
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Regioselective electrophilic aromatic borylation as a method for synthesising sterically hindered benzothiadiazole fluorophores†
Dominic Taylora,
Thomas Malcomson b,
Adilet Zhakeyevc,
Georgina M. Rosaira,
Martin J. Patersona,
Jose Marques-Huesoc,
Scott J. Dalgarno*a and
Filipe Vilela*a
b,
Adilet Zhakeyevc,
Georgina M. Rosaira,
Martin J. Patersona,
Jose Marques-Huesoc,
Scott J. Dalgarno*a and
Filipe Vilela*a
aInstitute of Chemical Sciences, School of Engineering and Physical Science, Heriot-Watt University, Riccarton, Edinburgh EH14 4AS, UK. E-mail: f.vilela@hw.ac.uk
bDepartment of Chemistry, Lancaster University, Lancaster, LA1 4YB, UK
cInstitute of Sensors, Signals and Systems, School of Engineering and Physical Science, Heriot-Watt University, Riccarton, Edinburgh, UK
First published on 16th February 2023
Abstract
Regioselective stepwise phenylation of 4,7-diarylbenzo[c][1,2,5]thiadiazole fluorophores has been achieved through a facile one-pot, three-step synthetic strategy involving sequential borylation, hydroxydechlorination and Suzuki–Miyaura cross-coupling reactions. Crucial to the selectivity was the use of BCl3 to regioselectively install a boronic acid group in the ortho-position of only one of the diaryl groups. The subsequent introduction of ortho-phenyl groups through Suzuki–Miyaura cross-coupling gave rise to twisted structures with hindered intramolecular rotation, providing a structural lever with which the fluorophore absorption and emission properties could be adjusted.
Introduction
In recent years, organic π-conjugated electron donor–acceptor (D–A) species have garnered significant interest due to their utility in organic light harvesting and emitting applications, such as organic photovoltaics,1 photocatalysts,2 and fluorophores.3 The successful design of an efficient light harvesting electron D–A system is underpinned by a fundamental understanding of the structure–property relationship of a particular combination of electron donor and acceptor building blocks. Amongst the various electron acceptor building blocks, the benzo[c][1,2,5]thiadiazole (BTZ) group has emerged as a promising candidate, mainly due to its strongly electron accepting nature in combination with its photostability.4–6 Combination of the BTZ group with various donor groups has led to light harvesting applications including photocatalysis,7–13 photovoltaic devices,14 and fluorescent sensing.5,15,16 BTZ offers a large scope for chemical derivatisation including replacing the sulfur atom with other chalcogens (O, Se and Te) or generating fused polycyclic systems, with such modifications allowing the photophysical and optoelectronic properties of a BTZ D–A system to be controlled.17,18Beyond direction modification to the acceptor group, derivatisation of the donor aryl groups, most often located in the 4- and 7-positions (4,7-diarylBTZ), has also been explored. Recently, we have reported on the effect that changing the aryl group has on the photophysical and photoredox properties of a 4,7-diarylBTZ D–A photocatalyst system.19 These photocatalysts were broadly assembled through the use of Suzuki–Miyaura cross-coupling, with the variation in structure achieved by varying the combination of coupling partners. A more direct approach towards aryl group modification has been realised by Zhang and coworkers, who have reported both the palladium catalysed regioselective C–H acyloxylation and halogenation of 4-aryl and 4,7-diaryl substituted BTZs using hypervalent iodine reagents (Fig. 1).20,21 Similarly, regioselective C–H borylation of 4,7-diarylBTZs has been reported by Ingleson and coworkers through the use of BCl3.22 This approach resulted in the ortho-regioselective installation of a BCl2 group that engaged in the formation of a dative bond with the BTZ nitrogen atom.23,24 This generated a rigid, planar structure that featured extended π-conjugation and reduced the separation between the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO). The susceptibility the B–Cl bond to reaction with water allowed for the formation of a boronic acid group, which Ingleson and coworkers took advantage of to synthesise thermally activated delayed fluorophores through Suzuki–Miyaura cross coupling reactions.25 Inspired by this approach, we herein report on the regioselective ortho-phenylation of 4,7-diphenylbenzo[c][1,2,5]thiadiazole (pH-BTZ) and 4,7-di(thiophen-2-yl)benzo[c][1,2,5]thiadiazole (Th-BTZ) through a similar one-pot, three step strategy. The regioselective installation of a phenyl group in the ortho-position introduced steric bulk that twisted the molecule around the electron donor–acceptor torsion angle, leading to hypsochromic shifts in the wavelengths of maximum absorption (λabs) and emission (λem), as well as changes in the lifetime of fluorescence (τf) and photoluminescence quantum yields (PLQY).
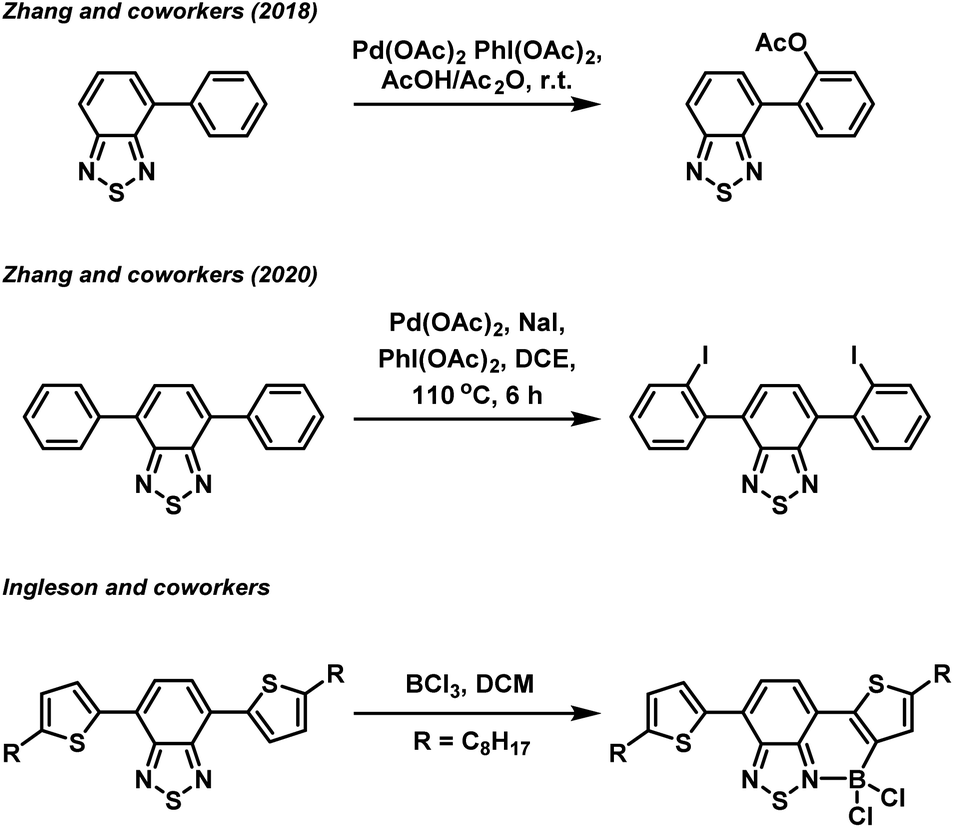 | ||
| Fig. 1 Examples of regioselective ortho-functionalisation of 4,7-diarylBTZs. | ||
Results and discussion
Fluorophore synthesis
Our previous studies conducted into BTZ compounds as fluorophores focused on the changes in light absorption and emission that could be made by derivatisation of pH-BTZ, which exhibited a λabs at 380 nm and a λem at 482 nm in chloroform.19 The most significant changes were observed upon substitution of the phenyl rings with heterocycles, leading to significant hypsochromic (e.g. 4-pyridyl groups) or bathochromic shifts (e.g. 2-thienyl, 2-pyrrolyl, 2-thiazoyl groups) for both absorption and emission. In particular, substitution of the phenyl groups with Th-BTZ resulted in the λabs and λem bathochromically shifting to 446 and 552 nm respectively, driven mainly by an increase in the energy of the HOMO. We were therefore interested in applying this borylation/Suzuki–Miyaura cross-coupling approach developed by Ingleson and co-workers to both pH-BTZ and Th-BTZ as representative examples of non-planar and planar D–A fluorophores respectively.The addition of BCl3 (1 M in DCM) to a solution of pH-BTZ or Th-BTZ in DCM resulted in the formation of a dative bond with the BTZ nitrogen atom and borylation of the aryl C–H bond ortho-to the BTZ group (Scheme 1).24 After allowing these solutions to stir overnight at room temperature, all of the volatiles were removed under reduced pressure to yield pure borylated products pH-BTZ-BCl2 and Th-BTZ-BCl2 in 95 and 90% yield respectively. UV-vis absorption spectra recorded for pH-BTZ-BCl2 and Th-BTZ-BCl2 in chloroform revealed large bathochromic shifts in absorption, with maxima located at 510 and 604 nm respectively, which can be rationalised by the formation of a rigid, planar structure with extensive π-conjugation (Fig. S10 and S14†).
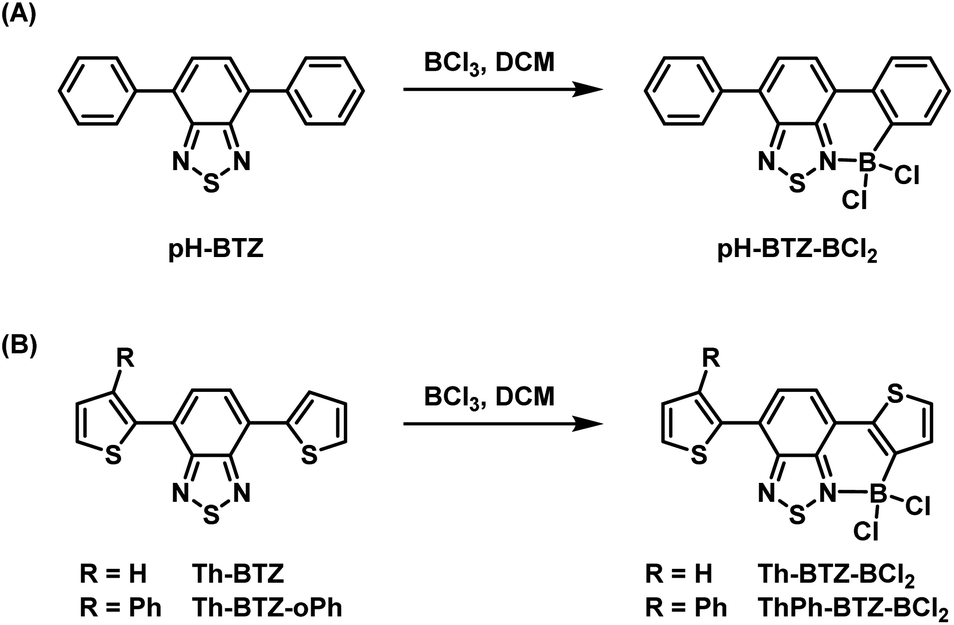 | ||
| Scheme 1 Regioselective ortho-borylation of 4,7-diarylBTZ containing (A) phenyl and (B) 2-thienyl groups using BCl3. | ||
Following the successful isolation of pH-BTZ-BCl2 and Th-BTZ-BCl2, the one-pot, three-step phenylation strategy described in Scheme 2 was investigated. After the initial borylation step, pH-BTZ-BCl2 and Th-BTZ-BCl2 were dissolved in a THF/water mixture to elicit formation of the boronic acid which was not isolated. Following degassing these mixture with nitrogen gas, bromobenzene (2 equivalents), Pd(PPh3)4 (5 mol%) and potassium carbonate (10 equivalents) were added and the mixture heated to 70 °C. Following completion of the reaction, pH-BTZ-oPh and Th-BTZ-oPh were isolated in 55 and 57% yields, respectively.
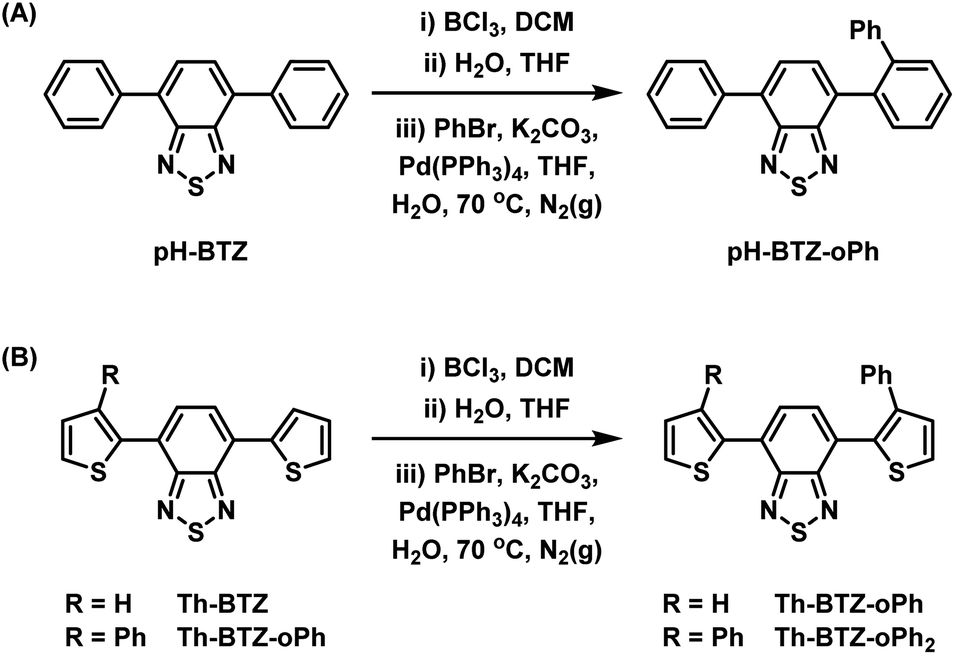 | ||
| Scheme 2 One pot, three-step approach towards regioselective ortho-phenylation of 4,7-diarylBTZ containing (A) phenyl and (B) 2-thienyl groups. | ||
Following the installation of the first ortho-phenyl group to Th-BTZ, the borylation/Suzuki–Miyaura cross coupling strategy was applied a second time to form Th-BTZ-oPh2 (Scheme 2B). This strategy was not extended to pH-BTZ-oPh as we have previously reported the synthesis of the symmetric molecule pH-BTZ-oPh2 directly via a single Suzuki–Miyaura cross-coupling reaction from 4,7-dibromoBTZ.19 However, pH-BTZ-oPh2 was also characterised in parallel with the fluorophores discussed in this report as an analogue of Th-BTZ-oPh2.
Fluorophore UV/vis absorption and emission properties
UV-vis absorption spectra were recorded for each of the fluorophores in chloroform solution. From an initial value of 380 nm, the absorption of pH-BTZ underwent modest hypsochromic shifts upon installing the ortho-phenyl groups (Table 1 and Fig. 2). This resulted in λabs of 377 and 373 nm for pH-BTZ-oPh and pH-BTZ respectively. Larger hypsochromic shifts in absorption were observed upon ortho-phenylation of Th-BTZ and Th-BTZ-oPh. From an initial value of 446 nm, the introduction of a single ortho-phenyl group resulted in a hypsochromic shift in λabs to 432 nm, with the addition of a second to form Th-BTZ-oPh2 further shifting the λabs to 423 nm. In both cases, the hypsochromic shift can be attributed to the sterically bulky ortho-phenyl groups increasing the donor–acceptor torsion angle, disrupting π-conjugation. This is consistent with previous observations made by Pathak et al., with regards the effect that introducing methyl groups had on the absorption profiles of 4,7-diarylBTZs.26 The hypsochromic shifts we observed were more pronounced for fluorophores bearing thiophene rings, as pH-BTZ was already non-planar in solution while Th-BTZ instead preferentially adopted a planar geometry and would therefore experience comparatively greater twisting.| Compound | λabsa/nm | λema,b/nm | Stokes' Shift/nm | εMc/× 103 M−1 cm−1 | PLQYa,d | τfa,e/ns | kfr/× 107 s−1 | knrf/× 107 s−1 |
|---|---|---|---|---|---|---|---|---|
| a In chloroform solution.b Excitation at the wavelength of maximum absorbance was used for each fluorophore.c Molar attenuation coefficient (εM) measured as the gradient of a Beer–Lambert plot.d Absolute PLQY.e Fluorescence lifetime was measured by time-resolved photoluminescence spectroscopy (Fig. S17).f kr and knr were estimated from the PLQY and τf 29. | ||||||||
| pH-BTZ | 380 | 482 | 102 | 7.5 ± 0.1 | 0.864 | 10.01 | 8.63 | 1.36 |
| pH-BTZ-oPh | 376 | 480 | 104 | 6.9 ± 0.1 | 1.00 | 12.69 | 7.88 | 0.00 |
| pH-BTZ-oPh2 | 373 | 486 | 113 | 6.5 ± 0.2 | 1.00 | 15.33 | 6.52 | 0.00 |
| Th-BTZ | 446 | 552 | 106 | 12.1 ± 0.4 | 0.881 | 13.13 | 6.71 | 0.91 |
| Th-BTZ-oPh | 432 | 543 | 111 | 9.3 ± 0.4 | 0.864 | 12.84 | 6.73 | 1.06 |
| Th-BTZ-oPh2 | 423 | 547 | 124 | 6.8 ± 0.4 | 0.795 | 13.01 | 6.11 | 1.58 |
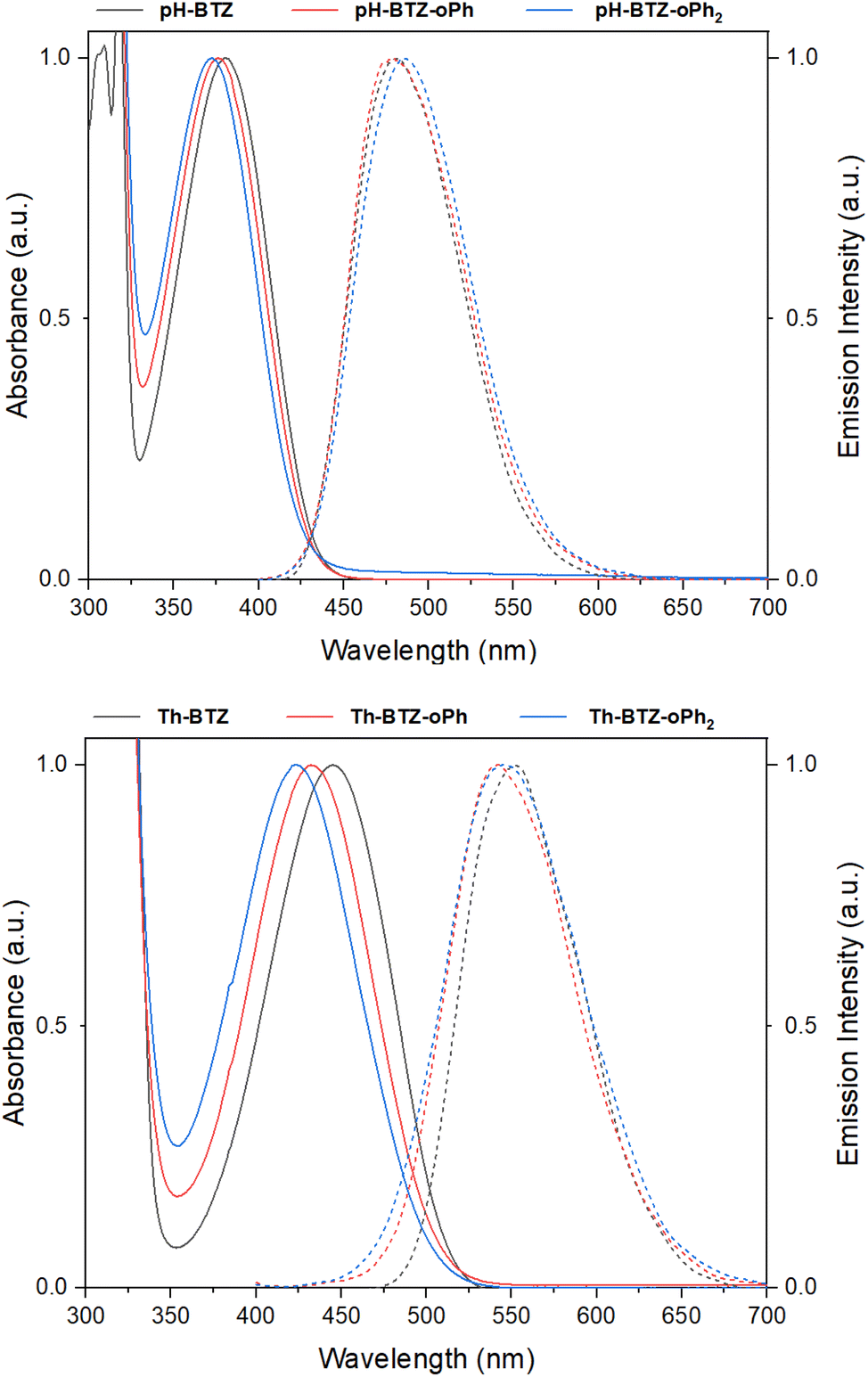 | ||
| Fig. 2 Normalised UV-vis absorption (solid lines) and emission (dashed lines) spectra for pH-BTZ (top) and Th-BTZ (bottom) derivatives in chloroform solution. | ||
Fluorescence lifetimes (τf) for each of the fluorophores involved in this study were measured using time-resolved photoluminescence spectroscopy (Table 1 and Fig. S17†). For the fluorophores based on pH-BTZ, successive ortho-phenylation led to an increase in τf from an initial value of 10.01 ns for pH-BTZ to 12.69 and 15.33 ns for pH-BTZ-oPh and pH-BTZ-oPh2 respectively. This is presumably due to the steric hinderance caused by introducing the ortho-phenyl substituents, preventing rotation around the donor–acceptor dihedral bond. This would eliminate a pathway for non-radiative relaxation of the excited state which could otherwise compete with radiative relaxation.27 This trend was also present in the measured photoluminescence quantum yields (PLQYs), which rose from 0.864 for pH-BTZ to 1.00 for both pH-BTZ-oPh and pH-BTZ-oPh2. This effect has previously been demonstrated in BTZ fluorophores by El-Zohry et al., where intermolecular hydrogen bonding between carboxylic acid substituent groups suppressed intramolecular rotation about the electron donor–acceptor dihedral angle, ultimately enhancing PLQY.28
The trends observed for the pH-BTZ series of fluorophores were not present in the series of fluorophores based on Th-BTZ, with neither τf nor the PLQYs correlating with the number of phenyl substituents introduced to the electron donor 2-thienyl groups. τf, in particular, was largely unaffected by introducing the phenyl substituents with values measured to be in the range of 12.84–13.13 ns. This would suggest that regioselective phenylation did not impact rotation of the thiophene group relative to the BTZ group to the same degree as the phenyl groups of pH-BTZ.
To gain further insight into the effect of the ortho-phenylation on pH-BTZ and Th-BTZ, the rate constants for radiative (kr) and non-radiative (knr) were estimated from the PLQY and τf using eqn (1) and (2).29
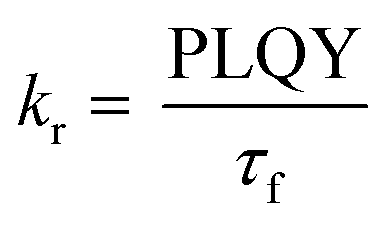 | (1) |
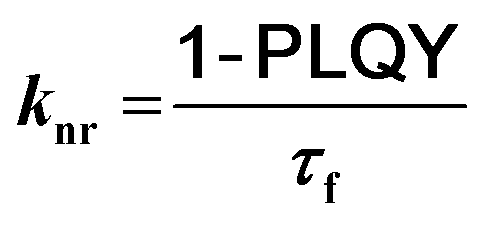 | (2) |
For radiative relaxation, the overall effect of ortho-phenylation of pH-BTZ was a modest reduction in kr from 8.63 × 107 s−1 to 7.88 × 107 s−1 and 6.52 × 107 s−1 for pH-BTZ-oPh and pH-BTZ-oPh2 respectively. The value of knr was essentially reduced to zero for both pH-BTZ-oPh and pH-BTZ-oPh2 by the increase in the PLQY to 1.00, highlighting the suppression of non-radiative relaxation pathways by ortho-phenylation. This can be directly contrasted with the effect that ortho-phenylation had on the Th-BTZ series of fluorophores, where kr and knr showed little variation in value, although kr still outweighed knr.
Computational studies
Further characterisation was conducted through density functional theory (DFT) and time dependant-DFT (TD-DFT) analysis. Investigation of the barriers to rotation of the electron donor moiety relative to the central acceptor moiety (Fig. S21 and S22†) shows a preferential deviation from planarity of 40° for the pH-BTZ structure which increases to a 60° deviation upon incorporation of the ortho-phenyl moiety. In contrast, Th-BTZ is shown to adopt a planar geometry with a 3 kJ mol−1 preference for the sulfur of the thiophene moiety to be orientated away from the central chalcogen. Planarity is lost upon incorporation of ortho-phenyl substituent, instead adopting a 40° deviation and inverting the orientation of the thiophene moiety, opting to place the bulkier phenyl substituent further from the central chalcogen.Each structure containing ortho-phenyl groups showed a substantially lower rotational barrier when moving the phenyl group away from the central chalcogen, whereas rotating the phenyl group past the central chalcogen produced a larger energy barrier. In the case of pH-BTZ-oPh and pH-BTZ-oPh2, the energy barrier to rotation is ca. 56 kJ mol−1, rendering this rotation unlikely (Fig. S21†). In comparison, rotation around the BTZ-thiophene bonds in Th-BTZ-oPh and Th-BTZ-oPh2 presents a substantially lower energy barrier of around 27 kJ mol−1 (Fig. S22†). The reason for this lower energy barrier is most likely due to the pentagonal shape of the thiophene groups, which would slightly orientate any ortho-phenyl substituent groups they bare away from the BTZ group and reduce the steric hinderance. The lower energy barriers to internal rotation that Th-BTZ-oPh and Th-BTZ-oPh2 present could also explain why τf and PLQY are not massively changed relative to Th-BTZ, as internal rotation is not suppressed to the same degree as pH-BTZ based fluorophores.
Additionally, TD-DFT spectra were obtained for each spectra (Fig. S19 and S20†) which show promising alignment with those observed from experimental data (Table S2†). Natural transition orbital (NTO) analysis of the excited state providing the major contribution to the 375 nm and 425 nm peaks of pH-BTZ and Th-BTZ, respectively, showed similar character across all structures, irrespective of ortho-functionalisation (Tables S5–S10†). This transition, shown in Fig. 3 for the Th-BTZ structure shows a π–π* transition involving the movement of charge from the outer, electron donor groups, onto the central BTZ electron acceptor group.
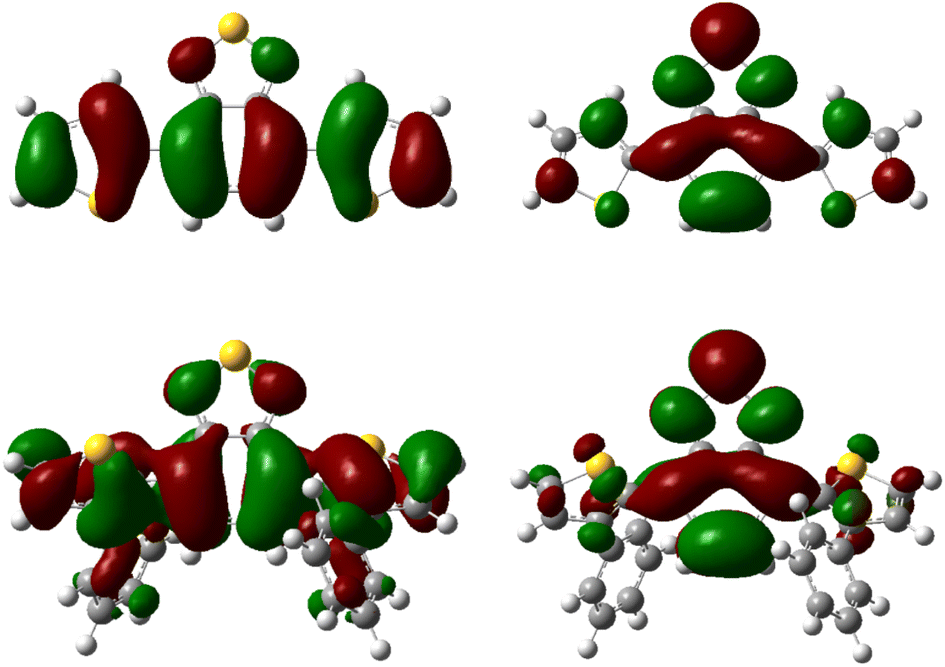 | ||
| Fig. 3 NTO representations of the hole (left) and particle (right) orbitals of the bright states responsible for the lowest energy peaks observed in the absorption spectra of Th-BTZ (top) and Th-BTZ-oPh2 (bottom). | ||
Single crystal X-ray structures
Single crystals of pH-BTZ-oPh, Th-BTZ-oPh and Th-BTZ-oPh2 suitable for X-ray diffraction were grown by slow evaporation from chloroform or acetone solution (Fig. S1–S8†). The structural twisting was best exemplified by the crystal structure of Th-BTZ-oPh in which the thiophene ring bearing a phenyl group was twisted to an angle of 50°, while the unmodified thiophene ring remained coplanar with the BTZ group (Fig. 4). This can be contrasted with the reported crystal structure of Th-BTZ previously reported by McCulloch and coworkers, in which both thiophene rings are coplanar with the BTZ group.30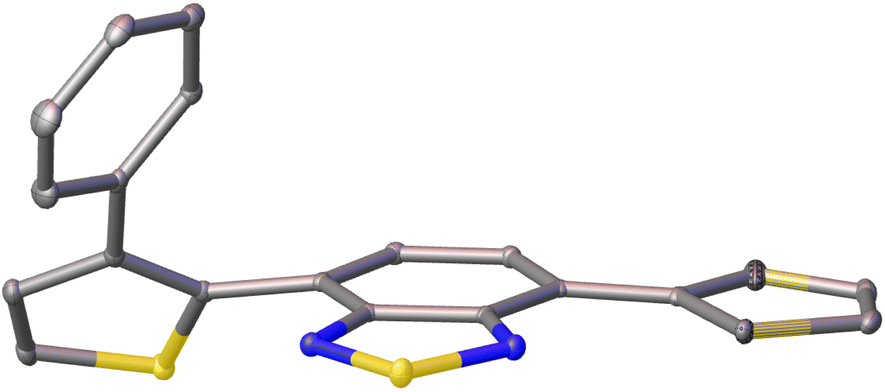 | ||
| Fig. 4 X-ray crystal structure of Th-BTZ-oPh. The atoms are shown as ellipsoids at 50% probability. H atoms omitted for clarity. | ||
Experimental
General methods and materials
4,7-Dibromobenzo[c][1,2,5]thiadiazole,31 palladium tetrakis(triphenylphosphine) (0) (Pd(PPh3)4),32 4,7-diphenylbenzo[c][1,2,5]thiadiazole (pH-BTZ), 4-([1,1′-biphenyl]-2 yl)-7-phenylbenzo[c][1,2,5]thiadiazole (oPh-BTZ) and 4,7-di(thiophen-2-yl)benzo[c][1,2,5]thiadiazole (Th-BTZ) were all synthesised according to literature procedures (see ESI† for details).19 All commercially available reagents were used as received and purchased from Sigma-Aldrich, Fluorochem, Apollo Scientific or Fischer Scientific. DCM was dried over 4 Å activated molecular sieves before using in borylation reactions.NMR spectra of synthetic products were recorded using a Bruker AVIII 300 MHz spectrometer using the residual solvent peak as an internal reference. All IR spectra were recorded on solid powder/crystals using a Nicolet™ iS™ 5 FTIR spectrometer. UV-vis absorption spectra for the synthesised fluorophores were obtained using a PerkinElmer Lambda 35 spectrometer in chloroform solution in quartz cuvettes with a path length of 1 cm. Emission spectra in the visible region were recorded using a PerkinElmer LS 55 fluorescence spectrometer in chloroform solution using quartz cuvettes with a path length of 1 cm. The excitation wavelength used was the wavelength of maximum absorption for each individual fluorophore. Single crystal X-ray structures were collected using a Bruker D8 venture using a Cu-Kα (λ = 1.5418 Å) IμS 3.0 microfocus source, using the APEX3 program suite, with the crystal kept at 100.0 K during data collection. The structures were solved using Olex2, using the SHELXT structure solution program using intrinsic phasing and refined with the SHELXL refinement package using least squares minimisation.33–35
Synthesis of ortho-functionalised BTZ fluorophores
![[small nu, Greek, macron]](https://www.rsc.org/images/entities/b_i_char_e0ce.gif) (cm−1) 3050 (w, C–H str.). UV-vis (CHCl3) λmax (nm) 377.
(cm−1) 3050 (w, C–H str.). UV-vis (CHCl3) λmax (nm) 377.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :hexane 1:4 as the eluent to yield red crystals (217 mg, 57%). 1H NMR (CDCl3, 300 MHz, 25.0 °C) δH 8.13 (dd, 1H, J = 3.9, 1.2 Hz), 7.74 (d, 1H, J = 7.5 Hz), 7.57 (d, 1H, J = 5.2 Hz), 7.47 (dd, 1H, J = 5.2, 1.1 Hz), 7.45 (d, 1H, J = 7.5 Hz), 7.30 (m, 2H), 7.29 (d, 1H, J = 5.2 Hz), 7.26 (m, 3H), 7.22 (dd, 1H, J = 5.2, 3.8 Hz). 13C NMR (CDCl3, 75.5 MHz, 25.0 °C) δC 154.2 (C), 152.3 (C), 141.1 (C), 139.3 (C), 136.8 (C), 133.1, 130.6 (CH), 130.0 (CH), 128.8 (CH), 128.5 (CH), 128.0 (CH), 127.7 (CH), 127.0 (CH), 127.0 (CH), 126.5 (CH), 126.2 (C), 125.4 (C). IR (cm−1) 3050 (w, C–H str.). UV-vis (CHCl3) λmax (nm) 432.:hexane 1:4 as the eluent to yield orange-red crystals (45 mg, 20%). 1H NMR (CDCl3, 300 MHz, 25.0 °C) δH 7.56 (d, 2H, J = 5.1 Hz), 7.27 (d, 2H, J = 5.1 Hz), 7.26 (s, 2H), 7.23 (m, 10H). 13C NMR (CDCl3, 75.5 MHz, 25.0 °C) δC 153.9 (C), 141.2 (C), 136.7 (C), 132.9 (C), 130.2 (CH), 130.0 (CH), 128.8 (CH), 128.4 (CH), 127.0 (CH), 126.7 (C), 126.6 (CH). IR (cm−1) 3050 (w, C–H str.). UV-vis (CHCl3) λmax (nm) 423.
:hexane 1:4 as the eluent to yield red crystals (217 mg, 57%). 1H NMR (CDCl3, 300 MHz, 25.0 °C) δH 8.13 (dd, 1H, J = 3.9, 1.2 Hz), 7.74 (d, 1H, J = 7.5 Hz), 7.57 (d, 1H, J = 5.2 Hz), 7.47 (dd, 1H, J = 5.2, 1.1 Hz), 7.45 (d, 1H, J = 7.5 Hz), 7.30 (m, 2H), 7.29 (d, 1H, J = 5.2 Hz), 7.26 (m, 3H), 7.22 (dd, 1H, J = 5.2, 3.8 Hz). 13C NMR (CDCl3, 75.5 MHz, 25.0 °C) δC 154.2 (C), 152.3 (C), 141.1 (C), 139.3 (C), 136.8 (C), 133.1, 130.6 (CH), 130.0 (CH), 128.8 (CH), 128.5 (CH), 128.0 (CH), 127.7 (CH), 127.0 (CH), 127.0 (CH), 126.5 (CH), 126.2 (C), 125.4 (C). IR (cm−1) 3050 (w, C–H str.). UV-vis (CHCl3) λmax (nm) 432.:hexane 1:4 as the eluent to yield orange-red crystals (45 mg, 20%). 1H NMR (CDCl3, 300 MHz, 25.0 °C) δH 7.56 (d, 2H, J = 5.1 Hz), 7.27 (d, 2H, J = 5.1 Hz), 7.26 (s, 2H), 7.23 (m, 10H). 13C NMR (CDCl3, 75.5 MHz, 25.0 °C) δC 153.9 (C), 141.2 (C), 136.7 (C), 132.9 (C), 130.2 (CH), 130.0 (CH), 128.8 (CH), 128.4 (CH), 127.0 (CH), 126.7 (C), 126.6 (CH). IR (cm−1) 3050 (w, C–H str.). UV-vis (CHCl3) λmax (nm) 423.PLQY and fluorescence lifetime measurements
PLQYs measurements were obtained following the conventions and considerations from Jones et al.,36 using a FLS920 spectrofluorometer (Edinburgh Instruments Ltd) equipped with an extended red-sensitive photon multiplier detector (R2658P, Hamamatsu) and an integrating sphere with a 102 mm inner diameter (Yobin Yvon) were used for all PLQY measurements. For the excitation, a 450 W xenon lamp (Xe2, Edinburgh Instruments Ltd) was employed. The samples consisting of 20 μM fluorophores diluted in CHCl3, were contained in a square quartz cuvette with a path length of 1 cm and placed in the centre of the integrating sphere. Pure CHCl3 was used as reference. The excitation wavelengths were set to the peak absorption of each sample with a bandwidth set to 5 nm. The excitation and emission regions were measured with a 0.25 nm step size. The associated measurement error is 3%.Lifetimes measurements were performed using the FLS920 spectrofluorometer, equipped with time-correlated single photon counting (TCSPC) electronics, and a pulsed light emitting diode (EPLED, from Edinburgh Instruments). The 405 nm excitation has an optical pulse approximately 1 ns long. The collection was recorded at the highest intensity emission, and the decay was fit with a single exponential.
Conclusions
In conclusion, we have demonstrated that regioselective ortho-borylation on the aryl groups of BTZ fluorophores can be achieved using BCl3. Through subsequent hydroxydechlorination and Suzuki–Miyaura cross-coupling reactions, phenyl groups could be installed that interfered with rotation around the electron D–A bond. Evidence of this was observed both in the non-planar X-ray crystal structures and DFT calculation of the energy barrier for rotation about the electron D–A bond. In the case of fluorophores based on pH-BTZ, ortho-phenylation resulted in increased fluorescence lifetimes and PLQYs of up to 1.00. While ortho-phenylation of Th-BTZ produced larger changes in the absorption and emission spectra, the fluorescence lifetimes and PLQYs were not as significantly impacted. This facile one-pot, three-step approach presents an additional structural modification lever with which the optoelectronic and photophysical properties of BTZ electron D–A fluorophores can be controlled. In future, we envisage that this approach could be extended beyond Suzuki–Miyaura cross-coupling strategies, by taking advantage of the versatile chemistry of boronic acids to generate bespoke BTZ electron D–A systems.Conflicts of interest
There are no conflicts to declare.Acknowledgements
FV and SJD thank the EPSRC funded (EP/L016419/1) CRITICAT centre for doctoral training. MJP thanks EPSRC for funding through grants EP/V006746 and EP/T021675. JMH and AZ thank the EPSRC for funding through grant EP/T013680/1.Notes and references
- X. Wan, C. Li, M. Zhang and Y. Chen, Chem. Soc. Rev., 2020, 49, 2828–2842 RSC.
- M. A. Bryden and E. Zysman-Colman, Chem. Soc. Rev., 2021, 50, 7587–7680 RSC.
- W. Che, Y. Xie and Z. Li, Asian J. Org. Chem., 2020, 9, 1262–1276 CrossRef CAS.
- B. A. D. Neto, A. A. M. Lapis, E. N. Da Silva Júnior and J. Dupont, Eur. J. Org. Chem., 2013, 228–255 CrossRef CAS.
- B. A. D. Neto, P. H. P. R. Carvalho and J. R. Correa, Acc. Chem. Res., 2015, 48, 1560–1569 CrossRef CAS PubMed.
- R. Li, J. Byun, W. Huang, C. Ayed, L. Wang and K. A. I. Zhang, ACS Catal., 2018, 8, 4735–4750 CrossRef CAS.
- R. Li, D. W. Gehrig, C. Ramanan, P. W. M. Blom, F. F. Kohl, M. Wagner, K. Landfester and K. A. I. Zhang, Adv. Synth. Catal., 2019, 361, 3852–3859 CrossRef CAS.
- K. Zhang, D. Kopetzki, P. H. Seeberger, M. Antonietti and F. Vilela, Angew. Chem., Int. Ed., 2013, 52, 1432–1436 CrossRef CAS PubMed.
- J. Shen, R. Steinbach, J. M. Tobin, M. Mouro Nakata, M. Bower, M. R. S. McCoustra, H. Bridle, V. Arrighi and F. Vilela, Appl. Catal. B Environ., 2016, 193, 226–233 CrossRef CAS.
- J. M. Tobin, T. J. D. McCabe, A. W. Prentice, S. Holzer, G. O. Lloyd, M. J. Paterson, V. Arrighi, P. A. G. Cormack and F. Vilela, ACS Catal., 2017, 7, 4602–4612 CrossRef CAS.
- A. Zhakeyev, J. Tobin, H. Wang, F. Vilela and J. Xuan, Energy Procedia, 2019, 158, 5608–5614 CrossRef CAS.
- S. Bhattacharyya, S. R. Ali, M. Venkateswarulu, P. Howlader, E. Zangrando, M. De and P. S. Mukherjee, J. Am. Chem. Soc., 2020, 142, 18981–18989 CrossRef CAS PubMed.
- E. Broumidis, C. M. S. Jones, M. Koyioni, A. Kourtellaris, G. O. Lloyd, J. Marques-Hueso, P. A. Koutentis and F. Vilela, RSC Adv., 2021, 11, 29102–29107 RSC.
- T. C. Parker, D. G. Patel, K. Moudgil, S. Barlow, C. Risko, J. L. Brédas, J. R. Reynolds and S. R. Marder, Mater. Horiz., 2015, 2, 22–36 RSC.
- B. A. D. Neto, J. R. Correa and J. Spencer, Chem. - Eur. J., 2021, 1–29 Search PubMed.
- Y. Tang, H. Huang, B. Peng, Y. Chang, Y. Li and C. Zhong, J. Mater. Chem. A, 2020, 8, 16542–16550 RSC.
- P. B. Pati, Org. Electron., 2016, 38, 97–106 CrossRef CAS.
- Y. Wang and T. Michinobu, J. Mater. Chem. C, 2016, 4, 6200–6214 RSC.
- D. Taylor, T. Malcomson, A. Zhakeyev, S.-X. Cheng, G. M. Rosair, J. Marques-Hueso, Z. Xu, M. J. Paterson, S. J. Dalgarno and F. Vilela, Org. Chem. Front., 2022, 9, 5473–5484 RSC.
- J. Guo, H. He, Z. Ye, K. Zhu, Y. Wu and F. Zhang, Org. Lett., 2018, 20, 5692–5695 CrossRef CAS PubMed.
- H. He, J. Guo, W. Sun, B. Yang, F. Zhang and G. Liang, J. Org. Chem., 2020, 85, 3788–3798 CrossRef CAS PubMed.
- D. L. Crossley, I. A. Cade, E. R. Clark, A. Escande, M. J. Humphries, S. M. King, I. Vitorica-Yrezabal, M. J. Ingleson and M. L. Turner, Chem. Sci., 2015, 6, 5144–5151 RSC.
- D. L. Crossley, I. Vitorica-Yrezabal, M. J. Humphries, M. L. Turner and M. J. Ingleson, Chem. - Eur. J., 2016, 22, 12439–12448 CrossRef CAS PubMed.
- D. L. Crossley, R. Goh, J. Cid, I. Vitorica-Yrezabal, M. L. Turner and M. J. Ingleson, Organometallics, 2017, 36, 2597–2604 CrossRef CAS.
- D. L. Crossley, P. Kulapichitr, J. E. Radcliffe, J. J. Dunsford, I. Vitorica-Yrezabal, R. J. Kahan, A. W. Woodward, M. L. Turner, J. J. W. McDouall and M. J. Ingleson, Chem. - Eur. J., 2018, 24, 10521–10530 CrossRef CAS PubMed.
- A. Pathak, K. R. Justin Thomas, M. Singh and J. H. Jou, J. Org. Chem., 2017, 82, 11512–11523 CrossRef CAS PubMed.
- G. J. Shi, Y. D. Wang, Z. X. Yu, Q. Zhang, S. Chen, L. Z. Xu, K. P. Wang and Z. Q. Hu, Dyes Pigm., 2022, 204, 110471 CrossRef CAS.
- A. M. El-Zohry, A. Alturki, J. Yin, A. Mallick, O. Shekhah, M. Eddaoudi, B. S. Ooi and O. F. Mohammed, J. Phys. Chem. C, 2019, 123, 5900–5906 CrossRef CAS.
- S. Dhar, D. K. Rana, S. Singha Roy, S. Roy, S. Bhattacharya and S. C. Bhattacharya, J. Lumin., 2012, 132, 957–964 CrossRef CAS.
- C. B. Nielsen, A. J. P. White and I. McCulloch, J. Org. Chem., 2015, 80, 5045–5048 CrossRef CAS PubMed.
- J. P. Heiskanen, P. Vivo, N. M. Saari, T. I. Hukka, T. Kastinen, K. Kaunisto, H. J. Lemmetyinen and O. E. O. Hormi, J. Org. Chem., 2016, 81, 1535–1546 CrossRef CAS PubMed.
- M. Ranger, D. Rondeau and M. Leclerc, Macromolecules, 1997, 30, 7686–7691 CrossRef CAS.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- C. M. S. Jones, A. Gakamsky and J. Marques-Hueso, Sci. Technol. Adv. Mater., 2021, 22, 810–848 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Characterisation of synthesised compounds, single crystal X-ray structures, computational calculations and fluorescence lifetime measurements. CCDC 2208644, 2208649–2208651. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2ra08319a |
| This journal is © The Royal Society of Chemistry 2023 |