Halogen acceptor |
| A:ALA213:O – 5A8Z:F19 | 3.07117 | Halogen | Halogen (fluorine) | A:ALA213:O | Halogen acceptor |
| A:SER214:C – 5A8Z:F18 | 3.4756 | Halogen | Halogen (fluorine) | A:SER214:C | Halogen acceptor |
| A:SER214:O – 5A8Z:F19 | 3.07211 | Halogen | Halogen (fluorine) | A:SER214:O | Halogen acceptor |
| A:PHE215:C – 5A8Z:F18 | 3.66881 | Halogen | Halogen (fluorine) | A:PHE215:C | Halogen acceptor |
| A:CYS220:SG – 5A8Z | 5.87877 | Other | Pi–sulfur | A:CYS220:SG | Sulfur |
| A:ALA213 – 5A8Z:C17 | 3.90034 | Hydrophobic | Alkyl | A:ALA213 | Alkyl |
| 5A8Z:C17 – A:VAL190 | 4.82975 | Hydrophobic | Alkyl | 5A8Z:C17 | Alkyl |
| 5A8Z – A:VAL216 | 4.17138 | Hydrophobic | Pi–alkyl | 5A8Z | Pi-orbitals |
3.2 ADMET studies
The ADMET study predicts the pharmacokinetic and pharmacodynamics properties of the designed compounds under study. The water solubility of a medicine is thought to be the most critical factor in determining its bioavailability. As indicated in Table 3, the anticipated ADMET solubility level ranged from 2 (low soluble) to 4 (high soluble) for synthesized compounds. At 25 °C, the produced chemicals and sivelestat are soluble in water. A logP value represents the partition co-efficient values (logarithm) of compounds in the octanol/water system and computes the lipophilicity of a molecule. All the compounds had logP values below five, which is acceptable. The polar surface area of the chemical is inversely proportional to human intestine absorption, and three compounds had values smaller than 150, indicating that they are likely to be absorbed well, as shown in Fig. 4.
| Compound | Solubilitya | BBBb | CYP2D6c | Hepatotoxicityd | PPBe | Absorptionf | Alogp9g | PSAh |
|---|---|---|---|---|---|---|---|---|
| a Solubility level: 0 extremely low, 1 no very low but possible, 2 yes low, 3 yes good, 4 yes optimal, 5 no too soluble.b BBB-blood–brain barrier level-0 very high, 1 high, 2 medium, 3 low, 4 undefined.c CYP2D6-cytochrome 450 inhibition.d Hepatotoxicity.e PPB-plasma–protein binding.f Absorption – 0 no absorption, 1 very poor, 2 poor, 3 medium, 4 good.g AlogP98-partition coefficient of octanol/water system N.h PSA-polar surface area. | ||||||||
| IBS | 3 | 3 | False | True | True | 0 | 1.234 | 69.203 |
| IBS1 | 3 | 3 | False | True | True | 0 | 1.628 | 85.365 |
| IBS2 | 3 | 3 | False | True | True | 0 | 1.806 | 85.365 |
| IBS3 | 2 | 3 | False | True | True | 0 | 2.459 | 85.365 |
| IBS4 | 2 | 3 | False | True | True | 0 | 2.915 | 85.365 |
| IDH1 | 4 | 3 | False | True | False | 0 | −0.184 | 91.252 |
| IDC2 | 3 | 3 | False | True | False | 0 | 0.367 | 104.062 |
| IDC4 | 2 | 4 | False | True | True | 0 | 2.784 | 115.630 |
| IDC7 | 2 | 4 | False | True | True | 1 | 2.724 | 131.669 |
| Sivelestat | 3 | 4 | False | True | True | 2 | 2.321 | 141.869 |
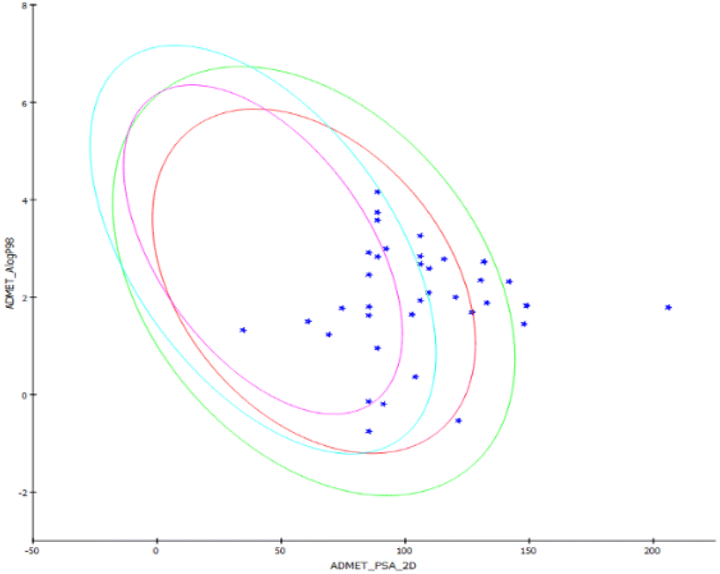 | ||
| Fig. 4 Plot of PSA versus logP with 95% and 99% confidence limit ellipses. | ||
A super family gene member of the human cytochrome P450 enzyme is versatile and could metabolize the various hydrophobic compounds through its oxidation and eventually remove the foreign compounds. Among this major family of CYPs, the three isoforms viz. 2D6, 2C9, and 3A4 are considered more important and responsible for the microsomal oxidation of most drugs in humans. CYP2D6 metabolizes more than 27.5% of drugs and is extensively considered a polymorphic drug-metabolizing enzyme isoform that could catalyze the bio-conversion of many xenobiotics. Consequently, we chose this isoform to predict the ADMET property of the synthesized compounds.28,29
For the CYP2D6 isoform, there are two ADMET classes for the prediction of the activity viz. non-inhibitor (false) and inhibitor (true). However, all the three compounds are predicted as non-inhibitors of CYP2D6 and a metabolic liver enzyme, which is one of the most important isoforms of CYP enzymes in drug metabolism, and none of them may be assumed to cause any serious drug interaction toxicity.30
This parameter also plays an essential role in the pharmacological and toxic properties of drugs. Consequently, the isoform was selected to predict the ADMET property of the synthesized compounds. For the CYP2D6 isoform, there are two ADMET classes for the prediction of the activity viz. non-inhibitor (false) and inhibitor (true). However, all the compounds are predicted to be non-inhibitors of CYP2D6, and a metabolic liver enzyme is the most isoforms of CYP enzymes in drug metabolism. None of them may be assumed to cause serious drug interaction toxicity.
The investigation predicts that all the synthesized compounds were metabolized by the liver. The hepatotoxicity value, which determines the extent of toxicity, is greater than −4.145 and holds for all the synthesized compounds.
Plasma protein binding affects the distribution of drugs and can be categorized as restrictive binding and permissive binding. It is also a means of measuring the efficiency of a drug. Compounds IDH 1 and IDC 2 have a Bayesian score less than −2.226 (false) and are considered to be weak or non-binder of plasma protein. The remaining compounds exhibited a Bayesian score higher than −2.226 (true), suggesting that they are likely to bound (≥90%) in blood with a carrier protein.31
Prediction study results indicate that the designed compounds can be metabolized by the liver. The hepatotoxicity value determines that the extent of toxicity is greater than −4.145 and holds for all the synthesized compounds. Plasma protein binding affects the distribution of drugs and can be categorized as restrictive binding and permissive binding, which is a means for measuring the efficiency of a drug. The ADME properties of indane-1,3-dione analogues are shown in Table 3.
3.3 TOPKAT values
The compounds were further screened to predict toxicity using the TOPKAT tool in DS for their safety. The toxicity parameters of the synthesized compounds were calculated using 9 descriptors, such as mouse NTP prediction, WOE prediction, rat oral LD50, chronic LOAEL, skin irritancy, skin sensitization, ocular irritancy, and aerobic biodegradability prediction. These parameters are essential for determining the toxic properties of drugs, and the results of our analysis indicate that the administration of these synthetic compounds does not pose any hazardous effects because all toxic parameters remain within the acceptable range. Furthermore, these predictions can also be used to evaluate intermediate metabolites and establish a suitable dose range for animal assays. The designed compounds and sivelestat have a non-carcinogenic effect on the mouse. Predictions from WOE indicate that rat oral level LD50 values for the synthesized compounds are more than 0.100 g kg−1 and hence seem to be less toxic. The lowest observed adverse effect level (LOAEL) was found to be below 0.3 g kg−1 and non-carcinogenic. The designed compounds did not show any skin irritancy but had strong sensitivity to the skin and mild ocular irritancy. The obtained TOPKAT values are presented in Table 4.| Comp. name | Mouse NTP prediction | Mouse NTP prediction | WOE prediction | Rat oral LD50 | Chronic LOAEL | Skin irritancy | Skin sensitization | Ocular irritancy | Aerobic biodegradability prediction |
|---|---|---|---|---|---|---|---|---|---|
| IDS | Non-carcinogen | Non-carcinogen | Non-carcinogen | 0.990727 | 0.041107 | None | Strong | Mild | Degradable |
| IBS1 | Non-carcinogen | Non-carcinogen | Non-carcinogen | 1.34912 | 0.021742 | None | Strong | Mild | Non-degradable |
| IBS2 | Non-carcinogen | Non-carcinogen | Non-carcinogen | 1.04843 | 0.023516 | None | Strong | Mild | Non-degradable |
| IBS3 | Non-carcinogen | Non-carcinogen | Non-carcinogen | 1.16231 | 0.014014 | None | Strong | Mild | Non-degradable |
| IBS-4 | Non-carcinogen | Non-carcinogen | Non-carcinogen | 1.02621 | 0.018742 | None | Strong | Mild | Non-degradable |
| IDH1 | Non-carcinogen | Non-carcinogen | Non-carcinogen | 1.18134 | 0.263751 | None | Strong | Mild | Degradable |
| IDC2 | Non-carcinogen | Non-carcinogen | Non-carcinogen | 0.31224 | 0.218569 | None | Strong | Mild | Non-degradable |
| IDC4 | Non-carcinogen | Non-carcinogen | Carcinogen | 0.778265 | 0.146458 | None | Strong | Mild | Non-degradable |
| IDC7 | Non-carcinogen | Non-carcinogen | Carcinogen | 0.895002 | 0.074102 | Mild | Strong | Mild | Non-degradable |
| Sivelestat | Non-carcinogen | Non-carcinogen | Non-carcinogen | 7.74267 | 0.109097 | None | Strong | Mild | Non-degradable |
3.4 Molecular dynamics simulations
Molecule IBS-4 was chosen based on the highest binding energy and subjected to molecular dynamic simulation studies for 50 nanoseconds against the 5A8Z protein. To understand the kinetics of ligand binding to the enzyme's active site, a molecular dynamics simulation was conducted for the same protein (apo) for a period of 50 nanoseconds. This simulation aimed to identify the stability and dynamic changes in the ligand–protein complex in a simulated biological environment. Thus, it was possible to obtain better insight into the ligand binding process.Apoprotein 5A8Z showed highly fluctuating root-mean-square-deviation (RMSD) values owing to its Cα atoms in the range of 1.0–1.35 Å during the first 10 nanoseconds (ns) of its simulation. However, the RMSD values for apo fluctuated in the range of 0.80–1.25 Å during the following 40 ns of the simulation, indicating a more stable state. In the case of the IBS-4–5A8Z complex, the RMSD for the ligand fluctuated in the range of 1.6–3.2 Å throughout the 50 ns simulation. This steady fluctuation of RMSD values shows that IBS-4 forms a stable complex with 5A8Z protein, as can be seen in Fig. 5a and b.
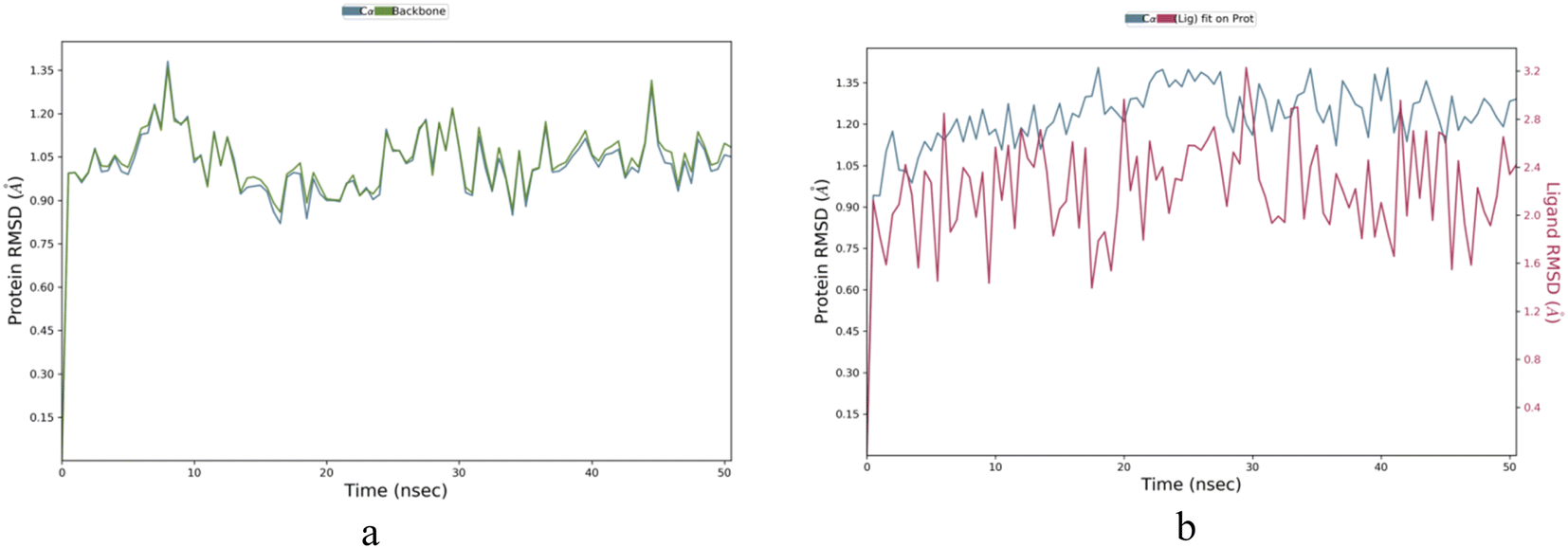 | ||
| Fig. 5 (a and b) RMSD apoprotein IBS-4 complex. | ||
Root Mean Square Fluctuation (RMSF) is a measure of the local changes in the protein chain that can be used to characterize fluctuations. It was observed that the RMSF value of the amino acid fluctuations for protein 5A8Z was in the range of 0.4–1.8 Å, while that of the IBS-4 complex with 5A8Z was in the range of 0.4–2.0 Å. Further, it was observed that the RMSF value of the amino acid fluctuations highly fluctuated in the range of 130–150 amino acids, but none of the active amino acids were present in that range. Thus, it can be concluded that the IBS-4 complex with 5A8Z had fewer fluctuations compared to the protein 5A8Z, as illustrated in Fig. 6a and b.
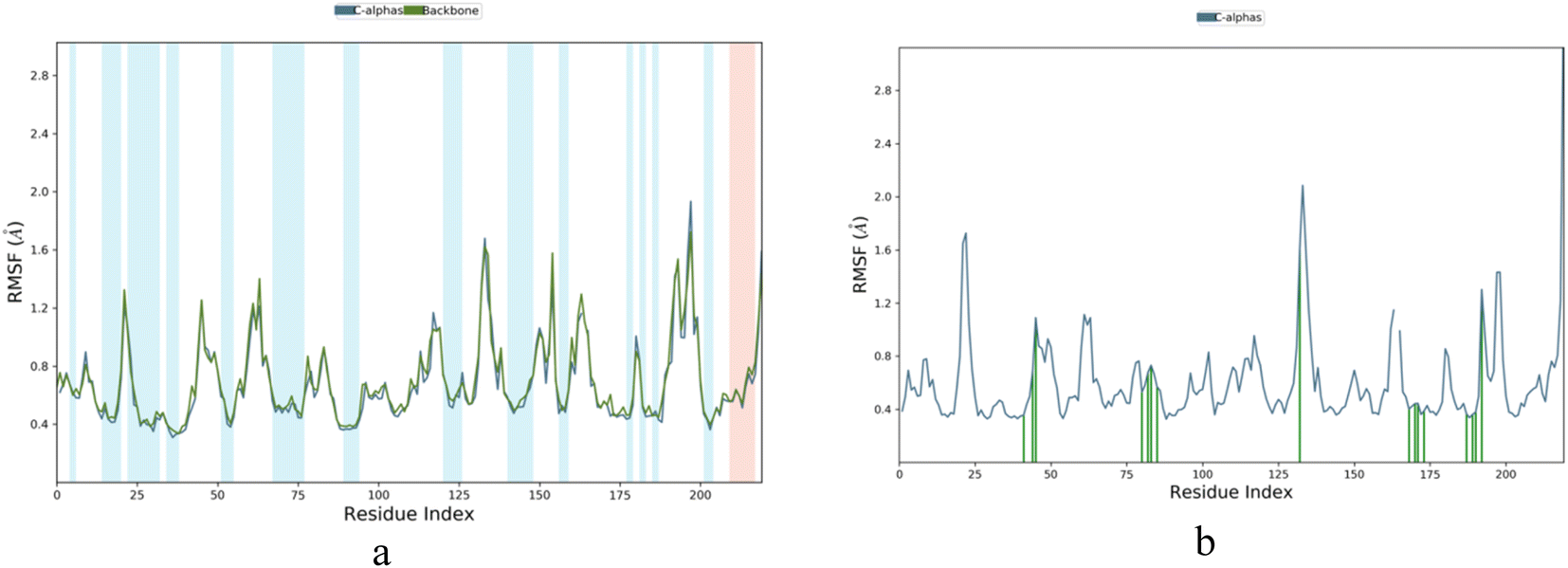 | ||
| Fig. 6 (a and b) RMSF for apoprotein and IBS-4 complex. | ||
The protein–ligand contacts of IBS-4 are highly specific and involve various types of interactions. Hydrophobic contacts are formed between IBS-4 and His 57, Leu 99, Arg 146, Val 190, Phe 192, Ala 213, and Phe 215, while hydrogen bonding interactions occur between IBS-4 and Tyr 94, Ser 195, and Val 216. These interactions are integral for a stable protein–ligand complex in molecular dynamic studies, as they are active in maintaining the stability of the system. Consequently, IBS-4 is shown to form a stable protein–ligand complex, allowing for further exploration of its structure and function, as depicted in Fig. 7a and b.
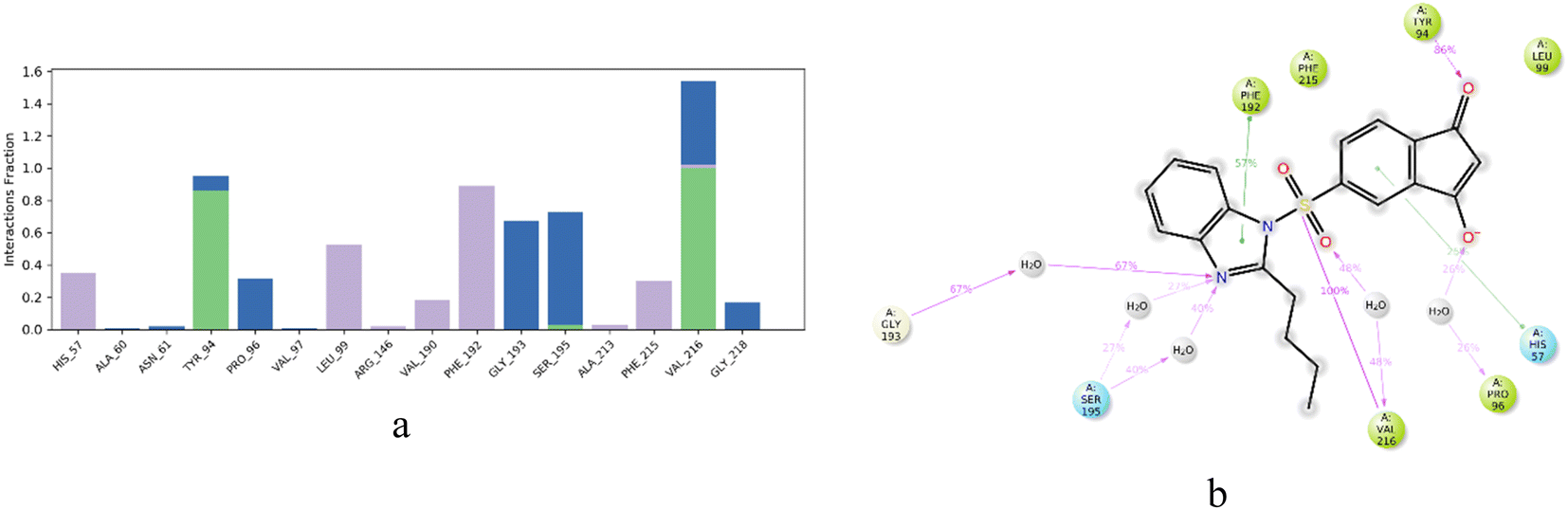 | ||
| Fig. 7 (a and b) Protein–ligand contacts for compound IBS-4 complex. | ||
3.5 Characterization of indanedione compounds
Compound 1,3-dioxo-1,2,3-trihydroindane-6-sulphonylchloride (IBS). Brown colored solid; retention factor (Rf) value = 0.45 (benzene
:acetone, 3:1, v/v developer, visualization: UV and I2), yield 68%. Mp > 360 °C. MF: C9H5ClO4S; MW: 244.65. FTIR (KBr, cm−1) 1712 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O str), 1201.6 (SO2Cl str), 1464 (cyclopentane CH band), 1363 (SO2 str), 597.9 (Cl str), and 742 (Ar C–H band). 1H NMR (400 MHz, DMSO-d6) 3.94 (s, 2H, CH2); 7.45 (m, 3H, Ar–H). 13C NMR (400 MHz, DMSO-d6) 44.95 (CH2); 124.41, 124.81, 126.01 (aromatic carbons); 141.59, 143.18; 143.25 (C–S), 177.76 (two carbonyl carbons); MS: (C9H5ClO4S) 246 (M + 1), (C9H6O2+) 146 (m/z). Elemental analysis calcd for C9H5ClO4S: C (44.18%), H (2.06%), Cl (14.49%), O (26.16%), and S (13.11%). Found: C (41.64%), H (2.2%), Cl (14.39%), O (26.92%), S (12.98%)
O str), 1201.6 (SO2Cl str), 1464 (cyclopentane CH band), 1363 (SO2 str), 597.9 (Cl str), and 742 (Ar C–H band). 1H NMR (400 MHz, DMSO-d6) 3.94 (s, 2H, CH2); 7.45 (m, 3H, Ar–H). 13C NMR (400 MHz, DMSO-d6) 44.95 (CH2); 124.41, 124.81, 126.01 (aromatic carbons); 141.59, 143.18; 143.25 (C–S), 177.76 (two carbonyl carbons); MS: (C9H5ClO4S) 246 (M + 1), (C9H6O2+) 146 (m/z). Elemental analysis calcd for C9H5ClO4S: C (44.18%), H (2.06%), Cl (14.49%), O (26.16%), and S (13.11%). Found: C (41.64%), H (2.2%), Cl (14.39%), O (26.92%), S (12.98%)
1,3-Dioxo-1,2,3-trihydro indane-6-sulphonyl benzimidazole (IBS 1). Dark green solid: retention factor (Rf) value = 0.79 (chloroform
:methanol, 9:1, v/v developer, visualization: UV and I2), yield 61%. Mp > 360 °C. MF: C16H12N2O4S; MW: 328.34. FTIR (KBr, cm−1) 1650 (CO str), 3393 (NH str), 2926 cm−1 (CH str), 1364 cm−1 (SO str), 924 cm−1 (C–H str). 1H NMR (400 MHz, DMSO-d6) 3.3 (s, 2H, CH2 indane); 7.5 (m, 3H, Ar–H indanedione); 8.6 (m, 4H, Ar–H, benzimidazole); 8.9 (s, 1H, CH benzimidazole). 13C NMR (400 MHz, DMSO-d6) 39.08 (CH2 indanedione); 114.67, 116.65, 117.53, 120.30, 120.39, 122.07, 122.53, 122.55, 125.45, 126.50, 132.83, (aryl and hetero aryl carbons); 140.65 (C–S); 177.76 (two carbonyl carbons). MS: (C16H12N2O4S) 328 (M), (C6H11NO2S+) 160 (m/z − 1), (C3H7N2O2S+) 132 (m/z − 3), (NO2S+) 76 (m/z − 2). Elemental analysis calcd for C16H12N2O4S: C (58.53%), H (3.68%), N (8.53%), O (19.49%), and S (9.77%). Found: C (59.18%), H (3.48%), N (8.69%), O (19.31%), S (9.42%)
1,3-Dioxo-1,2,3-trihydro indane-6-sulphonyl (2-methyl)benzimidazole (IBS 2). Pale green solid: retention factor (Rf) value = 0.72 (chloroform
:methanol, 9:1, v/v developer, visualization: UV and I2), yield 64%. Mp > 360 °C. MF:C17H14N2O4S; MW: 342.37. FTIR (KBr, cm−1) 1666 (CO str), 3384 (NH str), 2908 cm−1 (CH str), 1372 cm−1 (SO str), 912 cm−1 (C–H str). 1H NMR (400 MHz, DMSO-d6) 2.3 (s, 3H, CH3 benzimidazole); 3.3 (s, 2H, CH2 indanedione),7.2–7.3 (m, 3H, Ar–H),7.5–7.8 (m, 4H, Ar–H benzimidazole), 13C NMR (400 MHz, DMSO-d6) 27.78 (CH3 benzimidazole); 39.73 (CH2 indanedione); 115.45, 116.68, 117.53, 119.30, 122.53, 122.83, 124.67,126.50, 127.55, 130.65,133.17,155.76 (aryl and hetero aryl carbons); 144.52 (C–S); 177.76 (two carbonyl carbons); MS: (C17H14N2O4S) 343 (M+1), (C6H11NO2S+)160 (m/z − 1), (C3H7N2O2S+) 132 (m/z − 3), (NO2S+) 76 (m/z − 2). Elemental analysis calcd for C17H14N2O4S: C (59.64%), H (4.12%), N (8.18%), O (18.69%), and S (9.37%). Found: C (59.24%), H (3.77%), N (8.12%), O (18.59%), S (9.39%)
1,3-Dioxo-1,2,3-trihydro indane-6-sulphonyl (2-ethyl)benzimidazole (IBS 3). Pale green solid: retention factor (Rf) value = 0.63 (chloroform
:methanol, 9:1, v/v developer, visualization: UV and I2), yield 64%. Mp > 360 °C. MF:C18H16N2O4S; MW: 356.4 FTIR (KBr, cm−1) 1634 (CO str), 3352 (NH str), 2960 cm−1 (CH str), 1352 cm−1 (SO str), 918 cm−1 (C–H str). 1H NMR (400 MHz, DMSO-d6) 0.8 (t, 3H, CH3-ethyl); 1.2 (q, 2H, CH2-ethyl); 3.3 (s, 2H, CH2 indane); 7.6 (m, 3H, Ar–H indanedione); 8.8 (m, 4H, Ar–H, benzimidazole); 13C NMR (400 MHz, DMSO-d6), 25.87, 27.48 (CH2, CH3); 39.73 (CH2 indanedione); 143.25 (C–S); 102.83, 105.45, 110.65, 114.67, 124.35,129.30, 139.61, 126.66, 124.95, 127.55, 145.17, 157.74 (aryl and hetero aryl carbons); 177.76 (two carbonyl carbons); MS: (C18H16N2O4S)357 (M + 1), (C6H11NO2S+) 160 (m/z − 1), (C3H7N2O2S+) 132 (m/z − 3), (NO2S+) 76 (m/z − 2). Elemental analysis calcd for C18H16N2O4S: C (60.66%), H (4.52%), N (7.86%), O (17.96%), and S (9.00%). Found: C (59.61%), H (4.53%), N (7.91%), O (17.46%), S (8.92%)
1,3-Dioxo-1,2,3-trihydro indane-6-sulphonyl (2-propyl)benzimidazole (IBS 4). Pale green solid: retention factor (Rf) value = 0.60 (chloroform
:methanol, 9:1, v/v developer, visualization: UV and I2), yield 65%. Mp > 360 °C. MF: C19H18N2O4S; MW: 370.42. FTIR (KBr, cm−1) 1638 (CO str), 3370 (NH str), 2908 cm−1 (CH str), 1334 cm−1 (SO str), 934 cm−1 (C–H str). 1H NMR (400 MHz, DMSO-d6), 0.8 (t, 3H, CH3 propyl), 1.2 (m, 2H, CH2-propyl) 1.9 (t, 2H, CH2 propyl); 3.4 (s, 2H, CH2 indane); 7.5 (m, 3H, Ar–H indanedione); 8.6 (m, 4H, Ar–H, benzimidazole) 13C NMR (400 MHz, DMSO-d6) 26.78, 27.21, 28.90 (CH3, CH2, CH2) 39.73 (CH2 indanedione); 144.48 (C–S); 110.65, 124.45, 125.45, 122.83,126.63, 127.55, 129.30, 133.12,134.67, 137.26, 139.53, 143.25, (aryl and hetero aryl carbons); 177.76 (two carbonyl carbons). MS: (C19H18N2O4S) 371 (M + 1), (C6H11NO2S+) 160 (m/z − 1), (C3H7N2O2S+) 132 (m/z − 3), (NO2S+) 76 (m/z − 2). Elemental analysis calcd for C19H18N2O4S: C (61.61%), H (4.90%), N (7.56%), O (17.28%), and S (8.66%). Found: C (61.51%), H (4.87%), N (7.76%), O (17.11%), S (8.72%)
2-(1,3-Dioxo-2,3-dihydro-1H-inden-2-yl)acetohydrazide (IDH 1). Colorless solid: retention factor (Rf) value = 0.42 (ethyl acetate
:petroleum ether 1:4 v/v developer, visualization: UV and I2), yield 78%. Mp 150–152 °C. MF: C11H10N2O3; MW: 218.21. FT-IR (KBr, cm−1) 2854 (C–H str), 1628 (CO str), 3411 (NH2 str), 2926 (NH str) and 763 (CH str aromatic) group. 1H NMR (400 MHz, DMSO-d6): 2.4 (d, 2H, CH2); 3.1 (t, 1H, CH indanedione); 6.6 (s, 2H, NH2); 7.2 (m, 4H, Ar–H indanedione); 7.8 (s, 1H, NH); 13C NMR (400 MHz, DMSO-d6): 39.91 (CH2 hydrazide); 50.26 (CH indanedione); 119.79, 123.0, 128.47,135.10, 140.53,142.40, (aryl carbons); 145.33 (carbonyl carbon hydrazide); 162.96 (two carbonyl carbons indane-dione); MS: (C11H10N2O3) 218 (M), (C11H5O3+) 185 (m/z), (C9H6O2+) 145 (m/z − 1). Elemental analysis calcd for C11H10N2O3: C (60.55%), H (4.62%), N (12.84%), and O (22.00%). Found: C (58.71%), H (3.29%), N (12.96%), O (23.98%).
2-(2-(1,3-Dioxo-2,3-dihydro-1H-inden-2-yl)acetyl)hydrazine-1-carbothioamide (IDC 2). Brown colored solid: retention factor (Rf) value = 0.55 (ethyl acetate
:petroleum ether 1:4, v/v developer, visualization: UV and I2), yield 90%. Mp 110–112 °C. MF: C12H11N3O3S; MW: 277.30. FTIR (KBr, cm−1) 1635 (CO str), 3415 cm−1 (NH2 str), 2923 (NH str), and 1245 cm−1 (CS str) group. 1H NMR (400 MHz, DMSO-d6): 3.09 (d, 2H, CH2); 4.35 (t, 1H, CH indanedione); 7.65 (s, 2H, NH2); 8.05 (m, 4H, Ar–H indanedione); 9.10 (s, 1H, NH); 9.72 (s, 1H, NH);13C NMR (400 MHz, DMSO-d6): 39.41(CH2 hydrazide); 50.05 (CH indanedione); 118.79, 118.79, 128.37,128.37, 130.53, 130.53 (aryl carbons); 141.23 (carbonyl carbon hydrazide); 165.96 (carbonyl carbons indanedione); 181.65 (CS);. MS: (C12H11N3O3S) 278 (M + 1), (C11H7O3+) 188 (m/z + 1), (C9H6O2+) 147 (m/z + 1), (C3H6N3OS+) 131 (m/z − 1), (CH4N3S+) 92 (m/z + 2). Elemental analysis calcd for C12H11N3O3S: C (51.98%), H (4.00%), N (15.15%), O (17.31%), and S (11.56%). Found: C (49.97%), H (3.47%), N (15.43%), O (17.92%), S (12.93%)
4-2-(2-(1,3-Dioxo-2,3-dihydro-1H-inden-2-yl)acetyl)-N-(3,4,5-trimethoxybenzylidene)hydrazine-1-carbothioamide (IDC-4). Brown colored solid; retention factor (Rf) value = 0.64 (ethyl acetate
:petroleum ether 1:4 v/v developer, visualization: UV and I2), yield 91%. Mp 115–117 °C. MF: C22H21N3O6S; MW: 455.48. FTIR (KBr, cm−1) 1645 cm−1 (CO str), 1235 cm−1 (CS str), 2925 cm−1 (CH str), and 2855 cm−1 (OCH3 str). 1H NMR (400 MHz, DMSO-d6): 2.9 (t, 1H, CH indanedione); 3.1 (d, 2H, CH2 hydrazide); 3.9 (s, 9H, O–CH3); 6.8 (m, 4H, Ar–H indanedione); 7.5 (m, 2H, Ar–H benzaldehyde); 7.9 (s, 1H, NCH) 8.0 (s, 1H, NH); 9.1 (s, 1H, NH); 13C NMR (400 MHz, DMSO-d6): 33.09 (CH indanedione); 39.28(CH2 hydrazide); 50.48 (OCH3). 123.00, 123.00, 128.18, 130.62, 134.17, 134.17, 135.10, 135.10, 142.40, 142.40 156.22, 156.22, (aryl carbons); 160.62 (NCH); 165.32 (carbonyl carbon hydrazide); 170.92 (two carbonyl carbons indanedione); CS (174.16); MS: (C22H21N3O6S) 455 (M), (C12H9N3O3S+) 276 (m/z + 1) (C10H12O3N+) 195 (m/z + 1). Elemental analysis calcd for C22H21N3O6S: C (58.01%), H (4.65%), N (9.23%), O (21.08%), and S (7.04%). Found: C (56.97%), H (4.50%), N (9.43%), O (21.92%), S (7.13%).
2-(2-(1,3-Dioxo-2,3-dihydro-1H-inden-2-yl)acetyl)-N-(3-nitrobenzylidene)hydrazine-1-carbothioamide (IDC7). Brown colored solid; retention factor (Rf) value = 0.48 (ethyl acetate
:petroleum ether 1:4 v/v developer, visualization: UV and I2), yield 91%. Mp 110–112 °C. MF: C19H14N4O5S; MW: 410.40: FTIR (KBr, cm−1) 1647 (CO str), 2853 (CH str-indanedione), 1470 (NO2 str), 3404 (NH str). 1H NMR (400 MHz, DMSO-d6) 2.3 (t, 1H, CH indanedione); 2.9 (d, 2H, CH2 hydrazide); 7.4 (s, 1H,NCH); 7.7 (m, 4H, Ar–H indanedione); 7.9 (m, 4H, Ar–H benzaldehyde); 7.9 (s, 1H, NH); 8.3 (s, 1H, NH); 13C NMR (400 MHz, DMSO-d6): 38.86 (CH indanedione), 39.07 (CH2 hydrazide); 102.16, 110.22, 112.86, 111.91, 111.91, 122.68, 122.68, 126.60, 127.76, 135.60, 135.81,146.18, (aryl carbons); 153.55 (C–NO2); 160.28 (CS); 160.62 (NCH); 161.17 (carbonyl carbon hydrazide); 169.05 (carbonyl carbons indanedione); MS: (C19H14N4O5S) 410 (M), (C12H9N2O3S+) 257 (m/z − 4), (C11H9N2O3+) 213 (m/z − 4), (C11H7O3+)183 (m/z − 4), (C10H7O2+) 157 (m/z − 2). Elemental analysis calcd for C19H14N4O5S:C (55.61%), H (3.44%), N (13.65%), O (19.49%), and S (7.81%). Found C (54.60%), H (2.84%), N (14.24%), O (20.21%), and S (8.19%).
3.6 Neutrophil elastase inhibitor activity
The activity of the compound is a promising way to develop a better lead compound from the IC50 values. The assay results indicate that the type of inhibition is mechanism based. The primary evidence is the varying degree of inhibition by the compounds at a concentration of 0.1 μm. The degree of inhibition data is presented in Table 5 and Fig. 8. Transition state analogue inhibition is seldom observed at this concentration. Moreover, IC50 value of the most promising compound IBS is around the same value obtained by methoxysuccinyl–Ala–Ala–Pro–Val chloromethyl ketone (SPCK). SPCK is the most studied mechanism-based serine protease inhibitor. The development of drug molecules for a specific enzyme inhibitory activity involves several constraints. The constraints faced during the research are as follows: (a) during the inflammatory period, the concentration of the enzyme is several folds higher than the introduced drug; (b) innate immunity response triggers the release of specific proteins in which some of them may be enzymes; and (c) may interfere with biologically designed feedback mechanisms.| Compound | Concentration | Mean | SD |
|---|---|---|---|
| IBS | 100 μM | 91.07 | 4.21 |
| 50 μM | 53.47 | 3.47 | |
| 10 μM | 44.86 | 18.63 | |
| 1 μM | 19.81 | 1.05 | |
| 0.1 μM | 14.34 | 8.04 | |
| IBS1 | 100 μM | 35.16 | 4.63 |
| 50 μM | 31.95 | 4.31 | |
| 10 μM | 25.35 | 1.07 | |
| 1 μM | 25.25 | 7.11 | |
| 0.1 μM | 24.09 | 2.58 | |
| IBS2 | 100 μM | 70.08 | 3.78 |
| 50 μM | 68.83 | 2.61 | |
| 10 μM | 63.99 | 3.76 | |
| 1 μM | 52.62 | 4.85 | |
| 0.1 μM | 42.11 | 0.98 | |
| IBS 3 | 100 μM | 46.14 | 6.92 |
| 50 μM | 37.08 | 7.44 | |
| 10 μM | 29.17 | 1.14 | |
| 1 μM | 26.69 | 3.65 | |
| 0.1 μM | 25.35 | 3.02 | |
| IBS 4 | 100 μM | 74.38 | 2.51 |
| 50 μM | 71.33 | 4.14 | |
| 10 μM | 65.94 | 2.06 | |
| 1 μM | 54.38 | 3.10 | |
| 0.1 μM | 30.42 | 1.34 | |
| IDH | 100 μM | 36.49 | 3.16 |
| 50 μM | 30.15 | 5.8 | |
| 10 μM | 28.7 | 6.06 | |
| 1 μM | 21.6 | 4.54 | |
| 0.1 μM | 18.64 | 1.34 | |
| IDC 2 | 100 μM | 28.82 | 6.33 |
| 50 μM | 24.77 | 4.86 | |
| 10 μM | 21.48 | 5.11 | |
| 1 μM | 18.94 | 4.12 | |
| 0.1 μM | 16.32 | 3.47 | |
| IDC 4 | 100 μM | 34.97 | 4.88 |
| 50 μM | 28.15 | 2.2 | |
| 10 μM | 17.33 | 4.28 | |
| 1 μM | 17.28 | 2.14 | |
| 0.1 μM | 13.97 | 1.55 | |
| IDC 7 | 100 μM | 26.72 | 0.04 |
| 50 μM | 25.78 | 3.88 | |
| 10 μM | 24.64 | 0.73 | |
| 1 μM | 16.7 | 1.61 | |
| 0.1 μM | 12.03 | 0.47 | |
| Sivelestat | 100 μM | 99.77 | 1.2 |
| 50 μM | 98.48 | 0.78 | |
| 10 μM | 98.60 | 0.44 | |
| 1 μM | 98.47 | 1.02 | |
| 0.1 μM | 96.24 | 0.56 |
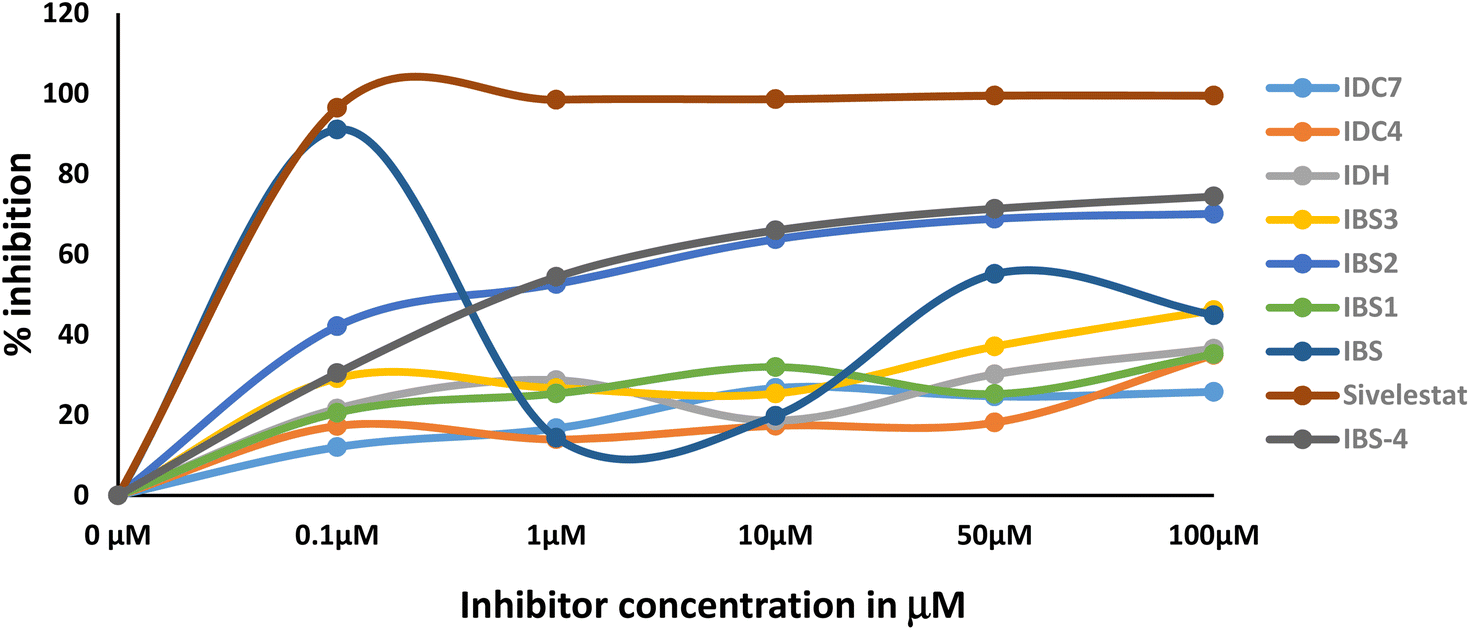 | ||
| Fig. 8 Degree of HNE inhibition by the synthesized compounds. | ||
4 Conclusion
Cyclic diketone derivatives are known for their broader synthetic applications and selective pharmacological responses. In this study, indanedione scaffolds were designed in silico, and the docking of compounds with HNE was done using Cdocker's protocol explorer. ADMET and carcinogenicity predication studies were performed using Discovery Studio 4.1. Two schemes were adopted: one to prepare indanedione benzimidazoles and the other to prepare indanedione hydrazides. The synthesized compounds were tested on HNE in vitro inhibition activity. Compared with sivelestat, the synthesized compounds seem to be mechanism-based inhibitors, while sivelestat acts as a transition base analogue inhibitor. The incorporation of benzimidazoles/substituted benzimidazoles via sulphonyl group linkage in the IBS series may be the reason for the increased activity. The hydrazine linker also imparts increased bioactivity. One of the most important concepts in drug design is the covalent conjugation of biologically active moieties, acting via various mechanisms that lead to a favorable case of synergism, resulting in compounds with increased activity and reduced toxicity. Based on this concept and considering the pharmacological activities shown by indane 1,3-dione, benzimidazoles and hydrazones, we turned our interest to study the incorporation of different benzimidazoles and hydrazones on the indane-1,3-dione via the sulphonyl chloride moiety to afford various new derivatives of our lead compound.Further processing of the raw data establishes the true nature of inhibition. All the compounds exhibited some degree of inhibition even at a concentration of 0.1 μM. Further redesigning of these scaffolds may lead to a higher degree of inhibition.
Conflicts of interest
The authors declare that there are no conflicts of interest.Acknowledgements
The authors extend their appreciation to the Deputyship for Research & Innovation, Ministry of Education in Saudi Arabia for funding this research work through the project number IFP 2021-082.References
- S. C. Yang, Y. F. Tsai, Y. L. Pan and T. L. Hwang, Biomed. J., 2021, 44, 439–446 CrossRef CAS PubMed.
- C. Johansson and F. C. Kirsebom, Mucosal Immunol., 2021, 14, 815–827 CrossRef CAS PubMed.
- C. T. Pham, Int. J. Biochem. Cell Biol., 2008, 40, 1317–1333 CrossRef CAS PubMed.
- F. Soualmia and C. El Amri, Expert Opin. Ther. Pat., 2018, 28, 93–110 CrossRef CAS PubMed.
- C. F. Koelsch and C. D. LeClaire, J. Org. Chem., 1941, 6, 516–533 CrossRef CAS.
- K. Mitka, P. Kowalski, D. Pawelec and Z. Majka, Croat. Chem. Acta, 2009, 82, 613–618 CAS.
- J. Karan Singh, Curr. Org. Synth., 2016, 13, 385–407 CrossRef.
- S. Asadi and G. Mohammadi Ziarani, Mol. Diversity, 2016, 20, 111–152 CrossRef CAS PubMed.
- R. Pluskota and M. Koba, Mini-Rev. Med. Chem., 2018, 18, 1321–1330 CrossRef CAS PubMed.
- S. Meena, D. Shankar, K. V. Ramseshu, D. Giles, M. S. Prakash, S. Venkataraman, D. Giles, M. S. Prakash and K. V. Ramseshu, Indian J. Chem., 2006, 45B, 1572 CAS.
- S. Jubie, S. Meena, K. V. Ramaseshu, N. Jawahar and S. Vijayakumar, Indian J. Chem., 2010, 49B, 1261 Search PubMed.
- H. S. Abd-Elghaffar, M. A. El-Hashash, S. F. Mohamed, A. A. Ibrahim, A. E. Amr, M. A. Al-Omar and E. S. Nossier, Russ. J. Gen. Chem., 2020, 90, 686–696 CrossRef CAS.
- T. C. Sanders, J. Med. Chem., 1999, 42(15), 2969–2976 CrossRef CAS PubMed , 10425106.
- N. D. Pearson, D. S. Eggleston, R. C. Haltiwanger, M. Hibbs, A. J. Laver and A. C. Kaura, Bioorg. Med. Chem. Lett., 2002, 12, 2359–2362 CrossRef CAS PubMed.
- F. Xue and C. T. Seto, Bioorg. Med. Chem. Lett., 2006, 14, 8467–8487 CrossRef CAS PubMed.
- J. C. Powers, J. L. Asgian, Ö. D. Ekici and K. E. James, Chem. Rev., 2002, 102, 4639–4750 CrossRef CAS PubMed.
- S. Sinha, W. Watorek, S. Karr, J. Giles, W. Bode and J. Travis, Proc. Natl. Acad. Sci. U. S. A., 1987, 84, 2228–2232 CrossRef CAS PubMed.
- W. Bode, E. Meyer Jr and J. C. Powers, Biochemistry, 1989, 28, 1951–1963 CrossRef CAS PubMed.
- L. Hedstrom, Chem. Rev., 2002, 102, 4501–4524 CrossRef CAS PubMed.
- J. Selvaraj, S. S. P, L. Rajan, D. Selvaraj, D. Palanisamy, K. N. PK and S. K. Mohankumar, RSC Adv., 2021, 11, 13051–13060 RSC.
- H. Balaji, A. Selvaraj, N. Saha, P. S. Sundar, S. Jubie and S. K. Mohankumar, Mini-Rev. Med. Chem., 2021, 21, 2612–2625 CrossRef CAS PubMed.
- S. Jubie, T. Prabha, S. Palanisamy, S. Latha and S. Ayyamperumal, Int. J. Pharma Sci. Res., 2020, 11, 4349 CAS.
- S. Jubie, U. Durai, S. Latha, S. Ayyamperumal, A. Wadhwani and T. Prabha, Curr. Drug Res. Rev., 2021, 13, 73–83 CrossRef CAS PubMed.
- A. Uniyal, M. K. Mahapatra, V. Tiwari, R. Sandhir and R. Kumar, J. Biomol. Struct. Dyn., 2022, 40, 3609–3625 CrossRef CAS PubMed.
- P. Naresh, A. Selvaraj, P. Shyam Sundar, S. Murugesan, S. Sathianarayanan, K. Namboori PK and S. Jubie, J. Biomol. Struct. Dyn., 2022, 40, 4866–4878 CrossRef CAS PubMed.
- V. Yele, M. A. Azam and S. Jupudi, Chem. Pap., 2020, 74, 4567–4580 CrossRef CAS.
- Z. Guo, U. Mohanty, J. Noehre, T. K. Sawyer, W. Sherman and G. Krilov, Chem. Biol. Drug Des., 2010, 75, 348–359 CrossRef CAS PubMed.
- Q. Qu, J. Qu, L. Han, M. Zhan, L. Wu, Y. Zhang, W. Zhang and H. Zhou, Acta Pharmacol. Sin., 2014, 35, 685–696 CrossRef CAS PubMed.
- D. R. McMasters, R. A. Torres, S. J. Crathern, D. L. Dooney, R. B. Nachbar, R. P. Sheridan and K. R. Korzekwa, J. Med. Chem., 2007, 50, 3205–3213 CrossRef CAS PubMed.
- P. Poulin, F. J. Burczynski and S. Haddad, J. Pharm. Sci., 2016, 105, 497–508 CrossRef CAS PubMed.
- S. Ekins, R. Pottorf, R. C. Reynolds, A. J. Williams, A. M. Clark and J. S. Freundlich, J. Chem. Inf. Model., 2014, 54, 1070–1082 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |