
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of symmetric bis-α-ketoamides from renewable starting materials and comparative study of their nucleating efficiency in PLLA†
Pietro Marchia,
Wei Wanga,
Cristián Puigb,
Ander Martinc,
Tullio Crovettoa,
Jalel Labidic,
Renata Riva a,
Dario Cavallo*a and
Lisa Moni*a
a,
Dario Cavallo*a and
Lisa Moni*a
aDepartment of Chemistry and Industrial Chemistry, University of Genoa, Via Dodecaneso 31, Genova, 16146, Italy. E-mail: lisa.moni@unige.it
bDepartamento de Ciencias de los Materiales, Universidad Simón Bolívar, Apartado 89000, Caracas, 1080A, Venezuela
cChemical & Environmental Engineering Department, Faculty of Engineering, University of the Basque Country UPV/EHU, Gipuzkoa, Plaza Europa 1, Donostia, 20018, Spain
First published on 8th February 2023
Abstract
An efficient and smart synthesis of bis-α-ketoamides has been disclosed. The desired products have been obtained through a Passerini multicomponent reaction using biobased aldehydes, acetic acid and bis-isocyanides (prepared from the corresponding biobased diamides), followed by a deprotection/oxidation step. The effect of the synthesized compounds on the crystallization behavior of poly(L-lactide) (PLLA) has been investigated by differential scanning calorimetry (DSC) in non-isothermal conditions. Among all the synthesized compounds, only a few are able to meaningfully enhance the nucleation of PLLA, as confirmed by a shift of the polymer crystallization peak temperature towards higher values. With the research of active polymer nucleating agents being mostly empirical, the combinatorial synthetic approach proposed herein, coupled with the possibility of a small scale mixing procedure, can potentially represent a useful strategy for the discovery of new efficient biobased polymer additives.
Introduction
Poly(L-lactide) (PLLA) is one of best polyester candidates to replace conventional petroleum-based polymers (like polypropylene) because it is bio-based, biodegradable and biocompatible. Even if its useful properties have already been exploited for specific applications, such as in biomedical devices or food packaging,1–4 its development as a large-scale commodity material is still limited by several drawbacks. Among them are the upper temperature boundary for practical applications of amorphous PLLA established by its low glass transition temperature (i.e., circa 60 °C) and its mechanically brittle behavior.5,6 Extending its temperature range of applications requires that the polymer crystallizes, but its crystallization rate is much slower than the cooling rates employed during processing.7,8 A successful strategy to enhance polymer crystallization rate is the use of particular additives, known as nucleating agents (NAs),9 which provide active seeds for the onset of the crystallization process. Due to its practical relevance, several compounds have been already tested as nucleating agents for PLLA.10,11In particular, a relatively small number of organic nucleating agents for PLLA is present in literature.12–17 Among these NAs, low molecular weight hydrazide compounds18–23 and bisoxalamides24 have gained increasing interest in recent years. Although these molecules are considered promising nucleating agents for biopolymers, their synthesis employs starting materials not from renewable resources and hazardous substances (hydrazines25 and several anilines are considered an environmental hazard and subjected to legislatively control by the European Economic Community (EEC) and the U.S. Environmental Protection Agency).
Moreover, the existing organic nucleating agents present a slight poor nucleation ability and the real effect of the structures and functional groups on the crystallization process has not yet been elucidated.
Thus, a combinatorial method to obtain bio-based organic nucleating agents with new structures through a sustainable synthetic process is highly required.
Multicomponent reactions (MCRs),26,27 reactions where three or more substrates are combined in a single step generating a product that contains all essential parts of the starting materials, play a central role in the toolbox of sustainable synthetic methodologies, being endowed with high atom and step economy, and operational simplicity.28
While MCRs have been widely exploited in medicinal chemistry,29–31 their use in other fields is still limited.32,33 Building on our experience in the development of new synthetic methodologies using MCRs,33–36 following our recent interest in the field of biopolymers,37 and inspired by the potential of the combination of MCRs with the use of renewable starting materials derived from biomass,35,38–40 we have planned to synthesize the symmetric bis-ketoamides 1 through an easy modulable multicomponent approach (Fig. 1).
 | ||
| Fig. 1 General structure of the novel nucleating agents obtained from biobased starting materials. | ||
In this paper we report the short and versatile synthesis of products 1, through a Passerini reaction41 using biobased aldehydes, acetic acid and bis-isocyanides (prepared from the corresponding biobased diamides), followed by a deprotection/oxidation step (Table 1).
|
||||
|---|---|---|---|---|
| Bis-α-ketoamides 1 | n | R1 | Yield % 3 | Yield % 1c |
| a Conditions for the synthesis of 3a: reaction has been performed using purified bis-isocyanide 4b, p-anisaldehyde, neat at rt for 5 days.b Isolated yields starting from bis-formamides 2a–c.c Isolated yields starting from 3a–g. | ||||
| 1a | 4 | pMeO-Ph | 44%a | 92% |
| 1b | 4 | iBu | 63%b | 52% |
| 1c | 4 | nBu | 75%b | 86% |
| 1d | 4 | Undecyl | 63%b | 63% |
| 1e | 4 | CH2CH2Ph | 79%b | 87% |
| 1f | 2 | nBu | 20%b | 78% |
| 1g | 6 | nBu | 71%b | 70% |

The combinatorial character of our synthesis allows a rapid modification of both the length of the spacer and the nature of the residue R1, thus the modulation of the NA properties, such as the interaction with PLLA, or self-assembly properties.42 A small scale dry-powder mixing approach was applied, and enabled the study of PLLA crystallization under the effect of the addition of the different synthesized molecules. Among the investigated compounds, few display a clear nucleating effect.
Results and discussion
Preparation of the bis-α-ketoamides
We first synthesized the bis-formamides 2 starting from different natural and non-natural aliphatic diamines. Although diamines such as putrescine (1,4-diaminobutane), cadaverine (1,5-diaminopentane), and 1,6-hexanediamine, are widely used for polyamides production, their preparations traditionally derived from oil or similar fossil feedstocks. However, recently, the production of the bio-based aliphatic diamines has been disclosed employing fermentation, whole cell bioconversion or enzymatic processes.43–45Following the standard formylation protocols, compounds 2a–c were obtained in gram-scale, in high yield and purity just after solvent evaporation and crystallization.
To overcome the typical drawbacks associated with the use of isocyanides, such as unpleasant smell and lability, we decided to generate bis-isocyanides in situ, using the crude preparation for the ensuing MCR, in a “one-pot” protocol. Based on our experience,46 we carried out the dehydration reaction employing Burgess reagent.47 To the unpurified isocyanide solution obtained in this way, acetic acid and the opportune aldehyde can be directly added, generating the Passerini adducts 3. The protocol was first tested using bis-formamide 2b and hydrocinnamaldehyde in CH2Cl2. After a brief optimization we found 8 h as the optimal time for the dehydration step, as the desired product 3e was isolated in 79% yield as a mixture of inseparable diastereomers (55% yield with 4 h). As expected, two sets of peaks were observed in both the 1H and 13C NMR spectra of 3e, corresponding to the dl and meso forms in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio. The high degree of efficiency of the protocol was verified, using different bis-isocyanides and aldehydes. Different terminal groups were introduced by employing alkyl aldehydes with short and long-chain (as fatty aldehydes), or aromatic aldehydes such as p-anisaldehyde and hydrocinnamaldehyde. All the bis-α-acetoxyamides were synthesized and fully characterized obtaining satisfactory yields, except for compound 3a, where p-anisaldehyde was employed (Table 1). Electron-rich aromatic aldehydes are known to be troublesome in Passerini reaction, indeed bis-α-acetoxyamide 3a was isolated in a poor yield (13%). Therefore, in this case we preferred to synthesize and isolate bis-isocyanide 4b and subject it to the Passerini reaction under harsher conditions, by increasing the temperature or by carrying out the reaction without solvent (Scheme 1), finding the last one the better compromise.
:1 ratio. The high degree of efficiency of the protocol was verified, using different bis-isocyanides and aldehydes. Different terminal groups were introduced by employing alkyl aldehydes with short and long-chain (as fatty aldehydes), or aromatic aldehydes such as p-anisaldehyde and hydrocinnamaldehyde. All the bis-α-acetoxyamides were synthesized and fully characterized obtaining satisfactory yields, except for compound 3a, where p-anisaldehyde was employed (Table 1). Electron-rich aromatic aldehydes are known to be troublesome in Passerini reaction, indeed bis-α-acetoxyamide 3a was isolated in a poor yield (13%). Therefore, in this case we preferred to synthesize and isolate bis-isocyanide 4b and subject it to the Passerini reaction under harsher conditions, by increasing the temperature or by carrying out the reaction without solvent (Scheme 1), finding the last one the better compromise.
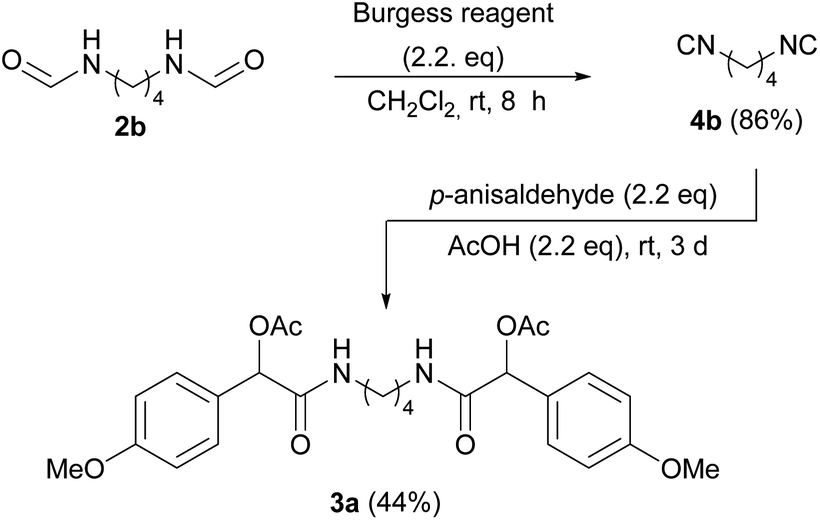 | ||
| Scheme 1 Synthesis of bis-α-acetoxyamide 3a. | ||
Then, products 3a–g were deacetylated to the corresponding diols under basic conditions, and the unpurified reaction mixtures were directly subjected to the final oxidation with Dess–Martin periodinane, leading to the bis-α-ketoamides 1a–g.
Screening of the nucleation efficiency of the compounds
To test the nucleating efficiency of the synthesized compounds, the latter needs to be physically mixed with the semicrystalline polymer. Typically, melt mixing in extruders or batch mixers is applied, but this method requires a large amount of material, which is not available in the present case for the synthesized compounds. To circumvent this problem, a novel mixing procedure, suitable for small amount of material, is developed and validated.Pellets of the polymer were submerged into liquid nitrogen for about 15 minutes and then grinded into powder with a rotatory grinder. Afterwards, about 200 mg of the polymer were placed in a small steel container with ca. 2 mg of the synthesized compound, and dry-mixed for 120 seconds in a vibratory mill for FT-IR spectroscopy sample preparation. The efficiency of the mixing procedure is verified by means of the good reproducibility of the crystallization behaviour obtained on different replica of the same mixture by differential scanning calorimetry.
Fig. 2(a) compares the DSC cooling curves of pure PLLA with those of the samples containing the various compounds of Table 1 at a concentration of 1 weight percent. For all the curves, an exothermic peak, representing the signature of the crystallization process, can be identified. The faster the crystallization rate of a nucleated sample, the higher the crystallization peak temperature. With the aid of the vertical dashed line, which indicates the crystallization temperature of pure PLLA (without any added molecule), we can notice that the compounds 1a, 1d, 1f and 1g barely affect the crystallization behaviour of PLLA, as no meaningful shift of the peak is observed. On the other hand, the molecules 1b, 1c and 1e present a well discernible nucleating activity. It is worth to note, however, that all the synthesised molecules perform worse than the known organic nucleating agent N1,N′1-(ethane-1,2-diyl)bis(N2-phenyl oxalamide) (OXA)13 added in Fig. 1(a) for the sake of comparison.
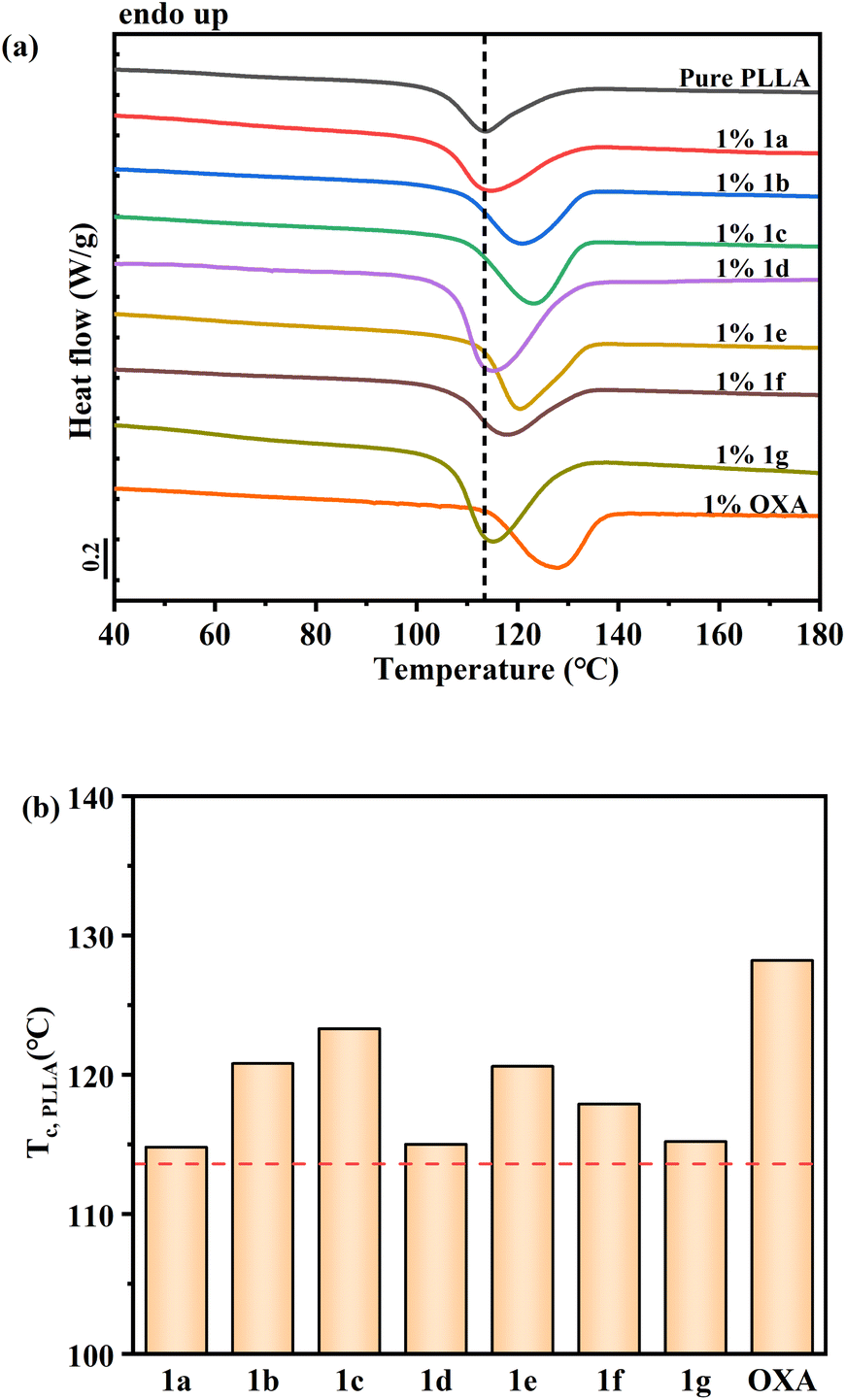 | ||
| Fig. 2 (a) DSC cooling curves for PLLA samples containing the molecules reported in Table 1 at a concentration of 1 wt%. Endothermic heat flow direction is upward; (b) measured crystallization peak temperature of PLLA in the different systems. Dashed lines in both (a) and (b) represent the crystallization peak temperature value for pure PLLA. The compound OXA, a known nucleating agent for PLLA is also added at the same concentration, for the sake of comparison. | ||
In Fig. 2(b), the crystallization peak temperature values of all the additivated PLLA samples are reported. The analyses showed compounds 1b and 1c displaying the best nucleating performance, as they are able to increase the neat PLLA crystallization peak temperature by approximately 5–7 °C. For compounds 1e and 1f a less marked increase is detected. However, the efficient nucleating agent OXA,13 provokes an increase of more than 10 °C. By looking at Table 1, it can thus be deduced that a better nucleating effect is afforded by an alkyl spacer length between ketoamide groups of 4 methylene units (rather than 2 or 6, see compounds 1c vs. 1f and 1g); and that relatively short linear or branched alkyl chains as R1 residues perform better than aromatic functionalities (see 1b and 1c vs. 1a and 1e).
Materials and methods
General remarks
NMR spectra were taken at room temperature in CDCl3 or d6-DMSO at 300 or 400 MHz (1H) and 75, 101 MHz (13C) by using as internal standards: in CDCl3 trimethylsilane (TMS) for 1H NMR spectroscopy and the central peak of CDCl3 (at δ = 77.16) for 13C NMR spectroscopy. Chemical shifts are reported in ppm (δ scale), coupling constants are reported in Hertz. Peak assignments were also made with the aid of gCOSY and gHSQC experiments. IR spectra were recorded with the ATR (attenuated total reflectance) technique. TLC analyses were carried out on silica gel plates and viewed at UV (λ = 254 nm) and developed with Hanessian stain (dipping into a solution of (NH4)4MoO4·4H2O (21 g) and Ce(SO4)2·4H2O (1 g) in H2SO4 (31 mL) and H2O (469 mL) and warming) or with potassium permanganate reagent (solution prepared dissolving potassium permanganate in distilled water until appearance of deep purple color). Rf values were measured after an elution of 7–9 cm. GC-MS analyses were performed on column HI-5 ms, 0.25 µm, 0.25 mm i.d., and 30 m. Analysis conditions are as follows: method A: solvent delay 2.5 min, mass range 35−400, injector temperature 250 °C, detector temperature 250 °C, MS temperature around 200 °C, starting temperature 150 °C, starting time 3 min, temperature gradient 25 °C min−1, final temperature 300 °C, flux through column 1.0 mL min−1, split ratio 10.0, sample concentration 0.1 mg mL−1, and amount injected 1 µL; method B: solvent delay 2.5 min, mass range 35−600, injector temperature 250 °C, detector temperature 250 °C, MS temperature around 200 °C, starting temperature 200 °C, starting time 3 min, temperature gradient 25 °C min−1, final temperature 330 °C, flux through column 1.0 mL min−1, split ratio 10.0, sample concentration 0.1 mg mL−1, and amount injected 1 µL.Column chromatographies were done with the “flash” methodology using 220–400 mesh silica.48 Petroleum ether (40–60 °C) is abbreviated as PE.
All reactions using dry solvents were carried out under a nitrogen atmosphere. Unless otherwise noted, analytical grade solvents and commercially available reactants were used without further purification. Common reagents were purchased from commercial sources and were used without further purification. OXA has been synthetized following the procedure reported in literature.49 All products were characterized by 1H,13C NMR, IR, GC-MS and elemental analysis.
The polymer, a PLLA kindly provided by Purac Biochem (Gorinchem, The Netherlands) has a molecular weight of 226 kg mol−1, a melt flow index of 6.9 g/10 min, and shows a nominal melting temperature of 175 °C. The polymer powder was produced via a Retsch rotatory grinder and mixed with the synthesized compounds in a PerkinElmer vibratory mill.
The possible nucleating effect of the various synthesized compounds has been scouted with a Mettler Toledo DSC1. About 3–5 mg of mixed powders was sealed in aluminium pan. The temperature was calibrated against the melting temperature of high purity Indium standard. The nucleated PLLA samples were first heated to 200 °C and kept at that temperature for 3 min to erase the previous thermal history. Subsequently the samples were cooled down to 30 °C, and finally heated again to a temperature of 200 °C, both the cooling and heating rates used are 10 °C min−1. All the measurements were carried out under a dry nitrogen flow of 20 mL min−1.
General procedure for the synthesis of bisformamides 2
A solution of the opportune diamine (30–40 mmol) in ethyl formate (1 M) was heated under reflux overnight. The reaction mixture was concentrated in vacuo, and the residue was purified by crystallization from iPrOH. Compounds 2a–c are a symmetric difunctional formamides. The 1H NMRs present two set of signals (one set for Major rotamer and one set for minor rotamer), while a more complex situation is present into 13C NMR spectrum, where the Major rotamer is symmetric, while the minor one is not more symmetric.:2); 1H NMR (300 MHz, d6-DMSO; 70 °C) (two rotamers 77:23) δ 8.03 (d, J = 1.7 Hz, 2H, 2 CHO major), 7.91 (d, J = 11.7 Hz, 2H, 2 CHO minor), 7.83 (bs, 2H, 2 NH major), 7.42 (bs, 2H, 2 NH minor), 3.22–3.08 (m, 8H, 4 CH2 major and minor); 13C NMR (75 MHz, d6-DMSO, 27 °C) δ 164.6 (CHO m), 161.4 (CHO m), 161.3 (2 CHO M), 40.4 (CH2 m), 38.5 (CH2 m), 36.9 (2 CH2 M); IR (cm−1) 3281, 3047, 2951, 2882, 2765, 1651, 1626, 1540, 1459, 1390, 1322, 1258, 1225, 1033, 981, 841, 744, 702; GC-MS (method A): Rt = 4.58 min; m/z 41 (6.9), 42 (20), 43 (22), 44 (6.4), 46 (6.1), 58 (25), 59 (75), 71 (100), 116 (0.04, M+); Anal. calcd for C4H8N2O2: C, 41.37; H, 6.94; N, 24.13; O, 27.56; found: C, 42.12; H, 7.02; N, 23.15; O, 27.41.:1 + 0.5% Et3N); 1H NMR (300 MHz, CDCl3, 27 °C) (two rotamers 86:14) δ 8.17 (d, J = 1.8 Hz, 2H, 2 CHO major), 8.04 (d, J = 12.0 Hz, 2H, 2 CHO minor), 5.98 (bs, 4H, 4 NH M and m), 3.40–3.20 (m, 8H, 4 CH2 M and m), 1.68–1.53 (m, 8H, 4 CH2 M and m); 13C NMR (75 MHz, CDCl3, 27 °C) δ 164.7 (CHO m), 161.6 (2 CHO M), 161.5 (CHO m), 41.4 (N–CH2 m), 37.8 (2 N–CH2 M), 37.6 (N–CH2 m), 28.5 (CH2 m), 26.9 (2 CH2 M), 26.7 (CH2 m); IR (cm−1) 3276, 3048, 2950, 2917, 2874, 2764, 2078, 1621, 1537, 1474, 1388, 1369, 1300, 1260, 1222, 1075, 1018, 776, 758, 691; GC-MS (method A): Rt = 6.37 min; m/z 39 (8.3), 41 (19), 42 (12), 43 (16), 44 (13), 45 (8.0), 46 (15), 54 (6.5), 56 (7.6), 58 (72), 59 (68), 70 (18), 71 (8.3), 72 (19), 73 (30), 81 (6.0), 86 (27), 87 (7.6), 98 (18), 99 (100), 100 (6.7) 143 (0.17, [M − H]+); Anal. calcd for C6H12N2O2: C, 49.99; H, 8.39; N, 19.43; O, 22.19; found: C, 50.01; H, 8.25; N, 18.43; O, 22.08.:2); 1H NMR (300 MHz, d6-DMSO, 27 °C) (two rotamers 88:12) δ 7.98 (d, J = 1.7 Hz, 2H, 2 CHO major), 7.96 (bs, 2H, 2 NH major), 7.92 (d, J = 11.7 Hz, 2H, 2 CHO minor), 7.66 (bs, 2H, 2 NH minor), 3.14–3.00 (m, 8H, 4 CH2 M and m), 1.43–1.32 (m, 8H, 4 CH2 M and m), 1.31–1.20 (m, 8H, 4 CH2 M and m); 13C NMR (75 MHz, CDCl3, 27 °C) δ 164.7 (CHO m), 161.5 (2 CHO M), 161.4 (CHO m), 41.7 (N–CH2 m), 38.0 (N–CH2 m), 37.8 (2 N–CH2 M), 31.3 (CH2 m), 29.6 (CH2 m), 29.4 (2 CH2 M), 26.4 (CH2 m), 26.1 (CH2 m), 26.0 (2 CH2 M); IR (cm−1) 3273, 3030, 2945, 2913, 2867, 2854, 2742, 2076, 1642, 1627, 1528, 1476, 1461, 1442, 1387, 1283, 1237, 1212, 1087, 1046, 1000, 777, 740, 705; GC-MS (method A): Rt = 7.51 min; m/z 39 (12), 41 (37), 42 (14), 43 (14), 44 (28), 45 (5.4), 46 (44), 54 (5.9), 55 (17), 56 (16), 58 (100), 59 (71), 60 (5.4), 67 (11), 69 (27), 70 (9.4), 72 (80), 73 (35), 82 (22), 83 (6.5), 84 (8.9), 86 (50), 87 (13), 98 (8.4), 99 (7.5), 100 (66), 101 (20), 114 (74), 115 (13), 116 (5.7), 126 (16), 127 (6.1), 172 (0.36, M+); Anal. calcd for C8H16N2O2: C, 55.79; H, 9.36; N, 16.27; O, 18.58; found: C, 56.32; H, 9.10; N, 17.27; O, 18.00.General procedure for the synthesis of compounds 3b–g
A solution of the opportune diformamide 2 (1 eq) in dry CH2Cl2 (0.4 M) was treated with Burgess reagent [(methoxycarbonylsulfamoyl)triethylammonium hydroxide, inner salt] (2.2 eq). After stirring for 8 h at room temperature, the appropriate aldehyde (2.4 eq) and acetic acid (2.4 eq) were added. The mixture was stirred at room temperature for 16 h, evaporated to dryness and the residue was purified by flash column chromatography (SiO2), eluting with the appropriate solvent, to give the Passerini products 3b–g as a mixture of diastereoisomers (a racemic mixture and a meso form). For this reason, two sets of peaks were observed in both the 1H and 13C NMR spectra.:3 + 2% MeOH). Yield from 2b: 63% as a mixture of diastereoisomers (50:50); 1H NMR (300 MHz, CDCl3, 27 °C) δ 6.31 (t, J = 5.9 Hz, 2H, 2 NH), 6.24 (t, J = 5.9 Hz, 2H, 2 NH), 5.18–5.09 (m, 4H, 4 C![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) OAc), 3.40–3.17 (m, 8H, 4 C2NH), 2.15 (s, 6H, 2 CH3 of Ac), 2.14 (s, 6H, 2 CH3), 1.80–1.63 (m, 12H, 4 CH and 4 CH2 iBu), 1.53 (m, 8H, 4 C2CH2NH), 0.96–0.90 (m, 24H, 8 CH3 iBu); 13C NMR (75 MHz, CDCl3, 27 °C) δ 170.69 (2 Cq), 170.67 (2 Cq), 170.3 (2 Cq), 170.2 (2 Cq), 73.12 (2 CH), 73.05 (2 CH), 40.96 (2 CH2NH), 40.94 (2 CH2NH), 38.8 (4
OAc), 3.40–3.17 (m, 8H, 4 C2NH), 2.15 (s, 6H, 2 CH3 of Ac), 2.14 (s, 6H, 2 CH3), 1.80–1.63 (m, 12H, 4 CH and 4 CH2 iBu), 1.53 (m, 8H, 4 C2CH2NH), 0.96–0.90 (m, 24H, 8 CH3 iBu); 13C NMR (75 MHz, CDCl3, 27 °C) δ 170.69 (2 Cq), 170.67 (2 Cq), 170.3 (2 Cq), 170.2 (2 Cq), 73.12 (2 CH), 73.05 (2 CH), 40.96 (2 CH2NH), 40.94 (2 CH2NH), 38.8 (4 ![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) H2CH), 26.76 (2 CH2), 26.71 (2 CH2), 24.69 (2 CH2), 24.67 (2 CH2), 23.23 (2 CH2), 23.21 (2 CH2), 21.91 (2 CH3), 21.89 (2 CH3), 21.14 (2 CH3), 21.12 (2 CH3); IR (cm−1) 3317, 2962, 2871, 1741, 1652, 1542, 1469, 1449, 1367, 1318, 1286, 1260, 1218, 1193, 1174, 1148, 1128, 1083, 1059, 1020, 992, 956, 921, 887, 754, 715, 665.:3). Yield from 2b: 75% as a mixture of diastereoisomers (50:50). 1H NMR (400 MHz, CDCl3, 25 °C) δ 6.27 (t, J = 5.7 Hz, 2H, 2 NH), 6.23 (t, J = 5.7 Hz, 2H, 2 NH), 5.12 (t, J = 5.4 Hz, 2H, 2 CHOAc), 5.10 (t, J = 5.4 Hz, 2H, 2 CHOAc), 3.39–3.23 (m, 8H, 4 C2NH), 2.17 (s, 12H, 4 CH3), 1.94–1.75 (m, 8H, 4 CH2), 1.58–1.51 (m, 8H, 4 CH2), 1.36–1.27 (m, 16H, 8 CH2), 0.89 (t, J = 6.9 Hz, 12H, 4 CH3); 13C NMR (101 MHz, CDCl3, 25 °C) δ 170.34 (2 Cq), 170.33 (2 Cq), 170.1 (2 Cq), 170.0 (2 Cq), 74.38 (2 CH), 74.33 (2 CH), 38.8 (4 CH2), 31.74 (4 CH2), 27.1 (4 CH2), 26.79 (2 CH2), 26.76 (2 CH2), 22.5 (4 CH2), 21.2 (4 CH3), 14.0 (4 CH3).:3). Yield from 2b: 63% as a mixture of diastereoisomers (50:50); 1H NMR (300 MHz, CDCl3, 27 °C) δ 6.28 (t, J = 5.9 Hz, 2H, 2 NH), 6.24 (t, J = 6.1 Hz, 2H, 2 NH), 5.14–5.06 (m, 4H, 4 CHOAc), 3.39–3.21 (m, 8H, 4 C2NH), 2.16 (s, 12H, 4 CH3), 1.93–1.72 (m, 8H, 4 CH2), 1.59–1.48 (m, 8H, 4 CH2), 1.25 (bs, 72H, 36 CH2), 0.86 (t, J = 6.9 Hz, 12H, 4 CH3); 13C NMR (75 MHz, CDCl3, 27 °C) δ 170.36 (4 Cq), 170.34 (2 Cq), 170.03 (2 Cq), 74.3 (4 CH), 38.8 (4 CH2), 32.0 (6 CH2), 29.85 (4 CH2), 29.76 (6 CH2), 29.70 (4 CH2), 29.6 (4 CH2), 29.5 (4 CH2), 29.4 (4 CH2), 26.8 (4 CH2), 25.0 (4 CH2), 22.8 (4 CH2), 21.2 (4 CH3), 14.3 (4 CH3); IR (cm−1) 3292, 3103, 2917, 2850, 1748, 1656, 1563, 1467, 1368, 1231, 1148, 1127, 1095, 1073, 1038, 989, 933, 901, 854, 758, 720, 695, 648, 604.:5 + 1% EtOH). Yield from 2b: 79% as a mixture of diastereoisomers (50:50). 1H NMR (300 MHz, CDCl3, 27 °C) δ 7.32–7.11 (m, 20H, 20 CH Ar), 6.30 (t, J = 6.0 Hz, 2H, 2 NH), 6.24 (d, J = 6.0 Hz, 2H, 2 NH), 5.13 (dd, J = 6.9, 5.2 Hz, 2H, 2 CH), 5.08 (dd, J = 6.6, 5.6 Hz, 2H, 2 CH), 3.35–3.23 (m, 8H, 4 CH2), 2.68 (t, J = 7.9 Hz, 8H, 4 CH2), 2.25–2.12 (m, 8H, 4 CH2), 2.11 (s, 6H, 2 CH3), 2.11 (s, 6H, 2 CH3), 1.62–1.47 (m, 8H, 4 CH2); 13C NMR (101 MHz, CDCl3, 27 °C) δ 170.17 (2 Cq), 170.04 (2 Cq), 170.01 (2 Cq), 169.98 (2 Cq), 140.8 (4 Cq), 128.6 (8 CH), 128.5 (8 CH), 126.3 (4 CH), 73.84 (2 CH), 73.81 (2 CH), 38.8 (4 CH2), 33.4 (4 CH2), 31.3 (4 CH2), 26.8 (2 CH2), 26.7 (2 CH2), 21.1 (4 CH3); IR (cm−1) 3292, 3105, 3028, 2931, 2865, 1740, 1653, 1603, 1562, 1497, 1454, 1442, 1370, 1266, 1229, 1197, 1084, 1044, 1028, 977, 948, 908, 878, 748, 694, 620, 604.:2). Yield from 2a: 20% as a mixture of diastereoisomers (50:50). 1H NMR (300 MHz, CDCl3, 27 °C) δ 6.76 (bs, 4H, 4 NH), 5.10 (dd, J = 4.1, 3.2 Hz, 2H, 2 CHOAc), 5.08 (dd, J = 4.6, 3.0 Hz, 2H, 2 CHOAc), 3.45–3.38 (m, 8H, 4 CH2), 2.17 (s, 12H, 4 CH3), 1.93–1.75 (m, 8H, 4 CH2), 1.35–1.26 (m, 16H, 8 CH2), 1.00–0.83 (m, 12H, 4 CH3); 13C NMR (75 MHz, CDCl3, 27 °C) δ 171.6 (2 Cq), 171.5 (2 Cq), 170.2 (2 Cq), 170.1 (2 Cq), 74.2 (2 CH), 74.1 (2 CH), 39.8 (4 CH2), 31.70 (2 CH2), 31.67 (2 CH2), 27.10 (2 CH2), 27.07 (2 CH2), 22.46 (4 CH2), 21.11 (2 CH3), 21.08 (2 CH3), 14.0 (4 CH3).:2). Yield from 2c: 71% as a mixture of diastereoisomers (50:50). 1H NMR (300 MHz, CDCl3, 27 °C) δ 6.18 (bt, J = 5.3 Hz, 4H, 4 NH), 5.23–5.10 (m, 4H, 4 CHOAc), 3.41–3.13 (m, 8H, 4 CH2), 2.16 (s, 12H, 4 CH3), 1.95–1.74 (m, 8H, 4 CH2), 1.61–1.43 (m, 8H, 4 CH2), 1.42–1.21 (m, 24H, 12 CH2), 1.00–0.79 (m, 12H, 4 CH3); 13C NMR (75 MHz, CDCl3, 27 °C) δ 170.08 (4 Cq), 169.94 (4 Cq), 74.4 (2 CH), 74.3 (2 CH), 38.8 (4 CH2), 31.7 (4 CH2), 29.4 (4 CH2), 27.0 (4 CH2), 25.9 (4 CH2), 22.5 (4 CH2), 21.16 (2 CH3), 21.10 (2 CH3), 14.0 (4 CH3).:1, to give 1,4-diisocyanobutane 4b (139 mg, 86%) as orange oil. Rf = 0.60 (EP/EtOAc 1:1). 1H NMR (300 MHz, CDCl3, 27 °C) δ 3.51–3.44 (m, 4H, 2 CH2), 1.88–1.81 (m, 4H, 2 CH2); 13C NMR (75 MHz, CDCl3, 27 °C) δ 157.1 (2 CN), 40.8 (2 CH2), 25.9 (2 CH2); a mixture of p-anisaldehyde (283 µL, 2.33 mmol), acetic acid (133 µL, 2.33 mmol) and 1,4-diisocyanobutane 4b (115 mg, 1.06 mmol) was stirred at room temperature for three days during which time it slowly solidifies. The residue was purified by flash column chromatography (SiO2), eluting with EtOAc, EtOAc + 2% MeOH, then EtOAc + 5% MeOH, to give 3a (237 mg, 44%) as a mixture of diastereoisomers (50:50). White solid; m.p. 121.1–122.0 °C (EtOAc); Rf = 0.31 (EtOAc); 1H NMR (300 MHz, CDCl3, 27 °C) δ 7.35 (dd, J = 9.0, 2.8 Hz, 8H, CH Ar), 6.89 (d, J = 8.9 Hz, 8H, CH Ar), 6.48 (t, J = 6.0 Hz, 2H, NH), 6.42 (t, J = 5.7 Hz, 2H, NH), 5.97 (s, 2H, CHOAc), 5.96 (s, 2H, CHOAc), 3.79 (s, 12H, 4 OCH3), 3.37–3.19 (m, 8H, 4 CH2NH), 2.16 (s, 6H, Ac), 2.14 (s, 6H, Ac), 1.55–1.50 (m, 8H, 4 CH2); 13C NMR (75 MHz, CDCl3, 27 °C) δ 169.9 (2 Cq), 169.8 (2 Cq), 169.1 (4 Cq), 160.1 (4 Cq), 129.0 (8 CH), 127.8 (2 Cq), 127.7 (2 Cq), 114.1 (8 CH), 75.4 (2 CH), 75.3 (2 CH), 55.3 (4 OCH3), 38.8 (4 CH2NH), 26.5 (4 CH2), 21.05 (2 CH3), 21.04 (2 CH3).
H2CH), 26.76 (2 CH2), 26.71 (2 CH2), 24.69 (2 CH2), 24.67 (2 CH2), 23.23 (2 CH2), 23.21 (2 CH2), 21.91 (2 CH3), 21.89 (2 CH3), 21.14 (2 CH3), 21.12 (2 CH3); IR (cm−1) 3317, 2962, 2871, 1741, 1652, 1542, 1469, 1449, 1367, 1318, 1286, 1260, 1218, 1193, 1174, 1148, 1128, 1083, 1059, 1020, 992, 956, 921, 887, 754, 715, 665.:3). Yield from 2b: 75% as a mixture of diastereoisomers (50:50). 1H NMR (400 MHz, CDCl3, 25 °C) δ 6.27 (t, J = 5.7 Hz, 2H, 2 NH), 6.23 (t, J = 5.7 Hz, 2H, 2 NH), 5.12 (t, J = 5.4 Hz, 2H, 2 CHOAc), 5.10 (t, J = 5.4 Hz, 2H, 2 CHOAc), 3.39–3.23 (m, 8H, 4 C2NH), 2.17 (s, 12H, 4 CH3), 1.94–1.75 (m, 8H, 4 CH2), 1.58–1.51 (m, 8H, 4 CH2), 1.36–1.27 (m, 16H, 8 CH2), 0.89 (t, J = 6.9 Hz, 12H, 4 CH3); 13C NMR (101 MHz, CDCl3, 25 °C) δ 170.34 (2 Cq), 170.33 (2 Cq), 170.1 (2 Cq), 170.0 (2 Cq), 74.38 (2 CH), 74.33 (2 CH), 38.8 (4 CH2), 31.74 (4 CH2), 27.1 (4 CH2), 26.79 (2 CH2), 26.76 (2 CH2), 22.5 (4 CH2), 21.2 (4 CH3), 14.0 (4 CH3).:3). Yield from 2b: 63% as a mixture of diastereoisomers (50:50); 1H NMR (300 MHz, CDCl3, 27 °C) δ 6.28 (t, J = 5.9 Hz, 2H, 2 NH), 6.24 (t, J = 6.1 Hz, 2H, 2 NH), 5.14–5.06 (m, 4H, 4 CHOAc), 3.39–3.21 (m, 8H, 4 C2NH), 2.16 (s, 12H, 4 CH3), 1.93–1.72 (m, 8H, 4 CH2), 1.59–1.48 (m, 8H, 4 CH2), 1.25 (bs, 72H, 36 CH2), 0.86 (t, J = 6.9 Hz, 12H, 4 CH3); 13C NMR (75 MHz, CDCl3, 27 °C) δ 170.36 (4 Cq), 170.34 (2 Cq), 170.03 (2 Cq), 74.3 (4 CH), 38.8 (4 CH2), 32.0 (6 CH2), 29.85 (4 CH2), 29.76 (6 CH2), 29.70 (4 CH2), 29.6 (4 CH2), 29.5 (4 CH2), 29.4 (4 CH2), 26.8 (4 CH2), 25.0 (4 CH2), 22.8 (4 CH2), 21.2 (4 CH3), 14.3 (4 CH3); IR (cm−1) 3292, 3103, 2917, 2850, 1748, 1656, 1563, 1467, 1368, 1231, 1148, 1127, 1095, 1073, 1038, 989, 933, 901, 854, 758, 720, 695, 648, 604.:5 + 1% EtOH). Yield from 2b: 79% as a mixture of diastereoisomers (50:50). 1H NMR (300 MHz, CDCl3, 27 °C) δ 7.32–7.11 (m, 20H, 20 CH Ar), 6.30 (t, J = 6.0 Hz, 2H, 2 NH), 6.24 (d, J = 6.0 Hz, 2H, 2 NH), 5.13 (dd, J = 6.9, 5.2 Hz, 2H, 2 CH), 5.08 (dd, J = 6.6, 5.6 Hz, 2H, 2 CH), 3.35–3.23 (m, 8H, 4 CH2), 2.68 (t, J = 7.9 Hz, 8H, 4 CH2), 2.25–2.12 (m, 8H, 4 CH2), 2.11 (s, 6H, 2 CH3), 2.11 (s, 6H, 2 CH3), 1.62–1.47 (m, 8H, 4 CH2); 13C NMR (101 MHz, CDCl3, 27 °C) δ 170.17 (2 Cq), 170.04 (2 Cq), 170.01 (2 Cq), 169.98 (2 Cq), 140.8 (4 Cq), 128.6 (8 CH), 128.5 (8 CH), 126.3 (4 CH), 73.84 (2 CH), 73.81 (2 CH), 38.8 (4 CH2), 33.4 (4 CH2), 31.3 (4 CH2), 26.8 (2 CH2), 26.7 (2 CH2), 21.1 (4 CH3); IR (cm−1) 3292, 3105, 3028, 2931, 2865, 1740, 1653, 1603, 1562, 1497, 1454, 1442, 1370, 1266, 1229, 1197, 1084, 1044, 1028, 977, 948, 908, 878, 748, 694, 620, 604.:2). Yield from 2a: 20% as a mixture of diastereoisomers (50:50). 1H NMR (300 MHz, CDCl3, 27 °C) δ 6.76 (bs, 4H, 4 NH), 5.10 (dd, J = 4.1, 3.2 Hz, 2H, 2 CHOAc), 5.08 (dd, J = 4.6, 3.0 Hz, 2H, 2 CHOAc), 3.45–3.38 (m, 8H, 4 CH2), 2.17 (s, 12H, 4 CH3), 1.93–1.75 (m, 8H, 4 CH2), 1.35–1.26 (m, 16H, 8 CH2), 1.00–0.83 (m, 12H, 4 CH3); 13C NMR (75 MHz, CDCl3, 27 °C) δ 171.6 (2 Cq), 171.5 (2 Cq), 170.2 (2 Cq), 170.1 (2 Cq), 74.2 (2 CH), 74.1 (2 CH), 39.8 (4 CH2), 31.70 (2 CH2), 31.67 (2 CH2), 27.10 (2 CH2), 27.07 (2 CH2), 22.46 (4 CH2), 21.11 (2 CH3), 21.08 (2 CH3), 14.0 (4 CH3).:2). Yield from 2c: 71% as a mixture of diastereoisomers (50:50). 1H NMR (300 MHz, CDCl3, 27 °C) δ 6.18 (bt, J = 5.3 Hz, 4H, 4 NH), 5.23–5.10 (m, 4H, 4 CHOAc), 3.41–3.13 (m, 8H, 4 CH2), 2.16 (s, 12H, 4 CH3), 1.95–1.74 (m, 8H, 4 CH2), 1.61–1.43 (m, 8H, 4 CH2), 1.42–1.21 (m, 24H, 12 CH2), 1.00–0.79 (m, 12H, 4 CH3); 13C NMR (75 MHz, CDCl3, 27 °C) δ 170.08 (4 Cq), 169.94 (4 Cq), 74.4 (2 CH), 74.3 (2 CH), 38.8 (4 CH2), 31.7 (4 CH2), 29.4 (4 CH2), 27.0 (4 CH2), 25.9 (4 CH2), 22.5 (4 CH2), 21.16 (2 CH3), 21.10 (2 CH3), 14.0 (4 CH3).:1, to give 1,4-diisocyanobutane 4b (139 mg, 86%) as orange oil. Rf = 0.60 (EP/EtOAc 1:1). 1H NMR (300 MHz, CDCl3, 27 °C) δ 3.51–3.44 (m, 4H, 2 CH2), 1.88–1.81 (m, 4H, 2 CH2); 13C NMR (75 MHz, CDCl3, 27 °C) δ 157.1 (2 CN), 40.8 (2 CH2), 25.9 (2 CH2); a mixture of p-anisaldehyde (283 µL, 2.33 mmol), acetic acid (133 µL, 2.33 mmol) and 1,4-diisocyanobutane 4b (115 mg, 1.06 mmol) was stirred at room temperature for three days during which time it slowly solidifies. The residue was purified by flash column chromatography (SiO2), eluting with EtOAc, EtOAc + 2% MeOH, then EtOAc + 5% MeOH, to give 3a (237 mg, 44%) as a mixture of diastereoisomers (50:50). White solid; m.p. 121.1–122.0 °C (EtOAc); Rf = 0.31 (EtOAc); 1H NMR (300 MHz, CDCl3, 27 °C) δ 7.35 (dd, J = 9.0, 2.8 Hz, 8H, CH Ar), 6.89 (d, J = 8.9 Hz, 8H, CH Ar), 6.48 (t, J = 6.0 Hz, 2H, NH), 6.42 (t, J = 5.7 Hz, 2H, NH), 5.97 (s, 2H, CHOAc), 5.96 (s, 2H, CHOAc), 3.79 (s, 12H, 4 OCH3), 3.37–3.19 (m, 8H, 4 CH2NH), 2.16 (s, 6H, Ac), 2.14 (s, 6H, Ac), 1.55–1.50 (m, 8H, 4 CH2); 13C NMR (75 MHz, CDCl3, 27 °C) δ 169.9 (2 Cq), 169.8 (2 Cq), 169.1 (4 Cq), 160.1 (4 Cq), 129.0 (8 CH), 127.8 (2 Cq), 127.7 (2 Cq), 114.1 (8 CH), 75.4 (2 CH), 75.3 (2 CH), 55.3 (4 OCH3), 38.8 (4 CH2NH), 26.5 (4 CH2), 21.05 (2 CH3), 21.04 (2 CH3).General procedure for the synthesis of compounds 1a–g
The opportune Passerini product (1 eq) was solubilized in MeOH (0.1 M) and treated with a solution of KOH in MeOH (2.5 eq, 0.5 M). The reaction mixture was stirred at room temperature for 30 min, then diluted with MeOH/CH2Cl2 1:1 and treated with previously washed (methanol) Amberlyst acid resin IRN-120 until pH = 4. The resin was filtered off and the solution evaporated to dryness. The crude was taken up in CH2Cl2 (0.1 M), cooled to 0 °C and treated with Dess–Martin periodinane (2.2 eq). The mixture was stirred at room temperature for 5 h, then treated with saturated aqueous solution of NaHCO3 and 0.5 M solution of Na2S2O3 and extracted with CH2Cl2 (3 times). The combined organic phases were dried (Na2SO4) and concentrated. The crude product was purified by crystallization.
:1 + 2% MeOH). Yield: 92%. 1H NMR (300 MHz, CDCl3, 27 °C) δ 8.42 (d, J = 9.1 Hz, 4H, 4 CH Ar), 7.22 (bt, J = 6.6 Hz, 2H, 2 NH), 6.94 (d, J = 9.2 Hz, 4H, 4 CH Ar), 3.89 (s, 6H, 2 CH3), 3.44 (q, J = 6.0, 5.5 Hz, 4H, 2 CH2), 1.70 (p, J = 3.5 Hz, 4H, 2 CH2); 13C NMR (75 MHz, CDCl3, 27 °C) δ 185.7 (2 Cq), 164.9 (2 Cq), 162.5 (2 Cq), 134.1 (4 CH), 126.5 (2 Cq), 114.0 (4 CH), 55.7 (2 CH3), 39.0 (2 CH2), 26.9 (2 CH2); IR (cm−1) 3362, 3074, 2986, 2940, 2872, 2847, 1675, 1643, 1591, 1564, 1514, 1480, 1465, 1454, 1444, 1423, 1373, 1322, 1308, 1294, 1264, 1252, 1206, 1187, 1166, 1115, 1085, 1022, 981, 923, 850, 822, 802, 772, 741, 724, 670, 649, 619; GC-MS (method B): Rt = 14.35 min; m/z 77 (8.6), 107 (6.0), 135 (100), 136 (9.0), 277 (15), 412 (0.53, M+); Anal. calcd for C22H24N2O6: C, 64.07; H, 5.87; N, 6.79; O, 23.27; found: C, 64.27; H, 6.27; N, 7.79; O, 23.20.:1). Yield: 52%. 1H NMR (300 MHz, CDCl3, 27 °C) δ 7.01 (bs, 2H), 3.35–3.27 (m, 4H, 2 CH2), 2.80 (d, J = 6.8 Hz, 4H, 2 CH2), 2.21–2.10 (m, 2H, 2 CH), 1.59 (d, J = 1.3 Hz, 4H, 2 CH2), 0.95 (d, J = 6.7 Hz, 12H, 4 CH3); 13C NMR (75 MHz, CDCl3, 27 °C) δ 199.0 (2 Cq), 160.5 (2 Cq), 45.4 (2 CH2), 38.9 (2 CH2), 26.8 (2 CH2), 24.5 (2 CH), 22.7 (4 CH3); IR (cm−1) 3330, 2958, 2930, 2901, 2874, 1719, 1659, 1518, 1473, 1453, 1378, 1362, 1301, 1269, 1196, 1170, 1147, 1104, 1041, 959, 921, 871, 802, 735, 656; GC-MS (method A): Rt = 8.91 min; m/z 41 (26), 43 (13), 55 (8.3), 57 (80), 58 (5.4), 70 (23), 85 (72), 86 (5.5), 98 (9.8), 115 (100), 116 (6.5), 227 (62), 228 (8.7), 312 (1.31, M+); Anal. calcd for C16H28N2O4: C, 61.51; H, 9.03; N, 8.97; O, 20.48; found: C, 62.43; H, 9.43; N, 8.94; O, 20.52.:2). Yield: 86%. 1H NMR (400 MHz, CDCl3, 27 °C) δ 7.01 (bs, 2H, NH), 3.33 (bq, J = 6.1 Hz, 4H, 2 CH2), 2.91 (t, J = 7.4 Hz, 4H, 2 CH2), 1.62–1.54 (m, 8H, 4 CH2), 1.36 (h, J = 7.3 Hz, 4H, 2 CH2), 0.92 (t, J = 7.3 Hz, 6H, 2 CH3); 13C NMR (101 MHz, CDCl3, 27 °C) δ 199.5 (2 Cq), 160.4 (2 Cq), 38.9 (2 CH2), 36.6 (2 CH2), 26.8 (2 CH2), 25.4 (2 CH2), 22.3 (2 CH2), 13.9 (2 CH3); IR (cm−1) 3308, 2955, 2893, 2874, 1717, 1683, 1660, 1529, 1470, 1450, 1436, 1408, 1382, 1368, 1352, 1320, 1287, 1250, 1230, 1191, 1134, 1100, 1033, 1002, 984, 922, 813, 777, 758, 738, 721, 661; GC-MS (method A): Rt = 9.32 min; m/z 41 (24), 55 (11), 57 (63), 70 (21), 85 (61), 86 (5.1), 98 (11), 115 (100), 116 (6.5), 227 (57), 228 (7.6), 312 (0.18, M+). Anal. calcd for C16H28N2O4: C, 61.51; H, 9.03; N, 8.97; O, 20.48; found: C, 62.62; H, 9.26; N, 9.04; O, 20.14.:1). Yield: 63%. 1H NMR (300 MHz, CDCl3, 27 °C) δ 7.00 (bt, J = 5.8 Hz, 2H, 2 NH), 3.32 (bq, J = 6.1 Hz, 4H, 2 CH2), 2.91 (t, J = 7.4 Hz, 4H, 2 CH2), 1.65–1.52 (m, 8H, 4 CH2), 1.35–1.19 (m, 32H, 16 CH2), 0.88 (t, J = 7.0 Hz, 6H, 2 CH3); 13C NMR (75 MHz, CDCl3, 27 °C) δ 199.5 (2 Cq), 160.4 (2 Cq), 38.9 (2 CH2), 36.9 (2 CH2), 32.1 (2 CH2), 29.7 (4 CH2), 29.6 (2 CH2), 29.5 (4 CH2), 29.2 (2 CH2), 26.8 (2 CH2), 23.3 (2 CH2), 22.8 (2 CH2), 14.3 (2 CH3); IR (cm−1) 3325, 2964, 2916, 2850, 1717, 1682, 1659, 1530, 1469, 1450, 1436, 1409, 1379, 1371, 1322, 1304, 1259, 1235, 1203, 1187, 1080, 1015, 984, 920, 868, 795, 756, 746, 728, 721, 660, 619; GC-MS (method B): Rt = 12.97 min; m/z 41 (11), 43 (26), 55 (13), 57 (32), 69 (5.4), 70 (23), 71 (15), 72 (6.6), 83 (5.5), 85 (9.4), 95 (8.3), 98 (8.2), 109 (5.6), 115 (77), 116 (5.7), 183 (9.3), 325 (100), 326 (22), 508 (0.45, M+); Anal. calcd for C30H56N2O4: C, 70.82; H, 11.09; N, 5.51; O, 12.58; found: C, 70.63; H, 11.00; N, 5.65; O, 12.02.:1). Yield: 87%. 1H NMR (400 MHz, CDCl3, 27 °C) δ 7.32–7.25 (m, 4H, 4 CH Ar), 7.23–7.16 (m, 6H, 6 CH Ar), 7.00 (bt, J = 6.1 Hz, 2H, 2 NH), 3.31 (bq, J = 5.0 Hz, 4H, 2 CH2), 3.27 (t, J = 7.5 Hz, 4H, 2 CH2), 2.94 (t, J = 7.5 Hz, 4H, 2 CH2), 1.59–1.55 (m, 4H, 2 CH2); 13C NMR (101 MHz, CDCl3, 27 °C) δ 198.4 (2 Cq), 160.2 (2 Cq), 140.5 (2 Cq), 128.7 (4 CH), 128.5 (4 CH), 126.4 (2 CH), 38.9 (2 CH2), 38.5 (2 CH2), 29.2 (2 CH2), 26.8 (2 CH2)); IR (cm−1) 3370, 3317, 3025, 2923, 2856, 1716, 1664, 1603, 1584, 1532, 1497, 1442, 1397, 1368, 1322, 1298, 1252, 1235, 1193, 1139, 1114, 1071, 1029, 1011, 988, 927, 845, 793, 753, 716, 701, 662, 627; GC-MS (method B): Rt = 10.42 min; m/z 55 (7.9), 65 (6.2), 70 (25), 77 (8.7), 79 (8.8), 91 (100), 92 (9.2), 98 (10), 103 (7.2), 104 (6.3), 105 (95), 106 (8.4), 115 (84), 116 (6.2), 133 (18), 143 (11), 257 (7.1), 275 (64), 276 (12), 408 (16, M+).; Anal. calcd for C24H28N2O4: C, 70.57; H, 6.91; N, 6.86; O, 15.67; found: C, 70.41; H, 7.02; N, 6.65; O, 15.42.:1). Yield: 70%. 1H NMR (300 MHz, CDCl3, 27 °C) δ 6.97 (bs, 2H, 2 NH), 3.28 (q, J = 6.8 Hz, 4H, 2 CH2), 2.91 (t, J = 7.4 Hz, 4H, 2 CH2), 1.63–1.51 (m, 8H, 4 CH2), 1.39–1.29 (m, 8H, 4 CH2), 0.91 (t, J = 7.3 Hz, 6H, 2 CH3); 13C NMR (75 MHz, CDCl3, 27 °C) δ 199.6 (2 Cq), 160.3 (2 Cq), 39.2 (2 CH2), 36.6 (2 CH2), 29.3 (2 CH2), 26.5 (2 CH2), 25.4 (2 CH2), 22.3 (2 CH2), 13.9 (2 CH3); IR (cm−1) 3291, 2957, 2930, 2873, 2852, 1716, 1687, 1662, 1529, 1462, 1445, 1410, 1381, 1370, 1350, 1306, 1282, 1271, 1246, 1213, 1169, 1133, 1102, 1066, 1027, 1015, 983, 927, 907, 892, 855, 815, 779, 742, 723, 661; GC-MS (method A): Rt = 10.15 min; m/z 41 (27), 55 (15), 56 (5.3), 57 (64), 85 (61), 86 (9.8), 98 (12), 100 (5.8), 143 (100), 144 (8.6), 255 (84), 256 (17), 340 (0.75, M+); Anal. calcd for C18H32N2O4: C, 63.50; H, 9.47; N, 8.23; O, 18.80; found: C, 63.74; H, 9.32; N, 8.00; O, 19.32.Conclusions
In conclusion, we have synthetized a new library of symmetric bis-ketoamides 1 using a simple and efficient three-steps procedure, involving a 3C Passerini reaction and employing biobased starting materials. Synthetizing only seven compounds, in a cost- and time-saving approach, we were able to evaluate both the length of the spacer and the nature of the terminal residues (aromatic, short or long alkyl chain, linear or branched alkyl chain). Moreover, we developed a material-efficient method to test the nucleating effect of these compounds towards PLLA. DSC in non-isothermal conditions showed significative differences on the nucleating effect, with compounds 1b and 1c being the best. Even though a known nucleating agent, as OXA, showed a slightly higher increment of the neat PLLA crystallization peak temperature, our results are noteworthy, as the bis-ketoamides 1b and 1c represent the first example of biobased synthetic nucleating agents for PLLA. Furthermore, the easy combinatorial synthesis, exploring both decoration and spacer length, allows the rapid expansion of the molecular diversity and the related NA properties. Having found two compounds with moderate nucleating ability opens the way to deepen the characterization of analogous molecules, also with the aid of computational efforts.Author contributions
P. M. and W. W. equally contributed to the study. P. M., T. C., A. M. and L. M. synthesized and characterized the compounds. L. M. and D. C. designed the study, analyzed the data, and wrote the manuscript. W. W. and C. P. performed and analyzed DSC experiments. J. L. and R. R. contributed to data discussion. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has received funding from the European Union's Horizon 2020 research and innovation programme under the Marie Sklodowska-Curie grant agreement No. 778092. The University of Genova is kindly acknowledged for the contribution to the acquisition of an NMR instrument (D.R. 3404 19/7/2018). We thank Mr. Walter Sgroi for his collaboration for IR spectra.Notes and references
- M. Malinconico, E. T. H. Vink and A. Cain, Applications of Poly(lactic Acid) in Commodities and Specialties in Industrial Applications of Poly(lactic acid), Advances in Polymer Science, ed. M. Di Lorenzo and R. Androsch, Springer, Cham, 2018, vol. 282, pp. 35–50, DOI:10.1007/12_2017_29.
- W. Hadasha and D. Bezuidenhout, Poly(lactic acid) as Biomaterial for Cardiovascular Devices and Tissue Engineering Applications, in Industrial Applications of Poly(lactic acid), Advances in Polymer Science, ed. M. Di Lorenzo and R. Androsch, Springer, Cham, 2017, vol. 282, pp. 51–77, DOI:10.1007/12_2017_27.
- S. Suzuki and Y. Ikada, Medical Applications, in Poly(Lactic Acid), ed. R. F. Grossman, D. Nwabunma, R. Auras, L.-T. Lim, S. E. M. Selke and H. Tsuji, 2010, pp. 443–456, DOI:10.1002/9780470649848ch.27.
- S. Obuchi and S. Ogawa, Packaging and Other Commercial Applications, in Poly(Lactic Acid), 2010, 28, DOI:10.1002/9780470649848.
- S. Domenek, S. Fernandes-Nassar and V. Ducruet, Rheology, Mechanical Properties, and Barrier Properties of Poly(lactic acid), in Synthesis, Structure and Properties of Poly(lactic acid), Advances in Polymer Science, ed. M. Di Lorenzo and R. Androsch, 2017, vol. 279, Springer, Cham, DOI:10.1007/12_2016_17.
- G. Perego and G. D. Cella, Mechanical Properties in Poly(Lactic Acid), ed. R. F. Grossman, D. Nwabunma, R. Auras, L.-T. Lim, S. E. M. Selke and H. Tsuji, 2010, pp. 141–153, DOI:10.1002/9780470649848.ch11.
- R. Androsch, C. Schick and M. L. Di Lorenzo, Kinetics of Nucleation and Growth of Crystals of Poly(L-lactic acid). in Synthesis, Structure and Properties of Poly(lactic acid), Advances in Polymer Science, ed. M. Di Lorenzo, R. Androsch, Springer, Cham, 2017, vol. 279, pp. 235–272, DOI:10.1007/12_2016_13.
- L. Fambri and C. Migliaresi, Crystallization and Thermal Properties, in Poly(Lactic Acid), ed. R. F. Grossman, D. Nwabunma, R. Auras, L.-T. Lim, S. E. M. Selke and H. Tsuji, 2010, DOI:10.1002/9780470649848.ch9.
- M. Gahleitner, J. Wolfschwenger and D. Mileva, Polymer Crystal Nucleating Agents, Reference Module in Materials Science and Materials Engineering, Elsevier, 2016, ISBN 9780128035818, DOI:10.1016/B978-0-12-803581-8.03773-5.
- L. Jiang, T. Shen, P. Xu, X. Zhao, X. Li, W. Dong, P. Ma and M. Chen, e-Polymers, 2016, 16, 1–13 CAS.
- S. Saeidlou, M. A. Huneault, H. Li and C. B. Park, Prog. Polym. Sci., 2012, 37, 1657–1677 CrossRef CAS.
- Z. Qiu and Z. Li, Ind. Eng. Chem. Res., 2011, 50, 12299–12303 CrossRef CAS.
- H. Bai, W. Zhang, H. Deng, Q. Zhang and Q. Fu, Macromolecules, 2011, 44, 1233–1237 CrossRef CAS.
- P. Pan, J. Yang, G. Shan, Y. Bao, Z. Weng and Y. Inoue, Macromol. Mater. Eng., 2012, 297, 670–679 CrossRef CAS.
- M. Weng and Z. Qiu, Thermochim. Acta, 2014, 577, 41–45 CrossRef CAS.
- Q. Xing, X. Zhang, X. Dong, G. Liu and D. Wang, Polymer, 2012, 53, 2306–2314 CrossRef CAS.
- N. Leoné, M. Roy, S. Saidi, G. de Kort, D. Hermida-Merino and C. H. R. M. Wilsens, ACS Omega, 2019, 4, 10376–10387 CrossRef.
- Q. Xing, R. Li, X. Dong, F. Luo, X. Kuang, D. Wang and L. Zhang, Macromol. Chem. Phys., 2015, 216, 1134–1145 CrossRef CAS.
- N. Kawamoto, A. Sakai, T. Horikoshi, T. Urushihara and E. Tobita, J. Appl. Polym. Sci., 2007, 103, 198–203 CrossRef CAS.
- Y. Fan, J. Zhu, S. Yan, X. Chen and J. Yin, Polymer, 2015, 67, 63–71 CrossRef CAS.
- T. Xu, A. Zhang, Y. Zhao, Z. Han and L. Xue, Polym. Test., 2015, 45, 101–106 CrossRef CAS.
- N. Kawamoto, A. Sakai, T. Horikoshi, T. Urushihara and E. Tobita, J. Appl. Polym. Sci., 2007, 103, 244–250 CrossRef CAS.
- X. Zheng, H. Luo, S. Chen, B. Zhang, B. Sun, M.-F. Zhu, B. Zhang, X.-K. Ren and J. Song, Cryst. Growth Des., 2020, 20, 653–660 CrossRef CAS.
- P. Ma, Y. Xu, D. Wang, W. Dong and M. Chen, Ind. Eng. Chem. Res., 2014, 53, 12888–12892 CrossRef CAS.
- H. N. Nguyen, J. A. Chenoweth, V. S. Bebarta, T. E. Albertson and C. D. Nowadly, Mil. Med., 2021, 186, e319–e326 CrossRef PubMed.
- E. Schaumann, M. J. Arévalo, M. Ayaz, L. Banfi, A. Basso, L. Bernardi, F. Berrée, B. Carboni, V. A. Chebanov, F. De Moliner, A. Dömling, N. Yu. Gorobets, Y. Huang, C. Hulme, G. N. Kaluđerović, R. Lavilla, E. Le Gall, G. Lemanski, C. J. Li, E. D. Matveeva, A. Mayence, R. S. Menon, G. A. Morales, M. C. Morejon, T. J. J. Müller, V. Nair, R. A. W. Neves Filho, A. Ricci, R. Riva, Y. V. Sedash, M. V. Shuvalov, J. J. Vanden Eynde, L. A. Wessjohann, W. J. Yoo, N. S. Zefirov, L. Zhao and T. Ziegler, Multicomponent Reactions 1: Reactions Involving a Carbonyl Compound as Electrophilic Component, 2014, ed: T. J. J. Müller, Georg Thieme Verlag, DOI:10.1055/b-003-125816.
- J. Zhu and H. Bienaymé, Multicomponent Reactions, John Wiley and Sons, 2005 Search PubMed.
- R. C. Cioc, E. Ruijter and R. V. A. Orru, Green Chem., 2014, 16, 2958–2975 RSC.
- H. A. Younus, M. Al-Rashida, A. Hameed, M. Uroos, U. Salar, S. Rana and K. M. Khan, Expert Opin. Ther. Pat., 2021, 31, 267–289 CrossRef CAS PubMed.
- D. Insuasty, J. Castillo, D. Becerra, H. Rojas and R. Abonia, Molecules, 2020, 25, 505 CrossRef PubMed.
- C. S. Graebin, F. V. Ribeiro, K. R. Rogerio and A. E. Kummerle, Curr. Org. Synth., 2019, 16, 855–899 CrossRef CAS PubMed.
- A. Pinna, A. Basso, C. Lambruschini, L. Moni, R. Riva, V. Rocca and L. Banfi, RSC Adv., 2020, 10, 965–972 RSC.
- L. Moni, C. F. Gers-Panther, M. Anselmo, T. J. J. Müller and R. Riva, Chem.–Eur. J, 2016, 22, 2020–2031 CrossRef CAS PubMed.
- S. Caputo, A. Kovtun, F. Bruno, E. Ravera, C. Lambruschini, M. Melucci and L. Moni, RSC Adv., 2022, 12, 15834–15847 RSC.
- L. Moni, L. Banfi, A. Basso, E. Martino and R. Riva, Org. Lett., 2016, 18, 1638–1641 CrossRef CAS PubMed.
- L. Moni, M. Denissen, G. Valentini, T. J. Muller and R. Riva, Chem.–Eur. J., 2015, 21, 753–762 CrossRef CAS PubMed.
- D. Lanteri, S. Quattrosoldi, M. Soccio, A. Basso, D. Cavallo, A. Munari, R. Riva, N. Lotti and L. Moni, Front. Chem., 2020, 8, 85 CrossRef CAS PubMed.
- C. Lambruschini, A. Basso, L. Moni, A. Pinna, R. Riva and L. Banfi, Eur. J. Org. Chem., 2018, 2018, 5445–5455 CrossRef CAS.
- L. Moni, L. Banfi, D. Cartagenova, A. Cavalli, C. Lambruschini, E. Martino, R. V. A. Orru, E. Ruijter, J. M. Saya, J. Sgrignani and R. Riva, Org. Chem. Front., 2020, 7, 380–398 RSC.
- G. Vitali Forconesi, L. Banfi, A. Basso, C. Lambruschini, L. Moni and R. Riva, Molecules, 2020, 25, 3227 CrossRef PubMed.
- L. Banfi, A. Basso, C. Lambruschini, L. Moni and R. Riva, Chem. Sci., 2021, 12, 15445–15472 RSC.
- W. Wang, A. Saperdi, A. Dodero, M. Castellano, A. J. Muller, X. Dong, D. Wang and D. Cavallo, Cryst. Growth Des., 2021, 21, 5880–5888 CrossRef CAS PubMed.
- F. Yang, K. Qiu, Y. Zhu, X. Zhang, T. Yang, G. Yi, M. Xu and Z. Rao, ACS Sustainable Chem. Eng., 2022, 10(44), 14407–14416 CrossRef CAS.
- X. Wang, S. Gao, J. Wang, S. Xu, H. Li, K. Chen and P. Ouyang, Chin. J. Chem. Eng., 2021, 30, 4–13 CrossRef CAS.
- H. Hui, Y. Bai, T.-P. Fan, X. Zheng and Y. Cai, Catalysts, 2020, 10, 947 CrossRef CAS.
- L. Moni, L. Banfi, A. Basso, L. Carcone, M. Rasparini and R. Riva, J. Org. Chem., 2015, 80, 3411–3428 CrossRef CAS PubMed.
- S. M. Creedon, H. Kevin Crowley and D. G. McCarthy, J. Chem. Soc. Perkin Trans. 1, 1998, 1015–1018, 10.1039/A708081F.
- W. C. Still, M. Kahn and A. Mitra, J. Org. Chem., 1978, 43, 2923–2925 CrossRef CAS.
- T. Shen, Y. Xu, X. Cai, P. Ma, W. Dong and M. Chen, RSC Adv., 2016, 6, 48365–48374 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ra07934h |
| This journal is © The Royal Society of Chemistry 2023 |