
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProton transfer network with luminescence display controls OFF/ON catalysis that generates a high-speed slider-on-deck†
Sohom Kundu‡
,
Isa Valiyev‡ ,
Debabrata Mondal,
Vishnu Verman Rajasekaran,
Abir Goswami and
Michael Schmittel*
,
Debabrata Mondal,
Vishnu Verman Rajasekaran,
Abir Goswami and
Michael Schmittel*
Center of Micro and Nanochemistry and (Bio)Technology, Organische Chemie I, Universität Siegen, Adolf-Reichwein-Str. 2, D-57068 Siegen, Germany. E-mail: schmittel@chemie.uni-siegen.de; Tel: +49 2717404356
First published on 9th February 2023
Abstract
A three-component network for OFF/ON catalysis was built from a protonated nanoswitch and a luminophore. Its activation by addition of silver(I) triggered the proton-catalyzed formation of a biped and the assembly of a fast slider-on-deck (k298 = 540 kHz).
Proton relays in living systems1 are often coupled to bio-machinery with enzymatic catalysis.2 Frequently, such proton transfer systems comprise intricate networks with information exchange3,4 amongst multiple components. Using manmade molecules,5 however, it is still arduous to create networks that approach the complexity6 and functions of biosystems.
Although activation via proton transfer plays an eminent role in molecular logic,7 switching,8–10 and machine operation,11–15 proton transfer networks in combination with the regulation of catalysis remain scarce.16,17 Beyond that, we present the in situ catalytic synthesis of a highly dynamic supramolecular device by a new proton transfer network requiring interference-free information handling,18 self-sorting19–21 and communication22 among multiple components.23–25
Recently, our group has developed various chemically actuated networks of communicating components and used them for the ON/OFF regulation of transition metal catalysis.26–28 Herein, we describe a proton relay system that reversibly toggles between two networked states and is useful for regulation of acid catalysis. It catalyzes an amine deprotection which was utilized to fabricate in situ a high-speed slider-on-deck,29,30 for the first time based on aniline → zinc-porphyrin31 (Nan → ZnPor) interactions.
The communicating network consists of nanoswitch 1 (ref. 32) and luminophore 2 (ref. 33) as receptors (Fig. 1). In the self-sorted networked state (NetState-I), the added proton is captured in the cavity of nanoswitch 1 resulting in [H(1)]+, while luminophore 2 remains free. NetState-I is catalytically inactive as the encapsulated proton is unable to catalyze the deprotection of the trityl protected slider biped 4. Upon addition of AgBF4 to NetState-I, the silver(I) coordinates to switch 1 and the proton is released onto luminophore 2 thus leading to formation of [Ag(1)]+ and [H(2)]+ in NetState-II (see Scheme 1), which may be followed by characteristic ratiometric emission changes. If NetState-II is formed in presence of 3 and 4, the catalytic trityl deprotection of 4 produces the free biped 5 that eventually binds to deck 3 furnishing the slider-on-deck [(3)(5)].
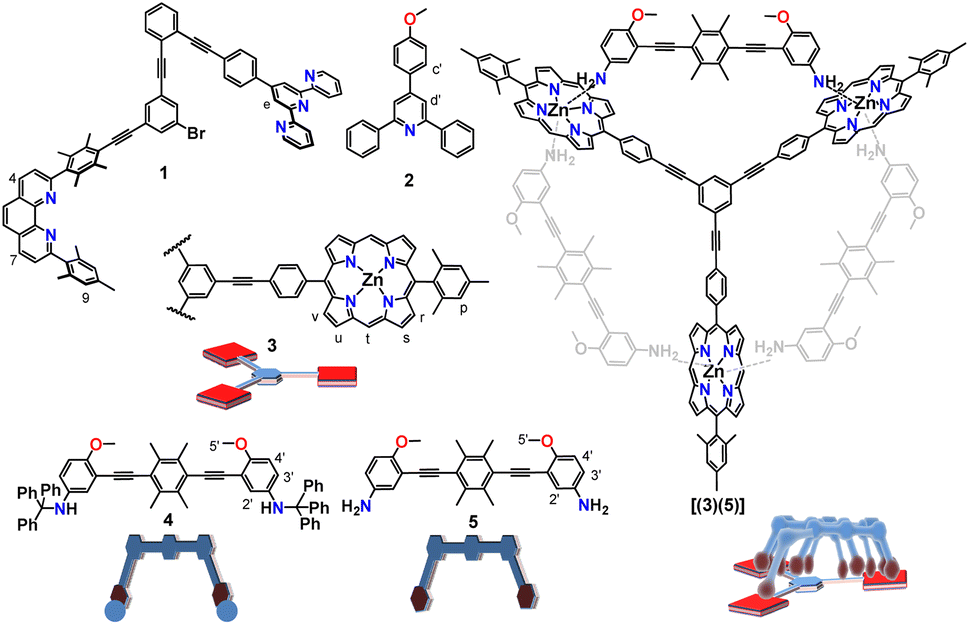 | ||
| Fig. 1 Chemical structures and cartoon representations of the ligands used in the study. | ||
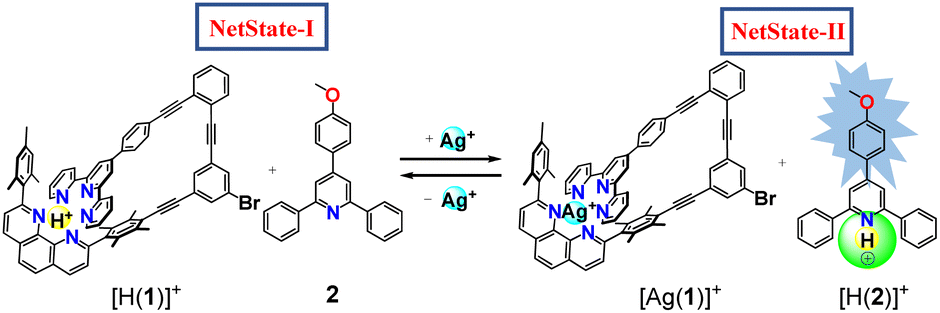 | ||
| Scheme 1 Schematic representation of the reversible proton relay network. Green circle represents catalytic site. | ||
The concept of the catalyzed biped formation through a proton transfer network relies on the following considerations: (a) the initial networked state should be catalytically inactive as expected from a self-sorting of the proton inside the nanoswitch leaving the luminophore free. (b) Addition of silver(I) ions should release the proton from the protonated switch thus forming the protonated luminophore which we anticipated to act as catalyst. (c) A di-protected biped should undergo acid catalyzed deprotection by the protonated luminophore and eventually form a slider-on-deck by coordination to a deck. (d) Finally, the product of the catalysis should not intervene in any state of the operation with the main system.
As proton receptor we chose the known phenanthroline–terpyridine nanoswitch 1 (ref. 32) in order to tightly capture the proton/silver(I) as HETTAP (HETeroleptic Terpyridine And Phenanthroline) complexes.34 Luminophore 2 was based on the 2,4,6-triarylpyridine scaffold which is known to fluoresce strongly upon protonation due to intramolecular charge transfer (ICT).35
To investigate the self-sorting within the network, we mixed nanoswitch 1, luminophore 2 and TFA in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 ratio in CD2Cl2 which furnished NetState-I = [H(1)]+ + 2. 1H NMR shifts of the 4/7-H proton signals to 8.43 and 8.69 ppm and of 9-H and e-H proton peaks to 6.60 and 8.48 ppm confirmed formation of [H(1)]+. Moreover, proton signals of c′-H and d′-H at 7.07 and 7.92 ppm corroborated presence of the free luminophore 2 (Fig. 2). Addition of one equiv. of AgBF4 to NetState-I instantaneously generated NetState-II = [Ag(1)]+ + [H(2)]+. In the 1H NMR of NetState-II, the 9-H and e-H proton signals shifted to 6.55 and 8.23 ppm, respectively, which substantiated the formation of [Ag(1)]+ whereas the shift of proton peaks c′-H and d′-H to 7.12 and 8.00 ppm, respectively, established the protonated luminophore [H(2)]+ (Fig. 2). Upon excitation at 300 nm, NetState-I exhibited emissions at λ = 346, 372 and 393 nm which represent an overlap of the emission peaks of the free luminophore 2 at λ = 346 and 372 nm and of [H(1)]+ at λ = 376 and 393 nm (Fig. 3c). In NetState-II, the fluorescence spectrum showed a single emission at λ = 461 nm, which is attributed to [H(2)]+ (Fig. 3). In contrast, [Ag(1)]+ in NetState-II is nonfluorescent as advocated by the full quenching of the emission of [H(1)]+ at λ = 376 and 393 nm upon titration with silver(I) ions (ESI, Fig. S18†). When one equiv. of tetra-n-butylammonium iodide (TBAI) was added to NetState-II, complete restoration of NetState-I was achieved as confirmed from 1H NMR data. TBAI acted as a scavenger for silver(I) by removing AgI through precipitation thus reversing the translocation sequence. Multiple switching of NetState-I ⇆ NetState-II was readily followed by 1H NMR (Fig. 2) and luminescence changes. Upon successive addition of AgBF4 to NetState-I, the emission changed to λ = 461 nm (sky blue emission of [H(2)]+) in NetState-II (Fig. 3a and b) in a clean ratiometric manner that allowed monitoring of the amount of liberated protons. Alternating switching between the NetStates was followed by emission spectroscopy over three cycles with a small decline in emission intensity (Fig. 3c and d), which may be due to the progressive accumulation of AgI.
:1:1 ratio in CD2Cl2 which furnished NetState-I = [H(1)]+ + 2. 1H NMR shifts of the 4/7-H proton signals to 8.43 and 8.69 ppm and of 9-H and e-H proton peaks to 6.60 and 8.48 ppm confirmed formation of [H(1)]+. Moreover, proton signals of c′-H and d′-H at 7.07 and 7.92 ppm corroborated presence of the free luminophore 2 (Fig. 2). Addition of one equiv. of AgBF4 to NetState-I instantaneously generated NetState-II = [Ag(1)]+ + [H(2)]+. In the 1H NMR of NetState-II, the 9-H and e-H proton signals shifted to 6.55 and 8.23 ppm, respectively, which substantiated the formation of [Ag(1)]+ whereas the shift of proton peaks c′-H and d′-H to 7.12 and 8.00 ppm, respectively, established the protonated luminophore [H(2)]+ (Fig. 2). Upon excitation at 300 nm, NetState-I exhibited emissions at λ = 346, 372 and 393 nm which represent an overlap of the emission peaks of the free luminophore 2 at λ = 346 and 372 nm and of [H(1)]+ at λ = 376 and 393 nm (Fig. 3c). In NetState-II, the fluorescence spectrum showed a single emission at λ = 461 nm, which is attributed to [H(2)]+ (Fig. 3). In contrast, [Ag(1)]+ in NetState-II is nonfluorescent as advocated by the full quenching of the emission of [H(1)]+ at λ = 376 and 393 nm upon titration with silver(I) ions (ESI, Fig. S18†). When one equiv. of tetra-n-butylammonium iodide (TBAI) was added to NetState-II, complete restoration of NetState-I was achieved as confirmed from 1H NMR data. TBAI acted as a scavenger for silver(I) by removing AgI through precipitation thus reversing the translocation sequence. Multiple switching of NetState-I ⇆ NetState-II was readily followed by 1H NMR (Fig. 2) and luminescence changes. Upon successive addition of AgBF4 to NetState-I, the emission changed to λ = 461 nm (sky blue emission of [H(2)]+) in NetState-II (Fig. 3a and b) in a clean ratiometric manner that allowed monitoring of the amount of liberated protons. Alternating switching between the NetStates was followed by emission spectroscopy over three cycles with a small decline in emission intensity (Fig. 3c and d), which may be due to the progressive accumulation of AgI.
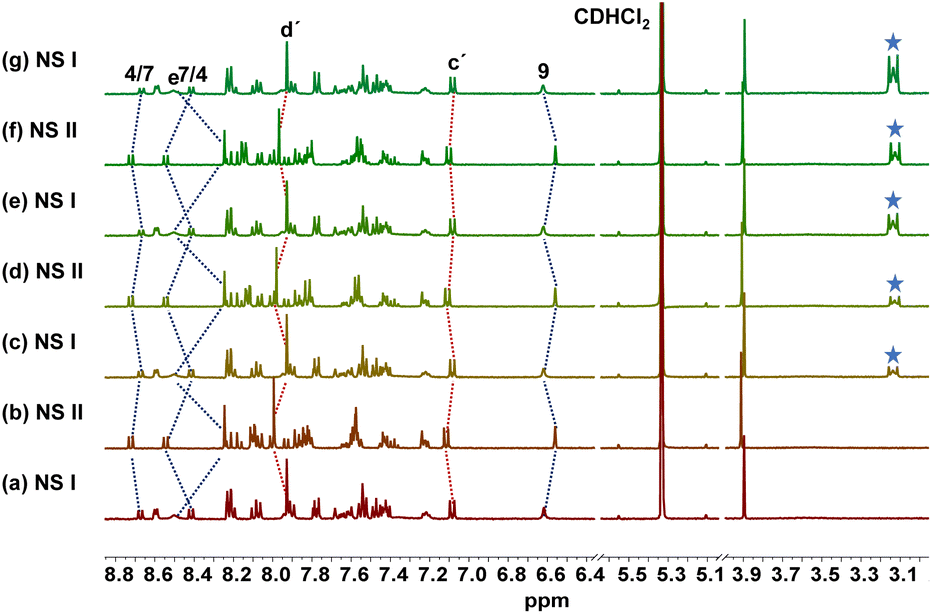 | ||
| Fig. 2 Partial 1H NMR spectra (500 MHz, CD2Cl2, 298 K) showing reliable switching between NetState-I and II over 3 cycles. (a) After mixing of TFA, 1, and 2 in 1:1:1 ratio; (b) after adding 1.0 equiv. of AgBF4 furnishing [Ag(1)]+ and [H(2)]+; (c) after addition of 1.0 equiv. of TBAI; (d) after adding 1.0 equiv. of AgBF4; (e) after addition of 1.0 equiv. of TBAI, (f) after adding 1.0 equiv. of AgBF4; (g) after addition of 1.0 equiv. of TBAI. Blue asterisk marked proton signals represent the tetra-butylammonium ion. | ||
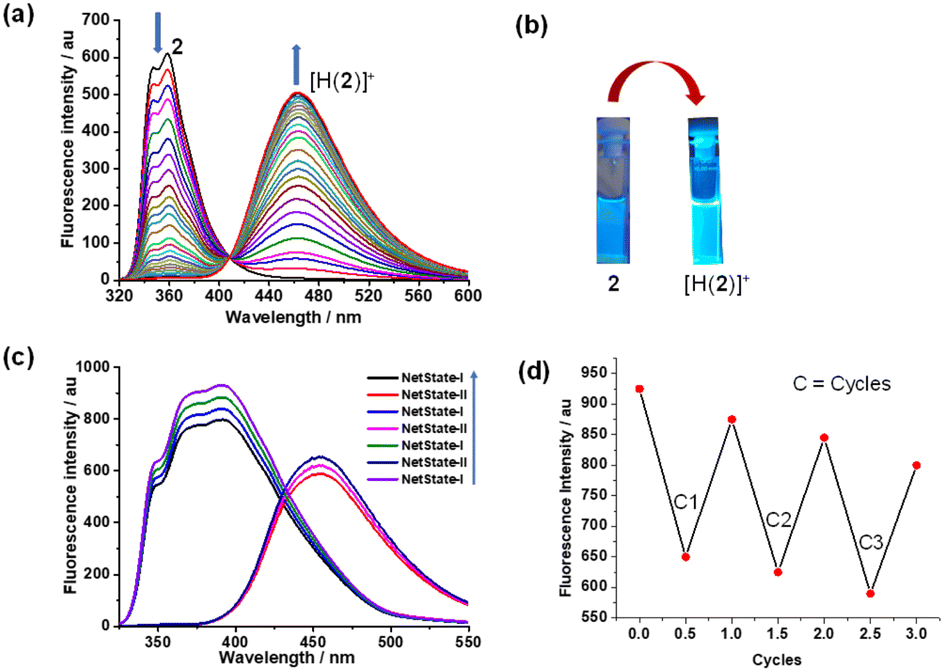 | ||
| Fig. 3 (a) Ratiometric emission titration of 2 (2.1 × 10−5 M) with TFA (5.3 × 10−3 M) in CH2Cl2. (b) Change of emission upon protonation of 2. (c) Reversibility of the network NetState-I ⇆ II over three cycles upon addition of silver(I) and TBAI (see text, blue arrow assigns direction of switching) monitored by luminescence changes at λ = 461 nm. (d) Multiple cycles monitored by the change of the fluorescence intensity. | ||
For probing the catalytic efficiency of each state towards a simple amine deprotection reaction we chose the trityl-protected aniline 6 (Fig. 4) performing two NetState-I ⇆ NetState-II cycles (Fig. 4b). We first prepared NetState-I by mixing 1, 2 and TFA (1:1:1) in CD2Cl2 and then added 6 and TMB (1,3,5-trimethoxybenzene as an internal standard) in a 10:10 ratio. After heating for 2 h at 40 °C, the 1H NMR spectrum revealed no product 7. Clearly, the proton in [H(1)]+ is catalytically inactive (ESI, Fig. S10b†). Addition of one equiv. of AgBF4 and probing the catalysis again for 2 h at 40 °C revealed formation of product 7 in (30 ± 2)% yield (ESI, Fig. S10c†). When the catalytic experiment was probed with only [H(2)]+ under identical reaction conditions, it provided the same yield (ESI, Fig. S16†). This suggested that the reversible proton relay network (Scheme 1) functioned reliably also in presence of 6 with complex [H(2)]+ as the catalytically active species. Addition of one equiv. of TBAI to the above NetState-II mixture and probing for catalysis, revealed no further product formation. Apparently, addition of TBAI translocated back the proton to nanoswitch 1 thus restoring NetState-I, which resulted in the OFF state for catalysis. Adding one equiv. of AgBF4 to the mixture and probing again the catalytic activity resulted in formation of further (29 ± 2)% of product 7, which was basically identical to the yield produced in the first cycle of NetState-II (ESI, Fig. S10e†). Adding TBAI and the consumed amount of substrate showed no further deprotection. In sum, the robust catalytic performance of the proton relay network as reflected in two successive catalytic cycles resulted in no significant decline of the catalytic activity (Fig. 4b).
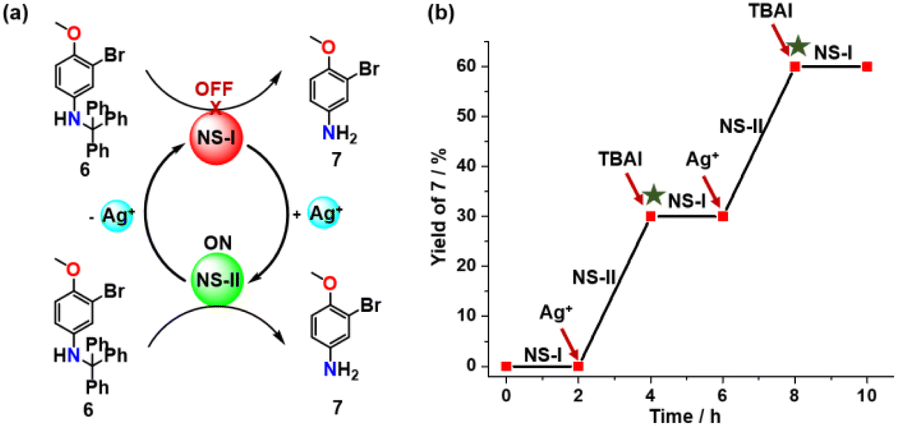 | ||
| Fig. 4 (a) Representation of the OFF/ON regulation of the trityl deprotection reaction in NetState-I and II. (b) Reversible switching between the network states furnished reproducible amounts of product 7 in NetState-II (two independent runs). Consumed amounts of substrates were added (green asterisk). | ||
The OFF–ON switching of the catalytic trityl deprotection of aniline 6 by the proton relay network inspired us to utilize the system to catalytically fabricate a slider-on-deck based on the Nan (= aniline) → ZnPor interaction. Deprotection of the protected biped 4 should afford the bis-aniline biped 5 and enable its attachment to the tris-zinc porphyrin deck 3 thus forming the slider-on-deck [(3)(5)].
Addition of one equiv. of biped 5 (for synthesis and characterization, see ESI†) to deck 3 (ref. 25) in CD2Cl2 quantitatively generated the slider-on-deck [(3)(5)], which was unambiguously characterized by 1H NMR, 1H DOSY NMR and elemental analysis (ESI, Fig. S5 and S6†). In the 1H NMR-spectrum, several diagnostic shifts of biped and deck proton signals attested the binding of biped 5 to the deck. Formation of the slider-on-deck was further confirmed from a single set in the 1H DOSY in CD2Cl2 (ESI, Fig. S9†).
The single set of protons (p-H, r-H, s-H, t-H, u-H, v-H) in the 1H NMR spectrum for all three-zinc porphyrin (ZnPor) stations of deck 3 unmistakably suggested rapid intrasupramolecular exchange29a of the aniline biped across all three ZnPor sites requiring fast Nan → ZnPor bond cleavage. At low temperature (−65 °C) the ZnPor p-H proton signals separated into two sets (1:2) (ESI, Fig. S7 and S8†). Kinetic analysis over a wide temperature range provided the exchange frequency (k) with k298 = 540 kHz at 298 K (Fig. 5a) and the activation data as ΔH‡ = 41.5 ± 0.7 kJ mol−1, ΔS‡ = 2.8 ± 0.7 J mol−1 K−1 and ΔG‡298 = 40.7 kJ mol−1 (Fig. 5).
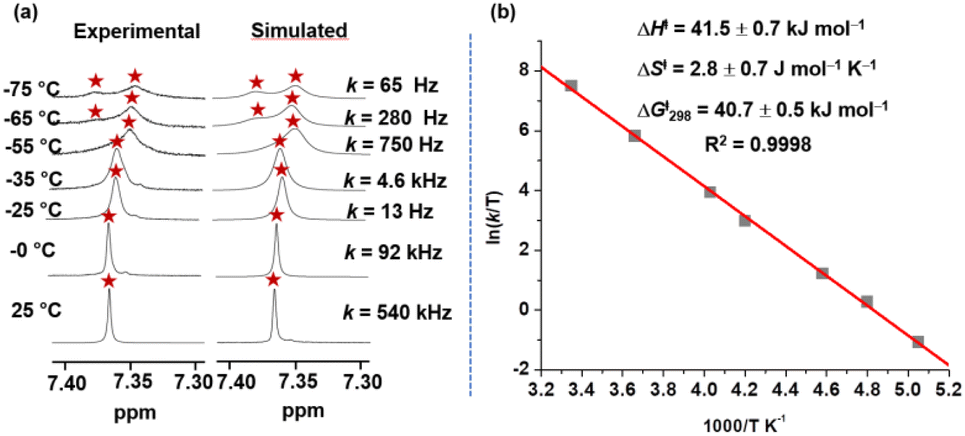 | ||
| Fig. 5 (a) Experimental and theoretical splitting of p-H proton signal of nanoslider [(3)(5)] in VT 1H-NMR (600 MHz) furnishing rate data in CD2Cl2. (b) Eyring plot providing kinetic parameters. | ||
After determining the kinetic parameters of the slider-on-deck [(3)(5)], we investigated its in situ catalyzed formation by the proton relay network. Therefore, we mixed nanoswitch 1, luminophore 2, TFA, trityl-protected biped 4, deck 3 and TMB in 1:1:1:5:5:5 ratio in CD2Cl2 and heated the reaction mixture for 12 h at 40 °C. The 1H NMR spectrum suggested no formation of any slider-on-deck in NetState-I (ESI, Fig. S13†). Addition of one equiv. of AgBF4 to the same mixture (converting NetState-I to NetState-II) and heating it at 40 °C for 12 h revealed full formation of the slider-on-deck as indicated by the disappearance of the 2′-H, 3′-H and 4′-H proton signals of biped 4 at 6.60, 6.31 and 6.51 ppm (ESI, Fig. S11†), respectively, and the quantitative emergence of 4′-H proton signal of biped 5 bound to deck 3 at 5.84 ppm (ESI, Fig. S12†). The upfield shift of the deck's meso t-H protons from 10.36 to 10.29 ppm allowed monitoring of the formation of [(3)(5)], e.g. by the gradually increased chemical shift difference of proton signal t-H (Δδt-H) (Fig. 6a; ESI, Fig. S11†). In sum, the protonated [H(2)]+ in NetState-II catalyzed the deprotection of the trityl-protected biped 4 with the effect that the resultant bis-aniline biped 5 would quantitatively bind to deck 3 affording the slider-on-deck [(3)(5)]. Furthermore, the OFF/ON switching of catalysis was probed for shortened time periods of 3 h. As illustrated in Fig. 6b, the process could be readily followed using the proton signal t-H (Δδt-H) (ESI, Fig. S14†) and the growth of proton signal 4′-H at 5.84 ppm of bound biped 5 (ESI, Fig. S15†). One clearly sees recurring OFF/ON regulation in NetState-I/II.
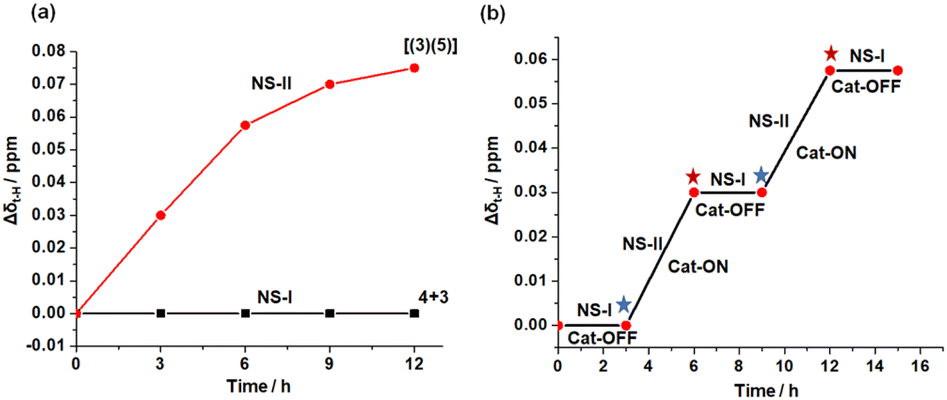 | ||
| Fig. 6 Plot of the shift difference of proton signal t-H (Δδt-H) vs. time for (a) the formation of [(3)(5)] from 3 and 4. Red curve: formation of [(3)(5)] over 12 h catalyzed by NetState-II. Black curve: OFF state of catalysis in NetState-I. (b) OFF/ON catalytic cycles by switching between NetState-I and NetState-II. Addition of Ag+, see blue asterisk; addition of TBAI: red asterisk. | ||
In conclusion, we have designed a supramolecular multicomponent network36 that acts as an OFF/ON proton relay with a luminescence display37 and is useful for switchable catalysis.38 The network is toggled by chemical input and intercomponent communication15,16 and as a result is a conceptual complement to photoacids driving networks.7,8
The release and capture of protons is demonstrated by the ON/OFF trityl deprotection of anilines. To demonstrate functioning in a complex setting, the network was utilized to catalyze formation of a high-speed slider-on-deck assembly based on Nan → ZnPor interactions (sliding frequency of k298 = 540 kHz). The robust operation of the proton relay furnishing dynamic machinery39 through acid catalysis followed by self-assembly is a valuable step mimicking sophisticated biological proton relays.2
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are indebted to the Deutsche Forschungsgemeinschaft for continued support under Schm 647/22-1 (No. 491092614) and to the University of Siegen. We thank Dr Paululat (Siegen) for measuring the VT-1H NMR.Notes and references
- D. Ray, J. T. Foy, R. P. Hughes and I. Aprahamian, Nat. Chem., 2012, 4, 757–762 CrossRef CAS PubMed.
- T. R. Li, F. Huck, G. M. Piccini and K. Tiefenbacher, Nat. Chem., 2022, 14, 985–994 CrossRef PubMed.
- M. Schmittel and P. Howlader, Chem. Rec., 2021, 21, 523–554 CrossRef CAS PubMed.
- J. Andréasson and U. Pischel, Coord. Chem. Rev., 2021, 429, 213695 CrossRef.
- F. R. Ludlow and S. Otto, Chem. Soc. Rev., 2008, 37, 101–108 RSC.
- J. L. Dempsey, J. R. Winkler and H. B. Gray, Proton-coupled electron flow in protein redox machines, Chem. Rev., 2010, 110, 7024–7039 CrossRef CAS PubMed.
- S. Silvi, E. C. Constable, C. E. Housecroft, J. E. Beves, E. L. Dunphy, M. Tomasulo, F. M. Raymo and A. Credi, Chem.–Eur. J., 2009, 15, 178–185 CrossRef CAS PubMed.
- F. M. Raymo and S. Giordani, Org. Lett., 2001, 3, 3475–3478 CrossRef CAS PubMed.
- M. Kadarkaraisamy, G. Caple, A. R. Gorden, M. A. Squire and A. G. Sykes, Inorg. Chem., 2008, 47, 11644–11655 CrossRef CAS PubMed.
- S. Yang, C.-X. Zhao, S. Crespi, X. Li, Q. Zhang, Z.-Y. Zhang, J. Mei, H. Tian and D.-H. Qu, Chem, 2021, 7, 1544–1556 CAS.
- J. A. Findlay and J. D. Crowley, Tetrahedron Lett., 2018, 59, 334–346 CrossRef CAS.
- A. W. Heard and S. M. Goldup, ACS Cent. Sci., 2020, 6, 117–128 CrossRef CAS PubMed.
- S. Krause and B. L. Feringa, Nat. Rev. Chem., 2020, 4, 550–562 CrossRef CAS.
- S. Amano, S. Borsley, D. A. Leigh and Z. Sun, Nat. Nanotechnol., 2021, 16, 1057–1067 CrossRef CAS PubMed.
- M. N. Tasbas, E. Sahin and S. Erbas-Cakmak, Coord. Chem. Rev., 2021, 443, 214039 CrossRef CAS.
- J. T. Foy, D. Ray and I. Aprahamian, Chem. Sci., 2015, 6, 209–213 RSC.
- S. Pramanik and I. Aprahamian, J. Am. Chem. Soc., 2017, 138, 15142–15145 CrossRef PubMed.
- Q. Wang, B. Lin, M. Chen, C. Zhao, H. Tian and D.-H. Qu, Nat. Commun., 2022, 13, 4185 CrossRef CAS PubMed.
- W. M. Bloch and G. H. Clever, Chem. Commun., 2017, 53, 8506–8516 RSC.
- S. Gaikwad, M. S. Özer, S. Pramanik and M. Schmittel, Org. Biomol. Chem., 2019, 17, 7956–7963 RSC.
- J.-F. Ayme, S. Dhers and J.-M. Lehn, Angew. Chem., Int. Ed., 2020, 59, 12484–12492 CrossRef CAS PubMed.
- P. Remon and U. Pischel, ChemPhysChem, 2017, 18, 1667–1677 CrossRef CAS PubMed.
- P. Remón, D. González, A. M. Romero, N. Basílio and U. Pischel, Chem. Commun., 2020, 56, 3737–3740 RSC.
- S. M. Wales, D. T. J. Morris and J. Clayden, J. Am. Chem. Soc., 2022, 144, 2841–2846 CrossRef CAS PubMed.
- S. Kundu, A. Ghosh, I. Paul and M. Schmittel, J. Am. Chem. Soc., 2022, 144, 13039–13043 CrossRef CAS PubMed.
- S. De, S. Pramanik and M. Schmittel, Angew. Chem., Int. Ed., 2014, 53, 14255–14259 CrossRef CAS PubMed.
- N. Mittal, S. Pramanik, I. Paul, S. De and M. Schmittel, J. Am. Chem. Soc., 2017, 139, 4270–4273 CrossRef CAS PubMed.
- A. Goswami, T. Paululat and M. Schmittel, J. Am. Chem. Soc., 2019, 141, 15656–15663 CrossRef CAS PubMed.
- (a) I. Paul, A. Goswami, N. Mittal and M. Schmittel, Angew. Chem., Int. Ed., 2018, 57, 354–358 CrossRef CAS PubMed; (b) D. Mondal, A. Ghosh, I. Paul and M. Schmittel, Org. Lett., 2022, 24, 69–73 CrossRef CAS PubMed.
- S. Saha, P. K. Biswas, I. Paul and M. Schmittel, Chem. Commun., 2019, 55, 14733–14736 RSC.
- M. Tanasova, Q. Yang, C. C. Olmsted, C. Vasileiou, X. Li, M. Anyika and B. Borhan, Eur. J. Org. Chem., 2009, 25, 4242–4253 CrossRef.
- A. Goswami, S. Saha, E. Elramadi, A. Ghosh and M. Schmittel, J. Am. Chem. Soc., 2021, 143, 14926–14935 CrossRef CAS PubMed.
- Z.-H. Ren, Z.-Y. Zhang, B.-Q. Yang, Y.-Y. Wang and Z.-H. Guan, Org. Lett., 2011, 13, 5394–5397 CrossRef CAS PubMed.
- S. Kundu, D. Mondal, V. V. Rajasekaran, A. Goswami and M. Schmittel, Inorg. Chem., 2022, 61, 17007–17011 CrossRef CAS PubMed.
- Y. Liu, M. Han, H.-Y. Zhang, L.-X. Yang and W. Jiang, Org. Lett., 2008, 10, 2873–2876 CrossRef CAS PubMed.
- G. Olivo, G. Capocasa, D. Del Giudice, O. Lanzalunga and S. Di Stefano, Chem. Soc. Rev., 2021, 50, 7681–7724 RSC.
- E. Spatola, F. Rispoli, D. Del Giudice, R. Cacciapaglia, A. Casnati, L. Marchiò, L. Baldini and S. Di Stefano, Org. Biomol. Chem., 2022, 20, 132–138 RSC.
- (a) L. van Dijk, M. J. Tilby, R. Szpera, O. A. Smith, H. A. P. Bunce and S. P. Fletcher, Nat. Rev. Chem., 2018, 2, 117 CrossRef CAS; (b) R. Parella, S. Jakkampudi, P. Bora, N. Sakkani and J. C.-G. Zhao, Org. Biomol. Chem., 2021, 20, 163–172 RSC.
- H. Inami, Y. Inagaki and W. Setaka, Org. Biomol. Chem., 2022, 20, 7092–7098 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, compound characterizations, spectral data, UV-vis titrations and computational data. See DOI: https://doi.org/10.1039/d3ra00062a |
| ‡ Sohom Kundu and Isa Valiyev contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |