
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSmall change for big improvement in the preparation of the key intermediate N1, N3-disubstituted 1,3,5-triazone of ensitrelvir†
Wei Hu‡
a,
Yuanchang Liu‡a,
Xiang Zhanga,
Panpan Zheng a,
Feifei Yanga,
Guangyang Guoa,
Xin Xieab,
Jiuzhong Huang*ab and
Weiming Chen*ab
a,
Feifei Yanga,
Guangyang Guoa,
Xin Xieab,
Jiuzhong Huang*ab and
Weiming Chen*ab
aSchool of Pharmaceutical Sciences, Gannan Medical University, Ganzhou 341000, P. R. China. E-mail: wader_chen@163.com
bKey Laboratory of Prevention and Treatment of Cardiovascular and Cerebrovascular Diseases of Ministry of Education, Gannan Medical University, Ganzhou 341000, P. R. China
First published on 25th January 2023
Abstract
In this study, the key intermediate N1, N3-disubstituted 1,3,5-triazone of ensitrelvir fumaric acid, approved in Japan for the treatment of SARS-CoV-2 infection under the emergency regulatory approval system, was produced from S-ethylisothiourea hydrobromide and aminomethyl triazole with CDI by four-step telescoped strategy including CDI-activated, condensation, CDI-cyclization, and N1-alkylation. The strategy with simple conditions and operations had a total yield of 53% on a gram scale. The strategy for synthesizing the key N1, N3-disubstituted 1,3,5-triazone intermediate of ensitrelvir might provide a new avenue for further research and development of ensitrelvir analogs.
Introduction
COVID-19 is an acute respiratory infectious disease caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2),1 which spreads rapidly. Ensitrelvir (S-217622, 1) was considered as the first orally active, noncovalent, nonpeptidic SARS-CoV-2 3C-like protease inhibitor, which solved the limitations of peptide covalent inhibitors and had favorable drug metabolism and pharmacokinetic (DMPK) profiles for the oral agents.2 On November 22nd, 2022, ensitrelvir fumaric acid was approved by the PMDA for the indication of SARS-CoV-2 infection under the emergency regulatory approval system.3According to the process followed by the originator (Scheme 1), N1, N3-disubstituted 1,3,5-triazone 2 was considered to be the key intermediate of ensitrelvir.2a Starting from S-ethylisothiourea hydrobromide 3, cyclization with tert-butylisocyanate 4 formed the 1,3,5-triazone core 5. After N1-alkylation with benzyl bromide 6 under basic conditions, intermediate 7 was obtained and then de-butylated to intermediate 8 under acidic conditions. Subsequently, N3-alkylation with chloromethyl-triazole 9 yielded the key intermediate 2 under basic conditions. Additionally, the yield of N3-alkylation was limited to 45%, causing a total yield of 28% for the four steps.
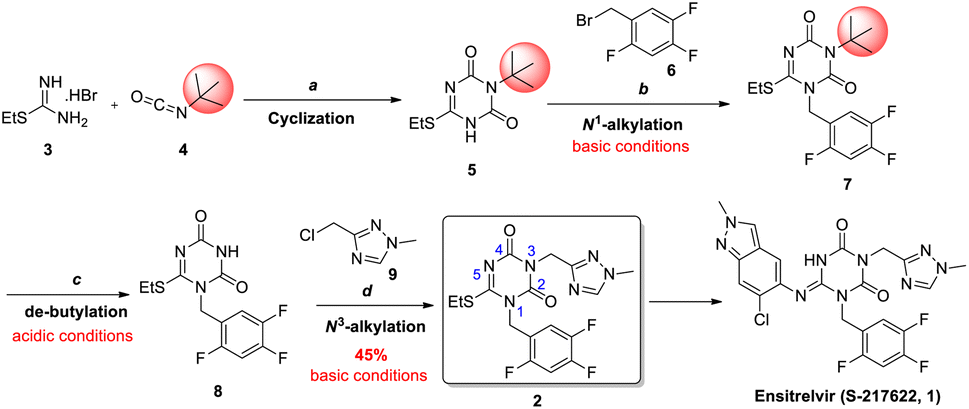 | ||
| Scheme 1 The strategy used for synthesizing intermediate 2 of ensitrelvir. Reagents and conditions: (a) DBU, 0 °C, 4 h, then CDI, DBU, 0 °C, 1 hour to r.t., overnight, 70%; (b) 1-(bromomethyl)-2,4,5-trifluorobenzene 6, K2CO3, MeCN, 80 °C, 93%; (c) TFA, r.t., 97%; (d) 3-(chloromethyl)-1-methyl-1H-1,2,4-triazole hydrochloride 9, K2CO3, DMF, 60 °C, 45%. | ||
Therefore, the industry-oriented process of ensitrelvir or its intermediates was highly desired and valuable, which attracted our and other researchers to develop novel synthetic strategies.4 However, these strategies had still several shortcomings, such as poor atom economy, cumbersome operation steps, and a large amount of waste.
To develop an atom-economical, green, and efficient synthetic strategy, we focused on the preparation of the key intermediate 2. The 1,3,5-triazone core could be constructed from fragments A, B, and C (Scheme 2), in which fragment C (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) was derived from a carbonyl reagent,5 such as 1,1′-carbonyldiimidazole (CDI).6 Fragments A and B might be combined by the reaction of isocyanate with S-ethylisothiourea, as described in the above work.2a,7 To improve the utilization of atoms and reduce environmental issues, herein we reported two novel strategies for synthesizing intermediate 2 based on the formation of 1,3,5-triazone using isocyanate or CDI, which provided –NH–CO– and –CO– respectively. In the CDI strategy, a small change with the combinations of fragments A and B presented a big improvement regardless of the total yield and efficiency.
O) was derived from a carbonyl reagent,5 such as 1,1′-carbonyldiimidazole (CDI).6 Fragments A and B might be combined by the reaction of isocyanate with S-ethylisothiourea, as described in the above work.2a,7 To improve the utilization of atoms and reduce environmental issues, herein we reported two novel strategies for synthesizing intermediate 2 based on the formation of 1,3,5-triazone using isocyanate or CDI, which provided –NH–CO– and –CO– respectively. In the CDI strategy, a small change with the combinations of fragments A and B presented a big improvement regardless of the total yield and efficiency.
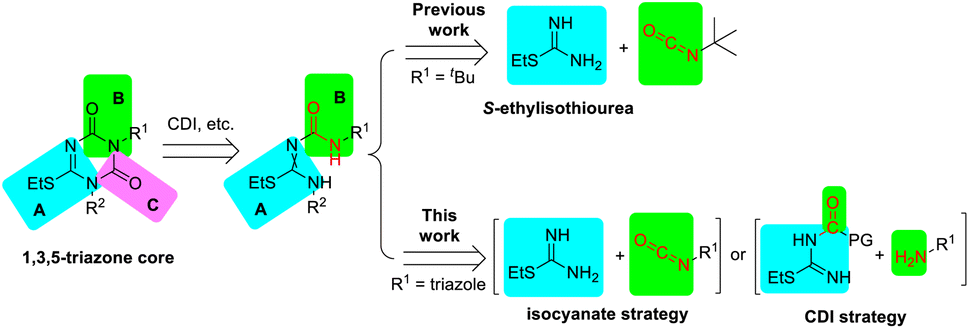 | ||
| Scheme 2 A retrosynthetic analysis for 1,3,5-triazone. | ||
Results and discussion
Investigation of the isocyanate strategy
Initially, to avoid the deprotection of the tert-butyl group in compound 7, isocyanate compound 11 containing 1,2,4-triazole scaffold was tried to use for cyclization with S-ethylisothiourea hydrobromide 3 (Scheme 3). Chloromethyl triazole 9 was ammonified with 25% ammonia water at 80 °C in quantity yield to produce desired aminomethyl triazole 10 as a hydrochloride salt, which was confirmed via 1H NMR and indicated that the alkalinity of the basic form of 10 was stronger than that of ammonia. Then, commercially unavailable triazole isocyanate 11 was obtained using bis(trichloromethyl)carbonate (BTC), which was used in the next step without further purification for the unstability.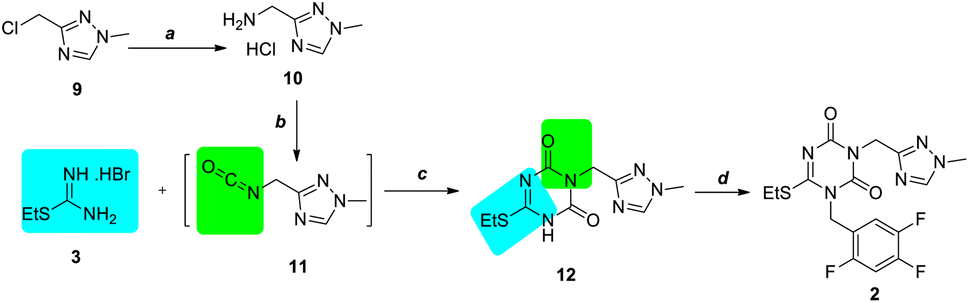 | ||
| Scheme 3 Preparation of intermediate 2 using the isocyanate strategy. Reagents and conditions: (a) 25% NH3·H2O, 80 °C, overnight, 90%; (b) TEA, BTC, DCM, 0 °C; (c) DBU, 0 °C, DMF; and DBU, CDI, 0 °C, 1h, then r.t., overnight, 30% for two steps; (d) 1-(bromomethyl)-2,4,5-trifluorobenzene 6, K2CO3, MeCN, 92%. | ||
Following a process similar to that described in as Scheme 1, triazole isocyanate 11 was condensed and cyclized with S-ethylisothiourea hydrobromide 3 to successfully obtain N3-substituted 1,3,5-triazone 12 with a yield of 30%, which was then alkylated to obtain N1, N3-disubstituted 1,3,5-triazone 2 in 92% yield. However, when our optimization was undergoing, the simultaneous work was reported in patent by Xu.4e So we changed to the other CDI strategy.
Investigation of the CDI strategy (strategy B)
Due to the poor efficiency of the isocyanate strategy, the CDI strategy was employed for the process (Scheme 4). When S-ethylisothiourea hydrobromide 3 and aminomethyl triazole hydrochloride 10 were mixed with 3.5 equiv. CDI and 6.5 equiv. of N, N-diisopropylethylamine (DIPEA), intermediate 12 was formed, which was isolated by column purification (10% yield). Then, following a process to that in the isocyanate strategy, key intermediate 2 was obtained by N1-alkylation in 90% yield using potassium carbonate in acetonitrile.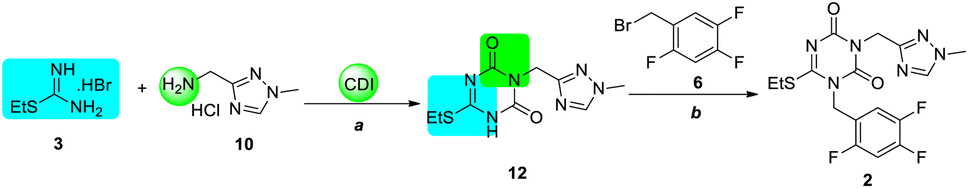 | ||
| Scheme 4 Preparation of intermediate 2 using the CDI strategy. Reagents and conditions: (a) CDI, DIPEA, DMF, r.t. to 110 °C, 10%; (b) K2CO3, MeCN, r.t., 90%. | ||
Preliminary optimization of the CDI strategy
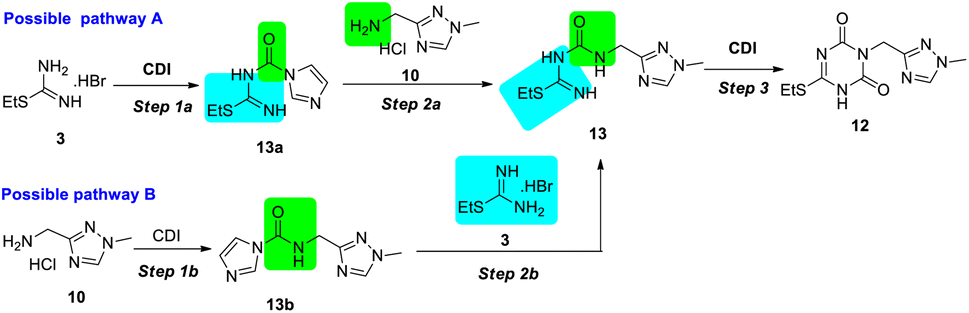 | ||
| Scheme 5 Possible pathways for the synthesis of intermediate 12 by the CDI strategy. | ||
In the trial of pathway A, a lightly excessive CDI (1.1 equiv.) was used to activate S-ethylisothiourea 3 in the mixture of triethylamine (TEA) and tetrahydrofuran (THF), and active intermediate 13a was isolated in 90% yield. The intermediate 13a was stable under common storage condition, but decomposed in methanol. Then the ammonolysis of intermediate 13a to 13 was performed using 2.2 equiv. TEA in different solvents. The results indicated that the reaction rate and yield in DMF and dimethyl sulfoxide (DMSO) were higher than that in acetonitrile (MeCN) and acetone, which might be due to the differences in the solubility of aminomethyl triazole 10 in solvents. The condensation step led to the formation of intermediate 13 (80% yield). During the trials of step 3, trace product 12 was observed in the mixture of 1.5 equiv. CDI and 1.5 equiv. DIPEA in DMF at room temperature. When the reaction temperature was raised to 110 °C for 3 hours, desired product 12 was isolated with 80% yield. Instead of DMF, the reaction could occur in MeCN at 85 °C in same reaction time, but more impurities were detected. Pathway B showed similar results regarding reaction time and yield. However, monitoring the intermediate 13b by thin layer chromatography (TLC) was difficult due to the weak colour rendering properties using common methods, and thus, for synthesizing intermediate 12, pathway A was considered to be the better method.
In pathway A, to achieve high efficiency of the stepwise strategy with convenient monitoring, further reaction optimization was performed. In consideration that each step was performed under basic conditions, unique base and solvent were expected to screen for the three steps.
First, while screening of the base, trace product 12 was detected using TEA (Table 1, entry 1), which was probably produced due to the weak basicity or low boiling point. This was speculated by the better results of DIPEA and 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) with stronger basicity or high boiling point (entries 2–3), but DBU presented a slightly lower yield. When K2CO3 was used as base, the approximate result was obtained as TEA (entry 4). After DIPEA was identified as a preferable base, the solvents were also investigated. As shown in entries 5–8, the solvents with good dissolving capacity of aminomethyl triazole 10 gave better results.
| Entry | Base (equiv.)a | Solvent | Intermediateb | ||
|---|---|---|---|---|---|
| 13a | 13 | 12 | |||
| a The common reaction conditions were shown unless otherwise mentioned. Base (1.5 equiv.) and CDI (1.1 equiv.) were added into the mixture of 3 in solvent and stirred. After 3 hours, 10 (1.0 equiv.) and base (2.2 equiv.) were added into the reaction, and stirred at room temperature overnight. Then CDI (1.2 equiv.) and base (2.2 equiv.) were added into the reaction and stirred at 110 °C for 3 hours.b Monitored by TLC and isolated yield by column purification.c Reaction temperature was 90 °C at boiling temperature of TEA.d “Well” meant that the reaction ran well.e “Trace” meant that trace product was observed in reaction mixture. | |||||
| 1 | TEA (5.5) | DMF | Well d | Well | Trace c,e |
| 2 | DIPEA (5.5) | DMF | Well | Well | 60% |
| 3 | DBU (5.5) | DMF | Well | Well | 55% |
| 4 | K2CO3 (5.0) | DMF | Well | Well | Trace |
| 5 | DIPEA (5.5) | DMSO | Well | Well | 59% |
| 6 | DIPEA (5.5) | MeCN | Well | Well | 50% |
| 7 | DIPEA (5.5) | THF | Well | Trace e | — |
| 8 | DIPEA (5.5) | Acetone | Well | Trace e | — |
By further optimization, starting from S-ethylisothiourea hydrobromide 3, CDI and DIPEA were added stepwise in DMF from r.t to 110 °C, and then the required K2CO3 was added to the reaction mixture to obtain the final product. Based on the four-step telescoped process (Scheme 6), the key intermediate N1, N3-disubstituted 1,3,5-triazone 2 was obtained on a gram scale in 53% yield, which was almost twice of the yield (28%) achieved by the originator.
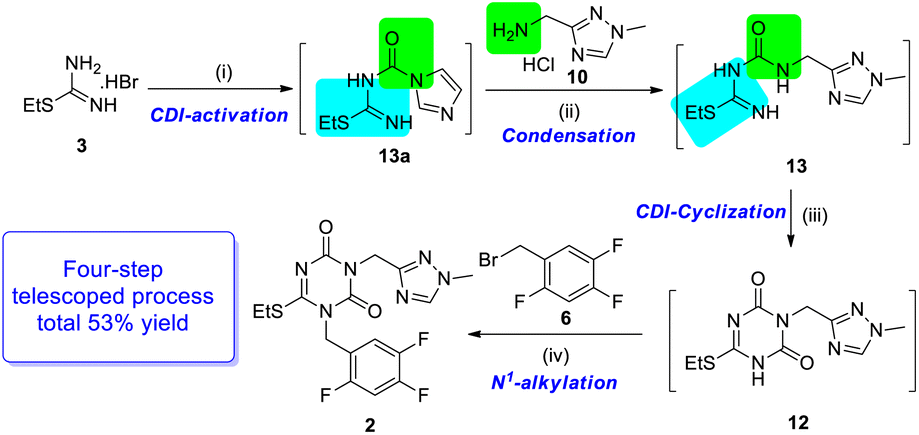 | ||
| Scheme 6 The four-step telescoped process for producing N1, N3-disubstituted 1, 3, 5-triazone 2 using the CDI strategy. Reagents and conditions: (i) CDI, DIPEA, DMF, r.t., 2 hours; (ii) r. t., overnight; (iii) CDI, 110 °C, 3 hours; (iv) K2CO3, r.t. 2 hours. | ||
Conclusion
To summarize, the isocyanate and CDI strategies were examined for producing the key intermediate N1,N3-disubstituted 1,3,5-triazone of ensitrelvir. Starting from S-ethylisothiourea hydrobromide, four-step telescoped process based on the CDI strategy with a small change provided a big improvement. The strategy gave a total yield of 53%, which was almost twice of the yield (28%) achieved by the originator. The process was atom-economical, convenient, and environmentally friendly, and conducive to further research and development.Experimental section
All commercially available materials and solvents were used directly without further purification unless otherwise noted. Thin-layer chromatography (TLC) was performed on 0.25 mm silica gel F-254 plates. 1H NMR and 13C NMR data were recorded with a Bruker spectrometer (400 MHz and 600 MHz) using TMS as internal standard and reported relative to residual solvent signals as follows: chemical shift (δ ppm), multiplicity, coupling constant (Hz), and integration. The multiplicities are denoted as follows: s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet; br, broad. The HRMS spectra were determined on a THERMO LTQ.Isocyanate strategy
Preparation of 6-(ethylthio)-3-[(1-methyl-1H-1,2,4-triazol-3-yl)methyl]-1-(2,4,5-trifluoro-benzyl)-1,3,5-triazine-2,4-(1H,3H)-dione (2).The reaction was evaporated under reduced pressure to afford a residue, which was purified by silica gel column chromatography (DCM: MeOH = 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to 20:1) to give product (54 mg, 0.202 mmol, 30%) as a white solid. 1H NMR (DMSO-d6, 400 MHz): δ 12.90 (s, 1H), 8.34 (s, 1H), 4.88 (s, 2H), 3.78 (s, 3H), 3.10 (q, J = 7.2 Hz, 2H), 1.29 (t, J = 7.3 Hz, 3H). The 1H NMR was slightly different from that in the publication.4e13C NMR (DMSO-d6, 150 MHz) δ 170.28, 169.25, 159.29, 145.57, 38.55, 36.20, 24.88, 14.80. HRMS (ESI+) m/z calcd for C9H13N6O2S+ [M + H]+: 269.0815, found 269.0820.
:1 to 20:1) to give product (54 mg, 0.202 mmol, 30%) as a white solid. 1H NMR (DMSO-d6, 400 MHz): δ 12.90 (s, 1H), 8.34 (s, 1H), 4.88 (s, 2H), 3.78 (s, 3H), 3.10 (q, J = 7.2 Hz, 2H), 1.29 (t, J = 7.3 Hz, 3H). The 1H NMR was slightly different from that in the publication.4e13C NMR (DMSO-d6, 150 MHz) δ 170.28, 169.25, 159.29, 145.57, 38.55, 36.20, 24.88, 14.80. HRMS (ESI+) m/z calcd for C9H13N6O2S+ [M + H]+: 269.0815, found 269.0820.
:1 to 1:2) to give product 2 (142 mg, 0.344 mmol, 92%) as a white solid. 1H NMR (CDCl3, 400 MHz): δ 7.95 (s, 1H), 7.17–7.09 (m, 1H), 7.00–6.90 (m, 1H), 5.24 (s, 2H), 5.17 (s, 2H), 3.85 (s, 3H), 3.21 (q, J = 7.4 Hz, 2H), 1.34 (t, J = 7.4 Hz, 2H). The 1H NMR data met with the publication.2a13C NMR (CDCl3, 100 MHz): δ 169.66, 159.27, 156.89–153.66 (m), 151.75, 150.61, 151.28–145.60 (m), 144.39, 118.09 (dt, J = 16.3, 4.7 Hz), 117.01–115.51 (m), 105.84 (dd, J = 27.6, 21.1 Hz), 41.15 (d, J = 4.4 Hz), 39.94, 36.20, 27.35, 13.59.CDI strategy (pathway A, step by step)
Preparation of 6-(ethylthio)-3-((1-methyl-1H-1,2,4-triazol-3-yl)methyl)-1,3,5-triazine-2,4(1H,3H)-dione (12).:1 to 1:1) to give ethyl N'-(1H-imidazole-1-carbonyl)carbamimidothioate 13a (96 mg, 0.49 mmol) as a white solid, yield: 90%. 1H NMR (CDCl3, 400 MHz) δ 9.50 (br s, 1H), 8.35 (s, 1H), 7.57 (s, 1H), 7.20 (br s, 1H), 7.04 (s, 1H), 3.15 (q, J = 7.4 Hz, 2H), 1.42 (t, J = 7.3 Hz, 3H). 13C NMR (CDCl3, 100 MHz) δ 176.24, 155.47, 137.52, 129.88, 117.11, 25.81, 14.77.:1 to 20:1) to give compound 13 (96 mg, 0.49 mmol) as a white solid, yield: 80%. 1H NMR (CDCl3, 600 MHz) δ 7.96 (s, 1H), 5.93 (s, 1H), 4.49 (d, J = 5.8 Hz, 2H), 3.87 (s, 3H), 2.97 (q, J = 7.4 Hz, 2H), 1.28 (t, J = 7.4 Hz, 3H). 13C NMR (CDCl3, 150 MHz) δ 169.06, 163.06, 161.98, 144.19, 37.96, 36.15, 24.96, 14.77. HRMS (ESI+) m/z calcd for C8H15N6OS+ [M + H]+: 243.1023, found 243.1029.:1 to 20:1) to give compound 12 (100 mg, 0.37 mmol) as a white solid, yield: 80%.To a solution of S-ethylisothiourea hydrobromide 3 (1.2 g, 6.73 mmol, 1.0 equiv.) and DIPEA (1.3 g, 10.10 mmol, 1.5 equiv.) in DMF (10 mL), CDI (1.2 g, 7.41 mmol, 1.1 equiv.) was added. The resulting mixture was stirred for 2 hours and monitored by TLC (PE: EA = 1:1, Rf value: ∼0.6).
Into the above reaction mixture, DIPEA (1.8 g, 14.14 mmol, 2.1 equiv.) and (1-methyl-1H-1,2,4-triazol-3-yl)methanamine hydrochloride 10 (1.0 g, 6.73 mmol, 1.0 equiv.) were added successively. The resulting mixture was stirred overnight and monitored by TLC (DCM: MeOH = 20:1, Rf value: ∼0.2).
DIPEA (2.6 g, 20.19 mmol, 3.0 equiv.) and CDI (1.64 g, 10.10 mmol, 1.5 equiv.) were added successively into the above mixture. The resulting mixture was stirred at 110 °C for 3 hours and monitored by TLC (DCM: MeOH = 20:1, Rf value: ∼0.15).
The reaction mixture was cooled to room temperature. K2CO3 (2.05 g, 14.81 mmol, 2.2 equiv.) and 1-(bromomethyl)-2,4,5-trifluorobenzene 6 (1.51 g, 6.73 mmol, 1.0 equiv.) were added successively. The resulting mixture was stirred at room temperature for 2 hours and monitored by TLC (PE: EA = 1:2, Rf value: ∼0.3). After the reaction was finished, crude product was separated out by adding saturated ammonium chloride (50 mL). After filtration, the crude residue was purified by silica gel column chromatography (PE/EA = 1:1) to give desired product 2 (1.47 g, 3.57 mmol) as a white solid in 53% yield.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Talent Project of Jiangxi Province (No. jxsq2019101065) and Jiangxi Provincial Natural Science Foundation (No. 20224BAB216005). The Start-up Fund of Gannan Medical University is gratefully acknowledged.Notes and references
- (a) D. R. Owen, C. M. N. Allerton, A. S. Anderson, L. Aschenbrenner, M. Avery, S. Berritt, B. Boras, R. D. Cardin, A. Carlo, K. J. Coffman, A. Dantonio, L. Di, H. Eng, R. Ferre, K. S. Gajiwala, S. A. Gibson, S. E. Greasley, B. L. Hurst, E. P. Kadar, A. S. Kalgutkar, J. C. Lee, J. Lee, W. Liu, S. W. Mason, S. Noell, J. J. Novak, R. S. Obach, K. Ogilvie, N. C. Patel, M. Pettersson, D. K. Rai, M. R. Reese, M. F. Sammons, J. G. Sathish, R. S. P. Singh, C. M. Steppan, A. E. Stewart, J. B. Tuttle, L. Updyke, P. R. Verhoest, L. Wei, Q. Yang and Y. Zhu, An oral SARS-CoV-2 Mpro inhibitor clinical candidate for the treatment of COVID-19, Science, 2021, 374, 1586–1593 CrossRef CAS PubMed; (b) L. Riva, S. Yuan, X. Yin, L. Martin-Sancho, N. Matsunaga, L. Pache, S. Burgstaller-Muehlbacher, P. D. D. Jesus, P. Teriete, M. V. Hull, M. W. Chang, J. F.-W. Chan, J. Cao, V. K.-M. Poon, K. M. Herbert, K. Cheng, T.-T. H. Nguyen, A. Rubanov, Y. Pu, C. Nguyen, A. Choi, R. Rathnasinghe, M. Schotsaert, L. Miorin, M. Dejosez, T. P. Zwaka, K.-Y. Sit, L. Martinez-Sobrido, W.-C. Liu, K. M. White, M. E. Chapman, E. K. Lendy, R. J. Glynne, R. Albrecht, E. Ruppin, A. D. Mesecar, J. R. Johnson, C. Benner, R. Sun, P. G. Schultz, A. I. Su, A. García-Sastre, A. K. Chatterjee, K.-Y. Yuen and S. K. Chanda, Discovery of SARS-CoV-2 antiviral drugs through large-scale compound repurposing, Nature, 2020, 586, 113–119 CrossRef CAS PubMed; (c) B. Hu, H. Guo, P. Zhou and Z.-L. Shi, Characteristics of SARS-CoV-2 and COVID-19, Nat. Rev. Microbiol., 2021, 19, 141–154 CrossRef CAS PubMed; (d) J. L. McKimm-Breschkin, A. J. Hay, B. Cao, R. J. Cox, J. Dunning, A. C. Moen, D. Olson, A. Pizzorno and F. G. Hayden, COVID-19, influenza and RSV: surveillance-informed prevention and treatment – meeting report from an isirv-WHO virtual conference, Antiviral Res., 2022, 197, 105227 CrossRef CAS PubMed; (e) WHO Solidarity Trial Consortium, Repurposed antiviral drugs for COVID-19 - interim WHO solidarity trial results. N. Engl. J. Med. 2021, 384, 497–511 Search PubMed.
- (a) Y. Unoh, S. Uehara, K. Nakahara, H. Nobori, Y. Yamatsu, S. Yamamoto, Y. Maruyama, Y. Taoda, K. Kasamatsu, T. Suto, K. Kouki, A. Nakahashi, S. Kawashima, T. Sanaki, S. Toba, K. Uemura, T. Mizutare, S. Ando, M. Sasaki, Y. Orba, H. Sawa, A. Sato, T. Sato, T. Kato and Y. Tachibana, Discovery of S-217622, a noncovalent oral SARS-CoV-2 3CL protease inhibitor clinical candidate for treating COVID-19, J. Med. Chem., 2022, 65, 6499–6512 CrossRef CAS PubMed; (b) J. D. A. Tyndall, S-217622, a 3CL protease inhibitor and clinical candidate for SARS-CoV-2, J. Med. Chem., 2022, 65, 6496–6498 CrossRef CAS PubMed; (c) H. Tan, Y. Hu, P. Jadhav, B. Tan and J. Wang, Progress and challenges in targeting the SARS-CoV-2 papain-like protease, J. Med. Chem., 2022, 65, 7561–7580 CrossRef CAS PubMed.
- From Shionogi's Website: https://www.shionogi.com/global/en/news/2022/11/e20221122.html.
- (a) W. Hu, X. Zhang, Y. Liu, T. Liu, J. Wen, X. Peng, X. Xie and W. Chen, Two-stage one-pot synthetic strategy for the key triazone-triazole intermediate of ensitrelvir (S-217622), an oral clinical candidate for treating COVID-19, RSC Adv., 2022, 12, 34808–34814 RSC; (b) C. Liu, Triazine compound as well as preparation method and application thereof. Invention Pat. CN114790198A, 2022 Search PubMed; (c) X. Xu; D. Zeng; Z. Bao; X. Leng; Z. Wang and X. Sun, Preparation method of oral medicine for treating new coronal pneumonia. Invention Pat. CN114751894A, 2022 Search PubMed; (d) X. Xu; D. Zeng; X. Leng, Z. Bao; J. Xue and D. Huang, Preparation method of novel coronavirus main protease inhibitor, Invention Pat. CN114853741A, 2022 Search PubMed; (e) X. Xu; D. Zeng; Z. Bao; X. Leng; J. Su and X. Gu, Preparation method of oral medicine S-217622 for treating new coronal pneumonia. Invention Pat. CN114644621A, 2022 Search PubMed; (f) S. Jiang; Y. Xiao; B. Guo; K. Zhang and C. Liu, Method for preparing anti-new coronavirus drug ensitrevir. Invention Pat. CN114874194A, 2022 Search PubMed; (g) Z. Chen; X. Tian; G. Liu; P. Wang and G. Qiang, Synthesis method of ensitrelvir. Invention Pat. CN114805314A, 2022 Search PubMed; (h) X. Liu; W. Li and C. Zhou, Synthesis method of protease inhibitor. Invention Pat. CN114805313A, 2022 Search PubMed; (i) W. Wei; J. Yang; X. Wu; T. Zhao; H. Wang; C. Li; L. Qu and B. Wang, High-purity compound and purification or preparation method of compound. Invention Pat. CN114591303A, 2022 Search PubMed; (j) S. Jiang; Y. Xiao; b. Guo; K. Zhang and C. Liu, Triazine compound or pharmaceutically acceptable salt, isomer, pharmaceutical composition and application thereof. Invention Pat. CN114539228A, 2022 Search PubMed; (k) X. Meng, Triazine compound and application thereof in preparation of antiviral drugs. Invention Pat. CN114507221A, 2022 Search PubMed.
- G. K. Mittapalli, K. R. Reddy, H. Xiong, O. Munoz, B. Han, F. D. Riccardis, R. Krishnamurthy and A. Eschenmoser, Mapping the landscape of potentially primordial informational oligomers: oligodipeptides and oligodipeptoids tagged with triazines as recognition elements, Angew. Chem., 2007, 119, 2522–2529 CrossRef.
- C. L. Meena, D. Singh, B. Kizhakeetil, M. Prasad, M. George, S. Tothadi and G. J. Sanjayan, Triazine-based janus G–C nucleobase as a building block for self-assembly, peptide nucleic acids, and smart polymers, J. Med. Chem., 2021, 86, 3186–3195 CAS.
- H. Kai, T. Horiguchi, T. Kameyama, K. Asahi, T. Endoh, S. Jikihara, T. Hasegawa, S. Tanaka, A. Nozu, C. Takeyama, M. Tomari, F. Takahashi, N. Tamura, S. Yagi, T. Itoh and Y. Isou, Dioxotriazine derivatives as a new class of P2X3 receptor antagonists: Identification of a lead and initial SAR studies, Bioorg. Med. Chem. Lett., 2021, 37, 127833 CrossRef CAS PubMed.
- K. H. Kong, C. K. Tan, X. Lin and Y. Lam, A versatile thiouronium-based solid-phase synthesis of 1,3,5-triazines, Chem.– Eur. J., 2012, 18, 1476–1486 CrossRef CAS PubMed.
- A. Madin and A. P. Owens, Triazolo-pyridazine derivatives as ligands for GABA receptors. Invention Pat. US6355798B1, 2002 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ra07844a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |