DOI:
10.1039/D2RA08280B
(Paper)
RSC Adv., 2023,
13, 2884-2895
Pyrazolone-type compounds (part II): in vitro and in silico evaluation of antioxidant potential; structure–activity relationship†
Received
28th December 2022
, Accepted 12th January 2023
First published on 18th January 2023
Abstract
The pyrazolone class comprises a variety of hybrid compounds displaying diverse biological actions. Although studied for decades, these compounds are still of interest due to their facile chemical transformations. In our previous work, we presented the synthetic route of functionalised pyrazolone derivatives. The presence of pyrazolone structural motif in many drugs, such as edaravone, prompted us to investigate the antioxidant features of the selected compounds. In this paper, we provide an extensive in vitro and in silico description of the antioxidant properties of selected pyrazolone analogues. The obtained in vitro results revealed their great antiradical potency against the DPPH radical (IC50 values in the 2.6–7.8 μM range), where the best results were obtained for analogues bearing a catechol moiety. Density functional theory (DFT) was used to assess their antioxidant capacity from the thermodynamic aspect. Here, good agreement with in vitro results was achieved. DFT was employed for the prediction of the most preferable radical scavenging pathway, also. In polar solvents, the SPLET mechanism is a favourable scavenging route, whereas in nonpolar solvents the HAT is slightly predominant. Furthermore, antioxidant mechanisms were studied in the presence of relevant reactive oxygen species. The obtained values of the reaction enthalpies with the selected radicals revealed that HAT is slightly prevailing in polar solvents, while the SPLET mechanism is dominant in nonpolar solvents. Regarding the well-known antioxidant features of the drug edaravone, these findings represent valuable data for this pyrazolone class and could be used as the basis for further investigations.
Introduction
Biological exploration of pyrazolone-endowed compounds has continued for more than a century. Ever since its introduction by Ludwig Knorr in 1883,1 the pyrazolone nucleus evolved into one of the most studied pharmacophores in medicinal chemistry.2 Such fruitful investigations uncovered diverse biological and therapeutical traits of pyrazolone-based compounds, where a number of them found multiple pharmaceutical roles (Fig. 1).3,4 Antipyrine (also known as phenazone) as one of the first synthetic drugs, alongside salicylate derivatives, i.e., aspirin, revolutionised the era of antipyretics and fever suppressants.5 Furthermore, the ampyrone sulfonate family representative, the drug metamizole, is a well-known pain reliever used in various acute/chronic conditions.6 Also, the pyrazolone skeleton is frequently embedded in many drugs (and drug candidates) for the treatment of various illnesses, such as eltrombopag, piperylone, nifenazone, morazone, propyphenazone, aminophenazone, etc (Fig. 1).4 Such an outcome was triggered by the diversity of pyrazolone-based compounds and their richness in beneficial biological virtues.2,7 Pyrazolone-cored derivatives mostly wield antimicrobial,8–13 anticancer,7,12,14,15 antiproliferative,14,16 and antiinflammatory potency,17–22 but they were also identified as analgesics,18,22,23 antivirals,24–26 hypoglycaemics,27 and antioxidants.11,28,29 The utilisation of pyrazolone synthon produced numerous hybrid molecules acting as multitarget agents.11,12,17,22,30,31 Biomodelling of pyrazolone structural unit emerged compounds with inhibitory activity on many enzymes as well, such as acetylcholinesterase (AChE) and butyrylcholinesterase (BuChE),30,32 α-amylase,33 α-glucosidase,34 cyclooxygenases (COXs),17,19,21,28,35 HIV-1 integrase,36 and even towards human telomerase.37 These accomplishments have enabled pyrazolone-type compounds to be considered in the treatment of complex illnesses, particularly in neurodegenerative diseases (NDDs), such as Alzheimer's and Parkinson's.30,32,38 Here, it is important to reference the FDA-approved pyrazolone drug edaravone.39 This drug is used in the treatment of acute cerebral infarction and has also exhibited a neuroprotective effect in amyotrophic lateral sclerosis (ALS) and Parkinson's disease.40 Edaravone is designated as a specific chemical entity, empowered for synthetic modifications and diverse usages.39 It was found that edaravone expresses preventive action in myocardial injury following ischemia and myocardial infarction,27 and it can be used in chronic obstructive pulmonary disease (COPD), Alzheimer's, rheumatoid arthritis, the treatment of cancer therapy side effects, and many other conditions.39 Such multipurpose potential of edaravone, particularly its role as a neuroprotective agent, is possible due to its antioxidant ability. Namely, edaravone acts as a potent free radical scavenger that prevents oxidative damage to the cell membrane, i.e., peroxidation of the cell membrane's fatty acids, and therefore, neuronal death and brain edema.2 The overproduction of free radicals leading to oxidative stress is designated as one of the leading causes of brain ischemic injury.41,42 Regarding this, the beneficial action of edaravone lies in its ability to quench ˙OH radicals, as well as other harmful reactive oxygen/nitrogen radical species (ROS and RNS).43
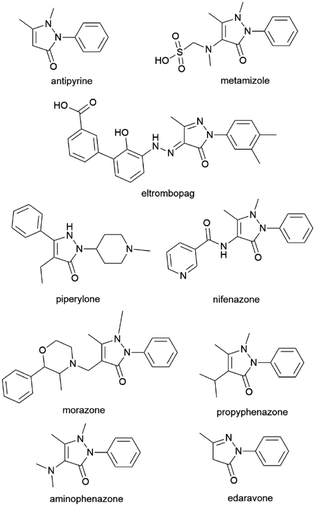 |
| | Fig. 1 Drugs containing pyrazolone structural fragment. | |
Although studied for decades, all these findings indicate the enormous potential of the pyrazolone entity and its almost “never-ending” exploitation in the field of medicinal chemistry. The development of novel edaravone-like and other functionalised pyrazolone scaffolds is still encouraged, as it's recognised as the essence of pharmacological and combinatorial chemistry.2
Herein, the versatility of pyrazolone-derived compounds, as well as the multifunctionality of the drug edaravone, inspired us to investigate the antioxidant potential of differently functionalised pyrazolone derivatives, using both in vitro and in silico methods. In our previous work, we reported the synthetic route for twenty pyrazolone analogues and investigated their potential antiviral activity against the SARS-CoV-2 virus.44 Generally, the biological investigations on these particular pyrazolone-type derivatives are scarce, i.e., the research is mainly focused on the development of novel synthetic procedures.45–52 To our knowledge, only one paper reported in vitro studies related to these compounds.53 Bearing this in mind, in this work, we have provided an extensive description of the antioxidant properties of the selected pyrazolone compounds. The insight into their radical scavenging features represents valuable data for this class of pyrazolones and could indicate the compounds with the prospect of neuroprotective application.
Results and discussion
Antioxidant activity of compounds a–t against DPPH radical
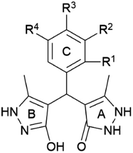
In our previous report,44 twenty pyrazolone derivatives a–t bearing different ring C substitution were synthesised and characterised (Table 1). The crystal structure analysis of compound p revealed the differences between rings A and B, i.e., the existence of pyrazolone and pyrazole moiety.44 In this work, compounds a–t were subjected to in vitro antioxidant screening using the 2,2-diphenyl-1-picrylhydrazyl (DPPH) assay (Table 2). This method was selected since it is noted as reliable for the prediction of the radical scavenging activity against reactive oxygen species present in the living cell.54,55 The structures of the investigated compounds a–t bearing different ring C substitution are presented in Table 1. Here, compound a represents a non-substituted analogue. Furthermore, derivatives b, c, and d possess one –OH group in R1, R2, and R3 positions (respectively). Analogues e, f, and g bear one halogen atom (chlorine or fluor), whereas h, i, and j carry the nitro group in different positions.
Table 1 Structural characteristics and ring C substitution of compounds a–t
| Entry |
R1 |
R2 |
R3 |
R4 |
Skeleton of compounds a–t |
| a |
H |
H |
H |
H |
 |
| b |
OH |
H |
H |
H |
| c |
H |
OH |
H |
H |
| d |
H |
H |
OH |
H |
| e |
H |
Cl |
H |
H |
| f |
H |
H |
Cl |
H |
| g |
H |
H |
F |
H |
| h |
NO2 |
H |
H |
H |
| i |
H |
NO2 |
H |
H |
| j |
H |
H |
NO2 |
H |
| k |
CH3 |
H |
H |
H |
| l |
H |
CH3 |
H |
H |
| m |
OH |
OH |
H |
H |
| n |
H |
OH |
OH |
H |
| o |
OH |
OCH3 |
H |
H |
| p |
H |
OCH3 |
OH |
H |
| q |
H |
OCH3 |
OH |
OCH3 |
| r |
H |
OH |
OCH3 |
OCH3 |
| s |
H |
OCH3 |
OCH3 |
OCH3 |
| t |
OH |
Cl |
H |
Cl |
Table 2 In vitro interactions of selected compounds a–t with DPPH radical
| Inhibition (%) |
IC50 (μM) |
SF |
| Concentration (μM) |
100 |
50 |
25 |
| Compound |
20 min |
60 min |
20 min |
60 min |
20 min |
60 min |
| a |
96.1 ± 1.1 |
97.4 ± 0.2 |
95.8 ± 0.4 |
96.6 ± 0.6 |
93.6 ± 0.4 |
94.4 ± 0.8 |
5.1 ± 0.1 |
2.5 |
| b |
91.6 ± 0.7 |
93.2 ± 1.5 |
91.5 ± 1.3 |
92.1 ± 2.2 |
86.9 ± 4.7 |
89.8 ± 4.6 |
6.2 ± 0.1 |
2.0 |
| c |
94.9 ± 0.1 |
95.4 ± 0.1 |
94.2 ± 0.2 |
95.3 ± 0.2 |
91.9 ± 0.7 |
93.9 ± 0.6 |
4.3 ± 0.1 |
2.9 |
| d |
95.7 ± 1.1 |
95.8 ± 0.5 |
95.1 ± 0.7 |
95.4 ± 0.3 |
93.1 ± 0.9 |
94.3 ± 1.0 |
4.9 ± 0.1 |
2.6 |
| e |
93.3 ± 2.1 |
94.4 ± 1.3 |
93.1 ± 1.9 |
94.3 ± 0.8 |
89.8 ± 0.5 |
93.8 ± 2.2 |
4.5 ± 0.1 |
2.8 |
| f |
95.8 ± 0.2 |
95.9 ± 0.2 |
93.4 ± 0.2 |
95.3 ± 0.5 |
92.2 ± 0.7 |
94.3 ± 0.2 |
5.4 ± 0.1 |
2.3 |
| g |
93.4 ± 1.3 |
94.7 ± 0.1 |
92.8 ± 1.0 |
93.9 ± 1.0 |
90.2 ± 0.3 |
92.6 ± 0.8 |
4.8 ± 0.1 |
2.6 |
| h |
95.6 ± 0.2 |
97.2 ± 0.6 |
87.9 ± 0.7 |
92.1 ± 0.3 |
80.5 ± 0.7 |
88.8 ± 0.6 |
7.8 ± 0.1 |
1.6 |
| i |
95.7 ± 0.1 |
95.8 ± 0.1 |
92.4 ± 1.0 |
94.3 ± 0.4 |
92.1 ± 0.8 |
94.2 ± 0.5 |
4.5 ± 0.1 |
2.8 |
| j |
95.1 ± 0.7 |
95.3 ± 0.1 |
92.1 ± 1.1 |
93.1 ± 1.1 |
85.1 ± 0.9 |
89.2 ± 0.2 |
5.1 ± 0.1 |
2.5 |
| k |
96.3 ± 1.0 |
98.3 ± 0.7 |
96.2 ± 0.2 |
96.3 ± 0.4 |
94.6 ± 0.6 |
96.2 ± 0.8 |
4.2 ± 0.1 |
3.0 |
| l |
95.0 ± 0.6 |
95.1 ± 0.5 |
94.9 ± 0.5 |
95.0 ± 0.6 |
92.2 ± 1.1 |
93.8 ± 0.8 |
3.5 ± 0.1 |
3.6 |
| m |
96.7 ± 0.3 |
97.2 ± 0.2 |
95.8 ± 0.2 |
96.4 ± 0.3 |
94.4 ± 0.6 |
94.9 ± 0.8 |
2.6 ± 0.1 |
4.8 |
| n |
94.9 ± 0.3 |
96.7 ± 0.3 |
94.3 ± 1.6 |
94.9 ± 0.3 |
94.2 ± 0.9 |
94.3 ± 1.1 |
2.9 ± 0.1 |
4.3 |
| o |
91.6 ± 1.1 |
91.7 ± 0.1 |
89.7 ± 0.7 |
90.4 ± 0.7 |
89.4 ± 0.9 |
89.6 ± 1.6 |
3.6 ± 0.1 |
3.5 |
| p |
93.6 ± 0.2 |
94.5 ± 0.2 |
92.3 ± 0.4 |
93.2 ± 1.3 |
91.7 ± 0.6 |
92.0 ± 0.6 |
6.1 ± 0.1 |
2.0 |
| q |
92.1 ± 0.4 |
93.9 ± 1.2 |
90.9 ± 0.8 |
91.7 ± 0.2 |
89.9 ± 0.6 |
90.7 ± 0.7 |
5.5 ± 0.1 |
2.3 |
| r |
95.1 ± 0.6 |
95.2 ± 0.1 |
94.4 ± 0.3 |
94.9 ± 0.9 |
90.5 ± 0.1 |
94.7 ± 0.2 |
4.4 ± 0.1 |
2.8 |
| s |
96.1 ± 0.1 |
96.8 ± 0.2 |
94.2 ± 0.8 |
94.8 ± 0.6 |
89.8 ± 1.1 |
92.8 ± 1.1 |
6.2 ± 0.1 |
2.0 |
| t |
88.5 ± 0.5 |
89.8 ± 0.7 |
88.1 ± 0.9 |
89.6 ± 0.7 |
84.2 ± 1.0 |
89.6 ± 0.9 |
6.4 ± 0.1 |
2.0 |
| Pyrazolone |
93.7 ± 0.1 |
95.5 ± 0.8 |
92.8 ± 1.2 |
94.5 ± 0.8 |
86.9 ± 1.6 |
92.3 ± 0.8 |
9.3 ± 0.1 |
1.3 |
| NDGA |
94.5 ± 0.2 |
94.1 ± 0.7 |
94.2 ± 0.7 |
94.2 ± 0.7 |
94.6 ± 0.7 |
94.6 ± 0.6 |
1.7 ± 0.1 |
7.4 |
| Quercetin |
95.1 ± 0.9 |
95.4 ± 0.8 |
96.8 ± 1.0 |
96.5 ± 0.9 |
95.3 ± 0.8 |
95.1 ± 0.9 |
1.9 ± 0.1 |
6.6 |
Methyl groups are introduced to the C ring in the case of compounds k and l, whereas derivatives m–s possess multiple –OH and/or –OCH3 groups. Finally, compound t bear R1–OH group on ring C, as well as two chlorine atoms.
Based on the obtained results, all analogues exhibited antioxidant activity against DPPH radical with IC50 values in the range of 2.6–7.8 μM. It is worth emphasizing that the parent pyrazolone compound (5-methyl-2,4-dihydro-3H-pyrazol-3-one) was also tested against the DPPH radical, with the expectation of enhanced antioxidant activity of compounds a–t. Based on the obtained results, all compounds expressed higher antiradical activity in comparison to their parent compound (Table 2). Furthermore, the antioxidant capacity of the investigated compounds was expressed through the values of stoichiometric factor (SF). Namely, if the value of SF is ≥2, such compound is designated as a good antioxidant. The results presented in Table 2 indicate that all compounds, except h (SF = 1.6), could be noted as good antioxidants (SF values in the range of 2.0–4.8). The obtained results could be compared to other findings, i.e., with compounds bearing pyrazolone fragment.27,56–60 Here, in comparison with the most similar edaravone-like set of compounds (and drug edaravone),27 compounds a–t expressed significantly higher antioxidant activity.
The ring C substitution effect on antioxidant activity. Besides the aim to investigate the antioxidant properties of compounds a–t, one of the objectives was to evaluate how the ring C substitution impacts the inhibition potency of the DPPH radical. Here, the non-substituted analogue a expressed good activity against DPPH (IC50 = 5.1 ± 0.1 μM). In comparison to a, among those bearing only one –OH group, the best result was obtained for derivative c (R2–OH; IC50 = 4.3 ± 0.1 μM). These results, i.e., the more favourable R2–substitution over R1– and R3–, are in agreement with literature data obtained for compounds with a similar set of rings.27 In the cases of halogen-substituted C ring, compounds e (R2–Cl) and g (R3–F) exhibited higher activity than a, whereas f (R3–Cl) was slightly lower. Here, the best activity expressed derivative e (IC50 = 4.5 ± 0.1 μM). Among those bearing nitro group, the R2–substituted derivative i expressed the highest inhibition activity compared to a (IC50 = 4.5 ± 0.1 μM), whereas the R1–substituted h the lowest (IC50 = 7.8 ± 0.1 μM). Such findings are in agreement with the literature data, also.27 Similarly, for compounds k and l, the best interaction with DPPH was achieved in the case of R2–CH3 substituted analogue l (IC50 = 3.5 ± 0.1 μM). For compounds m–s, the highest inhibition activity against the DPPH radical expressed analogue m (R1, R2–diOH), which was also the best result achieved among all tested compounds (IC50 = 2.6 ± 0.1 μM). It is worth emphasizing that analogues n, o, and r also exhibited noteworthy antioxidant activity (IC50 = 2.9 ± 0.1, 3.6 ± 0.1, and 4.4 ± 0.1 μM, respectively). Here, it is important to point out that derivatives n and r possess the R2–OH group, whereas compound o bears o-vanillin moiety. Finally, the antioxidant activity of derivative t was lower compared to compound a, i.e., similar to the activity of compound b. Here, both b and t possess R1–OH group, whereas t bear additional two chlorine atoms.
Based on the obtained results, several remarks could be noted:
(1) All compounds expressed higher inhibition activity than the parent pyrazolone compound, and FDA-drug edaravone (IC50 = 18.1 ± 0.5)27 as well;
(2) Ring C substitution, i.e., number, nature, and the position of specific groups, alters the antioxidant potency of the investigated compounds toward DPPH radical;
(3) The R2–substitution most significantly enhances the antioxidant activity in comparison to the other positions, as well as to non-substituted derivative a. This can easily be observed in the cases of derivatives c (R2–OH), e (R2–Cl), i (R2–NO2), and l (R2–CH3); the antioxidant activity is even more heightened when an additional group is neighbouring the R2–substituent. Such observations were made for the compounds m, n, o, and r which bear the –OH and neighbouring –OH or –OCH3 groups. This can be attributed to the formation of corresponding phenolic O˙ radical which is stabilised through resonance and electron-donating effects of the neighbouring group.61 Moreover, in the cases of compounds m and n, even better stabilisation can be achieved via intramolecular hydrogen bonding between the O˙ radical and the neighbouring –OH group, which is unique for catechol-like compounds.62–64 These statements are in the agreement with the obtained IC50 values for these derivatives;
(4) Besides the substituent position, the nature of the particular group is also influencing the antioxidant activity. This can be observed in the cases of compounds b (R1–OH), h (R1–NO2), and k (R1–CH3), where the groups with different electronic properties (electron-donating/withdrawal) are present. The results obtained for compound k suggest enhanced antioxidant activity if the R1-substituent possesses an electron-donating effect (such as the –CH3 group). In this respect, it is shown that the group in the R1–position with a strong electron-withdrawal effect (such as –NO2) is not favourable. Generally, the R1–substitution is the least favourable for antioxidant activity compared to R2– and R3–. However, by comparing the results obtained for R1–substituted compounds b, m, o, and t (R1–OH group), it was observed that the presence of an additional neighbouring group with an electron-donating effect improves antioxidant activity (compound m and o), whereas the presence of halogen groups has almost no effect (compound t). On the other hand, in the cases of R2–substituted derivatives c (R2–OH), e (R2–Cl), and i (R2–NO2), similar values of IC50 were obtained, which suggests that, in this position, the electronic properties of the particular group are not noticeably influencing the antioxidant activity. The slight exception to this trend is compound l (R2–CH3), which expressed somewhat higher activity than c, e, and i.
(5) Finally, the number of present groups also influences the antioxidant activity but is dependent on their position and nature. Here, compounds p (bearing vanillin moiety), q (possessing syringaldehyde fragment), and s (having three methoxy groups) can be compared. All three compounds expressed even lower activity than non-substituted derivative a. For derivatives p and s, similar results were obtained, whereas slightly higher activity was observed for derivative q. Such activity of analogue q can be appointed to the presence of the –OH group between two methoxy groups, resulting in the better stabilisation of the phenoxy radical. Also, the influence of the position of the –OH group could be observed in cases of q (R3–OH) and r (R2–OH), where r expressed higher activity. In this case, it was expected the opposite outcome since the –OH in q is neighboured by two –OCH3 groups. Similarly, analogue p should have expressed higher activity than d. Nevertheless, some authors suggest that the presence of an additional methoxy group might manifest a negative impact via steric effect.65
HOMO and LUMO
Energy values of the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) could be used for the general description of molecule reactivity, in our case, towards DPPH radical. For instance, a higher value of HOMO is indicating a better electron-donating ability of the molecule, which is an important feature for radical scavenging.66 On the other hand, the energy difference between HOMO and LUMO (HOMO–LUMO gap) is distinguishing the reaction prospect, i.e., the lower the energy gap, the reaction is more likely to happen, while the higher gap indicates higher molecule stability. The results for compounds a–t obtained in methanol, water, and benzene are presented in Table 3. Here, small but noticeable differences were observed. The highest energy values of HOMOs were calculated for compounds m–q. These findings are in good agreement with experimental results since m, n, and o expressed excellent antioxidant activity against DPPH. On the other hand, the HOMO values of other derivatives were quite similar (−0.224 to −0.227 range), where the lowest correspond to the nitro-substituted derivatives. Here, moderate agreement with the experimental results was achieved, particularly for the analogue h which expressed the lowest experimental radical scavenging activity among all tested compounds. Similar trends were obtained in all investigated solvents.
Table 3 Calculated energy values of HOMO and LUMO (eV) in methanol, water, and benzene for compounds a–t
| Compound |
HOMO (eV) |
LUMO (eV) |
HOMO–LUMO gap (eV) |
| Methanol |
Water |
Benzene |
Methanol |
Water |
Benzene |
Methanol |
Water |
Benzene |
| a |
−0.226 |
−0.226 |
−0.217 |
−0.026 |
−0.026 |
−0.035 |
0.200 |
0.200 |
0.182 |
| b |
−0.224 |
−0.225 |
−0.214 |
−0.026 |
−0.026 |
−0.031 |
0.198 |
0.198 |
0.183 |
| c |
−0.226 |
−0.226 |
−0.216 |
−0.025 |
−0.026 |
−0.034 |
0.200 |
0.200 |
0.182 |
| d |
−0.225 |
−0.226 |
−0.217 |
−0.027 |
−0.026 |
−0.034 |
0.198 |
0.199 |
0.183 |
| e |
−0.226 |
−0.227 |
−0.220 |
−0.028 |
−0.028 |
−0.037 |
0.198 |
0.199 |
0.183 |
| f |
−0.226 |
−0.227 |
−0.220 |
−0.029 |
−0.029 |
−0.038 |
0.196 |
0.198 |
0.183 |
| g |
−0.225 |
−0.226 |
−0.219 |
−0.029 |
−0.028 |
−0.037 |
0.197 |
0.198 |
0.183 |
| h |
−0.226 |
−0.228 |
−0.220 |
−0.106 |
−0.107 |
−0.100 |
0.120 |
0.120 |
0.121 |
| i |
−0.227 |
−0.228 |
−0.224 |
−0.113 |
−0.114 |
−0.102 |
0.115 |
0.114 |
0.122 |
| j |
−0.227 |
−0.228 |
−0.224 |
−0.112 |
−0.113 |
−0.100 |
0.115 |
0.115 |
0.125 |
| k |
−0.227 |
−0.228 |
−0.220 |
−0.024 |
−0.025 |
−0.034 |
0.203 |
0.203 |
0.187 |
| l |
−0.225 |
−0.226 |
−0.217 |
−0.026 |
−0.026 |
−0.034 |
0.199 |
0.200 |
0.183 |
| m |
−0.222 |
−0.223 |
−0.214 |
−0.024 |
−0.025 |
−0.032 |
0.198 |
0.198 |
0.182 |
| n |
−0.219 |
−0.220 |
−0.215 |
−0.025 |
−0.026 |
−0.034 |
0.194 |
0.195 |
0.181 |
| o |
−0.219 |
−0.220 |
−0.212 |
−0.024 |
−0.024 |
−0.029 |
0.195 |
0.196 |
0.184 |
| p |
−0.216 |
−0.217 |
−0.210 |
−0.025 |
−0.025 |
−0.034 |
0.192 |
0.192 |
0.176 |
| q |
−0.213 |
−0.214 |
−0.203 |
−0.025 |
−0.025 |
−0.035 |
0.187 |
0.189 |
0.168 |
| r |
−0.226 |
−0.227 |
−0.219 |
−0.025 |
−0.026 |
−0.034 |
0.201 |
0.201 |
0.185 |
| s |
−0.225 |
−0.226 |
−0.215 |
−0.026 |
−0.026 |
−0.035 |
0.199 |
0.200 |
0.180 |
| t |
−0.225 |
−0.227 |
−0.219 |
−0.037 |
−0.037 |
−0.041 |
0.188 |
0.191 |
0.178 |
The lowest HOMO–LUMO gaps were calculated for h, i, and j, which evidently deviate from the values obtained for other compounds. Such results were the consequence of their multiple times lower LUMO values in comparison to all other compounds. Although these results for h, i, and j, indicate their enhanced reaction probability with the DPPH radical, the parameters were not distinguished enough to explain their different experimental IC50 values. This outcome is suggesting that the activity of h, i, and j is not directly/only linked with the molecule reactivity, i.e., the driving force of the reaction may depend on other factors. On the other hand, if we exclude the results obtained for h, i, and j, a moderate agreement was achieved with the experimental results for other compounds. However, the results are implying the involvement of other factors responsible for the interaction of compounds a–t with the DPPH radical.
Stabilisation energies (ΔEiso)
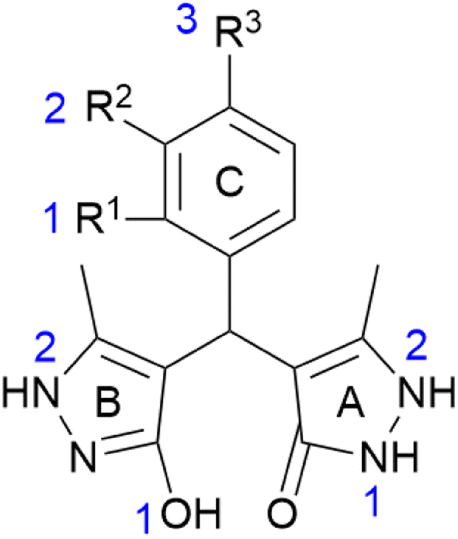
Stabilisation energies (ΔEiso) were calculated for all investigated compounds to evaluate the radical stability (O˙ and N˙) at specified positions (Fig. 2). The results obtained in methanol are presented in Table 4, whereas those in water and benzene are provided in the ESI (Tables S1 and S2†). Here, on pyrazolone ring (A), the formation of two N–radicals was observed (positions N1A and N2A), whereas on pyrazole ring (B) the stabilities of O˙ and N˙ radicals were studied (positions O1B and N2B). Depending on the substitution of the phenyl moiety (ring C), corresponding O–radicals were examined (positions O1C, O2C, and O3C). Based on the obtained results in methanol, the lowest ΔEiso values were calculated for the ring C radicals of compounds m–q (Table 4). These findings indicate the great involvement of the ring C –OH groups in the antioxidant activity, particularly in the cases m, n, and q, which is in good agreement with experimental results. Similarly, regarding the calculated ΔEiso values, it was observed that ring A also empowers the antioxidant capability of the investigated compounds, whereas the involvement of ring B could be considered negligible. In this respect, the –NH groups at position 2 of ring A participate significantly in the radical scavenging, whereas a minor contribution of the N1 was noted. In the case of ring B, high energy values suggest insufficient stabilisation of N2 radicals, and thus, no involvement of those –NH groups in the radical scavenging. The results obtained for ring B –OH groups (O1B position) indicated their insignificant contribution to the antioxidant activity, but they, mutually compared, diverse the most. Generally, the obtained results differentiate depending on the present substituents on the ring C. In comparison to the results obtained for a, it was observed that a particular substituent differently alters radical stabilisation at a different position. In this respect, considerably better stabilisation of ring N1A radical was noted for analogues k and l, bearing R1– and R2–CH3, respectively. Also, the stability of the O1B radical is most improved in the case of compound k. These statements are in the agreement with their experimental results, i.e., their enhanced activity against DPPH. On the other hand, the N1A is substantially less stable (compared to the non-substituted analogue a) if the –NO2 group is introduced to the ring C (compounds h, i, and j). This effect was also noticed for the N2A radical. Here, the results suggest that nitro-substitution is not favourable. However, compound i expressed slightly higher antioxidant activity toward DPPH. Nevertheless, literature reports variations between in silico and in vitro results regarding the influence of the particular group on radical scavenging.65 Interestingly, even in the case of the most active compounds m and n, N1A and N2A radicals are slightly less stable than those in the case of a. Still, in such a case, it is clear that the presence of catechol moiety on ring C could overcompensate these minor downsides since the best stabilisation was observed for these radicals.
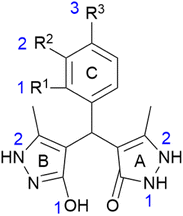 |
| | Fig. 2 Positions and labels of the investigated radical species (O˙ and N˙). A stand for pyrazolone, B for pyrazole, and C for phenyl ring. The specific positions of the radicals are marked separately for each ring (blue numbers). | |
Table 4 Calculated stabilisation energies (kJ mol−1) for compounds a–t in methanol
| Entry |
Ring A |
Ring B |
Ring C |
| N1 |
N2 |
O1 |
N2 |
O1 |
O2 |
O3 |
| a |
−5.83 |
−20.47 |
−0.58 |
28.53 |
— |
— |
— |
| b |
−5.10 |
−21.09 |
−3.55 |
25.12 |
−7.99 |
— |
— |
| c |
−5.74 |
−20.66 |
0.30 |
28.75 |
— |
−2.47 |
— |
| d |
−5.60 |
−20.20 |
0.34 |
29.33 |
— |
— |
−9.61 |
| e |
−5.03 |
−19.13 |
0.07 |
29.21 |
— |
— |
— |
| f |
−4.74 |
−19.00 |
0.69 |
29.76 |
— |
— |
— |
| g |
−5.06 |
−19.44 |
0.38 |
29.52 |
— |
— |
— |
| h |
−2.31 |
−15.80 |
−2.21 |
31.26 |
— |
— |
— |
| i |
−1.95 |
−15.76 |
3.90 |
32.38 |
— |
— |
— |
| j |
−3.72 |
−17.51 |
0.97 |
30.99 |
— |
— |
— |
| k |
−10.16 |
−19.18 |
−5.72 |
31.59 |
— |
— |
— |
| l |
−8.19 |
−20.66 |
0.45 |
28.51 |
— |
— |
— |
| m |
−3.79 |
−18.24 |
−4.22 |
29.84 |
−34.66 |
−31.42 |
— |
| n |
−5.04 |
−19.62 |
0.20 |
28.49 |
— |
−33.09 |
−35.89 |
| o |
−4.64 |
−19.54 |
−3.04 |
28.45 |
−25.59 |
— |
— |
| p |
−6.25 |
−20.90 |
0.57 |
27.74 |
— |
— |
−28.48 |
| q |
−5.21 |
−19.93 |
0.93 |
29.02 |
— |
— |
−44.34 |
| r |
−5.41 |
−19.58 |
0.82 |
28.85 |
— |
−17.06 |
— |
| s |
−5.04 |
−19.82 |
1.20 |
28.90 |
— |
— |
— |
| t |
−3.10 |
−18.05 |
−1.88 |
29.76 |
−12.84 |
— |
— |
The results obtained in water were similar to those in methanol, with some exceptions (Table S1†). Generally, the radical stabilisation is somewhat better in water since most values were slightly lower. Excellent agreement for the analogues b, c, and d was achieved. Nitro-substitution was the least favourable, particularly ortho analogue (compound h), which corresponds to the obtained experimental results. Moreover, the parameters obtained for compounds h, i, and j were mutually well distinguished. On the other hand, slightly lower stability of the N1A and N2A radicals was observed for compounds k, l, and m. In benzene as a solvent (Table S2†), parameters calculated for the most efficient radical scavengers l, m, n, and o were in good accordance with experimental results, as well as for –OH monosubstituted analogues b, c, and d. As in all previous solvents, nitro-substitution was, once again, designated as thermodynamically the least favourable.
Antioxidant mechanism analysis
The estimation of the most preferable radical scavenging pathway for compounds a–t was performed by mutual comparison of calculated thermodynamic parameters: bond dissociation enthalpy (BDE), ionisation potential (IP), proton affinity (PA), proton dissociation enthalpy (PDE), and electron transfer enthalpy (ETE). The results obtained in methanol are presented in Tables 5–7, while the others are presented in Tables S3–S8.† Here, several plausible antioxidant mechanisms were considered: hydrogen atom transfer (HAT), single electron transfer-proton transfer (SET-PT), and sequential proton loss electron transfer (SPLET).67 Namely, all these scavenging pathways lead to the inactivation of radical species, followed by the formation of corresponding radical from the antioxidant. The HAT route is a one-step process defined with the BDE parameter, which describes the ease of the X–H bond homolytic cleavage (X = O or N). Here, the radical species is inactivated with the direct transfer of the hydrogen atom.68 On the other hand, in the SET-PT mechanism, the transfer of hydrogen is observed as a two-stage process. The SET-PT occurs with electron transfer from the antioxidant to the radical species, after which the radical-cationic antioxidant is formed.65 This step is described with the value of IP since its related to the antioxidant's ability to ionise. The second step involves proton transfer from the radical-cationic antioxidant to radical species. Here, the PDE value defines this process since the dissociation of the proton is required. In the SPLET route, these two processes are observed oppositely, i.e., the reaction is initiated with the deprotonation of antioxidant, while in the second step the electrons are transferred.69 Hence, the possibility of the SPLET pathway is affected by the values of PA and ETE. All these processes could be influenced by the solvent's polarity; therefore, the calculations were performed in methanol, water, and benzene. The high values of IP indicated that the SET-PT can be eliminated as a scavenging route in all investigated solvents (Tables 5–7 and S3–S8†). In polar solvents, PA values were significantly lower than BDE, which designated the SPLET mechanism as a thermodynamically more favourable antioxidant pathway. On the other hand, in benzene as a solvent, the HAT mechanism is slightly predominant over the SPLET since the values of BDE were somewhat lower than PA. Generally, the BDEs were lower in benzene in comparison to the polar solvents, whereas PA values were higher. Such an outcome could be expected since the reactions involving charged intermediates are less likely to occur in a non-polar environment. Moreover, all parameters suggested an almost insignificant contribution of B-ring groups in antioxidant activity, especially –NH. This can be observed by the comparison of calculated parameters to other investigated positions. The most favourable involvement in antioxidant activity was noticed for C-ring positions of the most active compounds m, n, and o, which represents additional proof of their enhanced activity, as well as for the ring A –NH group in position 2. Also, the numbers suggested a significant influence of the ring A –NH group in position 1. All these observations pointed out ring A as most responsible for the antioxidant activity of compounds a–t, as well as ring C in the cases where its involvement is possible.
Table 5 Calculated bond dissociation enthalpies (kJ mol−1) for compounds a–t in methanol
| |
Bond dissociation enthalpy (BDE) kJ mol−1 |
| N1 (A) |
N2 (A) |
O1 (B) |
N2 (B) |
O1 (C) |
O2 (C) |
O3 (C) |
| a |
347 |
332 |
352 |
381 |
— |
— |
— |
| b |
347 |
331 |
349 |
378 |
344 |
— |
— |
| c |
347 |
332 |
353 |
381 |
— |
350 |
— |
| d |
347 |
332 |
353 |
382 |
— |
— |
343 |
| e |
347 |
333 |
352 |
382 |
— |
— |
— |
| f |
348 |
333 |
353 |
382 |
— |
— |
— |
| g |
347 |
333 |
353 |
382 |
— |
— |
— |
| h |
350 |
337 |
350 |
384 |
— |
— |
— |
| i |
350 |
337 |
356 |
385 |
— |
— |
— |
| j |
349 |
335 |
353 |
383 |
— |
— |
— |
| k |
342 |
333 |
347 |
384 |
— |
— |
— |
| l |
344 |
332 |
353 |
381 |
— |
— |
— |
| m |
349 |
334 |
348 |
382 |
318 |
321 |
— |
| n |
347 |
333 |
353 |
381 |
— |
319 |
317 |
| o |
348 |
333 |
349 |
381 |
327 |
— |
— |
| p |
346 |
332 |
353 |
380 |
— |
— |
324 |
| q |
347 |
333 |
353 |
381 |
— |
— |
308 |
| r |
347 |
333 |
353 |
381 |
— |
335 |
— |
| s |
347 |
333 |
354 |
381 |
— |
— |
— |
| t |
349 |
334 |
351 |
382 |
340 |
— |
— |
Table 6 Calculated IP and PDE values (kJ mol−1) for compounds a–t in methanol
| |
IP |
PDE |
| N1 (A) |
N2 (A) |
O1 (B) |
N2 (B) |
O1 (C) |
O2 (C) |
O3 (C) |
| a |
457 |
51 |
36 |
56 |
85 |
— |
— |
— |
| b |
456 |
53 |
37 |
54 |
83 |
50 |
— |
— |
| c |
458 |
50 |
35 |
56 |
85 |
— |
54 |
— |
| d |
466 |
43 |
28 |
49 |
78 |
— |
— |
39 |
| e |
460 |
49 |
35 |
54 |
83 |
— |
— |
— |
| f |
460 |
49 |
35 |
55 |
84 |
— |
— |
— |
| g |
459 |
50 |
35 |
55 |
84 |
— |
— |
— |
| h |
463 |
49 |
36 |
49 |
83 |
— |
— |
— |
| i |
465 |
47 |
34 |
53 |
82 |
— |
— |
— |
| j |
464 |
46 |
33 |
51 |
81 |
— |
— |
— |
| k |
457 |
47 |
38 |
51 |
88 |
— |
— |
— |
| l |
457 |
49 |
37 |
58 |
86 |
— |
— |
— |
| m |
461 |
49 |
35 |
49 |
83 |
19 |
22 |
— |
| n |
450 |
59 |
44 |
64 |
92 |
— |
31 |
28 |
| o |
454 |
56 |
41 |
57 |
89 |
35 |
— |
— |
| p |
444 |
64 |
50 |
71 |
98 |
— |
— |
42 |
| q |
437 |
72 |
57 |
78 |
106 |
— |
— |
33 |
| r |
459 |
50 |
36 |
56 |
84 |
— |
38 |
— |
| s |
439 |
70 |
56 |
77 |
104 |
— |
— |
— |
| t |
432 |
79 |
64 |
81 |
112 |
70 |
— |
— |
Table 7 Calculated PA and ETE values (kJ mol−1) for compounds a–t in methanol
| |
N1 (A) |
N2 (A) |
O1 (B) |
N2 (B) |
O1 (C) |
O2 (C) |
O3 (C) |
| PA |
ETE |
PA |
ETE |
PA |
ETE |
PA |
ETE |
PA |
ETE |
PA |
ETE |
PA |
ETE |
| a |
139 |
369 |
133 |
360 |
176 |
338 |
197 |
346 |
— |
— |
— |
— |
— |
— |
| b |
138 |
370 |
132 |
361 |
169 |
341 |
199 |
340 |
152 |
354 |
— |
— |
— |
— |
| c |
140 |
369 |
133 |
360 |
176 |
338 |
200 |
343 |
— |
— |
156 |
356 |
— |
— |
| d |
140 |
368 |
134 |
360 |
177 |
337 |
200 |
344 |
— |
— |
— |
— |
156 |
348 |
| e |
138 |
371 |
132 |
363 |
174 |
340 |
198 |
345 |
— |
— |
— |
— |
— |
— |
| f |
139 |
371 |
133 |
362 |
175 |
340 |
198 |
346 |
— |
— |
— |
— |
— |
— |
| g |
139 |
370 |
133 |
362 |
175 |
339 |
198 |
345 |
— |
— |
— |
— |
— |
— |
| h |
135 |
377 |
128 |
370 |
166 |
345 |
196 |
350 |
— |
— |
— |
— |
— |
— |
| i |
139 |
373 |
133 |
366 |
176 |
342 |
199 |
347 |
— |
— |
— |
— |
— |
— |
| j |
136 |
374 |
130 |
366 |
171 |
344 |
196 |
349 |
— |
— |
— |
— |
— |
— |
| k |
141 |
363 |
133 |
362 |
172 |
336 |
199 |
347 |
— |
— |
— |
— |
— |
— |
| l |
140 |
366 |
134 |
360 |
177 |
338 |
200 |
343 |
— |
— |
— |
— |
— |
— |
| m |
140 |
370 |
132 |
363 |
175 |
335 |
200 |
344 |
137 |
342 |
138 |
345 |
— |
— |
| n |
137 |
372 |
134 |
361 |
177 |
337 |
200 |
343 |
— |
— |
140 |
341 |
141 |
337 |
| o |
139 |
370 |
133 |
361 |
170 |
341 |
200 |
343 |
156 |
333 |
— |
— |
— |
— |
| p |
139 |
368 |
134 |
360 |
176 |
338 |
199 |
343 |
— |
— |
— |
— |
157 |
329 |
| q |
140 |
369 |
133 |
361 |
179 |
336 |
199 |
344 |
— |
— |
— |
— |
157 |
313 |
| r |
139 |
369 |
133 |
361 |
176 |
339 |
199 |
344 |
— |
— |
147 |
350 |
— |
— |
| s |
139 |
370 |
133 |
361 |
177 |
339 |
199 |
344 |
— |
— |
— |
— |
— |
— |
| t |
136 |
375 |
130 |
366 |
167 |
346 |
197 |
347 |
128 |
373 |
— |
— |
— |
— |
A more profound comprehension of the preferable radical quenching mechanism was reached by calculating the reaction enthalpies (ΔHBDE, ΔHIP and ΔHPDE, ΔHPA, and ΔHETE) for the reactions of a–t with the selected radicals. Here, seven radical species were chosen upon their cellular presence and behaviour (˙OCH3, ˙OC(CH3)3, ˙OH, ˙OOH, ˙OOCH3, ˙OO–CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2, and O2˙−),70 as well as DPPH. To estimate the influence of polar and non-polar surroundings, calculations were performed in different solvents (methanol, water, and benzene), and the electronic properties of selected radicals were accounted for as well.71 The results obtained in methanol for the most active derivative m are presented in Table 8, while all other results are provided in the ESI (Tables S9–S65†). The obtained results varied in dependence on radical species, reaction media (solvent), and investigated analogue, but some general trends were noticed. The SET-PT mechanism can be excluded in all investigated solvents. Here, the high positive values ΔHIP indicated that ionisation of a–t is an endothermic step with a high energy barrier, which eliminated the SET-PT as a preferable scavenging route. In benzene, energy requirements for ionisation were even higher than in polar solvents. The possibility of SET-PT could be noted for some derivatives only in the reaction with DPPH in polar solvents, where the values of ΔHIP were at least comparable with ΔHBDE and ΔHPA. In polar solvents, the competition between HAT and SPLET mechanisms was observed, although the HAT is slightly prevailing in most cases. Only in the case of ˙OH radical, the HAT is clearly predominant due to higher differences between ΔHBDE and ΔHPA. On the other hand, in benzene as a solvent, the SPLET pathway is prevailing in all cases, except in the reactions with DPPH where the HAT mechanism is more favourable. Mutual comparison of calculated reaction enthalpies within specified positions revealed more favourable engagement of certain groups. Here, the high engagement of ring C –OH groups (analogues m–q) in the antioxidant activity, as well as ring A –NH groups was noted, with the low contribution of ring B groups.
CH2, and O2˙−),70 as well as DPPH. To estimate the influence of polar and non-polar surroundings, calculations were performed in different solvents (methanol, water, and benzene), and the electronic properties of selected radicals were accounted for as well.71 The results obtained in methanol for the most active derivative m are presented in Table 8, while all other results are provided in the ESI (Tables S9–S65†). The obtained results varied in dependence on radical species, reaction media (solvent), and investigated analogue, but some general trends were noticed. The SET-PT mechanism can be excluded in all investigated solvents. Here, the high positive values ΔHIP indicated that ionisation of a–t is an endothermic step with a high energy barrier, which eliminated the SET-PT as a preferable scavenging route. In benzene, energy requirements for ionisation were even higher than in polar solvents. The possibility of SET-PT could be noted for some derivatives only in the reaction with DPPH in polar solvents, where the values of ΔHIP were at least comparable with ΔHBDE and ΔHPA. In polar solvents, the competition between HAT and SPLET mechanisms was observed, although the HAT is slightly prevailing in most cases. Only in the case of ˙OH radical, the HAT is clearly predominant due to higher differences between ΔHBDE and ΔHPA. On the other hand, in benzene as a solvent, the SPLET pathway is prevailing in all cases, except in the reactions with DPPH where the HAT mechanism is more favourable. Mutual comparison of calculated reaction enthalpies within specified positions revealed more favourable engagement of certain groups. Here, the high engagement of ring C –OH groups (analogues m–q) in the antioxidant activity, as well as ring A –NH groups was noted, with the low contribution of ring B groups.
Table 8 Calculated reaction enthalpies (kJ mol−1) for the reactions of compound m with the selected radical species in methanol
| m |
HAT |
SET-PT |
SPLET |
| Radical |
ΔHBDE |
ΔHIP |
ΔHPDE |
ΔHPA |
ΔHETE |
| ˙OCH3 |
N1 (A) |
−79 |
84 |
−162 |
−72 |
−7 |
| N2 (A) |
−93 |
−177 |
−79 |
−14 |
| N2 (B) |
−45 |
−129 |
−12 |
−33 |
| O1 (B) |
−79 |
−163 |
−37 |
−42 |
| O1 (C) |
−109 |
−193 |
−74 |
−35 |
| O2 (C) |
−106 |
−190 |
−74 |
−33 |
| ˙OC(CH3)3 |
N1 (A) |
−87 |
84 |
−171 |
−80 |
−7 |
| N2 (A) |
−101 |
−185 |
−88 |
−13 |
| N2 (B) |
−53 |
−137 |
−20 |
−33 |
| O1 (B) |
−87 |
−171 |
−46 |
−41 |
| O1 (C) |
−118 |
−202 |
−83 |
−35 |
| O2 (C) |
−114 |
−199 |
−82 |
−32 |
| ˙OH |
N1 (A) |
−150 |
5 |
−155 |
−64 |
−86 |
| N2 (A) |
−165 |
−169 |
−72 |
−93 |
| N2 (B) |
−116 |
−121 |
−4 |
−112 |
| O1 (B) |
−150 |
−155 |
−30 |
−121 |
| O1 (C) |
−181 |
−186 |
−67 |
−114 |
| O2 (C) |
−178 |
−183 |
−66 |
−111 |
| ˙OOH |
N1 (A) |
−10 |
106 |
−116 |
−26 |
16 |
| N2 (A) |
−25 |
−131 |
−33 |
9 |
| N2 (B) |
24 |
−83 |
34 |
−11 |
| O1 (B) |
−11 |
−117 |
9 |
−19 |
| O1 (C) |
−41 |
−147 |
−28 |
−13 |
| O2 (C) |
−38 |
−144 |
−28 |
−10 |
| ˙OOCH3 |
N1 (A) |
−3 |
115 |
−118 |
−27 |
24 |
| N2 (A) |
−17 |
−132 |
−35 |
17 |
| N2 (B) |
31 |
−84 |
33 |
−2 |
| O1 (B) |
−3 |
−118 |
8 |
−11 |
| O1 (C) |
−34 |
−148 |
−30 |
−4 |
| O2 (C) |
−30 |
−145 |
−29 |
−1 |
| ˙OO–CHCH2 |
N1 (A) |
−2 |
95 |
−98 |
−7 |
5 |
| N2 (A) |
−17 |
−112 |
−15 |
−2 |
| N2 (B) |
31 |
−64 |
53 |
−22 |
| O1 (B) |
−3 |
−98 |
28 |
−30 |
| O1 (C) |
−33 |
−129 |
−10 |
−24 |
| O2 (C) |
−30 |
−125 |
−9 |
−21 |
| DPPH |
N1 (A) |
26 |
83 |
−57 |
33 |
−7 |
| N2 (A) |
11 |
−72 |
26 |
−14 |
| N2 (B) |
60 |
−24 |
93 |
−34 |
| O1 (B) |
25 |
−58 |
68 |
−42 |
| O1 (C) |
−5 |
−88 |
31 |
−36 |
| O2 (C) |
−2 |
−85 |
31 |
−33 |
| O2˙− |
N1 (A) |
62 |
273 |
−211 |
46 |
16 |
| N2 (A) |
47 |
−225 |
39 |
9 |
| N2 (B) |
96 |
−177 |
106 |
−11 |
| O1 (B) |
62 |
−211 |
81 |
−19 |
| O1 (C) |
31 |
−242 |
44 |
−13 |
| O2 (C) |
34 |
−238 |
44 |
−10 |
Radical adduct formation (RAF) mechanism
Besides HAT, SET-PT, and SPLET antioxidant pathways, the formation of radical adduct was investigated for the reactions of compounds a–t with ˙OH radical. In this antioxidant route, the addition of the radical species to the antioxidant occurs, resulting in the formation of the radical adduct. To determine the most favourable sites for the radical attack, the Fukui functions were calculated for all compounds.72 Here, the values of f0nbo revealed multiple positions for radical attack (higher values indicate higher probability, Tables S68–S87†). Therefore, the structures of the formed radical adducts were optimised for each reaction site of each compound. The enthalpies of the reactions with ˙OH radical were calculated in methanol and benzene as solvents (Tables 9 and S68–S87†). Atom labelling is provided in Tables S68–S87.†
Table 9 Calculated reaction enthalpies (kJ mol−1) for the radical adduct formation of compounds a–t with ˙OH radicala
| Entry |
Position |
ΔHRAF (methanol) |
ΔHRAF (benzene) |
| Symbols * and # match equivalent sites for the radical attack for all compounds. |
| a + ˙OH |
C20* |
−90 |
−97 |
| b + ˙OH |
C19* |
−89 |
−85 |
| c + ˙OH |
C20* |
−89 |
−97 |
| d + ˙OH |
C20* |
−90 |
−97 |
| e + ˙OH |
C19* |
−89 |
−96 |
| f + ˙OH |
C20* |
−90 |
−96 |
| g + ˙OH |
C20* |
−90 |
−97 |
| h + ˙OH |
C9# |
−40 |
−42 |
| i + ˙OH |
C10# |
−102 |
−57 |
| j + ˙OH |
C10# |
−100 |
−57 |
| k + ˙OH |
C19* |
−88 |
−98 |
| l + ˙OH |
C20* |
−90 |
−98 |
| m + ˙OH |
C19* |
−87 |
−95 |
| C1 |
−84 |
−78 |
| C35 |
−84 |
−89 |
| n + ˙OH |
C22* |
−90 |
−98 |
| C2 |
−90 |
−80 |
| C3 |
−88 |
−92 |
| o + ˙OH |
C18* |
−88 |
−86 |
| C2 |
−81 |
−82 |
| C3 |
−88 |
−80 |
| p + ˙OH |
C20* |
−91 |
−97 |
| C34 |
−95 |
−85 |
| C35 |
−84 |
−79 |
| q + ˙OH |
C33 |
−106 |
−103 |
| C19* |
−91 |
−98 |
| r + ˙OH |
C17* |
−90 |
−97 |
| s + ˙OH |
C19* |
−87 |
−98 |
| t + ˙OH |
C4 |
−85 |
−77 |
| C32 |
−77 |
−72 |
Based on the obtained ΔHRAF values in methanol, in almost all cases (except derivatives h, i, j, and t), the formation of the radical adduct occurs on the pyrazolone ring A, where the carbon atom bearing methyl group (labelled with *, Table 9), was identified as the most reactive site for radical attack. Here, the influence of ring C substitution was noticed, i.e., in all these cases, all analogues bear an electron-donating group on ring C. On the other hand, when the electron-withdrawing group is introduced (derivatives h, i, j), the highest probability for radical adduct formation is on the pyrazole B ring, on the carbon neighbouring the methine group (labelled with #, Table 9). Additionally, for derivatives m–q, the formation of radical adduct could occur on ring C, initiated with the radical attack on carbon atoms bearing hydroxy or methoxy groups. Such an outcome could be linked to the proximity of oxygen atoms and their involvement in the stabilisation of the formed radical adduct.73 Similarly, in the case of t, the most reactive sites were identified on ring C, particularly on the C32, which is between two carbon atoms substituted with chlorine. An analogous trend was observed in benzene as a solvent.
Comparing the values of ΔHRAF obtained in methanol (Table 9) with ΔHBDE values obtained for the reactions with ˙OH radical (Tables 8 and S9–S27†), it could be observed that the HAT pathway is still predominant since ΔHRAF is significantly higher than ΔHBDE. On the other hand, in comparison to ΔHIP and ΔHPDE, ΔHPA, and ΔHETE values, the RAF mechanism is prevailing over SET-PT and SPLET in polar solvents.
Conclusions
In this work, the antioxidant potency of the selected pyrazolone derivatives a–t was assessed using in vitro and in silico methods. The obtained results revealed great radical scavenging abilities of the investigated compounds against the DPPH radical (IC50 values in the range of 2.6–7.8 μM). All compounds expressed higher antioxidant potency than the parent pyrazolone compound. The results indicated the dependence of the substituents on ring C, i.e., on their number, position, and nature. In comparison to the non-substituted analogue, enhanced antioxidant activity was noted for derivatives bearing R2-substituent on the ring C (compounds c (R2–OH), e (R2–Cl), i (R2–NO2), and l (R2–CH3)). Furthermore, the enhancement was even more noticeable in the cases of compounds m and n bearing catechol moiety, which exhibited the best antioxidant activity (IC50 = 2.6 and 2.9 μM, respectively). On the other hand, R1-substitution was generally identified as the least favourable for radical scavenging, especially when the electron-withdrawal group is introduced. Density functional theory calculations were in good agreement with experimental results. From the thermodynamical point of view, the calculated descriptors indicated compounds m, n, and o within the most prominent ones. The prediction of the most preferable antioxidant pathway was performed in the absence and presence of radical species, as well as in polar and non-polar surroundings. In the absence of free radicals, in polar solvents, the SPLET mechanism is a favourable scavenging route, whereas in nonpolar the HAT is slightly predominant. In the presence of radical species, the obtained values of the reaction enthalpies revealed that HAT is slightly prevailing in polar solvents, while the SPLET is prevalent in nonpolar. Additionally, the radical adduct formation (RAF) pathway was investigated with ˙OH radical. The obtained results revealed that RAF prevails over SPLET in polar solvents, whereas HAT is preferable radical scavenging pathway than RAF, and that the formation of the radical adduct is dependent on ring C substitution. The presence of electron-donating groups favours radical adduct formation on the pyrazolone ring A, while the electron-withdrawing group induce radical adduct formation on the pyrazole B ring. In the cases where ring C bears hydroxy or methoxy groups, the formation of radical adduct could occur on this ring. These findings represent valuable data for this pyrazolone class and could be used as the basis for further investigations.
Experimental section
Antioxidant activity of compounds a–t
The insight into the antioxidant potency of pyrazolones a–t was reached using the 2,2-diphenyl-1-picrylhydrazyl (DPPH) method.74 The samples were prepared by mixing the methanolic solution of DPPH radical (0.05 mM, 1 mL) with the tested compound (20 μL of different concentrations in dimethyl sulfoxide (DMSO) and 980 μL of methanol). After the incubation period (twenty and sixty minutes in a dark room at room temperature), the absorbance was determined spectrophotometrically at 517 nm. Quercetin and nordihydroguaiaretic acid were used as reference compounds, whereas methanol was a control solution. All measurements were made in triplicate. The IC50 values (which represent a minimal concentration of the assessed compound necessary for 50% of a maximum scavenging capacity) were determined for all compounds (regression equations are given in Table S88†) and presented as mean values ± standard deviation (SD) of three independent measurements. For all compounds, the stoichiometric factor (SF) was calculated using the equation:75,76| | |
SF = [DPPH]/(2× IC50)
| (1) |
Density functional theory (DFT) calculations
The density functional theory (DFT) method was used for the calculation of equilibrium geometries of pyrazolones a–t (Pyr) and all radical species, employing the Gaussian 09 program package.77 All calculations were performed using the B3LYP/6-311+g(d,p) level of theory.78–80 The SMD solvation model was applied for the calculations in different solvents (methanol, water, and benzene).81 Methanol was chosen since it was used in the in vitro experiments. Calculations were made in water to mimic the polar environment of the living cell, as well as in benzene to simulate nonpolar surroundings. Depending on the molecule state, charges and multiplicities were properly assigned to each species (for neutral molecules charge = 0/multiplicity = 1; for anions charge = −1/multiplicity = 1; for radicals charge = 0/multiplicity = 2; and for radical cations charge = 1/multiplicity = 2). Unrestricted spin calculations were performed for open-shell systems. Thermodynamic parameters required for the estimation of the preferable radical scavenging mechanism, i.e., bond dissociation enthalpy (BDE), ionisation potential (IP), proton affinity (PA), proton dissociation enthalpy (PDE), and electron transfer enthalpy (ETE) were calculated using the eqn (2)–(6):| | |
BDE = H(Pyr˙) + H(H˙) − H(Pyr)
| (2) |
| | |
IP = H(Pyr˙+) + H(e−) − H(Pyr)
| (3) |
| | |
PDE = H(Pyr˙) + H(H+) − H(Pyr˙+)
| (4) |
| | |
PA = H(Pyr−) + H(H+) − H(Pyr)
| (5) |
| | |
ETE = H(Pyr˙) + H(e−) − H(Pyr−)
| (6) |
For the estimation of the preferred antiradical pathway in the presence of selected radical species, reaction enthalpies defined with eqn (7)–(12) were calculated at 298 K.82 The values of the solvation enthalpies of electron and proton were acquired from the literature.83 Stabilisation energies (ΔEiso) were calculated to evaluate the radical stability at a particular position following the eqn (13),82 whereas the reaction enthalpies for the formation of radical adducts were acquired using the eqn (14):
| | |
ΔrHBDE = [H(Pyr˙) + H(R–OH)] − [H(Pyr) + H(R–O˙)]
| (7) |
| | |
ΔrHIP = [H(Pyr˙+) + H(R–O−)] − [H(Pyr) + H(R–O˙)]
| (8) |
| | |
ΔrHPDE = [H(Pyr˙) + H(R–OH)] − [H(Pyr˙+) + H(R–O−)
| (9) |
| | |
ΔrHPA = [H(Pyr−) + H(R–OH)] − [H(Pyr) + H(R–O−)]
| (10) |
| | |
ΔrHETE = [H(Pyr˙) + H(R–O−)] − [H(Pyr−) + H(R–O˙)]
| (11) |
| | |
ΔrHBDE = ΔrHIP + ΔrHPDE = ΔrHPA + ΔrHETE
| (12) |
| | |
ΔEiso= (H(Pyr˙) + H(Ph–OH)) − (H(Pyr) + H(Ph–O˙)
| (13) |
| | |
ΔrHRAF = H(Pyr–OH˙) − H(Pyr) − H(˙OH)
| (14) |
Author contributions
Conceptualisation, V. P.; methodology, J. B., and D. S.; validation, J. B., and V. M.; formal analysis, J. B., and V. P.; investigation, J. B., and V. M.; resources, V. P., and Z. P.; data curation, J. B., and V. P.; writing—original draft preparation J. B.; writing—review and editing, V. P., D. S., and Z. P; visualisation, J. B., and V. P.; supervision, V. P.; project administration, V. P. and Z.P.; funding acquisition, V. P., and Z. P. All authors have read and agreed to the published version of the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the Serbian Ministry of Education, Science and Technological Development (Agreement No. 451-03-68/2022-14/200122, 451-03-68/2022-14/200088, and 451-03-68/2022-14/200378).
References
- Z. Wang, in Comprehensive Organic Name Reactions and Reagents, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2010, pp. 331–334 Search PubMed.
- G. Mustafa, M. Zia-ur-Rehman, S. H. Sumrra, M. Ashfaq, W. Zafar and M. Ashfaq, J. Mol. Struct., 2022, 1262, 133044 CrossRef CAS.
- A. Kadam, B. Dawane, M. Pawar, H. Shegokar, K. Patil, R. Meshram and R. Gacche, Bioorg. Chem., 2014, 53, 67–74 CrossRef CAS PubMed.
- Z. Zhao, X. Dai, C. Li, X. Wang, J. Tian, Y. Feng, J. Xie, C. Ma, Z. Nie, P. Fan, M. Qian, X. He, S. Wu, Y. Zhang and X. Zheng, Eur. J. Med. Chem., 2020, 186, 111893 CrossRef CAS PubMed.
- W. Sneader, in Drug Discovery, John Wiley & Sons, Ltd, Chichester, UK, 2006, pp. 115–150 Search PubMed.
- I. Nikolova, V. Petkova, J. Tencheva, N. Benbasat, J. Voinikov and N. Danchev, Biotechnol. Biotechnol. Equip., 2013, 27, 3605–3619 CrossRef CAS.
- S. Adhikari, M. Singh, P. Sharma and S. Arora, J. Appl. Pharm. Sci., 2021, 11, 26–37 CAS.
- A. Indrasena, S. Riyaz, P. L. Mallipeddi, P. Padmaja, B. Sridhar and P. K. Dubey, Tetrahedron Lett., 2014, 55, 5014–5018 CrossRef CAS.
- S. Shubhalaxmi, L. Pathak, K. Ananda and K. S. Bhat, Cogent Chem., 2016, 2, 1141388 CrossRef.
- R. Al-Saheb, S. Makharza, F. Al-Battah, R. Abu-El-Halawa, T. Kaimari and O. S. Abu Abed, Biosci. Rep., 2020, 40, 1–13 CrossRef PubMed.
- N. Parmar, S. Teraiya, R. Patel, H. Barad, H. Jajda and V. Thakkar, J. Saudi Chem. Soc., 2015, 19, 36–41 CrossRef.
- S. Shamsuzzaman, A. Mashrai, A. Ahmad, A. M. Dar, H. Khanam, M. Danishuddin and A. U. Khan, Med. Chem. Res., 2014, 23, 348–362 CrossRef.
- S. K. Krishnasamy, V. Namasivayam, S. Mathew, R. S. Eakambaram, I. A. Ibrahim, A. Natarajan and S. Palaniappan, Arch. Pharm., 2016, 349, 383–397 CrossRef CAS PubMed.
- A. Dahal, M. Lo, S. Singh, H. Vo, D. ElHage, S. D. Jois and S. Murru, Chem. Biol. Drug Des., 2022, 99, 620–633 CrossRef CAS PubMed.
- K. M. Vyas, R. N. Jadeja, D. Patel, R. V. Devkar and V. K. Gupta, Polyhedron, 2013, 65, 262–274 CrossRef CAS.
- R. Pettinari, F. Marchetti, C. Pettinari, A. Petrini, R. Scopelliti, C. M. Clavel and P. J. Dyson, Inorg. Chem., 2014, 53, 13105–13111 CrossRef CAS PubMed.
- N. A. Khalil, E. M. Ahmed, K. O. Mohamed, Y. M. Nissan and S. A. B. Zaitone, Bioorg. Med. Chem., 2014, 22, 2080–2089 CrossRef CAS PubMed.
- G. Mariappan, B. P. Saha, L. Sutharson, A. Singh, S. Garg, L. Pandey and D. Kumar, Saudi Pharmaceut. J., 2011, 19, 115–122 CrossRef CAS PubMed.
- M. T. El Sayed, M. A. M. S. El-Sharief, E. S. Zarie, N. M. Morsy, A. R. Elsheakh, A. Voronkov, V. Berishvili and G. S. Hassan, Bioorg. Med. Chem. Lett., 2018, 28, 952–957 CrossRef CAS PubMed.
- R. V. Antre, Med. Chem., 2012, 02, 126–130 Search PubMed.
- A. A. Moneer, K. O. Mohammed and H. B. El-Nassan, Chem. Biol. Drug Des., 2016, 87, 784–793 CrossRef CAS PubMed.
- C. Dohutia, P. P. Kaishap and D. Chetia, Int. J. Pharm. Sci., 2013, 5, 86–90 CAS.
- N. Uramaru, H. Shigematsu, A. Toda, R. Eyanagi, S. Kitamura and S. Ohta, J. Med. Chem., 2010, 53, 8727–8733 CrossRef CAS PubMed.
- X. Fan, X. Zhang, L. Zhou, K. A. Keith, E. R. Kern and P. F. Torrence, Bioorg. Med. Chem. Lett., 2006, 16, 3224–3228 CrossRef CAS PubMed.
- V. A. Obakachi, N. D. Kushwaha, B. Kushwaha, M. C. Mahlalela, S. R. Shinde, I. Kehinde and R. Karpoormath, J. Mol. Struct., 2021, 1241, 130665 CrossRef CAS PubMed.
- N. Atatreh, S. Hasan, B. R. Ali and M. A. Ghattas, Acta Pharm., 2021, 71, 325–333 CrossRef CAS.
- J. E. Cadena-Cruz, L. M. Guamán-Ortiz, J. C. Romero-Benavides, N. Bailon-Moscoso, K. E. Murillo-Sotomayor, N. V. Ortiz-Guamán and J. Heredia-Moya, BMC Chem., 2021, 15, 38 CrossRef CAS PubMed.
- T. Anwar, H. Nadeem, S. Sarwar, H. Naureen, S. Ahmed, A. U. Khan and M. Arif, Drug Dev. Res., 2020, 81, 893–903 CrossRef CAS PubMed.
- E. A. Orabi, M. A. A. Orabi, M. H. Mahross and M. Abdel-Hakim, J. Saudi Chem. Soc., 2018, 22, 705–714 CrossRef CAS.
- Z. Zhang, M. Cheng, J. Guo, Y. Wan, R. Wang, Y. Fang, Y. Jin, S. S. Xie and J. Liu, J. Mol. Struct., 2022, 1254, 132319 CrossRef CAS.
- E. Çınar, E. Başaran, Ö. Erdoğan, R. Çakmak, M. Boğa and Ö. Çevik, J. Chin. Chem. Soc., 2021, 68, 2355–2367 CrossRef.
- M. Messaad, I. Dhouib, M. Abdelhedi and B. Khemakhem, J. Mol. Struct., 2022, 1263, 133105 CrossRef CAS.
- S. Yousuf, K. M. Khan, U. Salar, S. Chigurupati, M. T. Muhammad, A. Wadood, M. Aldubayan, V. Vijayan, M. Riaz and S. Perveen, Eur. J. Med. Chem., 2018, 159, 47–58 CrossRef CAS PubMed.
- F. Chaudhry, S. Naureen, S. Choudhry, R. Huma, M. Ashraf, M. al-Rashida, B. Jahan, M. Hyder Khan, F. Iqbal, M. Ali Munawar and M. Ain Khan, Bioorg. Chem., 2018, 77, 507–514 CrossRef CAS PubMed.
- R. C. Kulkarni, J. M. Madar, S. L. Shastri, F. Shaikh, N. S. Naik, R. B. Chougale, L. A. Shastri, S. D. Joshi, S. R. Dixit and V. A. Sunagar, Chem. Data Collect., 2018, 17–18, 497–506 CrossRef.
- V. Hadi, Y. H. Koh, T. W. Sanchez, D. Barrios, N. Neamati and K. W. Jung, Bioorg. Med. Chem. Lett., 2010, 20, 6854–6857 CrossRef CAS PubMed.
- Y. Kakiuchi, N. Sasaki, M. Satoh-Masuoka, H. Murofushi and K. Murakami-Murofushi, Biochem. Biophys. Res. Commun., 2004, 320, 1351–1358 CrossRef CAS PubMed.
- H. Ashtekar, N. Aggarwal, Z. Fernandes, A. Rao and N. Varghese, Res. Square, 2022 Search PubMed.
- C. Bailly, P. E. Hecquet, M. Kouach, X. Thuru and J. F. Goossens, Bioorg. Med. Chem., 2020, 28, 115463 CrossRef CAS PubMed.
- L. S. Zondagh, S. F. Malan and J. Joubert, J. Enzyme Inhib. Med. Chem., 2020, 35, 1596–1605 CrossRef CAS PubMed.
- S. Perrone, M. Szabó, C. V. Bellieni, M. Longini, M. Bangó, D. Kelen, A. Treszl, S. Negro, M. L. Tataranno and G. Buonocore, Pediatr. Neurol., 2010, 43, 236–240 CrossRef PubMed.
- H. Yoshida, H. Yanai, Y. Namiki, K. Fukatsu-Sasaki, N. Furutani and N. Tada, CNS Drug Rev., 2006, 12, 9–20 CrossRef CAS PubMed.
- J. R. Walker, K. E. Fairfull-Smith, K. Anzai, S. Lau, P. J. White, P. J. Scammells and S. E. Bottle, Medchemcomm, 2011, 2, 436–441 RSC.
- J. Branković, V. M. Milovanović, D. Simijonović, S. Novaković, Z. D. Petrović, S. S. Trifunović, G. A. Bogdanović and V. P. Petrović, RSC Adv., 2022, 12, 16054–16070 RSC.
- M. Zarghani and B. Akhlaghinia, RSC Adv., 2015, 5, 87769–87780 RSC.
- J. Safaei-Ghomi, B. Khojastehbakht-Koopaei and H. Shahbazi-Alavi, RSC Adv., 2014, 4, 46106–46113 RSC.
- J. Safaei-Ghomi, B. Khojastehbakht-Koopaei and S. Zahedi, Chem. Heterocycl. Compd., 2015, 51, 34–38 CrossRef CAS.
- A. Vafaee, A. Davoodnia and M. Pordel, Res. Chem. Intermed., 2015, 41, 8343–8354 CrossRef CAS.
- R. Ramesh, N. Nagasundaram, D. Meignanasundar, P. Vadivel and A. Lalitha, Res. Chem. Intermed., 2017, 43, 1767–1782 CrossRef CAS.
- S. Sadjadi, M. M. Heravi, V. Zadsirjan and V. Farzaneh, Res. Chem. Intermed., 2018, 44, 6765–6785 CrossRef CAS.
- Z. Zhou and Y. Zhang, Green Chem. Lett. Rev., 2014, 7, 18–23 CrossRef.
- N. G. Khaligh, S. B. A. Hamid and S. J. J. Titinchi, Chin. Chem. Lett., 2016, 27, 104–108 CrossRef CAS.
- G. Mohammadi Ziarani, F. Saidian, P. Gholamzadeh, A. Badiei, J. B. Ghasemi, E. Aghaee and A. Abolhasani Soorki, J. Iran. Chem. Soc., 2019, 16, 1401–1409 CrossRef CAS.
- M. Miliovsky, I. Svinyarov, Y. Mitrev, Y. Evstatieva, D. Nikolova, M. Chochkova and M. G. Bogdanov, Eur. J. Med. Chem., 2013, 66, 185–192 CrossRef CAS PubMed.
- S. Y. Li, X. B. Wang and L. Y. Kong, Eur. J. Med. Chem., 2014, 71, 36–45 CrossRef CAS PubMed.
- A. Jain, S. Yadav and P. Malhotra, J. Chem. Sci., 2021, 133, 1–8 CrossRef.
- E. S. S. Priya, R. P. Nayak, P. Saldanha, B. J. Mohan and A. Prabhu, Int. J. Curr. Res. Rev., 2020, 12(23), 68–75 CrossRef.
- S. R. Kang and Y. R. Lee, Mol. Divers., 2015, 19, 293–304 CrossRef CAS PubMed.
- V. W. Barajas-Carrillo, A. Estolano-Cobián, L. Díaz-Rubio, R. R. Ayllón-Gutiérrez, R. Salazar-Aranda, R. Díaz-Molina, V. García-González, H. Almanza-Reyes, I. A. Rivero, J. G. Marrero and I. Córdova-Guerrero, Med. Chem. Res., 2021, 30, 610–623 CrossRef CAS.
- O. G. Idemudia, A. I. Okoh, A. P. Sadimenko, E. C. Hosten and O. O. Okoh, J. Chem., 2017, 2017, 1–10 CrossRef.
- L. Valgimigli, R. Amorati, M. G. Fumo, G. A. DiLabio, G. F. Pedulli, K. U. Ingold and D. A. Pratt, J. Org. Chem., 2008, 73, 1830–1841 CrossRef CAS PubMed.
- A. Torres de Pinedo, P. Peñalver and J. C. Morales, Food Chem., 2007, 103, 55–61 CrossRef CAS.
- M. C. Foti, E. R. Johnson, M. R. Vinqvist, J. S. Wright, L. R. C. Barclay and K. U. Ingold, J. Org. Chem., 2002, 67, 5190–5196 CrossRef CAS PubMed.
- M. C. Foti, L. R. C. Barclay and K. U. Ingold, J. Am. Chem. Soc., 2002, 124, 12881–12888 CrossRef CAS PubMed.
- C. Y. Lee, A. Sharma, J. Semenya, C. Anamoah, K. N. Chapman and V. Barone, Antioxidants, 2020, 9, 1–14 CAS.
- S. Antonczak, J. Mol. Struct., 2008, 856, 38–45 CrossRef CAS.
- H. Zhou, X. Li, Y. Shang and K. Chen, Antioxidants, 2019, 8, 590 CrossRef CAS PubMed.
- L. Capaldo and D. Ravelli, Eur. J. Org. Chem., 2017, 2017, 2056–2071 CrossRef CAS PubMed.
- I. O. Alisi, A. Uzairu and S. E. Abechi, Bull. Natl. Res. Cent., 2020, 44, 137 CrossRef.
- A. Amić, Z. Marković, J. M. D. Marković, S. Jeremić, B. Lučić and D. Amić, Comput. Biol. Chem., 2016, 65, 45–53 CrossRef PubMed.
- J. Xie and K. M. Schaich, J. Agric. Food Chem., 2014, 62, 4251–4260 CrossRef CAS PubMed.
- R. G. Parr and W. Yang, Theory of Orientation and Steroselection, Springer-Verlag, 1984, vol. 106 Search PubMed.
- D. A. Milenković, D. S. Dimić, E. H. Avdović, A. D. Amić, J. M. Dimitrić Marković and Z. S. Marković, Chem. Eng. J., 2020, 395, 124971 CrossRef.
- C. Kontogiorgis and D. Hadjipavlou-Litina, J. Enzyme Inhib. Med. Chem., 2003, 18, 63–69 CrossRef CAS PubMed.
- D. Dimić, D. Milenković, J. Dimitrić Marković and Z. Marković, Phys. Chem. Chem. Phys., 2017, 19, 12970–12980 RSC.
- M. C. Foti, J. Agric. Food Chem., 2015, 63, 8765–8776 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. a. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. a. Petersson, H. Nakatsuji, X. Li, M. Caricato, a. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, a. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. a. Keith, R. Kobayashi, J. Normand, K. Raghavachari, a. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, GAUSSIAN 16 (Revision C.01), Gaussian Inc., Wallingford, CT, 2016 Search PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- T. H. Dunning, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef CAS.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 4538–4543 CrossRef CAS PubMed.
- Z. D. Petrović, J. Orović, D. Simijonović, V. P. Petrović and Z. Marković, RSC Adv., 2015, 5, 24094–24100 RSC.
- Z. Marković, J. Tošović, D. Milenković and S. Marković, Comput. Theor. Chem., 2016, 1077, 11–17 CrossRef.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Vesna M. Milovanović
a,
Vesna M. Milovanović