
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Collective synthesis of aspulvinone and its analogues by vinylogous aldol condensation of substituted tetronic acids with aldehydes†
Xiaotan Yu‡
 ,
Xiaoxia Gu‡,
Yunpeng Zhao,
Fengqing Wang,
Weiguang Sun,
Changxing Qi*,
Lianghu Gu* and
Yonghui Zhang*
,
Xiaoxia Gu‡,
Yunpeng Zhao,
Fengqing Wang,
Weiguang Sun,
Changxing Qi*,
Lianghu Gu* and
Yonghui Zhang*
Hubei Key Laboratory of Natural Medicinal Chemistry and Resource Evaluation, School of Pharmacy, Tongji Medical College, Huazhong University of Science and Technology, Wuhan 430030, People's Republic of China. E-mail: zhangyh@mails.tjmu.edu.cn; gulianghu@hust.edu.cn; qichangxing@hust.edu.cn
First published on 7th February 2023
Abstract
A mild, modular and efficient synthetic method with broad substrate scope was developed for aspulvinones. Nine natural aspulvinones were synthesized, six of which were for the first time. The aldol condensation delivered Z-configuration products predominantly in MeCN. Meanwhile, alkoxy exchange occurred in MeOH and EtOH. Aspulvinone O and E, and substrate 49, 50, and 51 exhibited modest anti-SARS-CoV-2 activity in a high-throughput screening and enzyme kinetics assay.
Introduction
Aspulvinones, mainly isolated from fungus metabolites, were first reported in the 19th century.1 Up to now, more than 40 aspulvinones have been reported. The core structure of aspulvinone is characterized by a tetronic acid ring, a 5-membered heterocycle connecting two substituted aromatic rings. Aspulvinones exhibit a wide range of bioactivities, such as antibacterial,2 anti-inflammatory,3 anti-DPPH radical,4 anti-fungal,5 and anti-α-glucosidase effects.6 Recently, a study showed that some aspulvinones could inhibit SARS-CoV-2 infection and reduce inflammatory reactions caused by SARS-CoV-2.7As part of our work, aspulvinone O and H were isolated from the rice culture of Aspergillus terreus and identified as potent bioactive inhibitors of GOT1 and novel anti-tumor agents for PDAC therapy.8,9 However, the poor isolated yield and limited structure variation restricted our further study of their pharmaceutical properties. The diverse activities of aspulvinone have attracted extensive attention from synthetic chemists. The synthetic approaches can be divided into three categories: noble metal catalysis,10–13 harsh conditions,14–16 and other conditions.2,17,18 Some of the methods reported after 2010 are listed in Scheme 1. However, noble metal catalysts often face the problem of high cost and scarce availability. The use of organolithium reagents requires low temperature (−78 °C) and the Schlenk technique, which is relatively hard to operate in labs and factories.
 | ||
| Scheme 1 Previous synthetic approaches for the aspulvinone. | ||
Our group aims to develop a new method which is economical and easy to operate in a short route. In Liu's work, a two-step method was developed to synthesize the precursor of aspulvinone in a mild condition (Scheme 2A).2 Besides, in synthesizing aspulvinone H, Brückner and co-workers obtained unexpected aspulvinone A after deprotection. They assumed that deprotection of methyl ether using BBr3 resulted in the cyclization of the prenyl chain with the neighboring phenol group (Scheme 2B).13 Based on their work, we modified the starting material to 4-(benzyloxy)-3-phenyl-5-furan-2(5H)-one, hoping to find a one-step method for the aldol condensation, which can also be used in a broad substrate scope (Scheme 2C).
 | ||
| Scheme 2 Synthetic Strategies of the aspulvinone. | ||
Results and discussion
Our work started from the synthesis of the substituted tetronic acid, 4-(benzyloxy)-3-(4-(benzyloxy) phenyl)furan-2(5H)-one (3). The commercially available p-benzyloxy phenylacetic acid was treated with ethyl chloroacetate and triethylamine to afford the acyclic ester (1), which then underwent an intramolecular cyclization by t-BuOK to afford the 4-hydroxy-3-(2-methoxyphenyl)furan-2(5H)-one (2). It was protected by the use of BnBr or BnOH to afford compound 3. Other substituted tetronic acids were synthesized using the same route from the different starting materials (Scheme 3).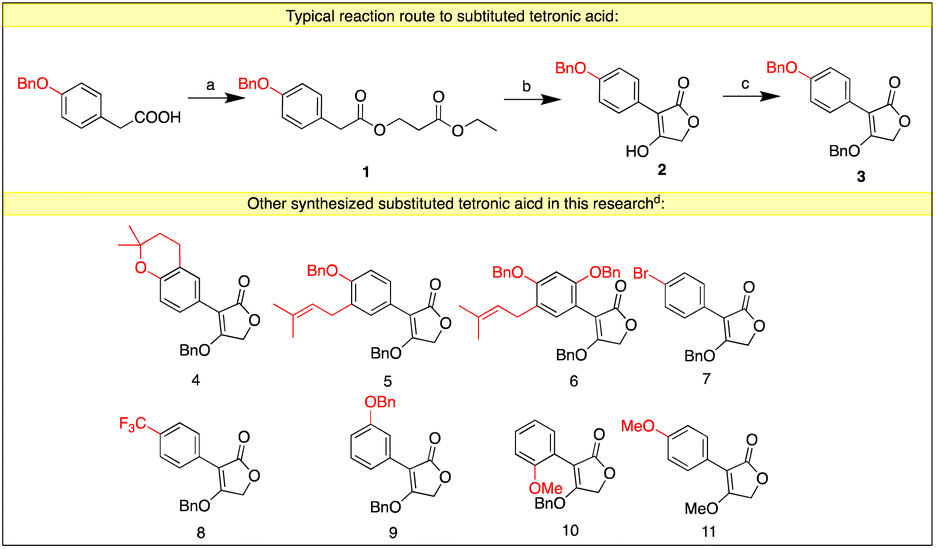 | ||
| Scheme 3 The synthesis of substituted tetronic acid. Reagents and conditions: aethyl chloroacetate, Et3N, THF, reflux, 12 h, 90%. bt-BuOK, DMF, 0 °C to rt, 2 h, 82%. cK2CO3, BnBr, DMF, rt, 12 h, 35% or BnOH, Ph3P, DEAD, rt, 12 h, 65%. dDetailed procedures and yields are in ESI.† | ||
Next, optimization of the aldol condensation between tetronic acid 3 and 4-(benzyloxy)benzaldehyde was performed. In Chopin's work, they proposed a method for aldol condensation using 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) as the base in acetonitrile (MeCN) at 65 °C.19 Initially, we used this method for aspulvinone's synthesis and successfully gained the desired product (Table 1, Entry 1). Then, the reactions were conducted at 65 °C employing DBU (2 eq.) as the base in various solvents, including tetrahydrofuran (THF), dichloromethane (DCM), methanol (MeOH) and isopropanol (i-PrOH) (Table 1, entry 2–5). With these tested solvents, MeOH afforded a methylated product, and i-PrOH afforded a mixture of Z/E products. MeCN showed the best yield in all solvents. To be noted, these results were quite different from Chopin's work, probably due to the difference in substrates. Next, several bases, including triethylamine (Et3N), 1,5-diazabicyclo[4.3.0]non-5-ene (DBN), 4-dimethylaminopyridine (DMAP), potassium tert-butoxide (t-BuOK), sodium hydroxide (NaOH), and potassium carbonate (K2CO3), were tested for this reaction (Table 1, entry 6–11), among which DBN showed a slight increase in yield. Then, different temperatures were tested (Table 1, entry 12–16), and the reaction at 30 °C gave the best yield. High and low temperatures lead to low yield, which is an interesting phenomenon that deserves more study. Finally, different times and equivalents of reactants and reagents were checked (Table 1, entry 17–20). It was found that extending reaction time, decreasing DBN, and increasing the aldehyde could increase the reaction yield (Table 1, entry 20). The reaction of 1.0 g scale was also performed in the same condition and gained the same yield.
|
||||||
|---|---|---|---|---|---|---|
| Entry | Temperature (°C) | Time (h) | Solvent | Base (eq.) | Yield (%)b | |
a Standard condition: tetronic acid (50 mg), aldehyde (57 mg, 2 eq.), solvent (1 ml), base (2 eq.).b Isolated yields.c Methylated product.d Z/E = 1.7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.e The equivalent of aldehyde is 3. :1.e The equivalent of aldehyde is 3. |
||||||
| 1 | 65 | 12 | MeCN | DBU (2) | 34 | |
| 2 | 65 | 12 | THF | DBU (2) | 21 | |
| 3 | 40 | 12 | DCM | DBU (2) | 6 | |
| 4 | 65 | 12 | MeOH | DBU (2) | 36c | |
| 5 | 65 | 12 | i-PrOH | DBU (2) | 28d | |
| 6 | 65 | 12 | MeCN | Et3N (2) | Trace | |
| 7 | 65 | 12 | MeCN | DBN (2) | 38 | |
| 8 | 65 | 12 | MeCN | DMAP (2) | Trace | |
| 9 | 65 | 12 | MeCN | t-BuOK (2) | 16 | |
| 10 | 65 | 12 | MeCN | NaOH (2) | 25 | |
| 11 | 65 | 12 | MeCN | K2CO3 (2) | 12 | |
| 12 | 80 | 12 | MeCN | DBN (2) | 10 | |
| 13 | 45 | 12 | MeCN | DBN (2) | 42 | |
| 14 | 30 | 12 | MeCN | DBN (2) | 46 | |
| 15 | 15 | 12 | MeCN | DBN (2) | 24 | |
| 16 | 0 | 12 | MeCN | DBN (2) | 5 | |
| 17 | 30 | 24 | MeCN | DBN (2) | 49 | |
| 18 | 30 | 48 | MeCN | DBN (2) | 55 | |
| 19 | 30 | 48 | MeCN | DBN (1) | 55 | |
| 20e | 30 | 48 | MeCN | DBN (1) | 65 | |
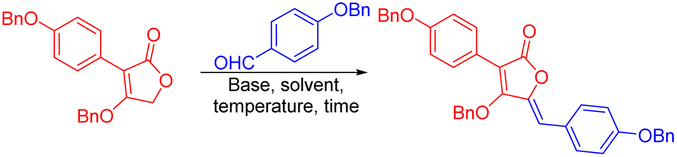
After optimization, the modular condensation between different substituted tetronic acids and aldehydes to afford various aspulvinone-skeleton products was investigated. Firstly, a series of fully methylated products (12–19) and partly ethylated ones (20–21) were synthesized using MeOH or EtOH as solvent. Then, some natural products protected by benzyl groups (22–30) were synthesized with similar conditions using MeCN as solvent. Some of them can be purified by filtration since precipitation of the products formed in the solvent. Next, more aspulvinone derivatives were synthesized from hetero cyclic aldehyde and alkyl ones (32–58). Most compounds are pure Z isomers except some derivatives from compound 10 (Scheme 4). Next, we conducted an investigation to gain insights into the factors controlling the reaction.
 | ||
| Scheme 4 Substrate scope. aIsolated yields. b60 °C. cCompound 31 is a natural product.20 | ||
In the substrate exploration, the reaction for synthesizing compound 58 is formed with by-product 59, which is the only reaction where an addition intermediate can be obtained. Then, the influence of temperatures was studied on the yield of 58 and 59 (Scheme 5, A). It was found that the yield of 58 was gradually increased from 0 to 30 °C and then decreased from 30 to 65 °C. While the best yield of 59 was at 0 °C, its yield decreased as the temperature increased. Additional controlled experiments using 59 further illustrated this (Scheme 5, Ba). However, no elimination product can be found in low temperatures, indicating the necessity of high-temperature for elimination. Interestingly, a unique equilibrium occurred at 30 °C, where both the aldol step and the elimination proceeded smoothly and afforded the best yield. Then, the alkoxy exchange mechanism was studied by using the starting material in MeOH condition (Scheme 5, Bb). We found that the exchange only took place in the starting material stage, thus proposing a mechanism containing both alkoxy exchange and aldol condensation (Scheme 6).
 | ||
| Scheme 5 The study of mechanism. | ||
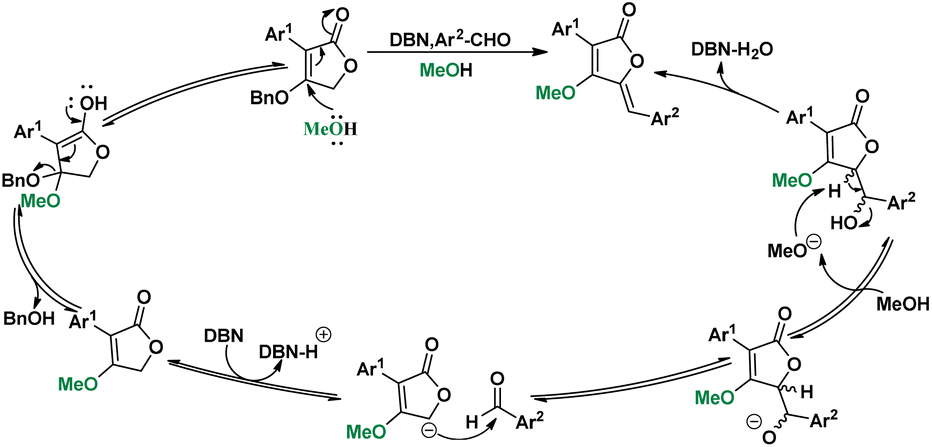 | ||
| Scheme 6 Plausible Mechanism. | ||
Next, the natural aspulvinone precursors were deprotected, and eight new natural aspulvinones were obtained. Six of them, including aspulvinone O, P, Q, R, compound 60 and 61, were synthesized for the first time. Pd/C and H2 were used for most compounds to cleave the O-benzyl bond. However, the deprotection of aspulvinone B precursor using this method invariably resulted in partial hydrogenation of the prenyl double bond. Finally, the Lewis acid BCl3 was used for deprotection and successfully delivered aspulvinone B (Scheme 7).13
 | ||
| Scheme 7 The deprotection and synthesis of natural aspulvinones in this study. a isolated yields. bUnnamed natural aspulvinones.26 cAspulvinone B use Cy2NMe and BCl3 for deprotection.13 | ||
In the optimization process, we found that the mixture of Z/E products can be formed in aspulvinone E precursor using i-PrOH in reflux condition. Through screening, two natural product precursors were formed with Z/E mixed configurations and transformed to natural aspulvinones of different configurations by deprotecting them. Isoaspulvinone have been observed in some researches.18,21–23 Our NMR data for isoaspulvinone E are consistent with those reported in Gao's work.21 The E configuration of aspulvinone E and P showed similar NMR changes compared to the Z configuration, which is consistent with Campbell's work.18
Mpro and PLpro are two important proteases for SARS-CoV-2 and other coronaviruses and essential for viral replication and transcription.24,25 These aspulvinone analogues exhibited modest antiviral activity in the high-throughput screening assay. Aspulvinone E and 49 inhibited SARS-CoV-2 Mpro activity with IC50 of 39.93 ± 2.42 μM and 28.25 ± 2.37 μM, respectively (Fig. S1B, Table S1†). 51 and 50 inhibited SARS-CoV-2 PLpro activity with IC50 of 23.05 ± 0.07 μM and 17.43 ± 2.60 μM, respectively (Fig. S1A, Table S2†). Notably, aspulvinone O exhibited dual-antiviral activity towards SARS-CoV-2 Mpro and PLpro, with IC50 of 12.41 ± 2.40 μM and 21.34 ± 0.94 μM, respectively (Fig. S1A and S1B, Table S1 and S2†).
Further enzyme kinetics assays revealed that aspulvinone E (Fig. S1C†) and 49 (Fig. 1A) showed uncompetitive inhibition against SARS-CoV-2 Mpro, with reduced Vmax and Km values. In contrast, aspulvinone O showed competitive inhibition against SARS-CoV-2 Mpro, with almost unchanged Vmax values and gradually increased Km values (Fig. 1A). The same assays were also performed against SARS-CoV-2 PLpro, which revealed that aspulvinone O, 51, and 50 all behaved as competitive inhibitors, consistent with GRL0617 (Fig. 1B, S1D†). These results gave substantial evidence for the docking analysis in Hawary's work.27
 | ||
| Fig. 1 Antiviral activity of aspulvinone analogues. (A) Mechanisms of action of 49 and aspulvinone O against SARS-CoV-2 Mpro. 49 inhibited Mpro in an uncompetitive way, while aspulvinone O in a competitive way. (B) The mechanism of action of 50 against SARS-CoV-2 PLpro, which inhibited PLpro in a competitive way. | ||
Conclusions
In conclusion, we have developed a method with high yield, mild conditions, and wide substrate tolerance to synthesize aspulvinones and their derivatives. This is a useful protocol for researchers who are interested in aspulvinones to quickly obtain their desired products. We found an interesting alkoxy exchange mechanism during the synthesis, further broadening the substrate scope. By using i-PrOH, we successfully synthesized aspulvinones with E configuration after separation by HPLC. In addition, our synthetic aspulvinone O, E, and derivatives 49, 50, and 51 exhibited modest anti-SARS-CoV-2 activity, which may provide new ideas for the design of lead compounds to treat COVID-19 in the future.Author contributions
Xiaotan Yu and Xiaoxia Gu contributed equally to this work. Xiaotan Yu completed the research of the organic synthesis part, and Xiaoxia Gu completed the research of the pharmacological part.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Program for Changjiang Scholars of Ministry of Education of the People's Republic of China (No. T2016088), the National Natural Science Foundation for Distinguished Young Scholars (81725021), the National Natural Science Foundation of China (81903461 and 82003633), and the Fundamental Research Funds for the Central Universities (2172019kfyXJJS166 and 2019kfyXJJS168). We thank the Medical Subcenter of HUST Analytical & Testing Center for data acquisition. We thank Prof. Chaomei Xiong at the School of Pharmacy, Huazhong University of Science and Technology, for NMR spectrum acquisition, and we thank Prof. Zimin Lv at the School of Pharmacy, Huazhong University of Science and Technology, for mass spectrum acquisition.References
- N. Ojima, S. Takenaka and S. Seto, New butenolides from Aspergillus terreus, Phytochemistry, 1973, 12, 2527–2529 CrossRef CAS.
- H.-W. Xu, C. Xu, Z.-Q. Fan, L.-J. Zhao and H.-M. Liu, A facile synthesis, antibacterial activity of pulvinone and its derivatives, Bioorg. Med. Chem. Lett., 2013, 23, 737–739 CrossRef CAS PubMed.
- G.-R. Hsiao, W.-C. Chi, C.-H. Chang, Y.-R. Chiang, Y.-J. Fu and T.-H. Lee, Bioactive pulvinones from a marine algicolous fungus Aspergillus terreus NTU243, Phytochemistry, 2022, 200, 113229 CrossRef CAS PubMed.
- P. Zhang, X.-M. Li, J.-N. Wang, X. Li and B.-G. Wang, New butenolide derivatives from the marine-derived fungus Paecilomyces variotii with DPPH radical scavenging activity, Phytochem. Lett., 2015, 11, 85–88 CrossRef CAS.
- N. Bunbamrung, C. Intaraudom, A. Dramae, S. Komwijit, T. Laorob, S. Khamsaeng and P. Pittayakhajonwut, Antimicrobial, antimalarial and anticholinesterase substances from the marine-derived fungus Aspergillus terreus BCC51799, Tetrahedron, 2020, 76, 131496 CrossRef CAS.
- C.-J. Wu, X. Cui, L.-Z. Sun, J.-J. Lu, F. Li, M.-H. Song, Y.-X. Zhang, X.-Q. Hao, C.-K. Tian, M.-P. Song and X.-M. Liu, Aspulvinones Suppress Postprandial Hyperglycemia as Potent alpha-Glucosidase Inhibitors From Aspergillus terreus ASM-1, Front. Chem., 2021, 9, 736070 CrossRef CAS PubMed.
- X.-X. Liang, X.-J. Zhang, Y.-X. Zhao, J. Feng, J.-C. Zeng, Q.-Q. Shi, J. S. Kaunda, X.-L. Li, W.-G. Wang, W.-L. Xiao and A.-H. Aspulvins, Aspulvinone Analogues with SARS-CoV-2 M-pro Inhibitory and Anti-inflammatory Activities from an Endophytic Cladosporium sp, J. Nat. Prod., 2022, 85, 878–887 CrossRef CAS PubMed.
- W.-G. Sun, S.-S. Luan, C.-X. Qi, Q.-Y. Tong, S. Yan, H. Li and Y.-H. Zhang, Aspulvinone O, a natural inhibitor of GOT1 suppresses pancreatic ductal adenocarcinoma cells growth by interfering glutamine metabolism, Cell Commun. Signal., 2019, 17, 111 CrossRef PubMed.
- S. Yan, C.-X. Qi, W. Song, Q.-Q. Xu, L.-H. Gu, W.-G. Sun and Y.-H. Zhang, Discovery of GOT1 Inhibitors from a Marine-Derived Aspergillus terreus That Act against Pancreatic Ductal Adenocarcinoma, Mar. Drugs, 2021, 19, 588 CrossRef CAS PubMed.
- A. Manchoju, R. A. Annadate, L. Desquien and S. V. Pansare, Functionalization of diazotetronic acid and application in a stereoselective modular synthesis of pulvinone, aspulvinones A-E, G, Q and their analogues, Org. Biomol. Chem., 2018, 16, 6224–6238 RSC.
- D. Hermann and R. Brückner, Silver-Catalyzed tert-Butyl 3-Oxopent-4-ynoate pi-Cyclizations: Controlling the Ring Size-Hydroxypyrone or Pulvinone Formation-by Counterion and Additive Optimization, Org. Lett., 2018, 20, 7455–7460 CrossRef CAS PubMed.
- Y. Sadamitsu, K. Komatsuki, K. Saito and T. Yamada, Access to Tetronic Acids via Silver-Catalyzed CO2 Incorporation into Conjugated Ynones, Org. Lett., 2017, 19, 3191–3194 CrossRef CAS PubMed.
- D. Bernier and R. Brückner, Novel synthesis of naturally occurring pulvinones: A heck coupling, transesterification, and Dieckmann condensation strategy, Synthesis, 2007, 15, 2249–2272 Search PubMed.
- K. C. Prousis, S. Katsamakas, J. Markopoulos and O. Igglessi-Markopoulou, A novel synthetic protocol for the synthesis of pulvinones, and naturally occurring Aspulvinone E, molecules of medicinal interest, Synth. Commun., 2022, 52, 117–128 CrossRef CAS.
- B. Nadal, J. Rouleau, H. Besnard, P. Thuery and T. Le Gall, Synthesis of pulvinones via tandem Dieckmann condensation-alkoxide beta-elimination, Tetrahedron, 2011, 67, 2605–2611 CrossRef CAS.
- N. Kaczybura and R. Brückner, Tandem claisen condensation/transesterification between arylacetate enolates and arylmethylene-substituted 2,2-dimethyl-1,3-dioxolan-4-ones: An improved synthesis of Z-configured pulvinones, Synthesis, 2007, 1, 118–130 Search PubMed.
- D. Bernier, F. Moser and R. Brückner, Synthesis and Cyclization of 3-aryl-2-(arylacetoxy)acrylates: A three-step access to pulvinones, Synthesis, 2007, 15, 2240–2248 Search PubMed.
- A. C. Campbell, M. S. Maidment, J. H. Pick and D. F. M. Stevenson, Synthesis of (E)- and (Z)-pulvinones, J. Chem. Soc., Perkin trans. 1, 1985, 1567–1576 RSC.
- N. Chopin, H. Yanai, S. Iikawa, G. Pilet, J. P. Bouillon and M. Medebielle, A Rapid Entry to Diverse gamma-Ylidenetetronate Derivatives through Regioselective Bromination of Tetronic Acid Derived gamma-Lactones and Metal-Catalyzed Postfunctionalization, Eur. J. Org. Chem., 2015, 2015, 6259–6269 CrossRef CAS.
- S. Chokpaiboon, P. Unagul, S. Kongthong, K. Danwisetkanjana, A. Pilantanapak, S. Suetrong and T. Bunyapaiboonsri, A pyrone, naphthoquinone, and cyclic urea from the marine-derived fungus Astrosphaeriella nypae BCC 5335, Tetrahedron Lett., 2016, 57, 1171–1173 CrossRef CAS.
- H.-Q. Gao, W.-Q. Guo, Q. Wang, L.-Q. Zhang, M.-L. Zhu, T.-J. Zhu, Q.-Q. Gu, W. Wang and D.-H. Li, Aspulvinones from a mangrove rhizosphere soil-derived fungus Aspergillus terreus Gwq-48 with anti-influenza A viral (H1N1) activity, Bioorg. Med. Chem. Lett., 2013, 23, 1776–1778 CrossRef CAS PubMed.
- E. Geib, M. Gressler, I. Viediernikova, F. Hillmann, I. Jacobsen, S. Nietzsche, C. Hertweck and M. Brock, A Non-canonical Melanin Biosynthesis Pathway Protects Aspergillus terreus Conidia from Environmental Stress, Cell Chem. Biol., 2016, 23, 587–597 CrossRef CAS PubMed.
- R. T. Dewi, S. Tachibana, S. Fajriah and M. Hanaf, α-Glucosidase inhibitor compounds from Aspergillus terreus RCC1 and their antioxidant activity, Med. Chem. Res., 2015, 24, 737–743 CrossRef CAS.
- Z.-M. Jin, X.-Y. Du, Y.-C. Xu, Y.-Q. Deng, M.-Q. Liu, Y. Zhao, B. Zhang, X.-F. Li, L.-K. Zhang, C. Peng, Y.-K. Duan, J. Yu, L. Wang, K.-L. Yang, F.-J. Liu, R.-D. Jiang, X.-L. Yang, T. You, X.-C. Liu, X.-N. Yang, F. Bai, H. Liu, X. Liu, L. Guddat, W.-Q. Xu, G.-F. Xiao, C.-F. Qin, Z.-L. Shi, H.-L. Jiang, Z.-H. Rao and H.-T. Yang, Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors, Nature, 2020, 582, 289–293 CrossRef CAS PubMed.
- D. Shin, R. Mukherjee, D. Grewe, D. Bojkova, K. Baek, A. Bhattacharya, L. Schulz, M. Widera, A. R. Mehdipour, G. Tascher, P. P. Geurink, A. Wilhelm, G. J. van der Heden van Noort, H. Ovaa, S. Müller, K.-P. Knobeloch, K. Rajalingam, B. A. Schulman, J. Cinatl, G. Hummer, S. Ciesek and I. Dikic, Papain-like protease regulates SARS-CoV-2 viral spread and innate immunity, Nature, 2020, 587, 657–662 CrossRef CAS PubMed.
- N. Ojima, S. Takenaka and S. Seto, Structures of pulvinone derivatives from Aspergillus terreus, Phytochemistry, 1975, 14, 573–576 CrossRef CAS.
- S. S. El-Hawary, R. Mohammed, H. S. Bahr, E. Z. Attia, M. H. El-Katatny, N. Abelyan, M. M. Al-Sanea, A. S. Moawad and U. R. Abdelmohsen, Soybean-associated endophytic fungi as potential source for anti-COVID-19 metabolites supported by docking analysis, J. Appl. Microbiol., 2021, 131, 1193–1211 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2183460 and 2204675–2204677. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2ra08133d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |