
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent progress of theoretical studies on electro- and photo-chemical conversion of CO2 with single-atom catalysts
Liyun Jiang a,
Qingqing Yanga,
Zhaoming Xiab,
Xiaohu Yua,
Mengdie Zhaoa,
Qiping Shia and
Qi Yu*ac
a,
Qingqing Yanga,
Zhaoming Xiab,
Xiaohu Yua,
Mengdie Zhaoa,
Qiping Shia and
Qi Yu*ac
aSchool of Physics and Telecommunication Engineering, School of Materials Science and Engineering, Shaanxi Laboratory of Catalysis, Shaanxi University of Technology, Hanzhong 723001, China. E-mail: qiyu@snut.edu.cn
bDepartment of Chemistry and Key Laboratory of Organic Optoelectronics & Molecular Engineering of Ministry of Education, Tsinghua University, Beijing, China
cGuangdong Provincial Key Laboratory of Catalysis, Southern University of Science and Technology, Shenzhen 518055, China
First published on 16th February 2023
Abstract
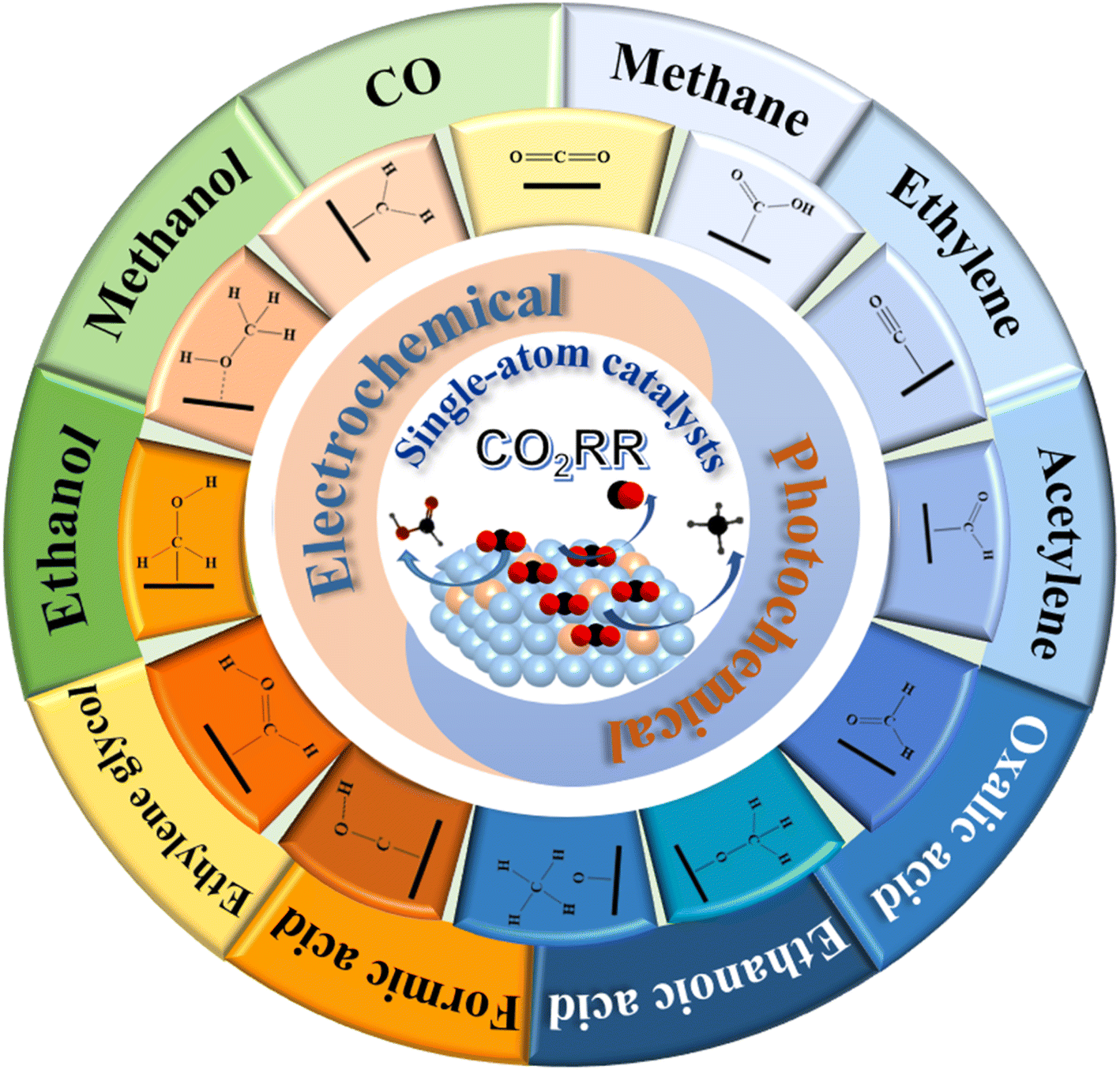
The CO2 reduction reaction (CO2RR) into chemical products is a promising and efficient way to combat the global warming issue and greenhouse effect. The viability of the CO2RR critically rests with finding highly active and selective catalysts that can accomplish the desired chemical transformation. Single-atom catalysts (SACs) are ideal in fulfilling this goal due to the well-defined active sites and support-tunable electronic structure, and exhibit enhanced activity and high selectivity for the CO2RR. In this review, we present the recent progress of quantum-theoretical studies on electro- and photo-chemical conversion of CO2 with SACs and frameworks. Various calculated products of CO2RR with SACs have been discussed, including CO, acids, alcohols, hydrocarbons and other organics. Meanwhile, the critical challenges and the pathway towards improving the efficiency of the CO2RR have also been discussed.
1. Introduction
Due to the increasing fossil fuel combustion and energy consumption, carbon dioxide (CO2) emission has led to serious global environmental problems such as global warming, sea level rising and ocean acidification.1 The shortage of fossil energy has become a serious problem which needs to be solved urgently. Fortunately, it has been found that CO2 can be used as a chemical feedstock to synthesize a variety of valuable chemical products or be converted into specific fuel substances such as methanol and methane. It will not only decrease the pressure on energy demand, but also reduce the amount of greenhouse gases.Many efforts have been devoted to the conversion of CO2 in recent years.2–5 The electrocatalytic CO2 reduction reaction (CO2RR) has been widely applied for both industry and academia because of its high efficiency and activity.6,7 The investigations normally focus on the electrochemical overpotential, current efficiency and products selectivity.8,9 Nowadays, due to the requirement of energy regeneration and environment protection, photocatalysts have been paid more and more attentions because of the significance in the future applications of CO2RR.10–15
The concept of single-atom catalysis (SACs), firstly proposed by Zhang, Li, Liu et al.,16 has been widely used in heterogeneous catalysis,17–20 which have shown the rosy prospects to achieve high activity, selectivity and ∼100% atom utilization. From then on, it had developed preternaturally fast on enabling heterogeneous reduction products.21–27 The 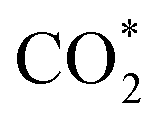 – radical intermediate formed from CO2 activation will affect the reaction pathways due to its instability and readily protonation, and further influence the activity and selectivity of electro- and photo-chemical reaction process. Some efforts have been taken to adjust the interaction between the intermediates and catalysts, such as improving the activities of catalyst surface and reaction environment.28 For SACs, the unsaturated coordination can effectively regulate the relevance of CO2 and correlative intermediates with particular active sites, which is promising for highly efficient CO2RR catalysts.29–31 However, the metal monatom has high surface energy that tends to aggregate particles with low surface energy during the reaction. Therefore, the preparation of stable SACs is still a great challenge. It has been proved that theoretical calculations methods such as density functional theory (DFT), molecular dynamics (MD) and Hartree–Fock (HF) are important for the data reliability. For instance, Qiu et al. revealed that Co–N5 formed by inducing electron delocalization of Co–N4 sites using nitrogen-rich carbon nitride as supporting material can improve the stability and conductivity by DFT calculations. The catalyst synthesized according to this prediction can remain for 40 h with excellent performance in CO2 conversion to CO with >99% selectivity.32 Metal–organic framework (MOF) materials, with periodic structure and designability of functional groups, have also been found an ideal carrier to stabilize metal monatom when coordination chelating sites were introduced. Hai et al. summarized theoretical studies of MOFs, in which the performances of electrocatalysts for CO2RR were involved. The MOFs showed the better catalytic performance than that in the respect of potential barriers, overpotentials, the energy barrier of C–C dimerization, selectivity and activity for CO2RR.33 Thus, it is promising to investigate CO2RR with SACs from the perspective of theoretical calculations, which can guide the catalysts design. The intermediate products of CO2RR include but not limit *COOH, *CO, *CHO, *OCH2, *OCH3, *COH, *CHOH, *CH2OH, *CH3OH, *CH2 (as shown in Fig. 1). The reduction of CO2 can generate CO, methane, ethylene, acetylene and other organics.
– radical intermediate formed from CO2 activation will affect the reaction pathways due to its instability and readily protonation, and further influence the activity and selectivity of electro- and photo-chemical reaction process. Some efforts have been taken to adjust the interaction between the intermediates and catalysts, such as improving the activities of catalyst surface and reaction environment.28 For SACs, the unsaturated coordination can effectively regulate the relevance of CO2 and correlative intermediates with particular active sites, which is promising for highly efficient CO2RR catalysts.29–31 However, the metal monatom has high surface energy that tends to aggregate particles with low surface energy during the reaction. Therefore, the preparation of stable SACs is still a great challenge. It has been proved that theoretical calculations methods such as density functional theory (DFT), molecular dynamics (MD) and Hartree–Fock (HF) are important for the data reliability. For instance, Qiu et al. revealed that Co–N5 formed by inducing electron delocalization of Co–N4 sites using nitrogen-rich carbon nitride as supporting material can improve the stability and conductivity by DFT calculations. The catalyst synthesized according to this prediction can remain for 40 h with excellent performance in CO2 conversion to CO with >99% selectivity.32 Metal–organic framework (MOF) materials, with periodic structure and designability of functional groups, have also been found an ideal carrier to stabilize metal monatom when coordination chelating sites were introduced. Hai et al. summarized theoretical studies of MOFs, in which the performances of electrocatalysts for CO2RR were involved. The MOFs showed the better catalytic performance than that in the respect of potential barriers, overpotentials, the energy barrier of C–C dimerization, selectivity and activity for CO2RR.33 Thus, it is promising to investigate CO2RR with SACs from the perspective of theoretical calculations, which can guide the catalysts design. The intermediate products of CO2RR include but not limit *COOH, *CO, *CHO, *OCH2, *OCH3, *COH, *CHOH, *CH2OH, *CH3OH, *CH2 (as shown in Fig. 1). The reduction of CO2 can generate CO, methane, ethylene, acetylene and other organics.
 | ||
| Fig. 1 The intermediate and final products of quantum-theoretical studies on electro- and photo-chemical CO2RR with SACs. | ||
Above all, we need to provide an updated review to investigate the theoretical research on electro- and photo-chemical conversion of CO2 with SACs. The framework is also described with reaction products, which will provide guidance for the experimental design of catalysts.
2. Electrochemical conversion of CO2
In Electrochemical CO2RR (eCO2RR), electrocatalyst plays a key role because of its promising application on selectively synthesizing various C1, C2, and other polycarbonate compounds. However, the kinetics of eCO2RR is limited due to the stable linear molecule structure of CO2. At the same time, the transfer of multiple electrons and protons causes intricate final products, including HCOOH,34 CO,35 HCOH,36 CH3OH,37 CH4,38,39 as well as competitive hydrogen evolution reaction (HER).40 In order to improve the activity, selectivity and stability of the catalyst, the theoretical study on eCO2RR can be commenced from two aspects, the design of the catalyst and the expand of the reaction mechanism.At present, most of catalyst designs focus on the final products or reaction characteristics with different types of catalyst supports and active sites. It is proposed two strategies to improve: (1) increase the number of active sites; (2) enhance the intrinsic activity of single active site.41 In addition, based on the theoretical calculations, the catalysts can be designed and predicted in microscopic level. The reaction energy barriers of intermediates for different catalysts are calculated to further investigate the reaction pathway and mechanism of CO2RR.
2.1 eCO2RR to CO
As one of the most fascinating conversion reaction methods, eCO2RR to CO has attracted lots of attentions due to the Fischer–Tropsch (FT) type reaction.42 Meanwhile, it's a key point to enhanced the selectivity for the eCO2RR to CO in liquid environment.43 Most of these works aimed at clarifying the reaction paths and selectivity trends, yet the mechanism of catalytic is still debatable which will not be further studied in this paper. eCO2RR to CO is a 2-electron reduction reaction consisted with two basic steps.44 CO2 reductively adsorbed on the surface of catalyst involved a proton coupled electron transfer process. As a result, *COOH intermediate is formed. The adsorbed intermediate further turned into *CO when another electron proton transfer, finally desorbed from the catalyst surface. Recently, SACs have been widely studied for eCO2RR to CO because of its superior activity and selectivity compared with traditional catalysts. Table 1 summarizes the key parameters of eCO2RR to CO with various SACs, including the reaction energy (ΔE) of the main steps, Faraday efficiency, over potential and max current density.| SACs | ΔE (eV) | Faraday efficiency (%) | Overpotential (V) | Max current density (mA cm−2) | TOF (h−1) | Calculation method | Ref. |
|---|---|---|---|---|---|---|---|
| FeN5 | 0.77 | 97.0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
0.35 | — | — | DFT | 45 |
| CoPc | −0.1 | 99 | −0.8 | — | — | DFT | 1 |
| Co–N5 | 0.67 | 99.2 | −0.73 | 4.5 | 480.2 | DFT | 46 |
| Ni–N3–C | 0.66 | 95.6 | −0.65 | 6.64 | 1425 | DFT | 47 |
| Fe–N4 | 0.94 | 86.5 | −0.75 | 5 | — | DFT | 7 |
| Pd–N4 | — | 55 | −0.5 | — | — | DFT | 48 |
| Cu–N4–NG | 1.28 | 80.6 | −1 | — | — | DFT | 49 |
| Zn SAs/N–C | ∼0.6 | 94.7 | 0.33 | 121.5 | 8190 | DFT | 50 |
| ZnO@ZIF-NiZn | — | 98 | −1 | 34.3 | 9366 | DFT | 51 |
| Cu–N4 | −0.9 | 96 | −0.7 | — | — | DFT | 52 |
| N–C–CoPc NR | −0.34 | 85.3 | −0.7 | 30(0.18 V) | — | DFT | 53 |
| Co@Pc/C | 0.89 | 84 | −0.9 | 28 | — | DFT | 54 |
| SA-Fe/NG | ∼0.5 | 97 | −0.5 | — | — | DFT | 55 |
| Fe–N4 | 0.29 | 94 | 0.58 | 4.59(−0.58 V) | 1630 | DFT | 56 |
| Fe–N/CNF | — | 95 | −0.53 | 4.71 | 3104 | DFT | 57 |
| F-CPC | — | 88.3 | −1 | 37.5 | — | DFT | 58 |
| Ni–N4 | 0.89 | 98 | −0.5 | 35.9(−1.35 V) | — | DFT | 59 |
| Ni/Fe–N–C | 0.47 | 98 | −0.7 | 23.7(−1.0 V) | 7682 | DFT | 60 |
| Ni–NG | 0.12 | 95 | 0.55 | 50 | 2100 | DFT | 61 |
| Ni–N–C | ∼1.7 | 85 | −1 | 200 | — | DFT | 62 |
| Ni–SA–NCs | — | 99 | −0.8 | 50(−1.0 V) | — | DFT | 63 |
| Ni@NC-900 | — | 96 | −1 | −17 | — | DFT | 64 |
| Ni–N@NPC | 1.53 | 98.44 | 0.67 | 30.96 | 2825 | DFT | 65 |
| NiPc–OMe | ∼0.4 | 99.5 | −0.61 | −150 | — | DFT | 66 |
Besides, the single-atom metal-composite materials have exhibited unique properties in eCO2RR. The metal centers dispersed atomically and coordination-unsaturated are favorable for providing active sites. Wang et al. synthesized a graphene-based Fe–N5 SCAs as seen in Fig. 2a. The weak binding energy of CO is conducive to dissociation from the adsorption site of the catalyst. The valence electrons of Fe atom are reduced due to the axial pyrrole nitrogen ligand at Fe–N5 site, which weaken the back donation from the 3d orbital of Fe atom to the π* orbital of CO to realize the rapid desorption. High CO-selectivity is achieved as a result. Fig. 2b shows the diverse rate determining steps of Fe–N4 and Fe–N5 catalysts. The plane charge density by O–C–Fe–N (Fig. 2c) indicated the decrease of charge density of Fe atom. It confirms that the oxidation state of Fe in Fe–N5 result in a weaker CO adsorption strength.45 Deng et al. have doped different transition metals (Mn, Fe, Co, Ni and Cu) in the M–N4 coordination phthalocyanine (MePcs). According to the results of DFT calculation (Fig. 2d), the rate determining steps for Co, Fe, and Mn reaction paths are the dissociation of *CO on the catalyst surface. But for Ni and Cu, the formation of *COOH exhibits the highest energy barriers among all the steps in CO2RR. Fig. 2e describes the linear relationship between the adsorption energies of *CO and the reaction energies of CO desorption or *COOH formation. The two linear relations reveal an inverted volcano curve, and CoPc is the closest to the peak of volcano. Thus, the Co center in CoPc is the optimist reaction site by calculation. Fig. 2f shows the intermediate from CO2 adsorption on the catalyst surface to the final continuous hydrogenation to CO*.1 Cao et al. reported a method of anchoring atomically dispersed Co on polymer-derived N-doped hollow porous C spheres. The Co–N5 plays a critical role in the CO2 activation, *COOH formation and CO dissociation. Its catalytic activity is 15.5 times by CoPc.46
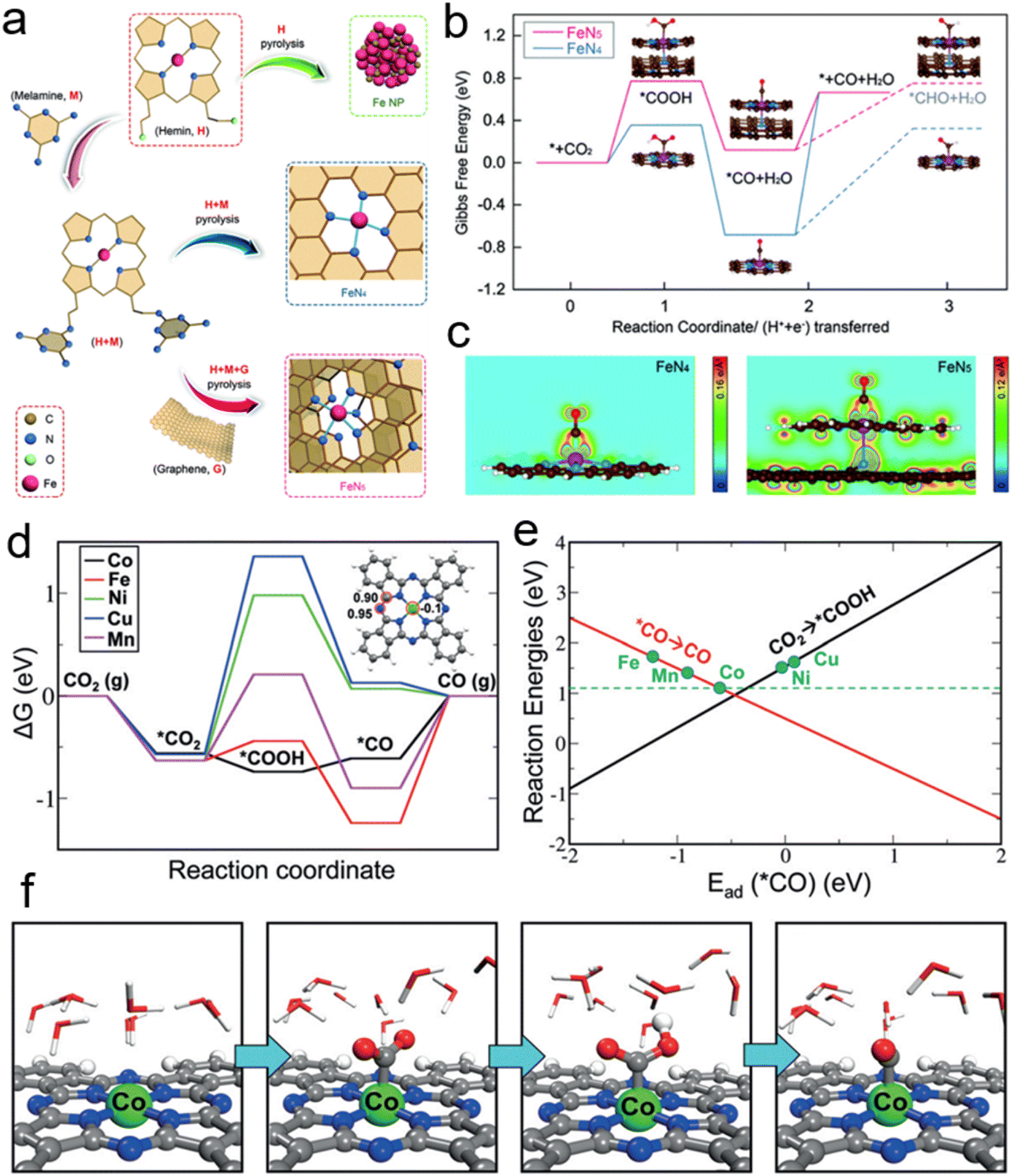 | ||
| Fig. 2 (a) The structures of single-atom FeN4 and FeN5 catalysts. (b) The free energy of the optimized intermediate for the electroreduction of CO2 to CO. (c) The plane charge density distribution of O–C–Fe–N. (d) The free energy of eCO2RR on several SACs. Illustration shows the adsorption energy of *COOH. (e) CO dissociation and *COOH formation tendency of catalysts doped with transition metals. (f) The adsorption configurations of intermediates in liquid environment. The balls in green, blue, grey, red and white represent Co, N, C, O and H atoms, respectively (reproduced from ref. 1 with permission from John Wiley and Sons, copyright 2018; reproduced from ref. 45 with permission from John Wiley and Sons, copyright 2019). | ||
Meanwhile, adjusting the coordination number can effectively influence the catalytic activity of eCO2RR. For instance, N-doped catalysts verified low coordination N could accelerate the formation of *COOH intermediate and promote eCO2RR to CO with the theoretical calculations;47 C-based SACs could exhibit low activation energy and favorable CO selectivity;7 Pd–N4 SACs also showed satisfactory reaction paths to the dissociation of CO.48 Recently, Qiao et al. successfully synthesized a N-doped graphene supported Cu SACs (Cu–N4-NG) which exhibited more favorable thermodynamics and kinetics than HER.49 They further studied the electrocatalytic activity of 3d transition metal doped M–N4–C (M = Mn, Fe, Co, Ni, Cu and Zn) catalysts for eCO2RR.67 Coincidentally, Lin et al. successfully prepared single atom Zn supported on NC nanofibers, and found that Zn–N4 could effectively reduce the free energy barrier of *COOH.50 Various Ni doped catalysts supporting have been extensively studied and demonstrated excellent performance in CO2RR.59–66 Wang et al. found that Ni–N@NPC with high dispersed Ni–N sites and desirable CO2 adsorption capacity exhibited high faradaic efficiency for CO (about 98.44%) and durable stability over 30 hours. The eCO2RR activity was indicated by DFT calculation which revealed that Ni–N sites can decrease the kinetic energy barriers for *CO2 to *COOH.65 Sun et al. reported a thermodynamically stable Ni single atom with a Ni–N3 structure and further wrapped in porous N-doped C shell. The free energy barrier for *COOH on Ni@N3 is much lower than that of Ni@N4.68
Moreover, Han et al. successfully prepared N, P-co-doped carbon aerogels, which could be used as electrocatalysts to efficiently reduce CO2 to CO. With high active area and conductivity, it provided a promising approach for non-metallic catalysts.69 Fig. 3a shows the configuration of N-/P-doped, and N, P-co-doped carbon. As shown in Fig. 3b, the free energy barrier is significantly reduced after doping with heteroatoms. Among them, the pyridinic N exhibited the lowest free energy barrier. Besides that, the adsorption of H* on both tri-pyridinic N and pyrrolic N is strong from the HER free energy in Fig. 3c. Thus, the pyridinic N is the active site in eCO2RR to CO. Moreover, the free energy changes of eCO2RR and HER on carbon Co doped with N–P were also calculated (Fig. 3d and e). N, P-co-doped carbon aerogels have considerable activity and selectivity for the eCO2RR to CO. Furthermore, they synthesized Sn4P3/reduced graphene oxide nanocomposites and Cu–Co bimetallic nanoparticles which exhibited excellent catalytic activity and selectivity of eCO2RR to CO.70–72
 | ||
| Fig. 3 (a) A series of N/P-doped, and N, P co-doped carbon configurations. (b) Gibbs free-energy (ΔG) diagrams for eCO2RR to CO. (c) ΔG for HER. (d) ΔG for eCO2RR to CO based on N, P-co-doped carbon. (e) ΔG for HER based N, P co-doped carbon (reproduced from ref. 69 with permission from John Wiley and Sons, copyright 2020). | ||
In addition, eCO2RR is thermodynamically more favorable compared with HER. For instance, N-doped carbon catalyst with Ni sites can exhibit nearly 100% CO selectivity and high activity.51 Atomically Fe3+ site facilitate high-efficiency CO2 electroreduction catalysis which catalyze eCO2RR to CO under a low overpotential. Zn supported on N-doped carbon catalyst can inhibit the generation of CO and increase the selectivity of CH4.29 Except for the use of carbon-based materials as the catalyst carrier, boron nitride is similar to graphene and has excellent physical and chemical properties. Therefore, transition metal-doped BN nanomaterials have been investigated in eCO2RR with SACs. Transition metals (TM = Sc to Zn, Mo, Ru, Rh, Pd and Ag) are doped on BN monolayers. The DFT calculation present that monoatomic TM–BN have excellent catalytic performance as catalysts for reducing CO2 to CH4.73
In recent years, the single atom alloy has developed catalytically active element composed of atoms dispersed in host metal with high catalytic selectivity. This catalyst combines the traditional advantages of alloy catalysts with the new features of SACs. The adsorption energy of *CO corresponds to the theoretical overpotential of methane or methanol, so as to achieve the screening of high-performance catalysts,74 further effectively reduce the activation barrier of CO2.75 The weak binding energy of CO greatly reduces the possibility of catalyst poisoning and further protonation, leading to perfect CO selectivity and stability.76
2.2 eCO2RR to alcohols
Another strategy to reduce CO2 is to generate safe and transportable liquid fuels. Alcohols has become an attractive product due to its core position in the fields of national defense, industry and agriculture.77 However, because of the complex reaction intermediates, high energy barrier and complicated mechanism of C–C coupling, the selectivity of alcohols is not optimistic.78By reducing the size of metal-based catalysts to single atom level,79,80 Wang et al. successfully prepared cobalt atom electrocatalysts/nanoparticles and carbon/selenium on N-doped porous carbon (CoSA–HC, CoNP–HC, Se@CoSA–HC, respectively). The catalyst exhibits distinguished coulombic efficiency, high reaction rate and strong stability. Fig. 4a shows the synthetic schematic diagram of Se@CoSA–HC, which is inserted selenium into carbon particles with hollow structure. The intermediate and free energy changes after structural optimization are observed in Fig. 4b. The transformation of 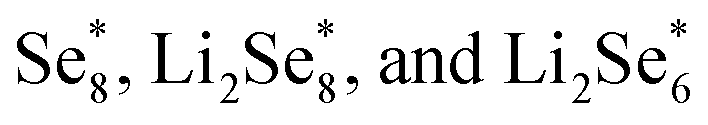 are exothermic process, while
are exothermic process, while 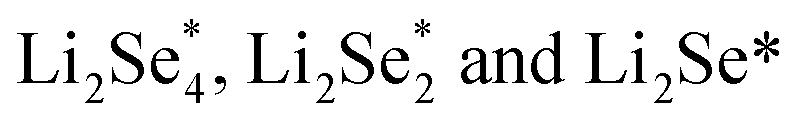 are endothermic. In addition, the free energy of reaction of
are endothermic. In addition, the free energy of reaction of 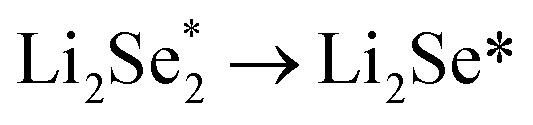 on Co–NC is lower than that of NC, which exhibits thermodynamically more favorable for the reduction of Se. By calculating the phase change barriers of
on Co–NC is lower than that of NC, which exhibits thermodynamically more favorable for the reduction of Se. By calculating the phase change barriers of  on Co–NC and NC (Fig. 4c and d), Co atoms are the active center of the catalyst.81 In addition, SACs with uniformly dispersed active metal centers are highly unsaturated coordinated and exhibit quantum confinement effect,82 resulting in a high electron density distribution near the Fermi level, which contributes to the rapid electron transfer in the catalytic reaction process.83,84 MXene anchored SACs by selectively etching hybrid A layers (Al and Cu) for eCO2RR have been studied by DFT calculation. The monoatomic Cu with unsaturated electronic structure effectively reduces the energy barrier of the rate determining step. The rate determining step of the reaction path is the conversion of HCOOH* to the intermediate of adsorbed CHO*, which was helpful for eCO2RR to CH3OH. As shown in Fig. 4e, through the charge density difference of SACs–Cu–MXene, Cu has a positively charged Cu–O3 structure and the designed reaction path. Based on DFT calculations (Fig. 4f), SACs can significantly reduce the reaction barrier of eCO2RR.85 Co@Cu SACs would also exhibit ideal performance in converting CO2 to CH3OH. The narrowed Co d-band stabilizes the adsorption of CO2 on the surface and further enhances the bonding of COH* to the active site. While stabilizing the intermediate, it reduces the activation energy of COH* for further hydrogenation. It effectively prevents the C–C coupling and enhanced the selectivity of the reaction to CH3OH.9
on Co–NC and NC (Fig. 4c and d), Co atoms are the active center of the catalyst.81 In addition, SACs with uniformly dispersed active metal centers are highly unsaturated coordinated and exhibit quantum confinement effect,82 resulting in a high electron density distribution near the Fermi level, which contributes to the rapid electron transfer in the catalytic reaction process.83,84 MXene anchored SACs by selectively etching hybrid A layers (Al and Cu) for eCO2RR have been studied by DFT calculation. The monoatomic Cu with unsaturated electronic structure effectively reduces the energy barrier of the rate determining step. The rate determining step of the reaction path is the conversion of HCOOH* to the intermediate of adsorbed CHO*, which was helpful for eCO2RR to CH3OH. As shown in Fig. 4e, through the charge density difference of SACs–Cu–MXene, Cu has a positively charged Cu–O3 structure and the designed reaction path. Based on DFT calculations (Fig. 4f), SACs can significantly reduce the reaction barrier of eCO2RR.85 Co@Cu SACs would also exhibit ideal performance in converting CO2 to CH3OH. The narrowed Co d-band stabilizes the adsorption of CO2 on the surface and further enhances the bonding of COH* to the active site. While stabilizing the intermediate, it reduces the activation energy of COH* for further hydrogenation. It effectively prevents the C–C coupling and enhanced the selectivity of the reaction to CH3OH.9
 | ||
| Fig. 4 (a) Schematic of Co/N doped hollow porous carbon. (b) The energy distribution of Li2Se compounds/cluster reduction on NC/Co–NC carriers. (c) The charge density of SACs–Cu–MXene. The green/orange area indicates the increase/decrease in electrons. The reaction path for the eCO2RR to CH3OH. (d) The free energy change trend of eCO2RR to CH3OH (Reproduced from ref. 81 with permission from Springer Nature, copyright 2020; reproduced from ref. 85 with permission from American Chemical Society, copyright 2021). | ||
Various efforts have been made on reducing the activation energy to promote eCO2RR to CH3OH. The normal reaction path for calculation is *CO2 → *CO → *CHO → *CH2O → *CH2OH and *CH3O → CH3OH.86 It could significantly enhance CO2 protonation ability by reducing the distance between adjacent monomers and catalysis.87 By DFT calculations, Ding et al. have discovered that TM (TM = Fe, Co, Ni, Cu, Ir and Pt) could effectively improve the catalytic activity and enhance the redistribution of electrons. Besides that, they could further activate the subsequent reaction intermediates, and finally selectively generate CH3OH.88
The instability safety limits the reserves of precious metal catalysts, which is the obstruction of industrial production, as well large-scale preparation. In addition, the stability is also an insurmountable gap for non-precious metals. Meanwhile, lots of attempts have been researched on metal catalysts, the main advantages of which catalysts are low overpotential, high current density, large specific surface area and less metal consumption. Cobalt phthalocyanine could be immobilized on carbon nanotubes to selectively catalyze eCO2RR to CH3OH. The electrocatalytic reaction followed domino process: CO2 → CO by two electrons, CO → CH3OH by four electrons.89 Meanwhile, TM dimer doped on graphene with adjacent single vacancies could reduce overpotential and enhanced current density in eCO2RR, which further improve the catalytic performance.2 By constructing the network inter-perforation and self-supporting structure, the probability of single atoms at the interface is greatly increased, high current density can be achieved at a lower metal single atom content as a result, which has certain practical application prospects.90 Besides that, new materials including 2D, 0D, and Janus material have been found to be promising as efficient electrocatalysts for eCO2RR. For instance, Chen et al. have developed tricycloquinazoline based 2D c-MOFs successfully, which showed high selectivity toward CH3OH with Faradaic efficiency up to 53.6%. The eCO2RR performance is even better than Cu- and MOFs-based electrocatalysts.91
In recent years, ethanol with higher energy density as an ideal fuel has also been a research hotspot.92 Copper clusters supported by catalyst had a mixed charge state of Cu0 and Cux+. The coverage of *CO on the surface was increased, making it possible to couple C–C on the Cut0–Cubx+ atomic interface, and finally generated C2H5OH.93 The construction of dual active sites by loading single atom on carrier provided a new attempt for the design of the catalyst through the synergistic effect of the single-atom center and substrates.94
At present, the design strategies of CO2 to alcohols monatomic catalysts mainly include by SACs in covalent organic frameworks. However, due to the complicated intermediates and reaction pathways, the mechanism of eCO2RR is still ambiguity. Meanwhile, varieties of by-products will be generated in the related reaction path. The reasonable design of the carrier, the regulation of the reaction conditions, and even the structure of the carrier are all significant effects on the selectivity of products.
2.3 eCO2RR to acids
Formic acid is one of the basic organic chemical raw materials. It is widely used in the industry of pesticide, leather, rubber, dyestuff and so on. It also has the potential application for hydrogen storage and fuel cells.95 In addition, formic acid is also an indispensable intermediate for the synthesis of higher alcohols. In fact, CO2 and H2 can generate formic acid relatively easily except the kinetic obstacles of reaction process that need to be resolved.96 Therefore, it is very necessary to find suitable, efficient and stable catalysts.As a very ideal electrocatalyst, indium (In) can efficiently reduce CO2 to formic acid, but it cannot be further promoted because of the high potential and low FE. In SACs based on N-doped carbon matrix derived from the metal–organic framework have found to be excellent in this regard. The catalyst has well-dispersed Inδ+–N4 atomic interface sites, which can efficiently catalyze eCO2RR to formic acid. Inδ+–N4 atomic interface has lower free energy for the formation of intermediate HCOO*, which is beneficial to improve the catalytic activity for eCO2RR. It provides a feasible strategy for precise adjustment of indium catalysts at the atomic level for efficient eCO2RR.97 Meanwhile, TM (TM = Sc, Ti, V, Cr, Mn, Fe, Co, Ni, Cu and Zn) SACs can stably be supported on porphyrin framework. This new catalyst designed strategy on eCO2RR to acids provides a different enlightenment for experimental design.21 Hai et al. found that, for some MOFs, the selectivity of HCOOH depended on the adsorption energy difference between *HCOO and *COOH by DFT calculations.98 Thus, the biggest obstacle is that HER would severely inhibit eCO2RR.99
Graphite carbonitride (g-C3N4) contains unique chemical composition, π-conjugated electronic structure and strong nucleophilic ability. Thus, it can be used as multifunctional catalyst in traditional organic catalytic reaction, such as green carrier and energy storage material. Based on first-principles calculations, systematic studies have been carried out on the geometric configuration, electronic structure and eCO2RR catalytic performance of graphdiyne-supported alkali metals which had great potential as SACs for eCO2RR. The conversion mechanism by constructing model of S-doped Cu has been calculated, which could effectively enhance the adsorption strength of *CO to further selectively generate HCOOH,100 as well as Ru SACs on layered double hydroxide.101 low-coordination active center and oxygen vacancies could selectively generate HCOOH while inhibiting HER.102
The heterogeneity of the catalyst support surface limits the further development of SACs, including synthesis and catalytic performance. In recent years, SACs have begun to construct active centers at the molecular level to improve the stability of the active centers and realize the “quasi-homogeneous” application. Zhang et al. successfully prepared a porous organic polymer containing pyridine-amide groups with Ir SACs on the porous organic polymer. It exhibits excellent catalytic activity in the CO2RR to formic acid with excellent cycle stability. The SACs have the same catalytic reaction mechanism as the homogeneous catalysis process.103 The solid micelle can increase the number of active sites of the carrier species, meanwhile, the reaction conditions are more tolerant, and the product can be easily separated.104
2.4 eCO2RR to other organics
In addition to C1 and C2, the products of eCO2RR also included olefins,105 alcohols, ketones,106 aldehydes,107 and even C3 and C4 products.108 Compared with the C1 product, the multi carbon products have more considerable energy density. Moreover, multi carbon products play an important role in the chemical industry.109 Ethylene is a basic raw material for the synthesis of plastics, rubber, fiber and ethanol, which occupies an important position in the petrochemical industry. However, eCO2RR to C2+ also faced with the obstacles of C–C coupling complex process, as well as the problem of adsorbate coverage on the surface.CO2 can be adsorbed on the surface with strong adsorption strength. Metal doped catalysts could show high selectivity and activity for the formation of CH3OH.110 Fig. 5a and b show the possible reaction pathway about C1 and C2.111 Recently. The reaction mechanism of C2H4 was studied by constructing asymmetric.112 Cu/C can significantly promote coupling and maintain a moderate binding energy between the intermediate and the carrier.113 For instance, Cu–C3N4 has been studied on the adsorption behavior of intermediates in the eCO2RR process. The catalyst can effectively reduce the onset potential with excellent catalytic activity. The generation rate of C2 is significantly better than that of Cu–NC.114 The excellent selectivity for C2H4 on the Cu-based MOFs for eCO2RR has been reported by Yang et al. with isolated Cu–S motifs in MOF based precatalysts.115 At present, the C3 and C4 products have also begun to arouse widespread concern on multi-carbon products. Dismukes et al. synthesized nickel phosphide compounds, which were used to reduce CO2 in aqueous solutions and studied the catalytic performance of eCO2RR. The product selectivity has a significant change by increasing the phosphorus content. They proposed a reaction pathway for the efficient synthesis of multi-carbon products from CO2 through formic acid and formaldehyde intermediates without CO intermediates.108
 | ||
| Fig. 5 The eCO2RR reaction pathways to (a) C1 products and (b) C2 products on Cu2@CN catalyst (reproduced from ref. 111 with permission from John Wiley and Sons, copyright 2020). | ||
Although lots of works have been done on eCO2RR in recent years, most of them only stayed in the laboratory stage. How to find the catalyst with high efficiency and selectivity becomes a challenge.116 Besides, most of the current catalyst designs have limitations due to that CO2RR has complicated reaction paths and produces intermediates.117 In addition, there is still lack of clear reaction mechanism. Many obstacles need to be overcome for SACs with excellent performance to be promoted and applied to industrial production.118 Above all, based on what has been achieved so far, we believe that there will be a qualitative breakthrough in the practicality of eCO2RR in the future.
3. Photochemical conversion of CO2
Investigating photochemical CO2RR (pCO2RR) with SACs is a pressing environmental issue due to the increasing concentration of CO2 in the atmosphere. Since Inous et al. first reported pCO2RR to organic compounds in gas–solid reaction systems, a great deal of researches have been devoted to the development of photocatalysts for pCO2RR.11 Photocatalysts mainly include the following types: (1) metal oxides, mainly composed of d0 structured (Ti4+, Zr4+, Nb5+, Ta5+, W6+, and Mo6+) and d10 (In3+, Ga3+, Ge4+, Sn4+, and Sb5+) metal cations,12,119 such as TiO2, ZrO2. XTaO3 (X = Li, Na, K), InVO4, ZnGa2O4, etc.;120–125 (2) metal sulfides such as XS (X = Cd, Zn);126–129 (3) metal nitrides such as GaN,130 TaON;131 (4) layered metal hydroxides (referred to as LDH), such as FeNi-LDH/g-C3N;132 (5) metal organic framework materials (MOFs), such as MOF-525-Co;133 (6) non-metallic semiconductors, such as g-C3N4,134 and graphene.135Moreover, metal oxides,16,136–138 MOFs,139,140 C-based materials,141,142 and LDH143,144 are the common carriers for SACs. It has been reported that TiO2,145–147 CeO2,148,149 MOFs,150–152 graphene,153,154 graphitic carbon nitride,155–157 LDHs158,159 have shown high activity, stability and selectivity in pCO2RR. The strong interaction between the single metal atom and the carriers is beneficial to improve the catalytic performance. Meanwhile, the application of pCO2RR with SACs is an effective way to improve the rate of photocatalysis and achieve carbon emission reduction.
However, the high recombination rate of electrons and holes, the narrow wavelength-range of light to achieve the exited state, and the small specific surface area of conventional semiconductor photocatalysts limit the catalytic rate.160,161 The photocatalytic performance of catalysts can be improved by co-catalysts and the noble metals, for example that Pt and Pd, are considered to be efficient co-catalysts.162,163 The high cost and low abundance of precious metals limited the industrial application. Therefore, large specific surface area, stability, high selectivity, ability to inhibit recombination are the critical issues to design practical photocatalysts.164–166
Single-atom photocatalysts are photoactive materials in which metals are present as SACs uniformly dispersed on the support. The catalytic performance of SACs depends largely on the interaction between the support and metal.167 For single atom photocatalysts, the introduction of SACs can effectively inhibit the electron–hole recombination, as well as increase the activity and selectivity.168,169 The favorable SACs dispersion effect and prevention of metal SACs aggregation play important roles in the final catalytic activity of monoatomic photocatalysts. CO2RR with SACs is a promising strategy for the conversion of CO2 to hydrocarbon fuels such as CO, CH4, and CH3OH.13,168,170
The pCO2RR requires the following steps (Fig. 6): (1) the photocatalyst is irradiated by light. It leads to the excitation of electron from valence band to conduction band, leaving a hole in the valence band, which is the photo-electron/hole pair;171 (2) the photogenerated electrons and holes diffuse and separate. The electrons/holes transfer to the reduction/oxidation sites, respectively; (3) CO2 is adsorbed onto the surface; (4) a redox reaction occurs on the photocatalyst surface, where the electrons reduce CO2 to CO,172,173 CH4,169,174,175 HCOOH,176–178 CH3OH179–181 or other hydrocarbons,170,182,183 and the holes oxidize water to molecular O2 by cavities;152,154,184 (5) products desorbed from the catalyst surface. The pCO2RR and potentials are also exhibited. Fig. 7 shows the band edges commonly used in photocatalysts.185 In this section, we summarize the theoretical studies of SACs in pCO2RR, including CO2 to CO, methane, and CH3OH.
 | ||
| Fig. 6 The mechanism and steps of pCO2RR. Including the electrons and reduction potential required for the pCO2RR to different products with respect to the standard hydrogen potential NHE at pH 7. | ||
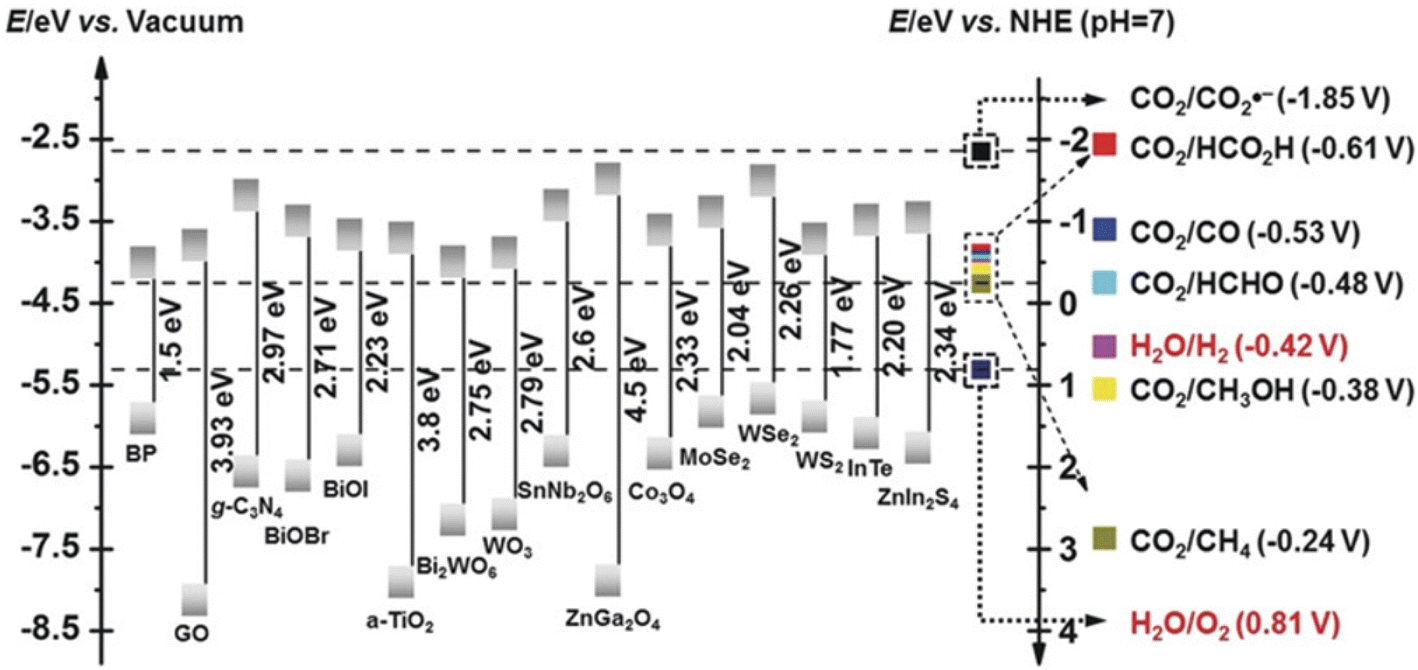 | ||
| Fig. 7 CB and VB potentials of common semiconductors with CO2 and water redox couples at PH 7 (reproduced from ref. 185 with permission from John Wiley and Sons, copyright 2018). | ||
3.1 pCO2RR to CO
Based on the thermodynamic principle of proton-assisted multi-electron transfer to reduce CO2, the specific pathway for CO2 reduction to CO is: CO2 + 2H+ + 2e− → CO + H2O,168,184,186 for pCO2RR relative to the Normal Hydrogen Electrode (NHE) in aqueous solution.187 The potential level of the conductive band of the photocatalyst determines the production of pCO2RR.188 In order to overcome the slow thermodynamic process and promote the reaction rate, the potential (vs. NHE) of the conduction band needs to be more negative than the reduction potential of CO2. The energy band structure of semiconductor photocatalysts is extremely important for the catalytic efficiency. How to abbreviate the residence time of CO on the catalyst surface and accelerate the desorption from the catalyst surface into the gas phase are the key points to improve the catalyst selectivity.189–191 The energy band structure of conventional semiconductor catalysts is far from ideal. In recent years, many researches have been conducted to improve the photogenerated electron–hole separation efficiency of photocatalysts by modifying the semiconductors to increase the light absorption rate and enhance the CO2 adsorption capacity.The combination of monatomic transition metals and polymeric carbon nitride has been an effective strategy for photocatalytic reactions. Li et al. reported the preparation of copper carbon nitride nanocatalysts (Cu–CCN) based on the doping of monatomic Cu into CN by molten salt and reflux methods to promote the photoreduction of CO2 to CO.192 Density functional theory calculations further clarified the mechanism of the pCO2RR process (Fig. 8a), where the CO2 adsorbed on the surface of the Cu–CCN catalyst was hydrogenated to *HCOO, *CO, successively. And CO desorbed from the surface, finally. Wang et al. analyzed the mechanism of CN-loaded Cu single-atom pCO2RR using DFT and found that the active center in the form of C–Cu–N2 not only promoted charge transfer but also reduced the energy potential energy for pCO2RR (Fig. 8b).165 Meanwhile, Ji et al. reported an atomic confinement and coordination design strategy applied to design various metal monatomic catalysts.193 Er monatomic catalysts (Er/CN-NT) loaded on carbon nitride nanotubes with tunable metal dispersion density were synthesized for pCO2RR. According to the computational hydrogen electrode (CHE) model, it is confirmed that the favorable production is CO, instead of CH4 on rare-earth erbium atom catalysts.
 | ||
| Fig. 8 (a) Schematic of the tentative photocatalytic mechanisms and reaction pathway of Cu–CCN for pCO2RR. (b) Schematic illustration of Cu/CN hybrid for pCO2RR. (c) Photocatalytic selective CO2RR on Ni-TpBpy. (d) Proposed reaction mechanism for the photoconversion of CO2 into CO on Ni-TpBpy (reproduced from ref. 192 with permission from American Chemical Society, copyright 2020; reproduced from ref. 165 with permission from American Chemical Society, copyright 2020; reproduced from ref. 194 with permission from American Chemical Society, copyright 2019). | ||
In addition to the above-mentioned design photocatalyst strategies, we have continuously developed single-atom photocatalyst design strategies that favor adjustable metal atom coordination and demonstrated their catalytic effects on the pCO2RR to CO. Zhong et al. synthesized Ni-rich single-atom 2,2′-bipyridine covalent organic frameworks of photocatalyst (Ni-TpBpy) (Fig. 8c and d).194 Ni-TpBpy was effective in improving the selectivity of CO generation in aqueous solution. Ni atom was stabilized by the dipyridine coordination unit based on the special structure. The hydrogen bonding between ketone node and Ni–CO2 reaction intermediate promoted the pCO2RR preferential to H2 formation. Meanwhile, the adsorption of CO2 by Ni-TpBpy was greatly improved due to the Lewis acid–base interaction between the loaded Ni ions and absorbed CO2 molecules. DFT calculations used to investigate the mechanism for pCO2RR confirmed that Ni-TpBpy could reduce the CO2 molecules and activate Ni site to Ni–COOH, which would combine with H+ to Ni–CO intermediate, followed by CO desorption from Ni–CO. Oxygen vacancies, as common defects in oxides or hydrogens carbide surfaces, has a great impact on pCO2RR.195,196 For instance, CO production rate of Co–Bi3O4Br was approximately 4 and 32 times than atomic layer Bi3O4Br and bulk Bi3O4Br.164 Ni atoms could occupy the oxygen vacancies of ZrO2, efficiently achieved pCO2RR to CO, which is 6 and 40 times by that of defective ZrO2 and complete ZrO2, respectively.63 Chen et al. have studied the adjustable catalytic performance and the modulation mechanism of Ni2+ doped NH2-MIL-125-Ti (NH2-MIL-125-Nix/Ti) with different Ni2+/Ti4+ molar ratio (x = 0.5–1.5%) via an in situ doping method. They found that Ni around O atoms of Ti oxo clusters of NH2-MIL-125 can change the electronic structure of the clusters thus lead to the improvements of electron transfer and charge separation. The highest CO2 conversion rate with 98.6% CO selectivity was exhibited with the molar ratio 1%.197
So far, researchers have focus on using atomic confinement strategy, coordination design strategy, and defect engineering strategy to increase the active sites with CN, metal–organic backbone, and photocatalysts of monatomic metal to achieve improving photocatalytic activity. Thus, the pCO2RR still have the obstacle in accelerating the rate of CO desorption, which is also the key to improve photocatalytic selectivity. So, it is also challenging to improve the CO2 photoreduction activity and product selectivity.
3.2 pCO2RR to CH4
CH4 is the main component of natural gas. Due to the limited petroleum resources, it will be a high-quality gas candidate fuel. pCO2RR to CH4 requires 8-electron reduction: CO2 + 8H+ + 8e− → CH4 + 2H2O.198 The CO2 molecule is very inert, which limits the catalytic conversion. At the same time, the products of CH4 are frequently accompanied by CO and H2, so that the efficiency and selectivity will be influenced. Thus, how to improve the quantum efficiency and increase the selectivity of pCO2RR become the key issue for photocatalysis.Zhang et al. realized the modular optimization of the metal–organic framework by injecting monoatomic Co SACs on porphyrin,133 so as to synthesize the catalyst with atom dispersion (Fig. 9a). The catalyst significantly improves the electron–hole separation efficiency of the porphyrin unit and the CO2 adsorption capacity, which realize the directional migration of photo-excitons from the porphyrin to Co center. It provides long-lived electrons for CO2 molecules adsorbed on the Co center. The addition of unsaturated Co SACs achieves the enhancement of pCO2RR, thus, greatly improves the selectivity of CH4.
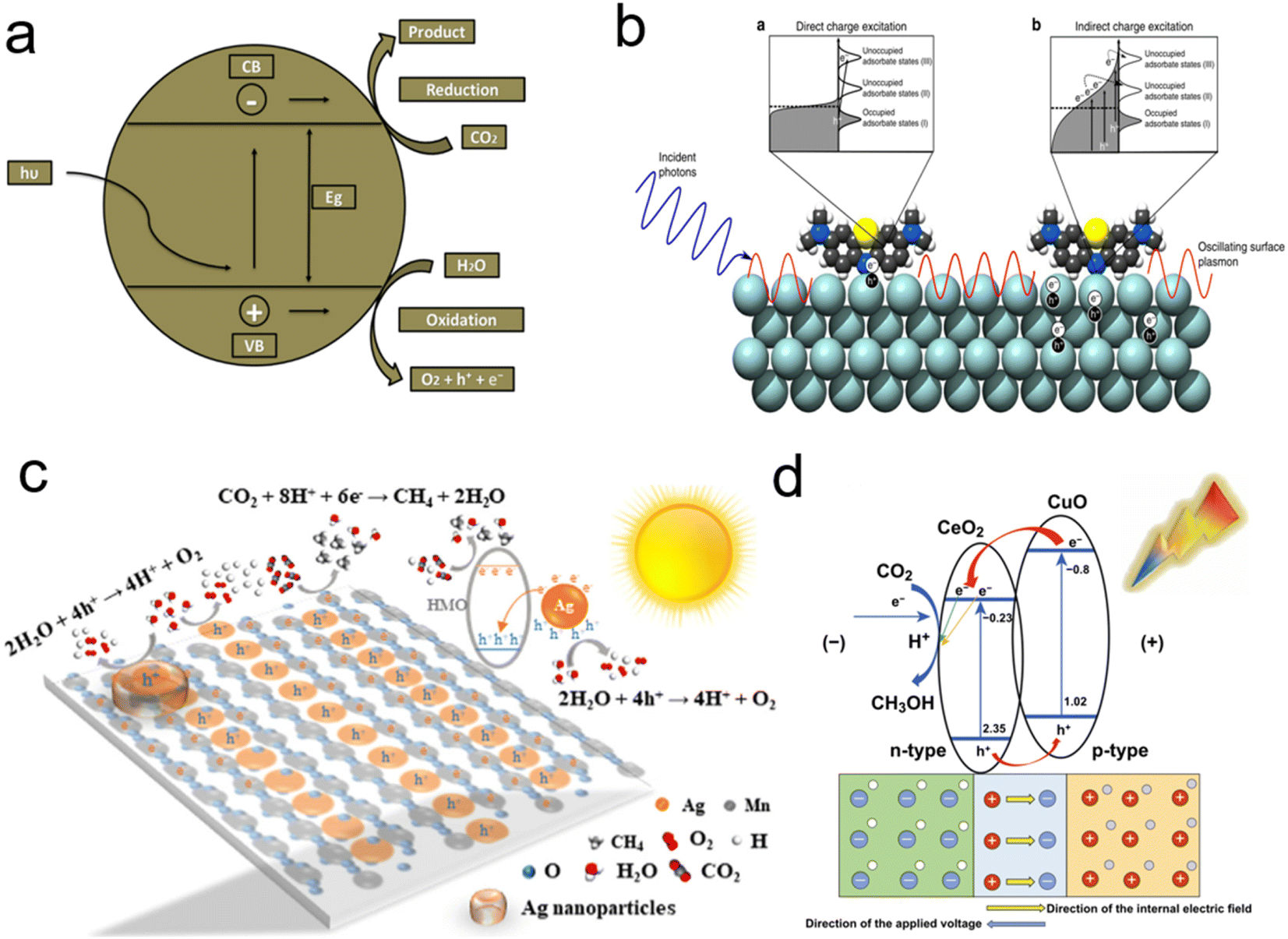 | ||
| Fig. 9 (a) Schematic representation for construction of Cu-based ZIF catalyst and representation of pCO2RR; (b) illustration of LSPR-mediated charge excitation mechanisms; (c) pCO2RR mechanisms of Ag-HMO; (d)Proposed mechanisms for PEC pCO2RR on the CeO2 NPs/CuO NPs composite catalyst (reproduced from ref. 133 with permission from MDPI, copyright 2018; reproduced from ref. 199 with permission from Springer Nature, copyright 2016; reproduced from ref. 200 with permission from Elsevier, copyright 2019; reproduced from ref. 201 with permission from Springer Nature, copyright 2020). | ||
Due to the heat generated by surface plasmon resonance of noble metals, the excited electrons can be easily transferred into the conduction band of the semiconductor,202 which could facilitate the separation of electrons and holes to increase pCO2RR to CH4. Liu et al. reported Au–Cu alloy catalysts which was adjusted the structure of co-catalysts and improved the electron capture capacity.203 Meanwhile, the A3Bi2I9 (A+ = Rb+, Cs+, CH3NH3+) were synthesized by Bhosale et al.,204 which has shown excellent photocatalytic activity for pCO2RR at the gas–solid interface. Besides, precious metal catalysts such as Ag, Au, and Pt loaded on metal-catalysts as SACs can effectively increase photoactivity. Ag nanoparticles were widely used to enhance photocatalytic reaction rates due to localized surface plasmon resonance (Fig. 9b). The “hot electrons” excited from surface plasmon resonance will be injected into the conduction band of the metal–semiconductor, thus enhancing photocatalytic activity.
Hydrothermal and impregnation methods have been used to develop Ag SACs supported on hollandite manganese dioxides (Ag-HMO) for pCO2RR to CH4.200 During the calcination process, the Ag atoms enter the HMO lattice tunnel through the anti-Oswald maturation mechanism to promote the formation of single-atom Ag chains, while effectively inhibiting Ag agglomeration to improve the catalytic efficiency. Recently, the heterostructured photocatalysts have been considered as an effective strategy to promote the spatial separation of photogenerated electrons and holes. The catalyst with NiMOF/CN heterojunction structure exhibited a photocatalytic activity 18 times larger than pure carbon–nitrogen.152 The Pd–Au hetero junction model co-catalyst have also been prepared,175 in which electron-deficient Pd massively inhibited the competition for H2 generation and enhanced the electron–hole separation, while the electron-rich Au promote the 8-electron reduction of CO2 to CH4 at the Pd–Au interface.
Even though the synthetic strategies are continuously innovative, CO and H2 are still formed along with the pCO2RR to CH4, which reduces the selectivity of CH4. The key for improving the pCO2RR activity and CH4 selectivity is preventing or reducing this phenomenon. Otherwise, this aspect of research would play a great potential in the field of theoretical studies of synthetic photocatalysts for pCO2RR, making it a viable technology for a carbon-neutral future.
3.3 pCO2RR to CH3OH
The two main reaction systems for pCO2RR by water reducing agent are liquid- and gas-phase systems.205,206 CH3OH as the reduced liquid product generally exists in liquid phase system. In addition, photocatalytic reduction of H2O produces relatively strong hydrogen side reactions, which can reduce the selectivity of the products. Therefore, the effects of different catalytic systems on the reaction products should be taken into account. Kavil et al. synthesized Cu/C–TiO2 nanocatalysts for pCO2RR to CH3OH in seawater using sol–gel method by co-doping Cu and C into TiO2.207 The pathway followed with thermodynamic principle is:| nTiO2 + 6hν → 6ecb− + 6hνb+ | (R-1) |
| 6H+ + 6ecb− → 6H˙ | (R-2) |
| CO2 + 6H˙ → CH3OH + H2O | (R-3) |
| 6OH− + 6hνb+ → 3/2 O2 + 3H2O | (R-4) |
The overall reaction is the following:
| 2CO2 + 4H2O → 2CH3OH + 3O2 | (R-5) |
It is shown that Cu doping into TiO2 can effectively trap carriers and inhibit electron–hole recombination, thus effectively improve the efficiency and selectivity for CH3OH generation. The effects of factors on the pCO2RR by Cu/C–TiO2 were investigated. Pan et al. used CeO2/CuO p–n junction photocatalyst with Cu cocatalysts for pCO2RR to CH3OH (Fig. 9d).201 Under light irradiation, the photoexcited electrons in CuO transfer to cerium oxide because the CB of cerium oxide is more positive than that of copper oxide, while the holes (h+) generated in the VB of CeO2 under light irradiation will migrate to the VB of CuO. By electron–hole exchange between CuO and CeO2, electron–hole pair complexation is suppressed and the effectiveness of electron reduction of CO2 on cerium dioxide nanoparticles is increased. The combination of p-type copper oxide and n-type cerium oxide make the carrier in the cerium oxide nanoparticles/CuO nanoparticles catalyst concentration is 108 times higher than that of the copper oxide nanoparticle catalyst, which greatly improves pCO2RR performance. Meanwhile simple catalyst modification (i.e. cerium dioxide electrodeposition) can significantly increase the carriers' concentration and electron availability, thus chemically reducing CO2 plasma to valuable commercial chemicals and providing a theoretical model for pCO2RR.
Cu-based catalysts have been widely applied for pCO2RR to CH3OH by different design strategies to increase the active sites and improve the catalytic efficiency. Meanwhile, MOFs are considered as potential light-driven catalysts, which have imidazolium or aromatic carboxylate-based linkers.208,209 Goyal et al. developed a catalyst (CuZrlm) with Cu combined with zirconium oxide loaded on imidazoline skeletons,13 which can effectively improve the photocatalytic performance of imidazolines as potential catalysts for pCO2RR to CH3OH. The authors stated that the presence of Cu2+ oxidation state and uniform dispersion of the active metal are the main reasons for the improved yield, because Zr introduction can distort the symmetry of the Cu2+ oxidation state thereby stimulating charge transfer.
Although pCO2RR research has made significant breakthroughs in artificial action and photosynthesis, the industrialization is still limited by high costs, carrier limitations, and imperfect analytical characterization techniques. To further improve the performance of pCO2RR, we should work on finding new carrier materials and designing different photocatalyst structures, as well as proposing new pathways for pCO2RR to design highly selective and stable photocatalysts.
4. Conclusions and outlook
In this review, we focus on the theoretical studies of electro- and photo-chemical CO2RR aspects with SACs. The conversion of CO2 to CO, CH4, alcohols, acids and other organics have been discussed in detail. The construction of SACs on different unique support, along with the detail catalytic mechanism and improvement of product selectivity were involved.In spite of gratifying results on the CO2RR, the challenges of how to improve the activity and selectivity of CO2RR products, as well as to find green mild conversion environment for large-scale industrial implementation are still the issues need to be solved in the future. The influences of electrode morphology, surface structure and reaction conditions on CO2RR are still required to be studied to improve the stability of catalysts. For the theoretical calculations, a multi-scale computational model covering all aspects, rather than a collection of theoretical models applicable to specific cases is also needed to achieve.
Author contributions
Q. Yu supervised the project and organized the collaboration. L. Jiang, Q Yang co-wrote the manuscript with input from all authors. Z. Xia and X. Yu summarized the electro-CO2RR, M. Zhao and Q. Shi summarized the photo-CO2RR.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 92061109), Natural Science Basic Research Program of Shaanxi (No. 2021JCW-20 and 2022KJXX-18), Open Project Program of Fujian Key Laboratory of Functional Marine Sensing Materials (Grant no. MJUKF-FMSM202002). The support by the Guangdong Provincial Key Laboratory of Catalysis (No. 2020B121201002) is also acknowledged.References
- Z. Zhang, J. Xiao, X. J. Chen, S. Yu, L. Yu, R. Si, Y. Wang, S. Wang, X. Meng, Y. Wang, Z. Q. Tian and D. Deng, Angew Chem. Int. Ed., 2018, 57, 16339–16342 CrossRef CAS PubMed.
- Y. Li, H. Su, S. H. Chan and Q. Sun, ACS Catal., 2015, 5, 6658–6664 CrossRef CAS.
- B. An, Z. Li, Y. Song, J. Zhang, L. Zeng, C. Wang and W. Lin, Nat. Catal., 2019, 2, 709–717 CrossRef CAS.
- Q. Liu, X. Yang, L. Li, S. Miao, Y. Li, Y. Li, X. Wang, Y. Huang and T. Zhang, Nat. Commun., 2017, 8, 1407 CrossRef PubMed.
- H. Liang, B. Zhang, P. Gao, X. Yu, X. Liu, X. Yang, H. Wu, L. Zhai, S. Zhao, G. Wang, A. Bavel and Y. Qin, Chem. Catal., 2022, 2, 610–621 CrossRef.
- Q. He, D. Liu, J. H. Lee, Y. Liu, Z. Xie, S. Hwang, S. Kattel, L. Song and J. G. Chen, Angew Chem. Int. Ed., 2020, 59, 3033–3037 CrossRef CAS PubMed.
- W. Ni, Z. Liu, Y. Zhang, C. Ma, H. Deng, S. Zhang and S. Wang, Adv. Mater., 2021, 33, 2003238 CrossRef CAS PubMed.
- M. Zhang, Z. Hu, L. Gu, Q. Zhang, L. Zhang, Q. Song, W. Zhou and S. Hu, Nano Res., 2020, 13, 3206–3211 CrossRef CAS.
- Z. Zhao and G. Lu, J. Phys. Chem. C, 2019, 123, 4380–4387 CrossRef CAS.
- S. Kanan, M. A. Moyet, R. B. Arthur and H. H. Patterson, Catal. Rev., 2019, 62, 1–65 CrossRef.
- T. Inoue, A. Fujishima, S. Konishi and K. Honda, Nature, 1979, 277, 637–638 CrossRef CAS.
- S. N. Habisreutinger, L. Schmidt-Mende and J. K. Stolarczyk, Angew Chem. Int. Ed., 2013, 52, 7372–7408 CrossRef CAS PubMed.
- S. Goyal, M. S. Shaharun, G. S. Jayabal, C. F. Kait, B. Abdullah and L. J. Wei, Catalysts, 2021, 11, 346 CrossRef CAS.
- E. A. Dolgopolova, A. M. Rice, C. R. Martin and N. B. Shustova, Chem. Soc. Rev., 2018, 47, 4710–4728 RSC.
- S. Saini, P. K. Prajapati and S. L. Jain, Catal. Rev., 2020, 64, 631–677 CrossRef.
- B. Qiao, A. Wang, X. Yang, L. F. Allard, Z. Jiang, Y. Cui, J. Liu, J. Li and T. Zhang, Nat. Chem., 2011, 3, 634–641 CrossRef CAS PubMed.
- X. F. Yang, A. Wang, B. Qiao, J. Li, J. Liu and T. Zhang, Acc. Chem. Res., 2013, 46, 1740–1748 CrossRef CAS PubMed.
- A. Wang, J. Li and T. Zhang, Nat. Rev. Chem., 2018, 2, 65–81 CrossRef CAS.
- H. Y. Zhuo, X. Zhang, J. X. Liang, Q. Yu, H. Xiao and J. Li, Chem. Rev., 2020, 120, 12315–12341 CrossRef CAS PubMed.
- P. Rong, Y.-F. Jiang, Q. Wang, M. Gu, X.-L. Jiang and Q. Yu, J. Mater. Chem. A, 2022, 10, 6231–6241 RSC.
- J.-H. Liu, L.-M. Yang and E. Ganz, J. Mater. Chem. A, 2019, 7, 11944–11952 RSC.
- Y. Wang, M. Wang, Z. Lu, D. Ma and Y. Jia, Nanoscale, 2021, 13, 13390–13400 RSC.
- P. Lv, D. Wu, B. He, X. Li, R. Zhu, G. Tang, Z. Lu, D. Ma and Y. Jia, J. Mater. Chem. A, 2022, 10, 9707–9716 RSC.
- Y. Wang, D. Wu, P. Lv, B. He, X. Li, D. Ma and Y. Jia, Nanoscale, 2022, 14, 10862–10872 RSC.
- D. Wu, B. He, Y. Wang, P. Lv, D. Ma and Y. Jia, J. Phys. D: Appl. Phys., 2022, 55, 203001 CrossRef.
- B. He, P. Lv, D. Wu, X. Li, R. Zhu, K. Chu, D. Ma and Y. Jia, J. Mater. Chem. A, 2022, 10, 18690–18700 RSC.
- D. Wu, P. Lv, J. Wu, B. He, X. Li, K. Chu, Y. Jia and D. Ma, J. Mater. Chem. A, 2023, 11, 1817–1828 RSC.
- X. Cui, W. Li, P. Ryabchuk, K. Junge and M. Beller, Nat. Catal., 2018, 1, 385–397 CrossRef CAS.
- J. Gu, C. S. Hsu, L. Bai, H. M. Chen and X. Hu, Science, 2019, 364, 1091–1094 CrossRef CAS PubMed.
- B. Zhang, J. Zhang, J. Shi, D. Tan, L. Liu, F. Zhang, C. Lu, Z. Su, X. Tan, X. Cheng, B. Han, L. Zheng and J. Zhang, Nat. Commun., 2019, 10, 2980 CrossRef PubMed.
- T. N. Nguyen, M. Salehi, Q. V. Le, A. Seifitokaldani and C. T. Dinh, ACS Catal., 2020, 10, 10068–10095 CrossRef.
- J. J. Masana, J. Xiao, H. Zhang, X. Lu, M. Qiu and Y. Yu, Appl. Catal. B Environ., 2023, 323, 122199 CrossRef CAS.
- G. Hai and H. Wang, Coord. Chem. Rev., 2022, 469, 214670 CrossRef CAS.
- K. R. Phillips, Y. Katayama, J. Hwang and Y. Shao-Horn, J. Phys. Chem. Lett., 2018, 9, 4407–4412 CrossRef CAS PubMed.
- S. Chen, Y. Li, Z. Bu, F. Yang, J. Luo, Q. An, Z. Zeng, J. Wang and S. Deng, J. Mater. Chem. A, 2021, 9, 1705–1712 RSC.
- D. Xue, H. Xia, W. Yan, J. Zhang and S. Mu, Nanomicro Lett., 2020, 13, 5 CrossRef PubMed.
- Q. H. Low, N. W. X. Loo, F. Calle-Vallejo and B. S. Yeo, Angew Chem. Int. Ed., 2019, 58, 2256–2260 CrossRef CAS PubMed.
- A. Guan, Z. Chen, Y. Quan, C. Peng, Z. Wang, T.-K. Sham, C. Yang, Y. Ji, L. Qian, X. Xu and G. Zheng, ACS Energy Lett., 2020, 5, 1044–1053 CrossRef CAS.
- Y. Liu, H. Zhu, Z. Zhao, N. Huang, P. Liao and X. Chen, ACS Catal., 2022, 12, 2749–2755 CrossRef CAS.
- Y. Sun, Z. Xue, Q. Liu, Y. Jia, Y. Li, K. Liu, Y. Lin, M. Liu, G. Li and C. Y. Su, Nat. Commun., 2021, 12, 1369 CrossRef CAS PubMed.
- J. D. Benck, T. R. Hellstern, J. Kibsgaard, P. Chakthranont and T. F. Jaramillo, ACS Catal., 2014, 4, 3957–3971 CrossRef CAS.
- C. Gao, S. Chen, Y. Wang, J. Wang, X. Zheng, J. Zhu, L. Song, W. Zhang and Y. Xiong, Adv. Mater., 2018, 30, 1704624 CrossRef PubMed.
- T. Zheng, K. Jiang and H. Wang, Adv. Mater., 2018, 30, 1802066 CrossRef PubMed.
- R. Kortlever, J. Shen, K. J. Schouten, F. Calle-Vallejo and M. T. Koper, J. Phys. Chem. Lett., 2015, 6, 4073–4082 CrossRef CAS PubMed.
- H. Zhang, J. Li, S. Xi, Y. Du, X. Hai, J. Wang, H. Xu, G. Wu, J. Zhang, J. Lu and J. Wang, Angew Chem. Int. Ed., 2019, 58, 14871–14876 CrossRef CAS PubMed.
- Y. Pan, R. Lin, Y. Chen, S. Liu, W. Zhu, X. Cao, W. Chen, K. Wu, W. C. Cheong, Y. Wang, L. Zheng, J. Luo, Y. Lin, Y. Liu, C. Liu, J. Li, Q. Lu, X. Chen, D. Wang, Q. Peng, C. Chen and Y. Li, J. Am. Chem. Soc., 2018, 140, 4218–4221 CrossRef CAS PubMed.
- Y. Zhang, L. Jiao, W. Yang, C. Xie and H. L. Jiang, Angew Chem. Int. Ed., 2021, 60, 7607–7611 CrossRef CAS PubMed.
- Q. He, J. H. Lee, D. Liu, Y. Liu, Z. Lin, Z. Xie, S. Hwang, S. Kattel, L. Song and J. G. Chen, Adv. Funct. Mater., 2020, 30, 2000407 CrossRef CAS.
- C. Xu, X. Zhi, A. Vasileff, D. Wang, B. Jin, Y. Jiao, Y. Zheng and S.-Z. Qiao, Small Structures, 2020, 2, 2000058 CrossRef.
- M. Fang, X. Wang, X. Li, Y. Zhu, G. Xiao, J. Feng, X. Jiang, K. Lv, Y. Zhu and W. F. Lin, ChemCatChem, 2020, 13, 603–609 CrossRef.
- Y. Hou, Y.-L. Liang, P.-C. Shi, Y.-B. Huang and R. Cao, Appl. Catal. B Environ., 2020, 271, 118929 CrossRef CAS.
- H. Cheng, X. Wu, X. Li, X. Nie, S. Fan, M. Feng, Z. Fan, M. Tan, Y. Chen and G. He, Chem. Eng. J., 2021, 407, 126842 CrossRef CAS.
- H.-L. Zhu, Y.-Q. Zheng and M. Shui, ACS Appl. Energy Mater., 2020, 3, 3893–3901 CrossRef CAS.
- C. He, Y. Zhang, Y. Zhang, L. Zhao, L. P. Yuan, J. Zhang, J. Ma and J. S. Hu, Angew Chem. Int. Ed., 2020, 59, 4914–4919 CrossRef CAS PubMed.
- J. Tuo, Y. Zhu, H. Jiang, J. Shen and C. Li, ChemElectroChem, 2020, 7, 4767–4772 CrossRef CAS.
- F. Pan, B. Li, E. Sarnello, Y. Fei, X. Feng, Y. Gang, X. Xiang, L. Fang, T. Li, Y. H. Hu, G. Wang and Y. Li, ACS Catal., 2020, 10, 10803–10811 CrossRef CAS.
- Q. Cheng, K. Mao, L. Ma, L. Yang, L. Zou, Z. Zou, Z. Hu and H. Yang, ACS Energy Lett., 2018, 3, 1205–1211 CrossRef CAS.
- W. Ni, Y. Xue, X. Zang, C. Li, H. Wang, Z. Yang and Y. M. Yan, ACS Nano, 2020, 14, 2014–2023 CrossRef CAS PubMed.
- X. Yang, J. Cheng, X. Xuan, N. Liu and J. Liu, ACS Sustainable Chem. Eng., 2020, 8, 10536–10543 CrossRef CAS.
- W. Ren, X. Tan, W. Yang, C. Jia, S. Xu, K. Wang, S. C. Smith and C. Zhao, Angew Chem. Int. Ed., 2019, 58, 6972–6976 CrossRef CAS PubMed.
- K. Jiang, S. Siahrostami, T. Zheng, Y. Hu, S. Hwang, E. Stavitski, Y. Peng, J. Dynes, M. Gangisetty, D. Su, K. Attenkofer and H. Wang, Energy Environ. Sci., 2018, 11, 893–903 RSC.
- T. Möller, W. Ju, A. Bagger, X. Wang, F. Luo, T. Ngo Thanh, A. S. Varela, J. Rossmeisl and P. Strasser, Energy Environ. Sci., 2019, 12, 640–647 RSC.
- H.-Y. Jeong, M. Balamurugan, V. S. K. Choutipalli, E.-s. Jeong, V. Subramanian, U. Sim and K. T. Nam, J. Mater. Chem. A, 2019, 7, 10651–10661 RSC.
- R. Daiyan, X. Lu, X. Tan, X. Zhu, R. Chen, S. C. Smith and R. Amal, ACS Appl. Energy Mater., 2019, 2, 8002–8009 CrossRef CAS.
- H. Wang, M. Li, G. Liu, L. Yang, P. Sun and S. Sun, New J. Chem., 2021, 45, 1063–1071 RSC.
- X. Zhang, Y. Wang, M. Gu, M. Wang, Z. Zhang, W. Pan, Z. Jiang, H. Zheng, M. Lucero, H. Wang, G. E. Sterbinsky, Q. Ma, Y.-G. Wang, Z. Feng, J. Li, H. Dai and Y. Liang, Nat. Energy, 2020, 5, 684–692 CrossRef CAS.
- C. Xu, A. Vasileff, Y. Zheng and S. Z. Qiao, Adv. Mater. Interfac., 2020, 8, 2001904 CrossRef.
- Q. Fan, P. Hou, C. Choi, T. S. Wu, S. Hong, F. Li, Y. L. Soo, P. Kang, Y. Jung and Z. Sun, Adv. Energy Mater., 2019, 10, 1903068 CrossRef.
- C. Chen, X. Sun, X. Yan, Y. Wu, H. Liu, Q. Zhu, B. B. A. Bediako and B. Han, Angew Chem. Int. Ed., 2020, 59, 11123–11129 CrossRef CAS PubMed.
- L. Lu, W. Guo, C. Chen, Q. Zhu, J. Ma, H. Wu, D. Yang, G. Yang, X. Sun and B. Han, Green Chem., 2020, 22, 6804–6808 RSC.
- W. Guo, J. Bi, Q. Zhu, J. Ma, G. Yang, H. Wu, X. Sun and B. Han, ACS Sustainable Chem. Eng., 2020, 8, 12561–12567 CrossRef CAS.
- X. Sun, C. Chen, S. Liu, S. Hong, Q. Zhu, Q. Qian, B. Han, J. Zhang and L. Zheng, Angew Chem. Int. Ed., 2019, 58, 4669–4673 CrossRef CAS PubMed.
- Q. Cui, G. Qin, W. Wang, L. Sun, A. Du and Q. Sun, Beilstein J. Nanotechnol., 2019, 10, 540–548 CrossRef CAS PubMed.
- Y. Feng, W. An, Z. Wang, Y. Wang, Y. Men and Y. Du, ACS Sustainable Chem. Eng., 2019, 8, 210–222 CrossRef.
- O. Mohan, R. Xu and S. H. Mushrif, J. Phys. Chem. C, 2021, 125, 4041–4055 CrossRef CAS.
- Y. Wang, H. Arandiyan, J. Scott, K.-F. Aguey-Zinsou and R. Amal, ACS Appl. Energy Mater., 2018, 1, 6781–6789 CrossRef CAS.
- H. Guzmán, N. Russo and S. Hernández, Green Chem., 2021, 23, 1896–1920 RSC.
- C. S. Lo, R. Radhakrishnan and B. L. Trout, Catal. Today, 2005, 105, 93–105 CrossRef CAS.
- M. M. Shi, D. Bao, B. R. Wulan, Y. H. Li, Y. F. Zhang, J. M. Yan and Q. Jiang, Adv. Mater., 2017, 29, 1606550 CrossRef PubMed.
- C. T. Dinh, T. Burdyny, M. G. Kibria, A. Seifitokaldani, C. M. Gabardo, F. P. Garcia de Arquer, A. Kiani, J. P. Edwards, P. De Luna, O. S. Bushuyev, C. Zou, R. Quintero-Bermudez, Y. Pang, D. Sinton and E. H. Sargent, Science, 2018, 360, 783–787 CrossRef CAS PubMed.
- H. Tian, H. Tian, S. Wang, S. Chen, F. Zhang, L. Song, H. Liu, J. Liu and G. Wang, Nat. Commun., 2020, 11, 5025 CrossRef CAS PubMed.
- X. Fang, Q. Shang, Y. Wang, L. Jiao, T. Yao, Y. Li, Q. Zhang, Y. Luo and H. L. Jiang, Adv. Mater., 2018, 30, 1705112 CrossRef PubMed.
- H. Yang, L. Shang, Q. Zhang, R. Shi, G. I. N. Waterhouse, L. Gu and T. Zhang, Nat. Commun., 2019, 10, 4585 CrossRef PubMed.
- J. Li, Q. Guan, H. Wu, W. Liu, Y. Lin, Z. Sun, X. Ye, X. Zheng, H. Pan, J. Zhu, S. Chen, W. Zhang, S. Wei and J. Lu, J. Am. Chem. Soc., 2019, 141, 14515–14519 CrossRef CAS PubMed.
- Q. Zhao, C. Zhang, R. Hu, Z. Du, J. Gu, Y. Cui, X. Chen, W. Xu, Z. Cheng, S. Li, B. Li, Y. Liu, W. Chen, C. Liu, J. Shang, L. Song and S. Yang, ACS Nano, 2021, 15, 4927–4936 CrossRef CAS PubMed.
- Z. Lu, Y. Cheng, S. Li, Z. Yang and R. Wu, Appl. Surf. Sci., 2020, 528, 147047 CrossRef CAS.
- H. Li, L. Wang, Y. Dai, Z. Pu, Z. Lao, Y. Chen, M. Wang, X. Zheng, J. Zhu, W. Zhang, R. Si, C. Ma and J. Zeng, Nat. Nanotechnol., 2018, 13, 411–417 CrossRef CAS PubMed.
- M. Li, B. Hua, L.-C. Wang, Z. Zhou, K. J. Stowers and D. Ding, Catal. Today, 2022, 388–389, 403–409 CrossRef CAS.
- Y. Wu, Z. Jiang, X. Lu, Y. Liang and H. Wang, Nature, 2019, 575, 639–642 CrossRef CAS PubMed.
- H. Yang, Y. Wu, G. Li, Q. Lin, Q. Hu, Q. Zhang, J. Liu and C. He, J. Am. Chem. Soc., 2019, 141, 12717–12723 CrossRef CAS PubMed.
- J. Liu, D. Yang, Y. Zhou, G. Zhang, G. Xing, Y. Liu, Y. Ma, O. Terasaki, S. Yang and L. Chen, Angew. Chem., Int. Ed., 2021, 60, 14473–14479 CrossRef CAS PubMed.
- T. Sun, S. Mitchell, J. Li, P. Lyu, X. Wu, J. Perez-Ramirez and J. Lu, Adv. Mater., 2021, 33, 2003075 CrossRef CAS PubMed.
- X. Bai, Q. Li, L. Shi, X. Niu, C. Ling and J. Wang, Small, 2020, 16, 1901981 CrossRef CAS PubMed.
- X. Ye, C. Yang, X. Pan, J. Ma, Y. Zhang, Y. Ren, X. Liu, L. Li and Y. Huang, J. Am. Chem. Soc., 2020, 142, 19001–19005 CrossRef CAS PubMed.
- T. Kiko, Y. Kashima, D. Kanno, T. Watanabe, Y. Tadano, T. Sugie, K. Kobayashi and T. Fujita, J. Am. Coll. Cardiol., 2016, 67, S224–S225 CrossRef.
- R. Sun, Y. Liao, S.-T. Bai, M. Zheng, C. Zhou, T. Zhang and B. F. Sels, Energy Environ. Sci., 2021, 14, 1247–1285 RSC.
- H. Shang, T. Wang, J. Pei, Z. Jiang, D. Zhou, Y. Wang, H. Li, J. Dong, Z. Zhuang, W. Chen, D. Wang, J. Zhang and Y. Li, Angew Chem. Int. Ed., 2020, 59, 22465–22469 CrossRef CAS PubMed.
- G. Hai, X. Xue, S. Feng, Y. Ma and X. Huang, ACS Catal., 2022, 12, 15271–15281 CrossRef CAS.
- Y. Meng, K. Li, D. Xiao, Y. Yuan, Y. Wang and Z. Wu, Int. J. Hydrogen Energy, 2020, 45, 14311–14319 CrossRef CAS.
- Z. Feng, G. Su, H. Ding, Y. Ma, Y. Li, Y. Tang and X. Dai, Mol. Catal., 2020, 494, 111142 CrossRef CAS.
- K. Mori, T. Taga and H. Yamashita, ACS Catal., 2017, 7, 3147–3151 CrossRef CAS.
- Z. Chen, T. Fan, Y.-Q. Zhang, J. Xiao, M. Gao, N. Duan, J. Zhang, J. Li, Q. Liu, X. Yi and J.-L. Luo, Appl. Catal. B Environ., 2020, 261, 118243 CrossRef CAS.
- X. Shao, X. Yang, J. Xu, S. Liu, S. Miao, X. Liu, X. Su, H. Duan, Y. Huang and T. Zhang, Chem, 2019, 5, 693–705 CAS.
- Q. Wang, S. Santos, C. A. Urbina-Blanco, W. Y. Hernández, M. Impéror-Clerc, E. I. Vovk, M. Marinova, O. Ersen, W. Baaziz, O. V. Safonova, A. Y. Khodakov, M. Saeys and V. V. Ordomsky, Appl. Catal. B Environ., 2021, 290, 120036 CrossRef CAS.
- H. Ning, Q. Mao, W. Wang, Z. Yang, X. Wang, Q. Zhao, Y. Song and M. Wu, J. Alloys Compd., 2019, 785, 7–12 CrossRef CAS.
- J. Yuan, M.-P. Yang, Q.-L. Hu, S.-M. Li, H. Wang and J.-X. Lu, J. CO2 Util., 2018, 24, 334–340 CrossRef CAS.
- A. Javier, B. Chmielowiec, J. Sanabria-Chinchilla, Y.-G. Kim, J. H. Baricuatro and M. P. Soriaga, Electrocatalysis, 2015, 6, 127–131 CrossRef CAS.
- K. U. D. Calvinho, A. B. Laursen, K. M. K. Yap, T. A. Goetjen, S. Hwang, N. Murali, B. Mejia-Sosa, A. Lubarski, K. M. Teeluck, E. S. Hall, E. Garfunkel, M. Greenblatt and G. C. Dismukes, Energy Environ. Sci., 2018, 11, 2550–2559 RSC.
- X. Wei, Z. Yin, K. Lyu, Z. Li, J. Gong, G. Wang, L. Xiao, J. Lu and L. Zhuang, ACS Catal., 2020, 10, 4103–4111 CrossRef CAS.
- Z. Wang, J. Zhao and Q. Cai, Phys. Chem. Chem. Phys., 2017, 19, 23113–23121 RSC.
- C. Wang, C. Zhu, M. Zhang, Y. Geng and Z. Su, Adv. Theory Simul., 2020, 3, 2000218 CrossRef CAS.
- X. F. Qiu, H. L. Zhu, J. R. Huang, P. Q. Liao and X. M. Chen, J. Am. Chem. Soc., 2021, 143, 7242–7246 CrossRef CAS PubMed.
- S. Fu, X. Liu, J. Ran, Y. Jiao and S.-Z. Qiao, Appl. Surf. Sci., 2021, 540, 148293 CrossRef CAS.
- Y. Jiao, Y. Zheng, P. Chen, M. Jaroniec and S. Z. Qiao, J. Am. Chem. Soc., 2017, 139, 18093–18100 CrossRef CAS PubMed.
- C. F. Wen, M. Zhou, P. F. Liu, Y. Liu, X. Wu, F. Mao, S. Dai, B. Xu, X. L. Wang, Z. Jiang, P. Hu, S. Yang, H. F. Wang and H. G. Yang, Angew. Chem., Int. Ed., 2022, 61, e202111700 CAS.
- D. Gao, T. Liu, G. Wang and X. Bao, ACS Energy Lett., 2021, 6, 713–727 CrossRef CAS.
- J. Xi, H. S. Jung, Y. Xu, F. Xiao, J. W. Bae and S. Wang, Adv. Funct. Mater., 2021, 31, 2008318 CrossRef CAS.
- B. Peng, H. Liu, Z. Liu, X. Duan and Y. Huang, J. Phys. Chem. Lett., 2021, 12, 2837–2847 CrossRef CAS PubMed.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC.
- L. Matějová, K. Kočí, M. Reli, L. Čapek, V. Matějka, O. Šolcová and L. Obalová, Appl. Surf. Sci., 2013, 285, 688–696 CrossRef.
- H. Zhang, T. Itoi, T. Konishi and Y. Izumi, J. Am. Chem. Soc., 2019, 141, 6292–6301 CrossRef CAS PubMed.
- J. Yang, J. Hao, S. Xu, Q. Wang, J. Dai, A. Zhang and X. Pang, ACS Appl. Mater. Interfaces, 2019, 11, 32025–32037 CrossRef CAS PubMed.
- S. Yan, J. Wang, H. Gao, N. Wang, H. Yu, Z. Li, Y. Zhou and Z. Zou, Adv. Funct. Mater., 2013, 23, 1839–1845 CrossRef CAS.
- X. Chen, S. Shen, L. Guo and S. S. Mao, Chem. Rev., 2010, 110, 6503–6570 CrossRef CAS PubMed.
- K. Teramura, S.-i. Okuoka, H. Tsuneoka, T. Shishido and T. Tanaka, Appl. Catal. B Environ., 2010, 96, 565–568 CrossRef CAS.
- X. F. Xie, X. Y. Dao, F. Guo, X. Y. Zhang, F. M. Wang and W. Y. Sun, ChemistrySelect, 2020, 5, 4001–4007 CrossRef CAS.
- K. Kočí, P. Praus, M. Edelmannová, N. Ambrožová, I. Troppová, D. Fridrichová, G. Słowik and J. Ryczkowski, J. Nanosci. Nanotechnol., 2017, 17, 4041–4047 CrossRef.
- D. V. Markovskaya, S. V. Cherepanova, E. Y. Gerasimov, A. V. Zhurenok, A. V. Selivanova, D. S. Selishchev and E. A. Kozlova, RSC Adv., 2020, 10, 1341–1350 RSC.
- M. Zhou, S. Wang, P. Yang, C. Huang and X. Wang, ACS Catal., 2018, 8, 4928–4936 CrossRef CAS.
- B. Zhou, X. Kong, S. Vanka, S. Cheng, N. Pant, S. Chu, P. Ghamari, Y. Wang, G. Botton, H. Cuo and Z. Mi, Energy Environ. Sci., 2019, 12, 2842–2848 RSC.
- N. T. P. Le Chi, N. T. Dieu Cam, D. Van Thuan, T. T. Truong, N. T. Thanh Truc, C. Van Hoang, T. T. Thu Phuong, T.-D. Pham, M. H. Thanh Tung, N. T. Minh Thu, N. M. Phuong and V. N. Nguyen, Appl. Surf. Sci., 2019, 467–468, 1249–1255 CrossRef CAS.
- N. Dewangan, W. M. Hui, S. Jayaprakash, A.-R. Bawah, A. J. Poerjoto, T. Jie, A. Jangam, K. Hidajat and S. Kawi, Catal. Today, 2020, 356, 490–513 CrossRef CAS.
- S. Goyal, M. Shaharun, C. Kait, B. Abdullah and M. Ameen, Catalysts, 2018, 8, 581 CrossRef.
- G. Gao, Y. Jiao, E. R. Waclawik and A. Du, J. Am. Chem. Soc., 2016, 138, 6292–6297 CrossRef CAS PubMed.
- D. Mateo, A. M. Asiri, J. Albero and H. Garcia, Photochem. Photobiol. Sci., 2018, 17, 829–834 CrossRef CAS PubMed.
- H. Wei, X. Liu, A. Wang, L. Zhang, B. Qiao, X. Yang, Y. Huang, S. Miao, J. Liu and T. Zhang, Nat. Commun., 2014, 5, 5634 CrossRef CAS PubMed.
- T. Yang, R. Fukuda, S. Hosokawa, T. Tanaka, S. Sakaki and M. Ehara, ChemCatChem, 2017, 9, 1222–1229 CrossRef CAS PubMed.
- Z. Luo, Z. Wang, J. Li, K. Yang and G. Zhou, Phys. Chem. Chem. Phys., 2020, 22, 11392–11399 RSC.
- J. Li, H. Huang, P. Liu, X. Song, D. Mei, Y. Tang, X. Wang and C. Zhong, J. Catal., 2019, 375, 351–360 CrossRef CAS.
- Y. N. Gong, L. Jiao, Y. Qian, C. Y. Pan, L. Zheng, X. Cai, B. Liu, S. H. Yu and H. L. Jiang, Angew Chem. Int. Ed., 2020, 59, 2705–2709 CrossRef CAS PubMed.
- M. R. Axet, J. Durand, M. Gouygou and P. Serp, Adv. Organomet. Chem., 2019, 71, 53–174 CrossRef CAS.
- H. Zhang, W. Tian, X. Duan, H. Sun, Y. Shen, G. Shao and S. Wang, Nanoscale, 2020, 12, 6937–6952 RSC.
- J. Jiang, F. Sun, S. Zhou, W. Hu, H. Zhang, J. Dong, Z. Jiang, J. Zhao, J. Li, W. Yan and M. Wang, Nat. Commun., 2018, 9, 2885 CrossRef PubMed.
- Y. Chen, H. Hong, J. Cai and Z. Li, ChemCatChem, 2020, 13, 656–663 CrossRef.
- M. Tahir and N. S. Amin, Appl. Catal., A, 2015, 493, 90–102 CrossRef CAS.
- X. Wang, Z. Zhang, Z. Huang, P. Dong, X. Nie, Z. Jin and X. Zhang, Mater. Res. Bull., 2019, 118, 110502 CrossRef CAS.
- H. She, Z. Zhao, W. Bai, J. Huang, L. Wang and Q. Wang, Mater. Res. Bull., 2020, 124, 110758 CrossRef CAS.
- Y. Ma, B. Chi, W. Liu, L. Cao, Y. Lin, X. Zhang, X. Ye, S. Wei and J. Lu, ACS Catal., 2019, 9, 8404–8412 CrossRef CAS.
- Z. Jiang, M. Jing, X. Feng, J. Xiong, C. He, M. Douthwaite, L. Zheng, W. Song, J. Liu and Z. Qu, Appl. Catal. B Environ., 2020, 278, 119304 CrossRef CAS.
- D. Sun, Y. Gao, J. Fu, X. Zeng, Z. Chen and Z. Li, Chem. Commun., 2015, 51, 2645–2648 RSC.
- C. Zheng, X. Qiu, J. Han, Y. Wu and S. Liu, ACS Appl. Mater. Interfaces, 2019, 11, 42243–42249 CrossRef CAS PubMed.
- L. Zhao, Z. Zhao, Y. Li, X. Chu, Z. Li, Y. Qu, L. Bai and L. Jing, Nanoscale, 2020, 12, 10010–10018 RSC.
- J. Low, J. Yu and W. Ho, J. Phys. Chem. Lett., 2015, 6, 4244–4251 CrossRef CAS PubMed.
- R. Zhou, Y. Yin, D. Long, J. Cui, H. Yan, W. Liu and J. H. Pan, Prog. Nat. Sci., 2019, 29, 660–666 CrossRef CAS.
- X.-Y. Dao, X.-F. Xie, J.-H. Guo, X.-Y. Zhang, Y.-S. Kang and W.-Y. Sun, ACS Appl. Energy Mater., 2020, 3, 3946–3954 CrossRef CAS.
- X. Hu, J. Hu, Q. Peng, X. Ma, S. Dong and H. Wang, Mater. Res. Bull., 2020, 122, 110682 CrossRef CAS.
- Z. Zhu, Z. Liu, X. Tang, K. Reeti, P. Huo, J. W.-C. Wong and J. Zhao, Catal. Sci. Technol., 2021, 11, 1725–1736 RSC.
- L. Tan, K. Peter, J. Ren, B. Du, X. Hao, Y. Zhao and Y.-F. Song, Front. Chem. Sci. Eng., 2020, 15, 99–108 CrossRef.
- S. Das and K. parida, Mater. Today Proc., 2021, 35, 275–280 CrossRef CAS.
- X. Liu, S. Inagaki and J. Gong, Angew Chem. Int. Ed., 2016, 55, 14924–14950 CrossRef CAS PubMed.
- Z. Guo, S. Cheng, C. Cometto, E. Anxolabehere-Mallart, S. M. Ng, C. C. Ko, G. Liu, L. Chen, M. Robert and T. C. Lau, J. Am. Chem. Soc., 2016, 138, 9413–9416 CrossRef CAS PubMed.
- C. Yang and Z.-Y. Zhao, Comput. Mater. Sci., 2018, 151, 160–173 CrossRef CAS.
- Y. Wang, Y. Chen, Q. Hou, M. Ju and W. Li, Nanomaterials, 2019, 9, 391 CrossRef PubMed.
- J. Di, C. Chen, S. Z. Yang, S. Chen, M. Duan, J. Xiong, C. Zhu, R. Long, W. Hao, Z. Chi, H. Chen, Y. X. Weng, J. Xia, L. Song, S. Li, H. Li and Z. Liu, Nat. Commun., 2019, 10, 2840 CrossRef PubMed.
- J. Wang, T. Heil, B. Zhu, C.-W. Tung, J. Yu, H. M. Chen, M. Antonietti and S. Cao, ACS Nano, 2020, 14, 8584–8593 CrossRef CAS PubMed.
- O. K. Varghese, M. Paulose, T. J. Latempa and C. A. Grimes, Nano Lett., 2009, 9, 731–737 CrossRef CAS PubMed.
- W. Tu, Y. Zhou and Z. Zou, Adv. Mater., 2014, 26, 4607–4626 CrossRef CAS PubMed.
- S. Zhu, S. Liang, Y. Tong, X. An, J. Long, X. Fu and X. Wang, Phys. Chem. Chem. Phys., 2015, 17, 9761–9770 RSC.
- Y. Liu, S. Zhou, J. Li, Y. Wang, G. Jiang, Z. Zhao, B. Liu, X. Gong, A. Duan, J. Liu, Y. Wei and L. Zhang, Appl. Catal. B Environ., 2015, 168–169, 125–131 CrossRef CAS.
- N. Li, Y. Li, R. Jiang, J. Zhou and M. Liu, Appl. Surf. Sci., 2019, 498, 143861 CrossRef CAS.
- X. Li, J. Wen, J. Low, Y. Fang and J. Yu, Sci. China Mater., 2014, 57, 70–100 CrossRef.
- M. Tahir and B. Tahir, Appl. Surf. Sci., 2016, 377, 244–252 CrossRef CAS.
- Y. Yang, F. Li, J. Chen, J. Fan and Q. Xiang, ChemSusChem, 2020, 13, 1979–1985 CrossRef CAS PubMed.
- Y. Ji and Y. Luo, ACS Catal., 2016, 6, 2018–2025 CrossRef CAS.
- X. Cai, J. Wang, R. Wang, A. Wang, S. Zhong, J. Chen and S. Bai, J. Mater. Chem. A, 2019, 7, 5266–5276 RSC.
- A. H. Chowdhury, A. Das, S. Riyajuddin, K. Ghosh and S. M. Islam, Catal. Sci. Technol., 2019, 9, 6566–6569 RSC.
- T. Li, C. Wang, T. Wang and L. Zhu, Appl. Catal. B Environ., 2020, 268, 118442 CrossRef CAS.
- J. He, P. Lv, J. Zhu and H. Li, RSC Adv., 2020, 10, 22460–22467 RSC.
- H. Guo, M. Chen, Q. Zhong, Y. Wang, W. Ma and J. Ding, J. CO2 Util., 2019, 33, 233–241 CrossRef CAS.
- F. Li, X. Chen, Q. Guo, D. Dai and X. Yang, J. Phys. Chem. C, 2020, 124, 8766–8774 CrossRef CAS.
- J. Li, B. Huang, Q. Guo, S. Guo, Z. Peng, J. Liu, Q. Tian, Y. Yang, Q. Xu, Z. Liu and B. Liu, Appl. Catal. B Environ., 2021, 284, 119733 CrossRef CAS.
- M. Mohamedali, O. Ayodele and H. Ibrahim, Renew. Sustain. Energy Rev., 2020, 131, 110024 CrossRef CAS.
- X. Yu, V. V. Ordomsky and A. Y. Khodakov, ChemCatChem, 2020, 12, 740–749 CrossRef CAS.
- X. Zhang, G. Ren, C. Zhang, R. Li, Q. Zhao and C. Fan, Catal. Lett., 2020, 150, 2510–2516 CrossRef CAS.
- Z. Sun, N. Talreja, H. Tao, J. Texter, M. Muhler, J. Strunk and J. Chen, Angew Chem. Int. Ed., 2018, 57, 7610–7627 CrossRef CAS PubMed.
- F. Almomani, R. Bhosale, M. Khraisheh, A. Kumar and M. Tawalbeh, Appl. Surf. Sci., 2019, 483, 363–372 CrossRef CAS.
- S. Nahar, M. F. M. Zain, A. A. H. Kadhum, H. A. Hasan and M. R. Hasan, Materials, 2017, 10, 629 CrossRef PubMed.
- N. Sasirekha, S. Basha and K. Shanthi, Appl. Catal. B Environ., 2006, 62, 169–180 CrossRef CAS.
- K. Mori, H. Yamashita and M. Anpo, RSC Adv., 2012, 2, 3165–3172 RSC.
- I. A. Shkrob, T. W. Marin, H. He and P. Zapol, J. Phys. Chem. C, 2012, 116, 9450–9460 CrossRef CAS.
- V. P. Indrakanti, H. H. Schobert and J. D. Kubicki, Energy Fuels, 2009, 23, 5247–5256 CrossRef CAS.
- Y. Li, B. Li, D. Zhang, L. Cheng and Q. Xiang, ACS Nano, 2020, 14, 10552–10561 CrossRef CAS PubMed.
- S. Ji, Y. Qu, T. Wang, Y. Chen, G. Wang, X. Li, J. Dong, Q. Chen, W. Zhang, Z. Zhang, S. Liang, R. Yu, Y. Wang, D. Wang and Y. Li, Angew Chem. Int. Ed., 2020, 59, 10651–10657 CrossRef CAS PubMed.
- W. Zhong, R. Sa, L. Li, Y. He, L. Li, J. Bi, Z. Zhuang, Y. Yu and Z. Zou, J. Am. Chem. Soc., 2019, 141, 7615–7621 CrossRef CAS PubMed.
- X. Xiong, C. Mao, Z. Yang, Q. Zhang, G. I. N. Waterhouse, L. Gu and T. Zhang, Adv. Energy Mater., 2020, 10, 2002928 CrossRef CAS.
- K. Xie, N. Umezawa, N. Zhang, P. Reunchan, Y. Zhang and J. Ye, Energy Environ. Sci., 2011, 4, 4211–4219 RSC.
- S. Chen, G. Hai, H. Gao, X. Chen, A. Li and X. Zhang, Chem. Eng. J., 2021, 406, 126886 CrossRef CAS.
- Y. Ji and Y. Luo, J. Am. Chem. Soc., 2016, 138, 15896–15902 CrossRef CAS PubMed.
- C. Boerigter, R. Campana, M. Morabito and S. Linic, Nat. Commun., 2016, 7, 10545 CrossRef CAS PubMed.
- J. Ding, X. Liu, M. Shi, T. Li, M. Xia, X. Du, R. Shang, H. Gu and Q. Zhong, Sol. Energy Mater. Sol. Cells, 2019, 195, 34–42 CrossRef CAS.
- Z. Pan, E. Han, J. Zheng, J. Lu, X. Wang, Y. Yin, G. I. N. Waterhouse, X. Wang and P. Li, Nanomicro Lett., 2020, 12, 18 CAS.
- S. Kawamura, H. Zhang, M. Tamba, T. Kojima, M. Miyano, Y. Yoshida, M. Yoshiba and Y. Izumi, J. Catal., 2017, 345, 39–52 CrossRef CAS.
- Q. Liu, Q. Chen, T. Li, Q. Ren, S. Zhong, Y. Zhao and S. Bai, J. Mater. Chem. A, 2019, 7, 27007–27015 RSC.
- S. S. Bhosale, A. K. Kharade, E. Jokar, A. Fathi, S. M. Chang and E. W. Diau, J. Am. Chem. Soc., 2019, 141, 20434–20442 CrossRef CAS PubMed.
- J. Hong, W. Zhang, Y. Wang, T. Zhou and R. Xu, ChemCatChem, 2014, 6, 2315–2321 CrossRef CAS.
- T. Ohno, N. Murakami, T. Koyanagi and Y. Yang, J. CO2 Util., 2014, 6, 17–25 CrossRef CAS.
- Y. N. Kavil, Y. A. Shaban, R. K. Al Farawati, M. I. Orif, M. Zobidi and S. U. M. Khan, Water, Air, Soil Pollut., 2018, 229, 236 CrossRef.
- S.-T. Gao, W.-H. Liu, N.-Z. Shang, C. Feng, Q.-H. Wu, Z. Wang and C. Wang, RSC Adv., 2014, 4, 61736–61742 RSC.
- Q. Liu, Z.-X. Low, L. Li, A. Razmjou, K. Wang, J. Yao and H. Wang, J. Mater. Chem. A, 2013, 1, 11563–11569 RSC.
| This journal is © The Royal Society of Chemistry 2023 |