
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceManipulating the formation of cesium lead bromide nanocrystals via oleic acid†
Miao Wanga,
Qiyu Yu *bc,
Tian Yua,
Sijie Zhang*ad,
Min Gonga and
Yuehui Liue
*bc,
Tian Yua,
Sijie Zhang*ad,
Min Gonga and
Yuehui Liue
aCollege of Physics, Sichuan University, Chengdu, 610065, China
bCollege of Material Science and Engineering, Sichuan University of Science and Engineering, Zigong, 643000, China. E-mail: yuqiyu2008@163.com
cKey Laboratory of Material Corrosion and Protection of Sichuan Province, Zigong, 643000, China
dSchool of Science, Guizhou University of Engineering Science, Guizhou 551700, China. E-mail: sijie.zhang@scu.edu.cn
eEngineering Research Center in Biomaterials, Sichuan University, Chengdu, 610065, China
First published on 9th February 2023
Abstract
To realize the precise modulation among the cesium lead halide perovskite-related phases is one of the most fascinating subjects and has motivated increasing research. The formation mechanisms of different phases of cesium lead halide have not been fully recognized. In this work, we reported the phase-selective synthesis of CsPbBr3 nanorods and Cs4PbBr6 nanocrystals (NCs) in amine-free systems, simply by adjusting the dosage of oleic acid (OA). By utilizing UV-visible absorption spectroscopy, we probed the evolution of the NCs and some lead bromide complex species during syntheses in reaction systems with different OA dosages. An OA-limited condition facilitated the formation of [PbBr4]2− and Cs4PbBr6 NCs. OA-rich conditions facilitated the formation of [PbBr3]−, [Pb2Br5]−, etc. at early stages and the subsequent generation of CsPbBr3 NCs. As the reaction systems equilibrated at a later stage, as-prepared CsPbBr3 or Cs4PbBr6 NCs would coexist with certain lead bromide complexes. OA dosage also greatly affected the kinetics of reactions toward CsPbBr3 NCs. A relatively large amount of OA would accelerate the formation of CsPbBr3 NCs. Our experimental results support two-step formation pathways of the cesium lead bromide NCs going through lead bromide complexes, and suggest that OA exerted all the influence by virtue of the lead bromide complexes. Our study presents a relatively clear picture of the formation of CsPbBr3 and Cs4PbBr6 NCs, which should be helpful in improving the preparation of lead halide perovskite-related NCs.
1. Introduction
All-inorganic cesium lead halide perovskite nanocrystals (NCs) have emerged as attractive materials for potential applications in photovoltaics, optoelectronics, etc., and for the fundamental science underlying synthesis–structure–property relationships.1–6 The synthesis of cesium lead halide perovskite NCs has been intensively investigated. To date, lead halide perovskite nanocrystals can be prepared with controlled size, uniform size distribution, and controllable surface.1,2,7,8 However, the synthesis technique is still far from mature, and many problems remained to be solved. This is especially true when compared with traditional chalcogenide-based II–VI and IV–VI semiconductor nanocrystals.9,10 The reasons for that may include but are not limited to the following. First, the perovskite-related NCs suffer from low stability against polar solvents, heat treatment, and ion–exchange reactions, which makes the manipulation of the NCs inconvenient. Second, the perovskite-related crystals grow rather fast and are difficult to control in the nanoscale regime. The fast growth also makes the nucleation and growth processes hardly accessible, and therefore, corresponding mechanistic study on the formation pathway, e.g. by probing the evolution of optical spectra, is rather challenging. Third, the phase control of the perovskites during synthesis is much more difficult than traditional semiconductor NCs. Based on the [PbBr6]4− octahedra units, the Cs–Pb–Br perovskites can exist in the forms of CsPbBr3 and Cs4PbBr6, depending on the different connecting modes of the units. Besides, there also exists the tetragonal phase of CsPb2Br5 with unusual [PbBr8]6− units.11 These perovskite-related NCs can be generated under very similar conditions and are liable to mutual transformation.5,12,13 Particularly, CsPbBr3 as a stoichiometric impurity often exists in the products of Cs4PbBr6.14–16 Moreover, the ready incorporation of protonated amine molecules into the inorganic structure to form two-dimensional organic–inorganic hybrid perovskites makes the phase control of the NCs even more complex.17,18 The above issues are more or less related to the intrinsic ionic nature of the perovskites. Such structural diversity and flexibility is one of the most fascinating aspects of this kind of material, due to the structure–property relationship. For the designed synthesis of lead halide perovskite-related NCs, the pursuit of precise control over phase is highly desirable yet challenging.CsPbBr3 and PbBr2-depleted Cs4PbBr6 are two stable ternary compounds in the phase diagram of a mixed system of CsBr–PbBr2. The modulation between CsPbBr3 and Cs4PbBr6 has emerged as a topic of great interest.12,13,18–23 According to reported results, several factors can be utilized to control the phase of the products. (i) A relatively high temperature was favorable for the formation of CsPbBr3 over Cs4PbBr6.13,18 (ii) The mixing ratio of the precursors is critical for the phase control of the perovskites. Generally, a low Cs/Pb ratio was favorable for the synthesis of CsPbBr3, and a relatively high Cs/Pb ratio was favorable for the generation of Cs4PbBr6.12,21 (iii) Most syntheses of perovskite NCs involves the use of binary ligand systems of aliphatic carboxylic acids and primary amines with various chain length.1,18,24,25 The acid–base equilibrium between the carboxylic acid and amine was found to have a great influence on the phase preference of perovskite products.18 Roughly, a relatively lower amine/acid ratio was favorable for the synthesis of CsPbBr3,21,22 while a high concentration of amine or a high amine/acid ratio will facilitate the generation of Cs4PbBr6 or the transformation of CsPbBr3 to Cs4PbBr6.13,22 Noteworthily, the relative concentration of the ligand system also have an effect.18,20 Despite these achievements, the underlying mechanisms of how the two phases form and by which these factors take effect are largely unknown. Take the role of acid–base equilibria for example. The amine-mediated phase transformation from CsPbBr3 to Cs4PbBr6 can usually be well explained by extracting PbBr2 from CsPbBr3, which shifts the 4CsPbBr3 ↔ Cs4PbBr6 + 3PbBr2 equilibrium.18,22,23 According to a recent report, however, a prerequisite for this effect was believed to be the protonation of the amine in moisture or a protic environment.26 Obviously, mechanistic study on the formation pathways of the two phases of cesium lead bromide is highly desirable.
In this work, we tried to investigate the formation pathways of CsPbBr3 and Cs4PbBr6 NCs. We realized the phase-selective synthesis by adjusting the dosage of oleic acid (OA). Based on the mechanistic study, we elucidated how OA takes effect during the formation of the cesium lead bromide NCs. In previous reports, the ligand effects on phase preference of cesium lead bromide were documented with an emphasis on the function of primary amines, while the underlying roles of fatty acids, such as OA, were not fully recognized. The reported roles of fatty acids during synthesis were mainly restricted to the acid–amine synergistic effect, and it includes: (i) solubilization of PbBr2 in a nonpolar solvent,18,20 (ii) controlling the size, shape, and phase of the NCs,18,24 and (iii) stabilization of the NCs.7,27,28 Herein, to avoid the interference of primary amines, amine-free reaction systems were adopted.21,29–31 Further, for the convenience of mechanistic study, we utilized a heating-up method and used 1-octadecene (ODE) as a noncoordinating solvent, since the NC growth in the frequently used hot-injection1 or reprecipitation synthesis25 was too fast to be accessible. Our experimental results revealed distinct two-step formation pathways going through lead bromide complexes for CsPbBr3 and Cs4PbBr6 NCs. We established a plausible relationship between the nature of lead bromide complexes and the phase of cesium lead bromide NCs, and presented that OA exerted the influence by controlling the lead bromide complexes.
2. Experimental
2.1. Materials
Commercial chemicals, including lead oxide (99.0%, Chengdu Kelong Chemical Reagent Factory), cesium carbonate (99.9%, Alfa Aesar), dimethyldioctadecylammonium bromide (DDOABr, ≥98%, Aldrich), oleic acid (OA, 90%, Aldrich), 1-octadecene (ODE, Aldrich) and hexanoic acid (99.5%, Aldrich), toluene (99.5%), cyclohexane (99.5%) and CDCl3 (99.8%, Adamas) were used as received without further purification.2.2. Preparation of Cs/Pb precursor solution
A mixture of PbO (0.022 g, 0.1 mmol), Cs2CO3 (0.008 g, 0.025 mmol), ODE (18.4 g), and a certain amount of OA, was heated to 150 °C and maintained for 20 minutes to give a clear Cs/Pb precursor solution. The solution was allowed to cool to room temperature.2.3. Synthesis of CsPbBr3 and Cs4PbBr6 NCs
CsPbBr3 and Cs4PbBr6 NCs were prepared via a heating-up method. Into the Cs/Pb precursor solution was added DDOABr (0.0946 g, 0.15 mmol). The mixture was then heated up to the desired temperature (about 10 °C min−1) in an oil bath for NC growth. For the typical synthesis of CsPbBr3 NCs, 2 mL of OA was used and the reaction system was heated up to 130 °C. For the typical synthesis of Cs4PbBr6 NCs, 0.1 mL of OA was used and the reaction system was heated up to 120 °C. After being heated to the desired temperature, the reaction vessel was removed from the oil bath. As-prepared NCs were precipitated from the reaction systems by centrifugation at 6000 rpm for 5 min, and washed twice with toluene. For detailed investigation, other medium amounts of OA were also used and all the reaction systems were heated up to 150 °C. Serial aliquots were taken from the reaction system during the heating-up process for optical measurement.2.4. Formation of lead bromide complexes
A series of control experiments without cesium were carried out. Only PbO was used to prepare the Pb precursor solution. Under otherwise the same conditions as above, the experiments were carried out with OA = 0.1, 0.2, 1, and 2 mL, respectively. The formation of the lead bromide complexes in these systems was also examined by UV-vis absorption spectroscopy.2.5. Cs4PbBr6 to CsPbBr3 transformation
First, a reaction system for the synthesis of Cs4PbBr6 NCs (0.1 mL of OA involved) was heated up to 120 °C to generate Cs4PbBr6 NCs. Then, at this temperature, 1.9 mL of OA was injected into the reaction mixture to induce the transformation to CsPbBr3 NCs. To follow the process, serial aliquots were taken from the reaction system for optical measurement.2.6. Formation of CsPbBr3 NCs from lead bromide complexes
First, a reaction system with the OA amount of 0.2 mL was heated to 90 °C to generate lead bromide complexes. Then, at this temperature, 1.8 mL of OA was injected into the reaction mixture to induce the formation of CsPbBr3 NCs. Other acids such as hexanoic acid were also tried to induce the formation of CsPbBr3 NCs.2.7. Observation of the degradation of cesium lead bromide NCs
2.8. Formation of [PbBr3]− from [PbBr4]2−
First, a reaction system without cesium and with the OA amount of 0.1 mL was heated to 90 °C to generate lead bromide complexes. At this temperature, 1.9 mL of OA was injected into the reaction mixture to induce the transition of the lead bromide complexes. Then the reaction system was kept at this temperature for 30 min. For comparison, another identical reaction system with the OA amount of 0.1 mL was heated to 90 °C and maintained for 30 min. Aliquots were taken from the two reaction systems for optical measurement.2.9. Characterization
Ultraviolet and visible (UV-vis) absorption spectra of the samples were collected using a UH4150 spectrophotometer (HITACHI). Photoluminescence data were collected using a fluorescence spectrophotometer F-380 (Tianjin Gangdong) with an excitation wavelength of 350 nm. For the absorption and photoluminescence measurement, all the samples were diluted in toluene unless otherwise specified. Transmission electron microscope (TEM) and high-resolution TEM images were taken with Tecnai G2 F20 S-TWIN. Scanning electron microscopy (SEM) images were taken on an FEI Quanta 600 field-emission microscope. X-ray diffraction patterns (XRD) of the samples were recorded on EMPYREAN (with Cu Kα) between 10 and 60°. Nuclear magnetic resonance (NMR) measurements were conducted on a Bruker Avance II 600 MHz spectrometer.3. Results and discussion
3.1. Synthesis and characterization of CsPbBr3 and Cs4PbBr6 NCs
Our synthesis of the CsPbBr3 and Cs4PbBr6 NCs was adjusted from previously reported amine-free methods.21,29–31 We adopted a heating-up technique and all experimental procedures were conducted in the open air. Typically, 22 mg of PbO (0.1 mmol), 8 mg of Cs2CO3 (0.025 mmol), 18.4 g of ODE, and a certain amount of OA were mixed and heated to form a clear Cs/Pb precursor solution. Then, the precursor solution was mixed with a quaternary ammonium salt of dimethyldioctadecylammonium bromide (DDOABr) at room temperature. Then the mixture was heated up to the desired temperature for the synthesis of the cesium lead bromide NCs. Typically, OA amounts of 2 and 0.1 mL were used for the synthesis of CsPbBr3 and Cs4PbBr6 NCs, respectively. Corresponding temperatures were 130 and 120 °C, respectively. In our amine-free reaction systems, the use of the quaternary alkylammonium salt as a bromide source eliminates possible amine generation via the deprotonation of alkylammonium.30The morphology, crystalline structures, and optical properties of as-prepared NCs were characterized by transmission electron microscopy (TEM), X-ray diffraction (XRD), UV-vis absorption, and photoluminescence (PL) spectra, as illustrated in Fig. 1. As-prepared CsPbBr3 NCs exhibit a rodlike shape, with diameters of a few to tens of nanometers and lengths up to about 100 nanometers (Fig. 1a and b, see also Fig. S1†). XRD analysis in Fig. 1c (see also Fig. S2†) shows that the CsPbBr3 NCs have an orthorhombic crystal structure (PDF #54-0752). The lattice spacing of 0.583 nm (indicated in Fig. 1b) agrees with the {101} facets of orthorhombic CsPbBr3. The anisotropic growth of the NCs may be attributed to the competition of Cs+ with DDOA+ at the surface of NCs during growth.18,32 In this research, the formation mechanism of the rodlike morphology will not be further discussed. The CsPbBr3 nanorods show absorption onset around 506 nm, and exhibit a symmetric sharp PL spectrum around 514 nm, with a full width at half-maximum of 17 nm (Fig. 1d). XRD analysis in Fig. 1c shows that the Cs4PbBr6 NCs have a hexagonal crystal structure (PDF #73-2478, see Fig. S3†). UV-vis absorption spectrum of the Cs4PbBr6 NCs exhibit a characteristic sharp peak at 313 nm (Fig. 1d). No obvious PL signal was detected for the Cs4PbBr6 NCs. TEM images of the Cs4PbBr6 NCs demonstrate that these NCs have an average size of about 40 nm (Fig. 1e). The high-resolution TEM image of Cs4PbBr6 NCs shows a lattice spacing of 0.686 nm, which agrees with the {110} facets of hexagonal Cs4PbBr6 NCs (Fig. 1f).
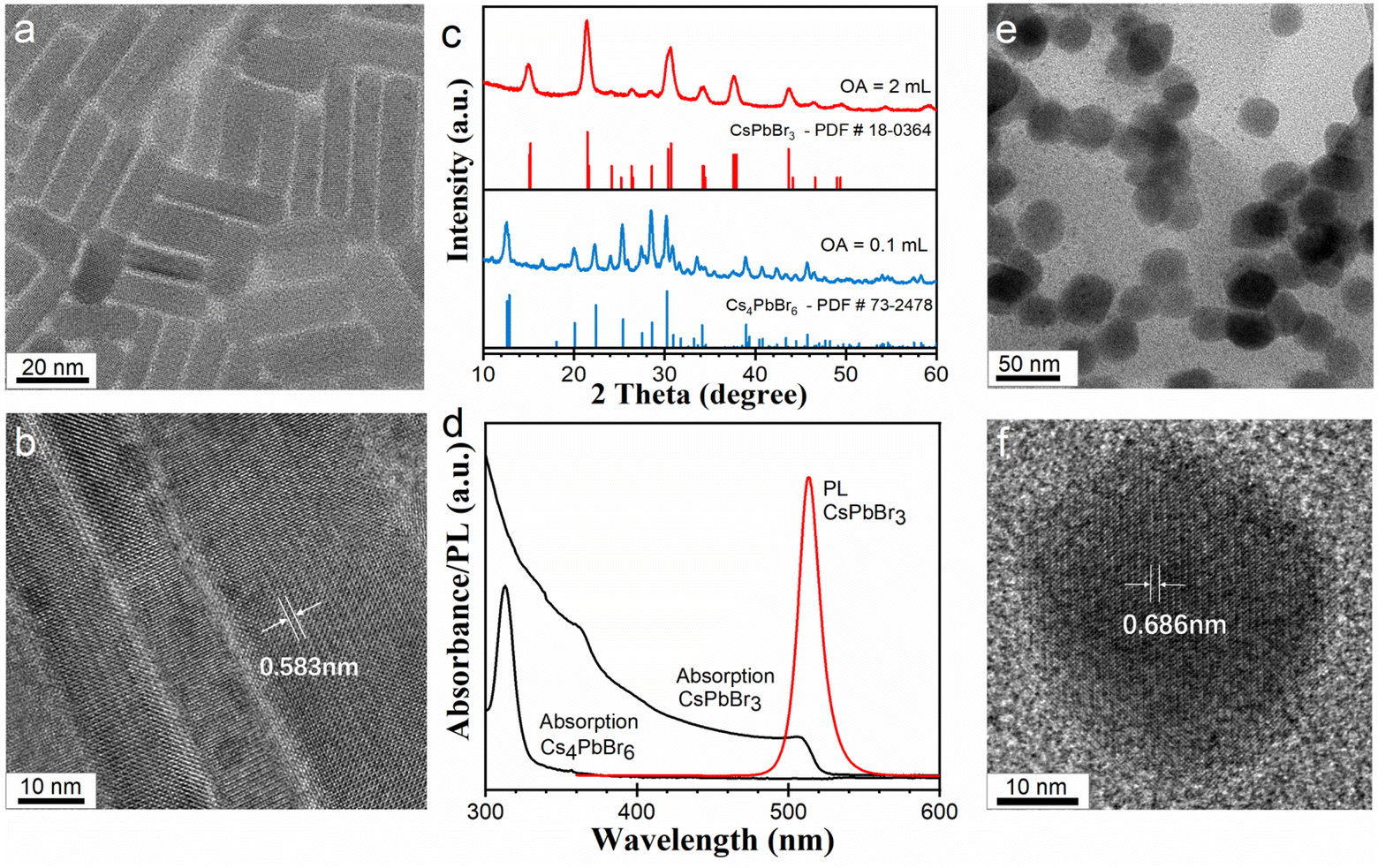 | ||
| Fig. 1 Typical CsPbBr3 and Cs4PbBr6 NCs prepared with 2 mL and 0.1 mL OA, respectively. (a and b) TEM and high-resolution TEM images of the rodlike CsPbBr3 NCs. (c) XRD pattern and (d) UV-vis absorption and PL spectra of the CsPbBr3 and Cs4PbBr6 NC dispersion in toluene. (e and f) Typical TEM and HRTEM images of the Cs4PbBr6 NCs. | ||
3.2. Syntheses with varying OA amounts
We carried out a series of syntheses with different OA amounts: 0.1, 0.2, 0.3, 0.5, 1, and 2 mL, respectively. Note that the OA amount of 0.1 mL is near the smallest amount needed to prepare a clear Cs/Pb precursor solution, and thus cannot be further reduced substantially. All the reaction systems were heated up to 150 °C. The evolution of these reaction systems was followed by UV-vis absorption spectroscopy, as illustrated in Fig. 2. Quite a few kinds of species can be observed in these absorption curves. The sharp absorption peak at 313 nm is characteristic of Cs4PbBr6 NCs (Fig. 2a),19 and the obvious absorption bands around 500 nm belong to regular CsPbBr3 NCs (Fig. 2b–f).1 The multiple absorption signals in between were somewhat complex. The absorption features at around 320 (Fig. 2b, c, and f) and 355 nm (Fig. 2b and c) belong to lead bromide complexes of [PbBr3]− and [PbBr4]2−, respectively.33 The absorption peak around 345 nm can be assigned to [Pb2Br5]− (Fig. 2c, d, and f).34 Other absorption signals including (but not limited to) those around 363 (Fig. 2b–f), 370 (Fig. 2c–e), 390 (Fig. 2d and e), and 400 nm (Fig. 2c) might also be assigned to lead bromide complexes. Another possibility was that these unidentified signals are arising from nanoclusters or nanoplates of cesium lead bromide. However, considering that these absorption signals are relatively wide and no related emission signals were observed, we estimate that they are more likely from the former. Compositional assignments of these absorption signals are lacking in the literature. As a matter of fact, the local signals around 363 and 400 nm were frequently observed in the reported absorption curves of cesium lead bromide samples,22,34,35 but received little attention. One might take it for granted that the absorption signals around 363 nm were arising from [PbBr4]2−. In this work, however, our experimental results suggest that this signal should be most likely from a distinct lead complex species. For convenience, these species are thereafter denoted as LB-363, LB-370, LB-390, and LB-400, respectively.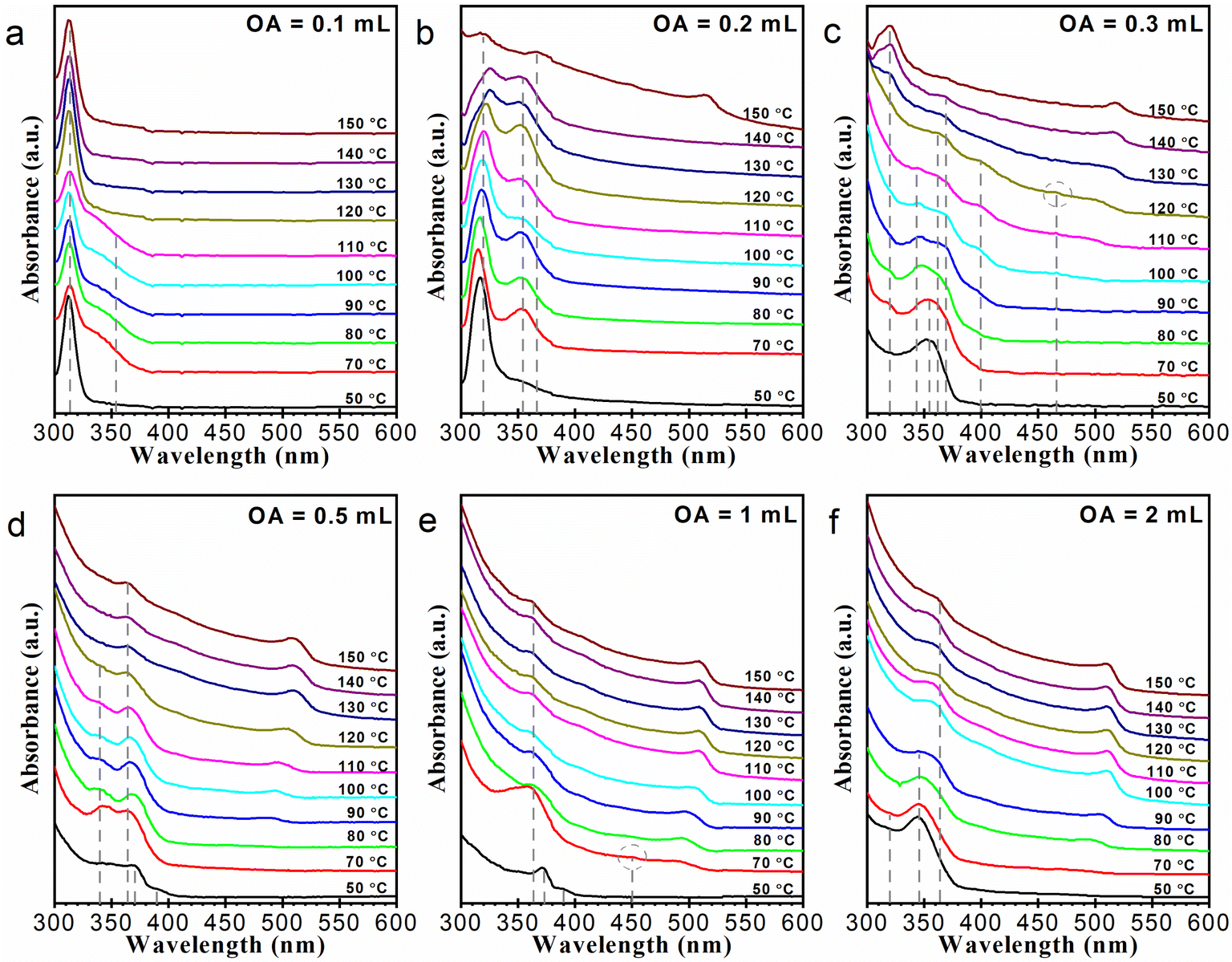 | ||
| Fig. 2 Evolution of the UV-vis absorption spectra during heating up the reaction systems with OA amounts of (a) 0.1, (b) 0.2, (c) 0.3, (d) 0.5, (e) 1 and (f) 2 mL, respectively. The absorption wavelengths of Cs4PbBr6 NCs (313 nm) and several lead bromide complex species (around 320, 345, 355, 363, 370, 390, and 400 nm, respectively) were indicated by vertical dashed lines. Two kinds of tiny peaks around 450 and 470 nm (possibly arising from quasi-2D CsPbBr3) were also indicated and were selectively circled. | ||
In the reaction system with a small OA amount of 0.1 mL (Fig. 2a), Cs4PbBr6 NCs (absorption at 313 nm) began to form below 50 °C. Then at 70 °C, an absorption tail around 355 nm appeared, while the absorption of Cs4PbBr6 NCs decreased. The evolution of the absorption spectrum between 50–120 °C appears to be a fluctuation between [PbBr4]2− and Cs4PbBr6 NCs. When the reaction system was heated up to 120 °C, the [PbBr4]2− species disappeared, and the absorption of Cs4PbBr6 NCs increased. The relative variation of the absorption of Cs4PbBr6 and [PbBr4]2− suggests the reversible conversion between the two species. Hui et al. demonstrated that Cs4PbBr6 NCs could be prepared by mixing cesium oleate and a precursor solution mainly containing [PbBr4]2−.31 Liu et al. observed an intermediate species of [PbBr4]2− in the duration of amine-induced transformation of CsPbBr3 into Cs4PbBr6.22 Our result was consistent with these reports and supports a [PbBr4]2− → Cs4PbBr6 formation pathway.
In the reaction system with 0.2 mL of OA (Fig. 2b), CsPbBr3 NCs instead could be produced. At the early stages of the heating-up process, there mainly exists [PbBr4]2− and [PbBr3]−. When the reaction system was heated up to about 150 °C, CsPbBr3 NCs were produced, accompanied by the appearance of LB-363, the disappearance of [PbBr4]2−, and the significant decrease of [PbBr3]−. In the reaction system with 0.3 mL of OA (Fig. 2c), CsPbBr3 NCs formed at about 100 °C. The gradual red shift of corresponding absorption bands indicates the slow growth of the CsPbBr3 NCs. At 50 °C, [PbBr4]2− and a tiny portion of [PbBr3]− formed initially, then [Pb2Br5]−, LB-370, and LB-400 were also generated. As temperature increased, the formation and growth of the CsPbBr3 NCs were mainly accompanied by the decrease of the complexes of [Pb2Br5]−, LB-370, and LB-400. In the range of 130–150 °C, the [PbBr3]− species gradually increased and coexisted with the CsPbBr3 NCs.
For the reaction systems with 0.5–2 mL of OA (Fig. 2d–f), the formation temperature of CsPbBr3 NCs further decreased. Also, the yield of as-prepared NCs obviously increased. The gradual red shift of the absorption band of CsPbBr3 NCs suggests that the NC growth proceeded in a well-controlled way. In the reaction system with 0.5 mL of OA, CsPbBr3 NCs formed at about 90 °C and the growth of the NCs was accompanied by the decrease of [Pb2Br5]− and LB-363. In the reaction system with 1 mL of OA (Fig. 2e), LB-370 and LB-390 initially formed, and then the two species were replaced by LB-363 and CsPbBr3 NCs at about 70 °C. Further growth of the NCs was accompanied by the decrease of LB-363. In the reaction system with 2 mL of OA (Fig. 2f), [Pb2Br5]− formed at first, and then CsPbBr3 NCs were generated also at about 70 °C. The growth of the NCs was accompanied by the decrease of [Pb2Br5]−. During the NC growing process, LB-365 species were generated. In the reaction systems with OA amounts of 0.5, 1, and 2 mL, the growth of CsPbBr3 NCs stopped at about 130, 110, and 100 °C, respectively. In the later stages of the three syntheses, the reaction systems would tend to equilibrate, and as-prepared CsPbBr3 NCs mainly coexisted with a portion of LB-363.
Taking a closer look at the absorption curves in Fig. 2, we can further find several tiny yet discernible peaks in the absorption curves. These peaks are rather weak and emerge only at persistent wavelengths on the blue side of the first absorption bands of the regular CsPbBr3 NCs. As indicated in Fig. 2c and e, the locations of these peaks are around 450 and 470 nm. These absorption signals may be assigned to quasi-2D CsPbBr3.20 For the highly fluorescent materials of lead halide perovskite NCs or traditional quantum dots, photoluminescence (PL) is generally more sensitive than absorption measurement. Indeed, the existence of these species is much more prominent in the PL curves. Corresponding PL signals persistent at about 450, 470 nm can also be identified. Note that the above-mentioned lead bromide complexes exhibit no PL signals. See Fig. S4† for PL spectra corresponding to Fig. 2. Besides, the PL spectra give relatively precise onset temperatures of the CsPbBr3 NCs. In reaction systems with 0.2 and 0.3 mL of OA, CsPbBr3 NCs emerged at about 100 and 80 °C, respectively. In reaction systems with 0.5, 1, and 2 mL of OA, CsPbBr3 NCs were already detected in the PL spectra of the first aliquot taken at 50 °C. Particularly, in the reaction system with 0.1 mL of OA, the formation of trace amounts of erratic CsPbBr3 NCs was detected in the samples of 130–150 °C.
As mentioned above, we realized the modulation of the perovskite phases between Cs4PbBr6 and CsPbBr3 by varying the dosage of OA. Cs4PbBr6 and CsPbBr3 NCs can be prepared under OA-limited and OA-rich conditions, respectively. In this regard, it appears that OA has an opposite control effect on the phase of CsPbBr3/Cs4PbBr6, as compared with that of primary amine.22 In our amine-free systems, the quaternary ammonia ligands cannot lose or acquire protons. Therefore, the possibility of phase control via acid–amine interaction can be eliminated. According to the literature, Cs/Pb ratio was generally utilized to control the phase of cesium lead halide NCs in amine-free systems.21,31 A high Cs/Pb ratio benefits the formation of Cs4PbBr6, while a low Cs/Pb ratio is favorable for the formation of CsPbBr3. Xu et al. reported that a Cs/Pb ratio < 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.5 (>2:1) gave a pure phase of CsPbBr3 (Cs4PbBr6).21 In this work, we adopted an unchanged Cs/Pb ratio of 1:2, which is supposed to yield a pure phase of CsPbBr3. Moreover, the generation of Cs4PbBr6 in OA-limited systems cannot be explained by considering the “effective” Cs/Pb ratio.21,36 Our experiment results demonstrate that OA dosage is also a key parameter influencing the phase preference of the cesium lead bromide NCs.
:1.5 (>2:1) gave a pure phase of CsPbBr3 (Cs4PbBr6).21 In this work, we adopted an unchanged Cs/Pb ratio of 1:2, which is supposed to yield a pure phase of CsPbBr3. Moreover, the generation of Cs4PbBr6 in OA-limited systems cannot be explained by considering the “effective” Cs/Pb ratio.21,36 Our experiment results demonstrate that OA dosage is also a key parameter influencing the phase preference of the cesium lead bromide NCs.
3.3. Effect of OA on the formation of lead bromide complexes
The chemistries of lead bromide complexes presented in Fig. 2 are complicated. The distribution of these species at the early stages of the heating-up process is labile, and usually could not be exactly reproduced in another synthesis. Despite this, it appears that the species are still closely correlated with the OA dosage of the reaction system. We performed blank experiments without a Cs source to investigate the effect of OA dosage on lead bromide complexes more directly, as illustrated in Fig. 3. Working with 0.1 mL of OA (Fig. 3a), the reaction system produced pure [PbBr4]2−, which greatly diminished when heated up to about 100 °C. This result is consistent with that in the presence of the Cs source (Fig. 2a). Working with 2 mL of OA (Fig. 3b), the reaction system generated lead bromide species with an absorption bump around 310–330 nm, which can probably be assigned to [PbBr3]−.34 In the presence of the Cs source, lead bromide complexes such as [Pb2Br5]3− and LB-363 were also detected in addition to [PbBr3]− (Fig. 2f). This variation may be caused by different chemical environments. Different from [PbBr4]2−, these [PbBr3]− species was relatively stable during the heating-up process. It is interesting to note that Cs4PbBr6 also exhibited little thermal stability against transformation into CsPbBr3 at high temperatures. Considering that Cs4PbBr6 and CsPbBr3 were generated in OA-limited and OA-rich systems, respectively, this result implies that the nature of lead bromide complexes and the phase preference of the cesium lead bromide NCs might be closely correlated.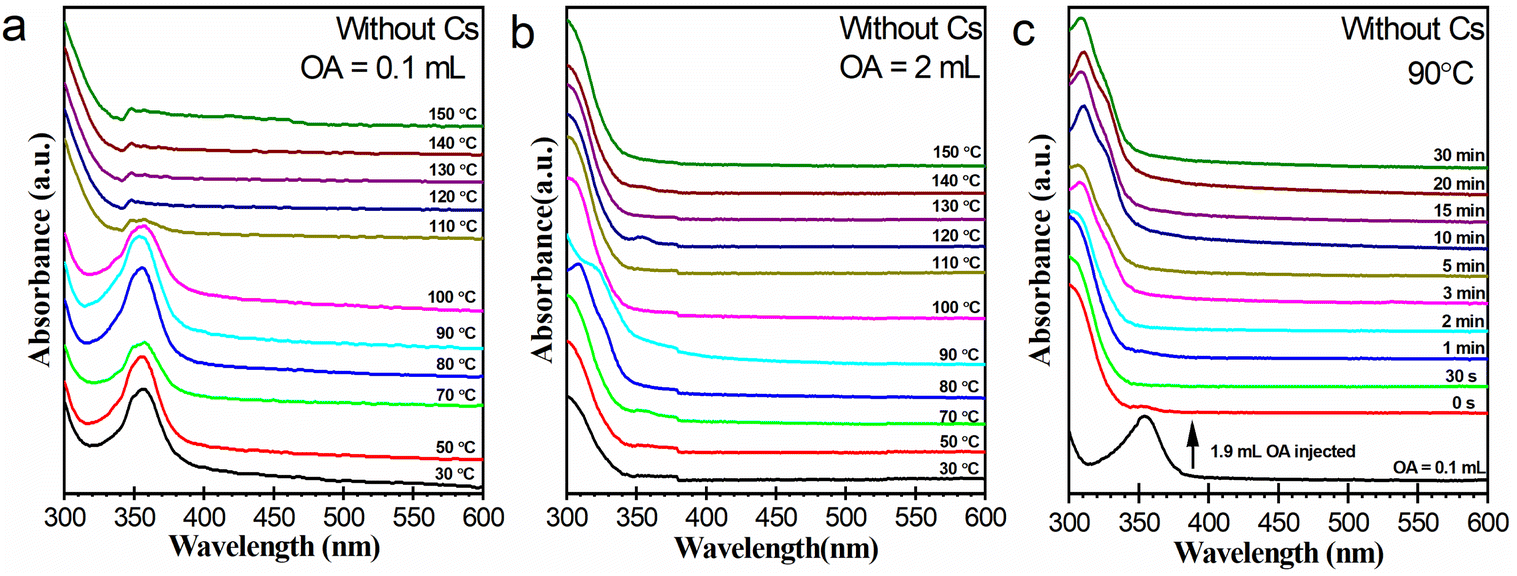 | ||
| Fig. 3 Evolution of the absorption spectra of reaction systems without Cs source (a and b) during the heating-up process and (c) at 90 °C. For the three reaction systems, the amounts of OA were (a) 0.1, (b) 2 mL, and (c) (0.1 + 1.9) mL (added in two batches), respectively. | ||
Another experiment was carried out to demonstrate the effect of OA concentration on the chemistry of lead bromide complexes. In this experiment, a total of 2 mL of OA was used but was added in two batches. At first, 0.1 mL of OA was used to prepare a reaction system containing [PbBr4]2−. Then into the system additional 1.9 mL was injected at 90 °C. As illustrated in Fig. 3c, most of the [PbBr4]2− species were immediately converted into [PbBr3]−. The conversion may take place via reaction (1). The possible generation of HBr from the interaction between lead bromide species and OA was also previously reported.20
| [PbBr4]2− + HOA → [PbBr3]− + OA− + HBr | (1) |
Considering the above protonation effect of OA, it is reasonable to postulate that lead bromide complexes with relatively low coordination states were readily generated in OA-rich reaction systems. Indeed, [PbBr4]2− prevails in reaction systems with OA amounts of 0.1, 0.2, and 0.3 mL (Fig. 2a–c), while [Pb2Br5]3− dominates the initial species in the reaction system with 2 mL of OA (Fig. 2f). The emergence of the species of [PbBr3]−, which possesses a medium coordination state, mainly took place in the reaction systems with OA amounts of 0.2 and 0.3 mL (Fig. 2b and c). Interestingly, these results coincide with the fact that CsPbBr3 possesses a lower Br/Pb stoichiometric ratio than that of Cs4PbBr6. Likely, the formation of CsPbBr3 (Cs4PbBr6) from lead bromide complexes with lower (higher) coordination states is energetically more favorable. Presumably, LB-363, LB-370, LB-390, and LB-400 might be assigned to some polynuclear lead bromide complexes with coordination states lower than [PbBr4]2−.37,38 Obviously, the chemistries of the lead bromide complexes were susceptible to the chemical environment. Likely, the complicated chemistries of the lead bromide complexes in the presence of a cesium source may be partly owing to the interaction of Br− with Cs+, which was reported to be important in the crystallization of Cs4PbBr6 and CsPbBr3.39
3.4. OA-induced CsPbBr3 formation from lead bromide complexes
The results presented in Fig. 2 suggest formation pathways of the NCs going through lead bromide complexes. In order to observe the formation of CsPbBr3 NCs from lead bromide complex species more intuitively, we designed a two-step experiment. At first, a reaction system with 0.2 mL of OA was heated up to 90 °C, to obtain a mixture of lead bromide complexes. As demonstrated in Fig. 4a, this mixture contains [PbBr3]−, [PbBr4]2−, and possibly a portion of LB-363. 1.8 mL of OA was added to the mixture swiftly. The solution turned yellow immediately, indicating the formation of CsPbBr3 NCs (see Video S1†). Besides, the absorption signals of LB-363 and LB-400 were also observed after OA addition. In the reaction system with 0.2 mL of OA, substantial formation of CsPbBr3 NCs occurred only when the temperature reached about 150 °C (Fig. 2b). Surely, the generation of the CsPbBr3 NCs in this experiment was induced by the replenishment of OA. This experimental result also suggests that OA dosage plays a more decisive role than temperature in influencing the kinetics of CsPbBr3 formation.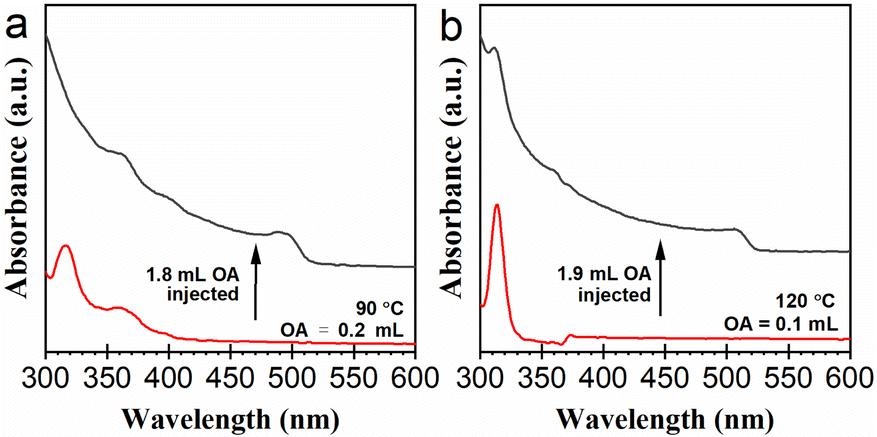 | ||
| Fig. 4 UV-vis absorption spectra illustrating the generation of CsPbBr3 NCs upon OA injection: (a) from lead bromide complex species made with 0.2 mL of OA, 1.8 mL of OA added; (b) from Cs4PbBr6 NCs made with 0.1 mL of OA, 1.9 mL of OA added. The spectra were collected before and immediately after the injection of OA. | ||
3.5. OA-induced Cs4PbBr6 to CsPbBr3 transformation
We further examined the feasibility of the postsynthetic transformation of Cs4PbBr6 to CsPbBr3 NCs by OA addition. A reaction system containing 0.1 mL of OA was heated up to 120 °C to obtain a colloidal system mainly containing Cs4PbBr6 NCs. Then 1.9 mL of OA was added swiftly. The solution turns yellow immediately, indicating the formation of CsPbBr3 NCs (see Video S2†). As demonstrated in Fig. 4b, the characteristic absorption of Cs4PbBr6 NCs at 311 nm fell off a cliff immediately, and another obvious absorption band at about 500 nm appeared. This OA-induced transformation of Cs4PbBr6 to CsPbBr3 NCs might occur through a dissolution–recrystallization process, intermediated by some lead bromide complex species.22,34 Interestingly, we found that the addition of OA into a purified Cs4PbBr6 dispersion could not induce such transformation. We estimate that the purification process removed the DDOA+ ligands in the solution, and therefore the dissolution of the Cs4PbBr6 NCs into DDOA+-ligated lead bromide complexes could not be triggered.263.6. Observation of the degradation of Cs4PbBr6 and CsPbBr3 NCs
The above postsynthetic transformation result and the fluctuation between Cs4PbBr6 and [PbBr4]2− in Fig. 2a imply that Cs4PbBr6 NCs can be degraded back into [PbBr4]2−. Also, a similar possibility might exist in CsPbBr3 NCs. Considering this, we checked the postsynthetic stability of the Cs4PbBr6 and CsPbBr3 NCs without being isolated from the original reaction systems. After synthesis, we treated three typical reaction systems containing Cs4PbBr6 or CsPbBr3 NCs by slowly reducing the temperature to 50 °C (see Experimental). Corresponding results are presented in Fig. 5. The reaction system with 0.3 mL of OA turned white after the temperature was lowered to 50 °C. Corresponding absorption spectra in Fig. 5a indicate the disappearance of the signal around 516 nm and the concomitant growth of the absorption at 320 nm after treatment. This result suggests that the CsPbBr3 NCs were partially degraded into [PbBr3]−. The reaction system with 2 mL of OA is relatively stable. However, after about 24 h treatment at 50 °C, we detected a decay of the 510 nm peak with the concomitant growth in the absorption around 363 nm (Fig. 5b), indicating the partial degradation of the CsPbBr3 NCs into LB-363. The Cs4PbBr6 NCs prepared with 0.1 mL of OA were also stable at low temperatures. However, interestingly, after diluting the sample in cyclohexane (instead of toluene), we detected a decay of the 313 nm peak with the concomitant emergence of an absorption signal around 355 nm, suggesting the partial degradation of the Cs4PbBr6 NCs into [PbBr4]2−. The substitution of cyclohexane for toluene has little effect on the CsPbBr3 samples prepared in the reaction systems with 0.3 and 2 mL of OA (Fig. S5†). Likely, the transitions demonstrated in Fig. 5 originated from shifts in the dynamic equilibria between the cesium lead bromide NCs and certain lead bromide complexes. These equilibria can be described as CsPbBr3 ↔ [PbBr3]− (Fig. 5a), CsPbBr3 ↔ LB-363 (Fig. 5b), and Cs4PbBr6 ↔ [PbBr4]2− (Fig. 5c), respectively.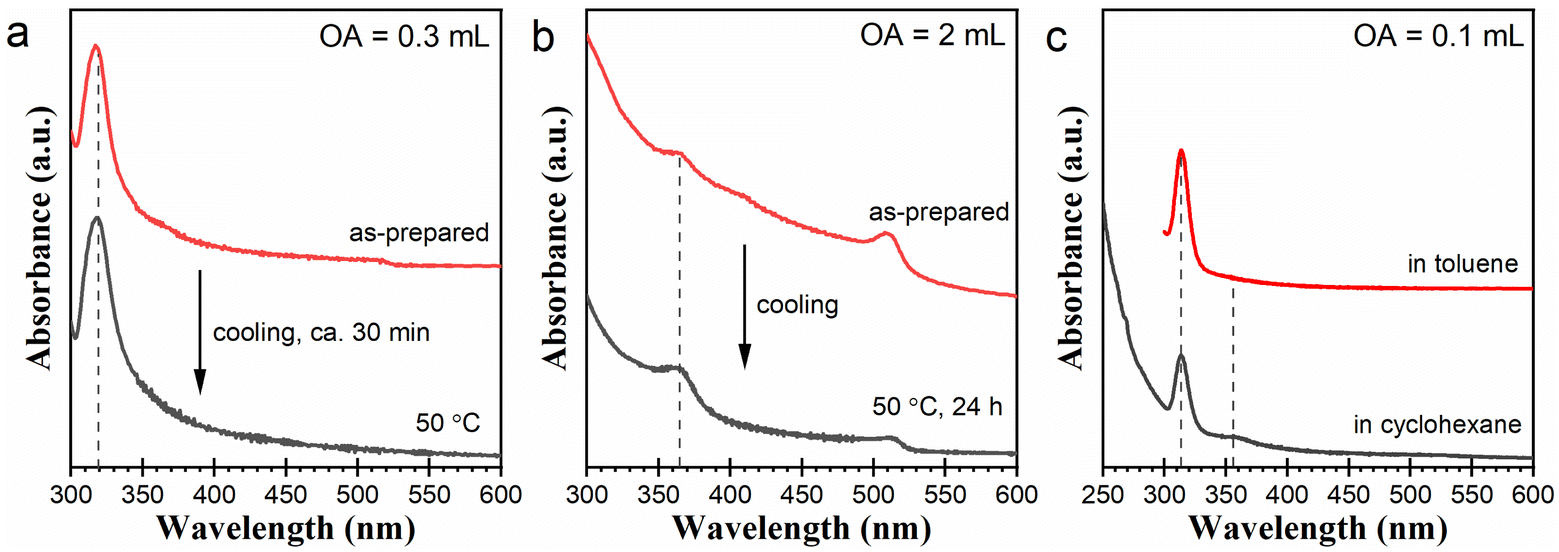 | ||
| Fig. 5 UV-vis absorption spectra illustrating the degradation of CsPbBr3 or Cs4PbBr6 NCs into lead bromide complexes: (a) from CsPbBr3 NCs to [PbBr3]2−; (b) from CsPbBr3 NCs to LB-363, and (c) from Cs4PbBr6 NCs to [PbBr4]2−. All the samples were diluted in toluene for measurement unless otherwise specified. | ||
Our explanation was also consistent with the results demonstrated in Fig. 2. Mutual conversions of Cs4PbBr6 NCs and [PbBr4]2− were observed in the reaction system with 0.1 mL of OA (Fig. 2a). In the reaction systems with 0.2 and 0.3 mL of OA, [PbBr3]− appeared or reappeared at later stages and coexisted with the CsPbBr3 NCs (Fig. 2b and c). As the reaction systems with 0.5, 1, and 2 mL of OA equilibrated at later stages, CsPbBr3 NCs coexisted with LB-363 (Fig. 2d–f). In a previous report about the transformation of CsPbBr3 into CsPb2Br5, Balakrishnan et al. recorded a spectral change exhibiting the decay of the CsPbBr3 NCs and the concurrent formation of [PbBr3]2− and another species obviously absorbing in 360–370 nm wavelength region (Fig. S6†).34 In their work, the [PbBr3]2− species was believed to account for the final generation of CsPb2Br5, while the other species was not discussed. The recorded species in 360–370 nm should be the same as LB-363 presented in this work. Our findings were consistent with their report.
3.7. Pathway-controlled formation of Cs4PbBr6 and CsPbBr3
Yoon et al. demonstrated that the complexation between Pb2+ and Br−/I− ions was the primary step in determining the final halide composition in CH3NH3PbBrxI3−x.33 In this work, we established reasonable correlations between the nature of lead bromide complexes and the phase preference of the cesium lead bromide NCs thereof. Based on the results presented above, we formulated the formation pathways of CsPbBr3 and Cs4PbBr6 NCs as simply depicted in Fig. 6. We proposed that lead bromide complexes acted as critical intermediates during the formation of perovskite-related cesium lead bromide NCs. During the heating-up synthesis of CsPbBr3 NCs, the formation at an early stage was kinetically controlled, and then at later stages, a thermodynamic equilibrium could be achieved. At early stages, CsPbBr3 NCs were favorably generated from [PbBr3]−, [Pb2Br5]− et al. As the reaction system equilibrated, CsPbBr3 NCs coexisted [PbBr3]− or LB-363, depending on the OA dosage. Alteration of the temperature or chemical environment would shift the equilibria. The formation of Cs4PbBr6 NCs was relatively fast and followed a similar pathway of [PbBr4]2−↔ Cs4PbBr6. The two formation pathways can be switched by adjusting OA dosage. Also, the Cs4PbBr6 NCs can be transformed into CsPbBr3 NCs upon OA addition, via lead bromide complex intermediates.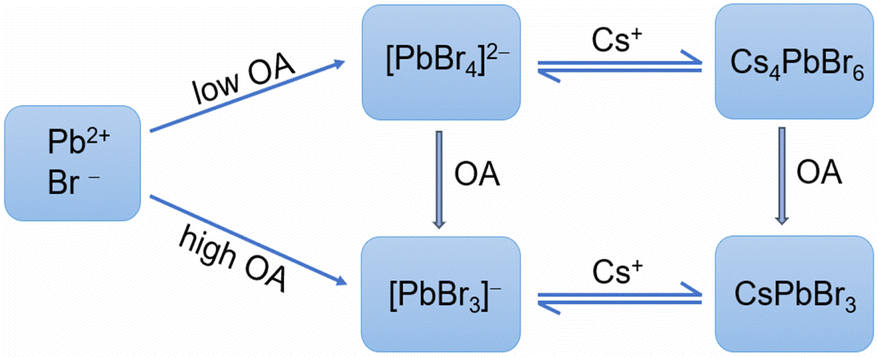 | ||
| Fig. 6 Schematic illustration of the proposed formation pathways for CsPbBr3 (exemplified by a reaction channel passing through [PbBr3]−) and Cs4PbBr6 NCs, which can be modulated by controlling the dosage of OA. | ||
The proposed mechanism can explain some reported results about the phase modulation between Cs4PbBr6 and CsPbBr3. Jing et al. reported that low Pb/Br ratios were favorable for the generation of Cs4PbBr6, while high Pb/Br ratios would benefit the formation of CsPbBr3.21 The Pb/Br ratio might have exerted the influence via lead bromide complexes: a low Pb/Br ratio favors the formation of [PbBr4]2−, and a high Pb/Br ratio favors [PbBr3]−, [Pb2Br5]−, etc.33,40 Besides, the better thermal stability of [PbBr3]− than [PbBr4]2− coincides with the preferential formation of CsPbBr3 over Cs4PbBr6 at high temperatures.18 Particularly, the mechanism is also consistent with the effect of acid-amine equilibrium on phase preference. Amine-rich reaction systems are well known to be favorable for the preparation of Cs4PbBr6 by direct synthesis or postsynthetic transformation from CsPbBr3. In this work, we reported that OA-rich systems can promote the formation of CsPbBr3 over Cs4PbBr6. The opposite effect of OA and primary amine can be well explained by their distinct effects in altering the equilibria between various lead bromide complexes (e.g., reaction (1)).40 It is necessary to point out that the factors influencing the chemistries of lead bromide complexes are complicated. Some species of lead bromide complexes still need to be definitely assigned. Thus, further comprehensive study should be carried out to reveal precise and immediate correlations between the complexes and the cesium lead bromide NCs.
3.8. Kinetics-controlled formation of CsPbBr3
As presented above, the kinetics of CsPbBr3 formation can be accelerated by increasing OA in the reaction systems. This acceleration may be attributed to the interaction of OA with the DDOA+ ligand.36 Owing to this interaction, the growth of CsPbBr3 NCs was able to proceed in a controlled way (Fig. 2 and S4†). In the nonpolar reaction media, the lead bromide complexes are surrounded and stabilized by the quaternary alkylammonium ligand. OA can solubilize DDOA+ and thus free the lead complexes to generate CsPbBr3 NCs. The interaction of DDOA+ with OA was verified by NMR results. As illustrated in Fig. 7, the addition of DDOABr to OA would shift the characteristic signals of OA to lower ppm. The OA-promoted formation of CsPbBr3 NCs could be schematized by reactions (2) and (3) (exemplified by a reaction pathway going through [PbBr3]−).| DDOA+[PbBr3]− + HOA → DDOA+OA− + HPbBr3 | (2) |
| CsOA + HPbBr3 → CsPbBr3 + HOA | (3) |
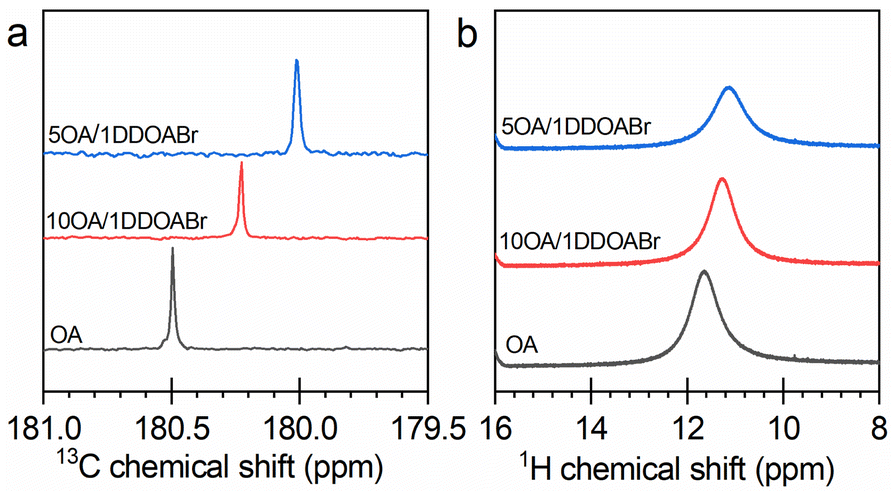 | ||
| Fig. 7 (a) 13C and (b) 1H NMR spectra of OA and two OA–DDOABr mixtures in CDCl3 at room temperature. | ||
4. Conclusions
In this work, we reported the phase-selective synthesis of CsPbBr3 nanorods and Cs4PbBr6 NCs in an amine-free reaction system via a facile heating-up approach. Cs4PbBr6 and CsPbBr3 NCs can be prepared under OA-limited and OA-rich conditions, respectively. Our experimental results support a two-step formation pathway going through lead bromide complexes. Furthermore, our work established reasonable correlations between the nature of the lead bromide complexes and the phase preference of the cesium lead bromide NCs thereof. Based on this mechanistic study, we revealed three roles of OA in this work: (i) manipulating the phase of cesium lead bromide NCs, (ii) influencing the dynamic equilibria between the cesium lead bromide NCs and certain lead bromide complexes, and (iii) controlling the kinetics of reactions toward CsPbBr3 NCs. It is straightforward to decide that other aliphatic acids should have a similar effect to OA (Fig. S7†).By using a simplified, amine-free reaction system, we excluded the existence of acid-amine equilibria, which have profound effects on the synthesis and phase control of lead halide perovskite NCs. Also, the utilization of the quaternary alkylammonium/OA binary ligand system and the heating-up synthetic method enables us to follow the synthetic system at an early stage. The lead bromide complexes were frequently observed in related studies on lead halide perovskites, but were generally ignored. Our study suggests that the investigation of these photoactive species by absorption spectroscopy is a convenient and powerful way to explore the formation mechanisms of the lead halide perovskite NCs, as was done to magic size clusters in the study of II–VI semiconductor nanocrystals.31,41,42 By doing all these, we were able to provide further insights into formation mechanisms and the phase manipulation of the lead halide perovskite-related materials.
Author contributions
M. W.: synthesis conduction, characterization, and writing–original draft. Q. Y.: methodology, writing–original draft, writing–review &editing, supervision, funding acquisition, and project administration; T. Y.: supervision, discussion, and project administration; S. Z.: funding acquisition, supervision, discussion, and project administration; M. G.: supervision and discussion; Y. L.: methodology and discussion.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No: 61871451), Sichuan Science and Technology Program (No: 2022YFG0166), and the opening project of State Key Laboratory of Polymer Materials Engineering (Sichuan University) (Grant No. sklpme2019-4-38). S. Z. also thanks the Joint Fund of Bijie City and Guizhou University of Engineering Science (Bijie Science and Technology Union Contract [2023] No.16). Q. Y. and M. W. sincerely thank Mr Jiaxin Song for valuable help in structural analysis and cartoon making. Q. Y. and M. W. also sincerely thank Prof. Kui Yu for valuable discussion and guidance.Notes and references
- L. Protesescu, S. Yakunin, M. I. Bodnarchuk, F. Krieg, R. Caputo, C. H. Hendon, R. X. Yang, A. Walsh and M. V. Kovalenko, Nano Lett., 2015, 15, 3692–3696 CrossRef CAS PubMed.
- J. Shamsi, A. S. Urban, M. Imran, L. De Trizio and L. Manna, Chem. Rev., 2019, 119, 3296–3348 CrossRef CAS PubMed.
- Y. Wu, H. Wei, L. Xu, B. Cao and H. Zeng, J. Appl. Phys., 2020, 128, 050903 CrossRef CAS.
- Q. Chen, J. Wu, X. Ou, B. Huang, J. Almutlaq, A. A. Zhumekenov, X. Guan, S. Han, L. Liang and Z. Yi, Nature, 2018, 561, 88–93 CrossRef CAS PubMed.
- S. Huang, S. Yang, Q. Wang, R. Wu, Q. Han and W. Wu, RSC Adv., 2019, 9, 42430–42437 RSC.
- R. Jin, J. Wang, K. Shi, B. Qiu, L. Ma, S. Huang and Z. Li, RSC Adv., 2020, 10, 43225–43232 RSC.
- G. Almeida, I. Infante and L. Manna, Science, 2019, 364, 833–834 CrossRef CAS PubMed.
- T. Qiao and D. H. Son, Acc. Chem. Res., 2021, 54, 1399–1408 CrossRef CAS PubMed.
- A. L. Efros and L. E. Brus, ACS Nano, 2021, 15, 6192–6210 CrossRef CAS PubMed.
- K. Yu, X. Liu, T. Qi, H. Yang, D. M. Whitfield, Q. Y. Chen, E. J. Huisman and C. Hu, Nat. Commun., 2016, 7, 1–11 Search PubMed.
- G. Li, H. Wang, Z. Zhu, Y. Chang, T. Zhang, Z. Song and Y. Jiang, Chem. Commun., 2016, 52, 11296–11299 RSC.
- H. Yang, Y. Zhang, J. Pan, J. Yin, O. M. Bakr and O. F. Mohammed, Chem. Mater., 2017, 29, 8978–8982 CrossRef CAS.
- H. Ding, H. Jiang and X. Wang, J. Mater. Chem. C, 2020, 8, 8999–9004 RSC.
- L. N. Quan, R. Quintero-Bermudez, O. Voznyy, G. Walters, A. Jain, J. Z. Fan, X. Zheng, Z. Yang and E. H. Sargent, Adv. Mater., 2017, 29, 1605945 CrossRef PubMed.
- J. Xu, W. Huang, P. Li, D. R. Onken, C. Dun, Y. Guo, K. B. Ucer, C. Lu, H. Wang and S. M. Geyer, Adv. Mater., 2017, 29, 1703703 CrossRef PubMed.
- N. Riesen, M. Lockrey, K. Badek and H. Riesen, Nanoscale, 2019, 11, 3925–3932 RSC.
- W. Huang, Y. Wang and S. K. Balakrishnan, Chem. Commun., 2018, 54, 7944–7947 RSC.
- G. Almeida, L. Goldoni, Q. Akkerman, Z. Dang, A. H. Khan, S. Marras, I. Moreels and L. Manna, ACS Nano, 2018, 12, 1704–1711 CrossRef CAS.
- Q. A. Akkerman, S. Park, E. Radicchi, F. Nunzi, E. Mosconi, F. De Angelis, R. Brescia, P. Rastogi, M. Prato and L. Manna, Nano Lett., 2017, 17, 1924–1930 CrossRef CAS PubMed.
- R. Grisorio, E. Fanizza, I. Allegretta, D. Altamura, M. Striccoli, R. Terzano, C. Giannini, V. Vergaro, G. Ciccarella and N. Margiotta, Nanoscale, 2020, 12, 623–637 RSC.
- Q. Jing, Y. Xu, Y. Su, X. Xing and Z. Lu, Nanoscale, 2019, 11, 1784–1789 RSC.
- Z. Liu, Y. Bekenstein, X. Ye, S. C. Nguyen, J. Swabeck, D. Zhang, S.-T. Lee, P. Yang, W. Ma and A. P. Alivisatos, J. Am. Chem. Soc., 2017, 139, 5309–5312 CrossRef CAS.
- F. Palazon, G. Almeida, Q. A. Akkerman, L. De Trizio, Z. Dang, M. Prato and L. Manna, Chem. Mater., 2017, 29, 4167–4171 CrossRef CAS PubMed.
- A. Pan, B. He, X. Fan, Z. Liu, J. J. Urban, A. P. Alivisatos, L. He and Y. Liu, ACS Nano, 2016, 10, 7943–7954 CrossRef CAS.
- S. Sun, D. Yuan, Y. Xu, A. Wang and Z. Deng, ACS Nano, 2016, 10, 3648–3657 CrossRef CAS PubMed.
- F. Zaccaria, B. Zhang, L. Goldoni, M. Imran, J. Zito, B. van Beek, S. Lauciello, L. De Trizio, L. Manna and I. Infante, ACS Nano, 2022, 16, 1444–1455 CrossRef CAS PubMed.
- J. De Roo, M. Ibáñez, P. Geiregat, G. Nedelcu, W. Walravens, J. Maes, J. C. Martins, I. Van Driessche, M. V. Kovalenko and Z. Hens, ACS Nano, 2016, 10, 2071–2081 CrossRef CAS PubMed.
- V. K. Ravi, P. K. Santra, N. Joshi, J. Chugh, S. K. Singh, H. Rensmo, P. Ghosh and A. Nag, J. Phys. Chem. Lett., 2017, 8, 4988–4994 CrossRef CAS.
- S. Wei, Y. Yang, X. Kang, L. Wang, L. Huang and D. Pan, Chem. Commun., 2016, 52, 7265–7268 RSC.
- E. Yassitepe, Z. Yang, O. Voznyy, Y. Kim, G. Walters, J. A. Castañeda, P. Kanjanaboos, M. Yuan, X. Gong and F. Fan, Adv. Funct. Mater., 2016, 26, 8757–8763 CrossRef CAS.
- J. Hui, Y. Jiang, O. O. Gokcinar, J. Tang, Q. Yu, M. Zhang and K. Yu, Chem. Mater., 2020, 32, 4574–4583 CrossRef CAS.
- D. Amgar, A. Stern, D. Rotem, D. Porath and L. Etgar, Nano Lett., 2017, 17, 1007–1013 CrossRef CAS PubMed.
- S. J. Yoon, K. G. Stamplecoskie and P. V. Kamat, J. Phys. Chem. Lett., 2016, 7, 1368–1373 CrossRef CAS PubMed.
- S. K. Balakrishnan and P. V. Kamat, Chem. Mater., 2018, 30, 74–78 CrossRef CAS.
- K. Wang, L. Wu, L. Li, H. Yao, H. Qian and S. Yu, Angew. Chem., Int. Ed. Engl., 2016, 55, 8328–8332 CrossRef CAS PubMed.
- J.-R. Wen, F. A. Rodríguez Ortiz, A. Champ and M. T. Sheldon, ACS Nano, 2022, 16, 8318–8328 CrossRef CAS PubMed.
- X.-W. Lei, C.-Y. Yue, J.-C. Wei, R.-Q. Li, Y. Li and F.-Q. Mi, Dalton Trans., 2016, 45, 19389–19398 RSC.
- C.-Q. Jing, J.-Z. Li, T. Xu, K. Jiang, X.-J. Zhao, Y.-F. Wu, N.-T. Xue, Z.-H. Jing and X.-W. Lei, CrystEngComm, 2021, 23, 292–298 RSC.
- J.-R. Wen, B. J. Roman, F. A. Rodriguez Ortiz, N. Mireles Villegas, N. Porcellino and M. Sheldon, Chem. Mater., 2019, 31, 8551–8557 CrossRef CAS.
- D. Ferri, F. Salvatore and E. Vasca, J. Coord. Chem., 1989, 20, 11–20 CrossRef CAS.
- Q. Yu and C.-Y. Liu, J. Phys. Chem. C, 2009, 113, 12766–12771 CrossRef CAS.
- J. Zhang, X. Hao, N. Rowell, T. Kreouzis, S. Han, H. Fan, C. Zhang, C. Hu, M. Zhang and K. Yu, J. Phys. Chem. Lett., 2018, 9, 3660–3666 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: More results including SEM, XRD and UV-vis absorption spectra (PDF). Two videos displaying the immediate generation of CsPbBr3 NCs upon OA injection (MP4). See DOI: https://doi.org/10.1039/d2ra06491j |
| This journal is © The Royal Society of Chemistry 2023 |