DOI:
10.1039/D2RA07807D
(Paper)
RSC Adv., 2023,
13, 3147-3154
Visible light mediated organocatalytic dehydrogenative aza-coupling of 1,3-diones using aryldiazonium salts†
Received
7th December 2022
, Accepted 3rd January 2023
First published on 20th January 2023
Abstract
An efficient protocol for diazenylation of 1,3-diones under photoredox conditions is presented herein. C–N bond forming Csp3–H functionalization of cyclic and alkyl diones by unstable aryl diazenyl radicals is achieved through reaction with aryldiazonium tetrafluoroborates by organocatalysts under visible light irradiation. The reaction has wide substrate scope, gives excellent yields, and is also efficient in water as a green solvent. This method provides an easy access to aryldiazenyl derivatives that are useful key starting materials for the synthesis of aza heterocycles as well as potential pharmacophores.
Introduction
Finding newer methods for a carbon–heteroatom bond, in particular, C–N bond formation reactions is pivotal in organic synthesis because of the rich abundance of C–N functionalization in a vast number of natural products, drugs, and other important molecules.1 Among the huge number of nitrogenous molecules, azo compounds are of great importance due to their rich abundance in naturally occurring bioactive compounds, drug molecules containing nitrogenous heterocycles, their applications in dye industry, and others.2 Many azo compounds are known to mediate site-specific drug delivery.3 Therefore, the development of various synthetic routes to azo compounds has attracted the attention of synthetic organic chemists at large.4 But till now only a limited number of synthetic routes to such azo compounds could be devised.5 Among them diazenylazation is considered to be a convenient and efficient method. Diazenylazation is the incorporation of diazo alkyl or aryl group into an active methylene compound6 or any C-nucleophile.7 These diazenylated products can be obtained from aryl diazonium salts, triazenes8 and hydrazine salts,9 which often require the use of heavy metal catalysis, difficult reaction conditions, and other complicated techniques. However, the generation of unstable diazenyl radical is very less known because of its short half-life period in solution,10 which loses a nitrogen molecule to produce a more stable phenyl radical.11 Therefore, mainly three types of reactions, viz. diazenylation, carbodiazenylation,12 and most widely arylation13 reactions are observed (Scheme 1).
 |
| | Scheme 1 Different types of reaction with aryldiazonium salts. | |
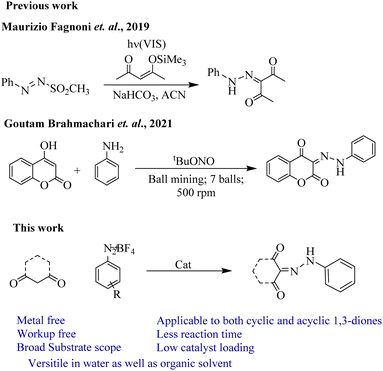
Aryl diazonium salts are easily accessed and synthesized in one-step by diazotization of readily available corresponding anilines,14 of which are mostly useful for arylation15 or less frequently for diazenylation16 depending upon the condition used. In the last few years, resourceful researches have been carried out using aryl diazonium salts as substrates under organophotocatalysed, visible light irradiation for arylation or diazenylation of different substituted heteroaryl moieties which resulted in the synthesis of drug scaffolds.17 But only a handful of diazenylazation type of reactions are being reported, where Fagnoni et al. (Fig. 1) method provides one-pot diazenyl compounds. However, the method requires stable arylazo sulfones and excess silyl enol ether, and maintaining the pH of the solution.6c Recently, Brahmachari et al. reported one-step diazenylation of 4-hydroxycoumarins with in situ generated aryl diazonium cation in ball milling process. However, this method is limited to the diazenylation of 4-hydroxycoumarin derivatives.18 In this regard, the advancement of the synthesis of diazenyl compound is still a challenging area for chemists which demand cheap and efficient synthetic techniques. Compared to aryl diazenyl compounds, the alkyl diazenyl compounds are least tolerant and unstable towards functional groups.19 But they are found to possess anti-inflammatory and antibiotic drug like properties. Over the last two decades, visible light photocatalysis has emerged as a greener approach in organic synthesis.20 Also, the replacement of volatile organic compounds (VOCs) as solvents with greener media21 like water22 has become a need of the day. In line with our current research interest in green chemistry and photoredox catalysis, herein we report an efficient and inexpensive visible light-mediated organocatalysed synthesis of diazenyl compounds using 1,3-diones and aryl diazonium salts without the use of any metal activator or ligand. Also, we have carried out the reactions in water obtaining equally excellent yields.
 |
| | Fig. 1 Dehydrogenative diazenylation of diones. | |
Results and discussion
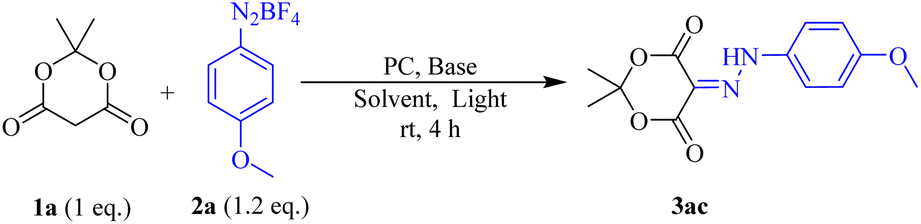
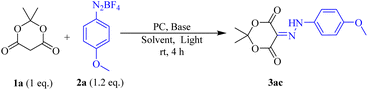
Considering our current interest in the synthesis of diazenyl compounds, we started our investigation using Meldrum's acid (1a, 0.2 mmol) and p-methoxy aryldiazonium salt (2a, 0.24 mmol) as model substrates. Taking into account the solubility of aryldiazonium tetrafluoroborate salts, we chose acetonitrile (ACN) as the preferred solvent.23 We then irradiated the reaction mixture with a 40 W Kessil Blue LED bulb in the presence of 5 mol% Eosin Y24 as a photocatalyst and 2 equivalent of diisopropyl ethyl amine (DIPEA). The reaction was carried out at room temperature in acetonitrile (0.1 M, 2 mL). Air cooling was maintained to dissipate the little heat generated in such visible light-catalysed reactions. We were delighted to isolate the corresponding diazenyl compound (3ac) in 80% yield after 3 hours. Remarkably, the reaction was very clean furnishing 3ac as a single product, and no significant amount of undesired products was observed. Then we embarked upon finding the optimum reaction condition (Table 1). First, we explored other polar solvents such as DMF, 1,4-dioxane, dichloromethane, and non-polar solvent like toluene were also examined. But we found that the reaction furnished optimum yield in ACN compared to other solvents (entries 1–5, Table 1). Noticeably, the reaction in DCM produced a significant number of undesired products (entry 5, Table 1). Hence, we carried out all the reactions in ACN as our solvent of choice. We then screened various bases such as triethyl amine (Et3N), diisopropyl ethyl amine (DIPEA), and K3PO4, and found that 1 equivalent of DIPEA was optimum for completion of the reaction (entries 7–12, Table 1). When we carried out the reaction in the absence of a base, the desired product was obtained in just 25% yield. Hence, it was inferred that DIPEA was needed for the regeneration of the catalyst in the catalytic cycle. However, increasing the equivalent of aryl diazonium salt from 1.2 to 2 equivalents, no improvement in the yield% was observed. The optimum catalyst loading was determined to be 2 mol% (entry 7, Table 1). Again, the same reaction was carried out in the presence of DIPEA and Eosin Y but in the absence of visible light for 24 h (entry 13, Table 1). Only 20% conversion was observed. But the same when done in the absence of either base or photocatalyst and absence of both, no conversion was observed. It established that both DIPEA and Eosin Y as the photocatalyst are essential for success of the diazenylation. With all the optimized conditions in our hand, we then started reacting different aryl diazonium salts with Meldrum's acid to furnish the corresponding diazenylated products (Scheme 2). The advantage of this reaction is that it does not require any external workups or quenching. The reaction mixture is directly concentrated and purified by column chromatography (CC).
Table 1 Optimization of reaction condition for 1,3 diones and aryl diazonium saltsa
|
| Entry |
Catalyst (mol%) |
Base (equivalent) |
Solvent |
Yieldb (%) |
| Reaction condition: 1,3-dione 1a (0.2 mmol), aryl diazonium salts (0.24 mmol), solvent (2.0 mL), room temperature, under 40 W Blue LEDs irradiation for 4 h. Isolated yield. Without base. 1,3-Dione 1a (0.2 mmol), aryl diazonium salts (0.24 mmol), solvent (2.0 mL), room temperature in dark condition. |
| 1 |
Eosin Y (5 mol%) |
DIPEA (2) |
ACN |
80 |
| 2 |
Eosin Y (5 mol%) |
DIPEA (2) |
DMF |
65 |
| 3 |
Eosin Y (5 mol%) |
DIPEA (2) |
Toluene |
ND |
| 4 |
Eosin Y (5 mol%) |
DIPEA (2) |
1,4-Dioxane |
70 |
| 5 |
Eosin Y (5 mol%) |
DIPEA (2) |
DCM |
ND |
| 6 |
Eosin Y (2 mol%) |
DIPEA (2) |
ACN |
80 |
| 7 |
Eosin Y (2 mol%) |
DIPEA (1) |
ACN |
92 |
| 8 |
— |
DIPEA (1) |
ACN |
20 |
| 9c |
Eosin Y (2 mol%) |
— |
ACN |
25 |
| 10 |
Eosin Y (2 mol%) |
K3PO4 (1) |
ACN |
58 |
| 11 |
Ru(bpy)3Cl2 (2 mol%) |
Et3N (1) |
ACN |
71 |
| 12 |
Eosin Y (2 mol%) |
Et3N (1) |
ACN |
25 |
| 13d |
Eosin Y (2 mol%) (in dark, 8 h) |
DIPEA (1) |
ACN |
20 |
| 14 |
Eosin Y (10 mol%) |
DIPEA (1) |
ACN |
84 |
| 15 |
Methylene Blue (2 mol%) |
DIPEA (1) |
ACN |
67 |
| 16 |
Rose Bengal (2 mol%) |
DIPEA (1) |
ACN |
74 |
| 17 |
Methylene Blue (2 mol%) |
DIPEA (1) |
ACN |
NR |
| 18 |
Rose Bengal (2 mol%) |
DIPEA (1) |
H2O |
85 |
| 19 |
Rose Bengal (2 mol%) |
— |
H2O |
NR |
| 20 |
Eosin Y |
DIPEA (1) |
H2O |
70 |
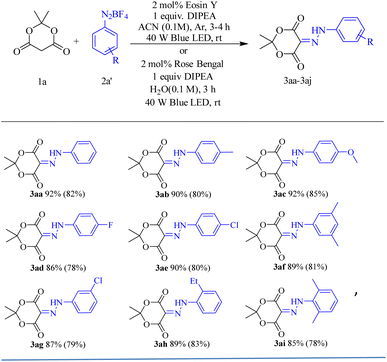 |
| | Scheme 2 Substrate scope of Meldrum's acid. a Isolated yield; reaction condition: 1a (0.20 mmol), 2 (0.24 mmol), Eosin Y (2 mol%), DIPEA (0.20 mmol), ACN (2 mL), 40 W Kessil Blue LED bulb and stirring at room temperature. | |
Use of volatile organic compounds (VOCs) significantly impacts our environment. Avoidance of such VOCs is one of the major goals of modern green chemistry. In this regard, water has evolved into a preferable solvent for green chemistry approach. Increasingly, visible light catalyzed reactions are being performed in water.25 Major reason behind it is that many of the dyes used as organophotocatalysts are soluble in water, like Rose Bengal, Eosin Y, Eosin B, Rhodamine B, Methylene Blue, various Flavonoids, etc. Therefore, we repeated the reaction of 1a and 2a in the presence of 2 mol% of Eosin Y and 1 equivalent of DIPEA in water (2 mL) and irradiated with a 40 W Blue LED bulb resulting in a little lower yield of 3ac (70%, entry 21, Table 1). Then we tried the same reaction with 2 mol% of different water soluble photocatalysts such as Ru(bpy)3Cl2, Methylene Blue, and Rose Bengal (Table 1). Gratifyingly, Rose Bengal in water gave 3ac in excellent yield (85%, entry 18, Table 1). Though the yield was slightly lower than in ACN using Eosin Y. Encouraged by this success, we decided to repeat each reaction with optimized conditions in both ACN and water. Obtained yields in water are reported in parentheses in each case.
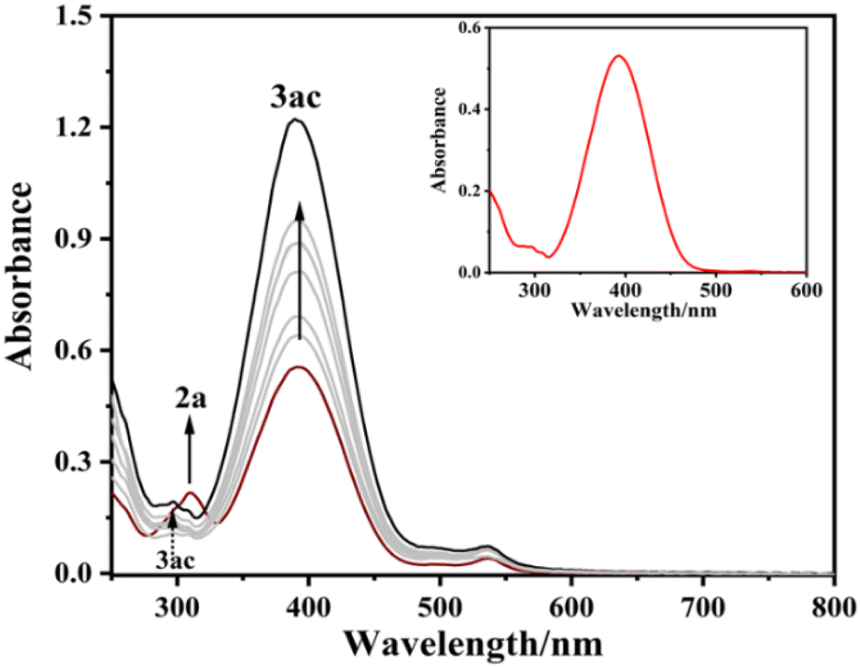
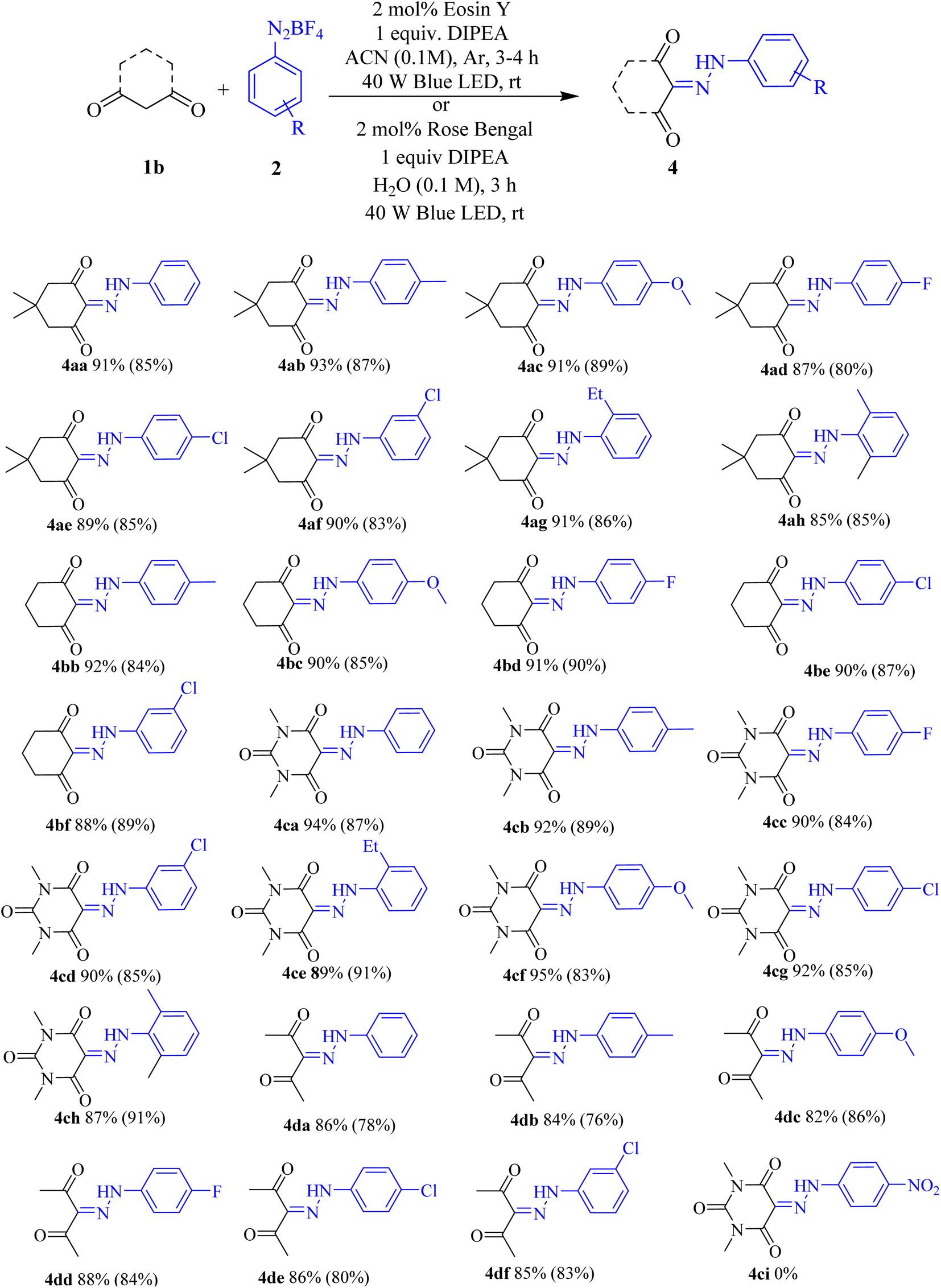
To shed light on the efficiency of the reaction, UV-Vis spectroscopy technique was used. We observed that p-methoxy aryldiazonium tetrafluoroborate salt (2a) exhibits a UV-vis absorption band at 310 nm whereas catalyst Eosin Y exhibits a notable UV-visible absorption band at 530 nm (Fig. S1, ESI†). The above reported reaction was performed using Meldrum's acid (1a), p-methoxy aryldiazonium salt (2a), Eosin Y, and DIPEA in 2 mL ACN irradiated with 40 W Blue LED light and UV-Vis spectra were recorded in a periodic time interval of 15 minutes. It was seen that two new peaks were generated at 295 nm and 390 nm (Fig. 2). We have confirmed these peaks were responsible for the formation of our product 3ac after purification (Fig. 2 inset). Further, these peaks gradually intensified and reached maximum at 105 minutes. It was also observed that the peak of aryl diazonium salt simultaneously kept decreasing and the peak of the catalyst (Eosin Y) at 530 nm remained practically static which confirms the regeneration of the catalyst in every cycle. To further evaluate the efficacy of the reaction, we examined different cyclohexane 1,3-dione derivatives and barbiturates. Altogether, thirteen diazenylated cyclohexane 1,3-dione derivatives were synthesized in excellent yields (85–93% in ACN and 80–90% in water). Next, eight diazenylated barbiturate derivatives were also prepared in excellent yields (87–95% in ACN and 83–91% in water) (Scheme 4). We found that barbituric acid derivatives (4ca–4ch, Scheme 4) are the most reactive among all the diones used. On the other hand, aryl diazonium salts having an electron donating group (EDG) were found to be more reactive than those having an electron withdrawing group (EWG). Interestingly, chloro and fluoro-aryl diazonium salts furnished expected products, but p-nitro aryldiazonium salts failed to give the desired product (Scheme 3). Alkylation, arylation, or diazenylation reactions with acyclic 1,3-diones are rarely achieved. We were pleasantly surprised when we carried out the reaction of p-methoxy aryl diazonium salt with acetylacetone in ACN, the reaction was completed in 4 hours and the desired diazenyl product was isolated in 80% yield. The acetylacetone was carried out in ACN and Eosin Y with five other aryl diazonium salts. All five other aryl diazonium salts were also found to be well tolerated and furnished desired products (4da–4df, Scheme 4) in good yields. These alkyl diazenes are a very important class of biologically active scaffolds.26
 |
| | Fig. 2 Time profile UV-vis spectroscopic analysis. | |
 |
| | Scheme 3 Reaction of barbiturate with p-nitro aryldiazonium salt. | |
 |
| | Scheme 4 Substrate scope for other 1,3 diones. a Isolated yield; reaction condition: 1a (0.20 mmol), 2 (0.24 mmol), Eosin Y (2 mol%), DIPEA (0.20 mmol), ACN (2 mL), Ar, 40 W Kessil 160WE Blue LED bulb and stirring at room temperature. b Reaction condition: 1a (0.20 mmol), 2 (0.24 mmol), Rose Bengal (2 mol%), DIPEA (0.20 mmol), water (2 mL), 40 W Kessil 160WE Blue LED bulb and stirring at room temperature. | |
Lastly, to understand the mechanism, a series of controlled potential cyclic voltammetry (CV) experiment were conducted to rationalize the step propagation. From CV experiments, it was found that the redox potential of Eosin Y (E1/2 = −1.1 V vs. NHE in ACN), would likely reduce the p-methoxy aryldiazonium salts (E1/2 = −1.0 V vs. NHE in ACN) and itself would be oxidized to Eosin Y cationic radical (Eosin Y+˙). Keeping all the factors in mind, a plausible mechanism is proposed in Fig. 3. First, in the presence of Blue LED light (455 nm), Eosin Y will be excited to generate Eosin Y* which will reduce the p-methoxy aryldiazonium salts to p-methoxy aryldiazonium radical and itself get oxidized to Eosin Y cationic radical (Eosin Y+˙). The generated p-methoxy aryldiazonium radical will then combine with Meldrum's acid to give the radical intermediate 5. To complete the catalytic cycle, the Eosin Y cationic radical (Eosin Y+˙) will accept an electron from the intermediate 5 to give the cationic intermediate 6, and Eosin Y* reverts to ground state. The base present, DIPEA will abstract the acidic proton to give diazo enol 7. The intermediate 7 will then tautomerize to the more stable diazenyl compound 3ac. To rationalize the reaction mechanism, cyclic voltammetry (CV) was performed in acetonitrile with p-methoxy aryldiazonium salt (2a) and p-nitro aryldiazonium salt (2c) using glassy carbon as the working electrode. Results are depicted in Fig. 4 and 5, respectively. From Fig. 4 it is observed that 2a exhibits an irreversible reduction wave at −1.0 V vs. NHE at 0.1 V s−1 scan rate. On the other hand, in Fig. 5, a quasi-reversible redox wave is observed for 2c with Ep,c = −0.35 V vs. NHE at 0.7 V s−1 scan rate. We were unable to detect any redox wave for 2c at a lower scan rate which suggests that after reduction 2c produced a very short-lived reduced species. But the quasi-reversible peak at a higher scan rate strongly suggests that the reduced species are stable enough and the available electron density on the radical undergoes delocalization into the conjugated π orbitals. On the contrary, another CV of p-fluoro aryldiazonium salt showed an irreversible reduction wave at −0.5 V vs. NHE at 0.1 V s−1 scan rate (Fig. 6) which suggests that the p-fluoro aryldiazonium radical is different from p-nitro aryldiazonium. Hence the reactivity of both the electron withdrawing groups are different.
 |
| | Fig. 3 Plausible mechanism for C–N bond forming Csp3–H functionalization of cyclic diones. | |
 |
| | Fig. 4 Cyclic voltammogram of p-methoxy aryldiazonium tetrafluoroborates salt 2a (red, 1 mM) and blank (black). Supporting electrolyte LiClO4 (0.1 M); scan rate 0.1 V s−1. | |
 |
| | Fig. 5 Cyclic voltammogram of p-nitro aryldiazonium tetrafluoroborates salt 2c (red, 1 mM) and blank (black). Supporting electrolyte LiClO4 (0.1 M); scan rate 0.7 V s−1. | |
 |
| | Fig. 6 Cyclic voltammogram of p-fluoro aryldiazonium tetrafluoroborates salt 2d (red, 1 mM) and blank (black). Supporting electrolyte LiClO4 (0.1 M); scan rate 0.1 V s−1. | |
Gram scale synthesis
We also tried the same model substrate 1a and 2a for gram scale synthesis of 3ac which furnished a yield of 68%. Hence the reaction is practical for reasonably large-scale synthesis (Scheme 5).
 |
| | Scheme 5 Gram scale synthesis. | |
Conclusion
In conclusion, we have developed a mild, visible light catalyzed atom economic approach towards the synthesis of diazenyl compounds. The reaction does not require use of any ligand, metal catalyst, work-up, or expensive starting materials. Here, we were able to modulate the activity of the diazonium cation and trap the unstable, somewhat elusive diazenyl radical by the diones and avoid arylation through loss of nitrogen. Here we have synthesized various chromophores of alkyl diazenes efficiently in a single step without heating or hydrolysis. Altogether 37 derivatives have been prepared in 80–94% (ACN); 76–91% (water) yields. Notably, all these reactions were completed within 3–4 hours compared to 20 hours reported earlier using other aryl azo derivatives. We have done mechanistic studies using cyclic voltammetry to explain differential reactivities of various aryldiazonium cations in terms of their reduction potentials. This strategy gives an effective method of C–N bond formation through Csp3–H functionalization giving access to diazenyl compounds useful for synthesis of aza substituted heterocycles and interesting bioactive scaffolds. Herein reported method shows high substrate scope and sufficient green chemistry quotient as the reactions were almost equally efficient in water compared to the optimized organic solvent acetonitrile.
Experimental
Method A
0.2–0.3 mmol of 1,3-diones, 2 mol% of Eosin Y, and 1.2 eq. of aryldiazonium tetrafluoroborate were dissolved in 2 mL of acetonitrile in a 20 mL oven-dried glass vial and argon were purged through a needle to the solution for 5 min, 1 eq. of diisopropyl ethyl amine was added slowly to the mixture and again argon was purged for another 5 min and sealed with a cap. The glass vial was irradiated with a 40 W Blue LED Kessil bulb with continuous stirring for 3–4 hours. The progress of the reaction was monitored by TLC.
Method B
0.2–0.3 mmol of 1,3-diones, 2 mol% of Rose Bengal, and 1.2 eq. of aryldiazonium tetrafluoroborate were dissolved in 2 mL of water in a 20 mL oven dried glass vial. 1 eq. of diisopropyl ethyl amine was added slowly to the mixture and sealed with a cap. The glass vial was irradiated with a 40 W Blue Led Kessil bulb with continuous stirring for 3–4 hours. The progress of the reaction was monitored by TLC.
After the completion of the reaction, the solvent was evaporated under reduced pressure in a rotary evaporator without any workup procedure. The resulting crude was purified in a column chromatography using petroleum ether and ethyl acetate as eluent.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
NIT Sikkim is acknowledged for its infrastructure facilities and a fellowship to RD. Dr Biswajit G. Roy (Sikkim University) is thanked for various helps, and Mr Srijan N. Chowdhury (NIT Sikkim) is thanked for helping with cyclic voltammetry analyses.
References
-
(a) A. A. Beharry and G. A. Woolley, Chem. Soc. Rev., 2011, 40, 4422–4437 RSC;
(b) W. Szymański, J. M. Beierle, H. A. V. Kistemaker, W. A. Velema and B. L. Feringa, Chem. Rev., 2013, 113, 6114–6178 CrossRef PubMed;
(c) M. Dong, A. Babalhavaeji, S. Samanta, A. A. Beharry and G. A. Woolley, Acc. Chem. Res., 2015, 48, 2662–2670 CrossRef CAS PubMed;
(d) J. F. Hartwig, Nature, 2008, 455, 314–322 CrossRef CAS PubMed;
(e) A. K. Yudin and J. F. Hartwig, Catalysed Carbon-Heteroatom Bond Formation, Wiley-VCH, Weinheim, 2010 CrossRef;
(f) N. Yasukawa, M. Kuwata, T. Imai, Y. Monguchi, H. Sajiki and Y. Sawama, Green Chem., 2018, 20, 4409–4413 RSC;
(g) J. Shi, T. Yuan, R. Wang, M. Zheng and X. Wang, Green Chem., 2021, 23, 3945–3949 RSC.
-
(a) Y. Yu, M. Nakano and T. Ikeda, Nature, 2003, 425, 145 CrossRef CAS PubMed;
(b) S. M. Sondhi, M. Dinodia and A. Kumar, Bioorg. Med. Chem., 2006, 14, 4657–4663 CrossRef CAS PubMed;
(c) T. Fehrentz, M. Schönberger and D. Trauner, Angew. Chem., Int. Ed., 2011, 50, 12156–12182 CrossRef CAS PubMed;
(d) A. A. Beharry and G. A. Woolley, Chem. Soc. Rev., 2011, 40, 4422–4437 RSC;
(e) B. K. Ho, Z. Ngaini, P. Matthew Neilsen, S. S. Hwang, R. Entigu Linton, E. L. Kong and B. K. Lee, J. Chem., 2017, 2017, 1–7 CrossRef;
(f) S. H. Lee, E. Moroz, B. Castagner and J. C. Leroux, J. Am. Chem. Soc., 2014, 136, 12868–12871 CrossRef CAS PubMed;
(g) M. A. Sofan, A. El-Mekabaty, A. M. Hasel and S. B. Said, J. Heterocycl. Chem., 2021, 58, 1645–1655 CrossRef CAS;
(h) G. S. Kumar and D. C. Neckers, Chem. Rev., 1989, 89, 1915–1925 CrossRef CAS.
-
(a) M. Roldo, E. Barbu, J. F. Brown, D. W. Laight, J. D. Smart and J. Tsibouklis, Expert Opin. Drug Delivery, 2007, 4, 547–560 CrossRef CAS PubMed;
(b) R. Cassano, S. Trombino, A. Cilea, T. Ferrarelli, R. Muzzalupo and N. Picci, Chem. Pharm. Bull., 2010, 58, 103–105 CrossRef CAS PubMed;
(c) A. Jain, Y. Gupta and S. K. Jain, Crit. Rev. Ther. Drug Carrier Syst., 2006, 23, 349–400 CrossRef CAS PubMed.
-
(a) S. Benkhaya, S. M’rabet and A. El Harfi, Heliyon, 2020, 6, e03271 CrossRef PubMed;
(b) S. M. Koshti, J. P. Sonar, A. E. Sonawane, Y. A. Pawar, P. S. Nagle, P. P. Mahulikar and D. H. More, Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem., 2008, 47, 329–331 Search PubMed;
(c) R. Zhao, C. Tan, Y. Xie, C. Gao, H. Liu and Y. Jiang, Tetrahedron Lett., 2011, 52, 3805–3809 CrossRef CAS;
(d) A. Grirrane, A. Corma and H. García, Science, 2008, 322, 1661–1664 CrossRef CAS PubMed;
(e) R. Z. Qiao, Y. Zhang, X. P. Hui, P. F. Xu, Z. Y. Zhang, X. Y. Wang and Y. L. Green, Chem, 2001, 3, 186–188 CAS;
(f) C. Zhang and N. Jiao, Angew. Chem., 2010, 122, 6310–6313 CrossRef.
-
(a) B. Deng, C. Li, J. Yuan, C. B. Rao, Y. Zhao, R. Zhang and D. Dong, Tetrahedron, 2019, 75, 2273–2279 CrossRef CAS;
(b) G. B. Raju, M. Mahesh, G. Manjunath and P. V. Ramana, Chem. Sci. Trans., 2016, 5, 125–136 CAS;
(c) J. M. dos Santos Filho, D. M. A. de Queiroz e Silva, T. S. Macedo, H. M. P. Teixeira, D. R. M. Moreira, S. Challal, J. L. Wolfender, E. F. Queiroz and M. B. P. Soares, Bioorg. Med. Chem., 2016, 24, 5693–5701 CrossRef CAS PubMed;
(d) D. Pranali and J. Kale, Chem. Pharm. Res., 2013, 5, 130–134 Search PubMed.
-
(a) E. S. Zhilin, L. L. Fershtat, D. M. Bystrov, A. S. Kulikov, A. O. Dmitrienko, I. V. Ananyev and N. N. Makhova, Eur. J. Org. Chem., 2019, 2019, 4248–4259 CrossRef CAS;
(b) C. G. T. Oliveira, F. F. Miranda, V. F. Ferreira, C. C. Freitas, R. F. Rabello, J. M. Carballido and L. C. D. Corrêa, J. Braz. Chem. Soc., 2001, 12, 339–345 CrossRef CAS;
(c) H. Othman Abdulla, S. Scaringi, A. A. Amin, M. Mella, S. Protti and M. Fagnoni, Adv. Synth. Catal., 2020, 362, 2150–2154 CrossRef.
-
(a) W.-C. Gao, Y.-F. Cheng, Y.-Z. Shang, H.-H. Chang, X. Li, R. Zhou, Y. Qiao and W.-L. Wei, J. Org. Chem., 2018, 83, 11956–11962 CrossRef CAS PubMed;
(b) H. Jiang, Y. Chen, B. Chen, H. Xu, W. Wan, H. Deng, K. Ma, S. Wu and J. Hao, Org. Lett., 2017, 19, 2406–2409 CrossRef CAS PubMed;
(c) G. Feng, M. Zhu, L. Liu and C. Li, Green Chem., 2019, 21, 1769–1776 RSC;
(d) A. Chakraborty, S. Jana, G. Kibriya, A. Dey and A. Hajra, RSC Adv., 2016, 6, 34146–34152 RSC;
(e) M. Lv, J. Ma, Q. Li and H. Xu, Bioorg. Med. Chem. Lett., 2018, 28, 181–187 CrossRef CAS PubMed.
- C. Liu, J. Lv, S. Luo and J. P. Cheng, Org. Lett., 2014, 16, 5458–5461 CrossRef CAS PubMed.
-
(a) D. S. Barak, S. U. Dighe, I. Avasthi and S. Batra, J. Org. Chem., 2018, 83, 3537–3546 CrossRef CAS PubMed;
(b) G. Shukla, T. Alam, H. K. Srivastava, R. Kumar and B. K. Patel, Org. Lett., 2019, 21, 3543–3547 CrossRef CAS PubMed;
(c) H. P. Zhou, J. B. Liu, J. J. Yuan and Y. Y. Peng, RSC Adv., 2014, 4, 25576–25579 RSC.
- M. N. Weaver, S. Z. Janicki and P. A. Petillo, J. Org. Chem., 2001, 66, 1138–1145 CrossRef CAS PubMed.
- C. Galli, Chem. Rev., 1988, 88, 765–792 CrossRef CAS.
-
(a) O. Blank, A. Wetzel, D. Ullrich and M. R. Heinrich, Eur. J. Org. Chem., 2008, 3179–3189 CrossRef CAS;
(b) O. Blank and M. R. Heinrich, Eur. J. Org. Chem., 2006, 4331–4334 CrossRef CAS;
(c) M. R. Heinrich, O. Blank and A. Wetzel, J. Org. Chem., 2007, 72, 476–484 CrossRef CAS PubMed.
-
(a) F. Mo, D. Qiu, L. Zhang and J. Wang, Chem. Rev., 2021, 121, 5741–5829 CrossRef CAS PubMed;
(b) J. Chen, X. Xie, J. Liu, Z. Yu and W. Su, React. Chem. Eng., 2022, 7, 1247–1275 RSC;
(c) S. Mahouche-Chergui, S. Gam-Derouich, C. Mangeney and M. M. Chehimi, Chem. Soc. Rev., 2011, 40, 4143–4166 RSC;
(d) F. Mo, G. Dong, Y. Zhang and J. Wang, Org. Biomol. Chem., 2013, 11, 1582–1593 RSC.
-
(a) F. X. Felpin and E. Fouquet, Adv. Synth. Catal., 2008, 350, 863–868 CrossRef CAS;
(b) A. K. Singh and J. Kandasamy, Org. Biomol. Chem., 2018, 16, 5107–5112 RSC;
(c) P. S. Gribanov, M. A. Topchiy, Y. D. Golenko, Y. I. Lichtenstein, A. V. Eshtukov, V. E. Terekhov, A. F. Asachenko and M. S. Nechaev, Green Chem., 2016, 18, 5984–5988 RSC.
-
(a) L. Wang, J. Shen, S. Yang, W. Liu, Q. Chen and M. He, Green Chem., 2018, 20, 1290–1296 RSC;
(b) S. S. Babu, M. Shahid and P. Gopinath, Chem. Commun., 2020, 56, 5985–5988 RSC;
(c) S. Witzel, J. Xie, M. Rudolph and A. S. K. Hashmi, Adv. Synth. Catal., 2017, 359, 1522–1528 CrossRef CAS;
(d) D. P. Hari, P. Schroll and B. König, J. Am. Chem. Soc., 2012, 134, 2958–2961 CrossRef CAS PubMed;
(e) E. Tatunashvili, B. Chan, P. E. Nashar and C. S. P. McErlean, Org. Biomol. Chem., 2020, 18, 1812–1819 RSC;
(f) R. C. Silva, L. F. Villela, T. J. Brocksom and K. T. De Oliveira, RSC Adv., 2020, 10, 31115–31122 RSC.
-
(a) R. A. Angnes, C. Potnis, S. Liang, C. R. D. Correia and G. B. Hammond, J. Org. Chem., 2020, 85, 4153–4164 CrossRef CAS PubMed;
(b) X. L. Yu, J. R. Chen, D. Z. Chen and W. J. Xiao, Chem. Commun., 2016, 52, 8275–8278 RSC;
(c) S. Kindt, K. Wicht and M. R. Heinrich, Org. Lett., 2015, 17, 6122–6125 CrossRef CAS PubMed;
(d) M. Wang, B. C. Tang, J. G. Wang, J. C. Xiang, A. Y. Guan, P. P. Huang, W. Y. Guo, Y. D. Wu and A. X. Wu, Chem. Commun., 2018, 54, 7641–7644 RSC;
(e) J. Rong, H. Jiang, S. Wang, Z. Su, H. Wang and C. Tao, Org. Biomol. Chem., 2020, 18, 3149–3157 RSC.
-
(a) S. S. Babu, P. Muthuraja, P. Yadav and P. Gopinath, Adv. Synth. Catal., 2021, 363, 1782–1809 CrossRef CAS;
(b) Ł. W. Ciszewski, K. Rybicka-Jasińska and D. Gryko, Org. Biomol. Chem., 2019, 17, 432–448 RSC;
(c) M. E. Trusova, K. V. Kutonova, V. V. Kurtukov, V. D. Filimonov and P. S. Postnikov, Resour. Technol., 2016, 2, 36–42 Search PubMed;
(d) Q. Zhang, M. Yu, J. Yuan, R. Zhang, Y. Liang, J. Tian and D. Dong, J. Org. Chem., 2016, 81, 6036–6041 CrossRef CAS PubMed;
(e) X. Zhang, Y. Mei, Y. Li, J. Hu, D. Huang and Y. Bi, Asian J. Org. Chem., 2021, 10, 453–463 CrossRef CAS;
(f) N. Kvasovs and V. Gevorgyan, Chem. Soc. Rev., 2021, 50, 2244–2259 RSC.
- G. Brahmachari, I. Karmakar and P. Karmakar, Green Chem., 2021, 23, 4762–4770 RSC.
-
(a) S. S. Dhaneshwar, N. Gairola, M. Kandpal, G. Vadnerkar, L. Bhatt, B. Rathi and S. S. Kadam, Eur. J. Med. Chem., 2009, 44, 3922–3929 CrossRef CAS PubMed;
(b) D. A. Kennedy, N. Vembu, F. R. Fronczek and M. Devocelle, J. Org. Chem., 2011, 76, 9641–9647 CrossRef CAS PubMed;
(c) P. S. Engel, C. Wang, Y. Chen, C. Rüchardt and H. D. Beckhaus, J. Am. Chem. Soc., 1993, 115, 65–74 CrossRef CAS.
-
(a) C. R. J. Stephenson, T. P. Yoon and D. W. C. MacMillan, Visible Light Photocatalysis in Organic Chemistry, Wiley-VCH, Weinheim, 2018 CrossRef;
(b) T. Xiong and Q. Zhang, Chem. Soc. Rev., 2016, 45, 3069–3087 RSC;
(c) K. L. Skubi, T. R. Blum and T. P. Yoon, Chem. Rev., 2016, 116, 10035–10074 CrossRef CAS PubMed;
(d) A. K. Bagdi, M. Rahman, D. Bhattacherjee, G. V. Zyryanov, S. Ghosh, O. N. Chupakhin and A. Hajra, Green Chem., 2020, 22, 6632–6681 RSC;
(e) S. Sharma and A. Sharma, Org. Biomol. Chem., 2019, 17, 4384–4405 RSC;
(f) V. Srivastava and P. P. Singh, RSC Adv., 2017, 7, 31377–31392 RSC;
(g) D. Ravelli, S. Protti and M. Fagnoni, Chem. Rev., 2016, 116, 9850–9913 CrossRef CAS PubMed;
(h) Y. Qin, L. Zhu and S. Luo, Chem. Rev., 2017, 117, 9433–9520 CrossRef CAS PubMed;
(i) P. G. Karmaker, M. A. Alam and F. Huo, RSC Adv., 2022, 12, 6214–6233 RSC.
-
(a) C. J. Clarke, W. C. Tu, O. Levers, A. Bröhl and J. P. Hallett, Chem. Rev., 2018, 118, 747–800 CrossRef CAS PubMed;
(b) F. P. Byrne, S. Jin, G. Paggiola, T. H. M. Petchey, J. H. Clark, T. J. Farmer, A. J. Hunt, C. Robert McElroy and J. Sherwood, Sustainable Chem. Processes, 2016, 4, 1–24 CrossRef;
(c) C. Capello, U. Fischer and K. Hungerbühler, Green Chem., 2007, 9, 927–993 RSC;
(d) T. Welton, Proc. R. Soc. A, 2015, 471 Search PubMed;
(e) N. Winterton, Clean Technol. Environ. Policy, 2021, 23, 2499–2522 CrossRef PubMed;
(f) R. Chércoles, RESEÑA LIBRO: Greener Solvents in Conservation: An Introductory Guide, 2022, vol. 21 Search PubMed.
-
(a) T. Kitanosono, K. Masuda, P. Xu and S. Kobayashi, Chem. Rev., 2018, 118, 679–746 CrossRef CAS PubMed;
(b) T. Kitanosono and S. Kobayashi, Chem.–Eur. J., 2020, 26, 9408–9429 CrossRef CAS PubMed;
(c) B. M. Sahoo and B. K. Banik, Solvent-less reactions: Green and sustainable approaches in medicinal chemistry, Elsevier, 2020 Search PubMed;
(d) M. Cortes-Clerget, J. Yu, J. R. A. Kincaid, P. Walde, F. Gallou and B. H. Lipshutz, Chem. Sci., 2021, 12, 4237–4266 RSC.
- I. U. Hoque, S. R. Chowdhury and S. Maity, J. Org. Chem., 2019, 84, 3025–3035 CrossRef CAS PubMed.
-
(a) A. K. Yadav and L. D. S. Yadav, Green Chem., 2016, 18, 4240–4244 RSC;
(b) H. K. Singh, A. Kamal, S. Kumari, D. Kumar, S. K. Maury, V. Srivastava and S. Singh, ACS Omega, 2020, 5, 29854–29863 CrossRef CAS PubMed;
(c) D. M. Yan, Q. Q. Zhao, L. Rao, J. R. Chen and W. J. Xiao, Chem.–Eur. J., 2018, 24, 16895–16901 CrossRef CAS PubMed;
(d) W. Wei, H. Cui, D. Yang, H. Yue, C. He, Y. Zhang and H. Wang, Green Chem., 2017, 19, 5608–5613 RSC;
(e) J. Y. Tao, Y. X. Wang, Q. H. Zhang, K. Ni, T. H. Zhu and K. Zhao, Green Chem., 2022, 24, 4004–4011 RSC;
(f) G. Kumar, G. Bhargava, Y. Kumar and R. Kumar, J. Chem. Sci., 2022, 134, 44 CrossRef CAS.
-
(a) A. D. A. Bartolomeu, R. C. Silva, T. J. Brocksom, T. Noël and K. T. De Oliveira, J. Org. Chem., 2019, 84, 10459–10471 CrossRef CAS PubMed;
(b) A. Das and K. R. Justin Thomas, Green Chem., 2022, 24, 4952–4957 RSC;
(c) M. Bu, C. Cai, F. Gallou and B. H. Lipshutz, Green Chem., 2018, 20, 1233–1237 RSC;
(d) C. Russo, F. Brunelli, G. C. Tron and M. Giustiniano, J. Org. Chem., 2022 DOI:10.1021/acs.joc.2c00805.
-
(a) H. Bin Yang, Y. Z. Zhao, R. Sang and M. Shi, J. Org. Chem., 2014, 79, 3519–3528 CrossRef PubMed;
(b) F. R. Dietz, A. Prechter, H. Gröger and M. R. Heinrich, Tetrahedron Lett., 2011, 52, 655–657 CrossRef CAS;
(c) A. Prechter, H. Gröger and M. R. Heinrich, Org. Biomol. Chem., 2012, 10, 3384–3387 RSC;
(d) C. J. Patil, D. S. Talele, S. P. Talele, P. R. Pohekar and D. S. Kolhe, J. Pharm. Sci. Res., 2019, 11, 2213–2219 CAS;
(e) M. Stasevych, V. Zvarych, V. Lunin, N. Kopak, O. Komarovska-Porokhnyavets, N. G. Deniz, C. Sayil, M. Ozyurek, K. Guclu, M. Vovk and V. Novikov, Monatsh. Chem., 2018, 149, 1111–1119 CrossRef CAS;
(f) A. Budeev, G. Kantin, D. Dar’in and M. Krasavin, Molecules, 2021, 26, 2530 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence