
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Alternative and facile production pathway towards obtaining high surface area PtCo/C intermetallic catalysts for improved PEM fuel cell performance†
Philipp A. Heizmann‡
 ab,
Hien Nguyen‡ac,
Miriam von Holstac,
Andreas Fischbacha,
Mitja Kostelecd,
Francisco Javier Gonzalez Lopezde,
Marjan Beled,
Luka Pavkod,
Tina Đukićd,
Martin Šalaf,
Francisco Ruiz-Zepedad,
Carolin Kloseac,
Matija Gatalode,
Nejc Hodnikd,
Severin Vierrathabc and
Matthias Breitwieser*ac
ab,
Hien Nguyen‡ac,
Miriam von Holstac,
Andreas Fischbacha,
Mitja Kostelecd,
Francisco Javier Gonzalez Lopezde,
Marjan Beled,
Luka Pavkod,
Tina Đukićd,
Martin Šalaf,
Francisco Ruiz-Zepedad,
Carolin Kloseac,
Matija Gatalode,
Nejc Hodnikd,
Severin Vierrathabc and
Matthias Breitwieser*ac
aElectrochemical Energy Systems, IMTEK – Department of Microsystems Engineering, University of Freiburg, Georges-Koehler-Allee 103, 79110 Freiburg, Germany. E-mail: matthias.breitwieser@hahn-schickard.de
bInstitute and FIT – Freiburg Center for Interactive Materials and Bioinspired Technologies, University of Freiburg, Georges-Köhler-Allee 105, 79110 Freiburg, Germany
cHahn-Schickard, Georges-Koehler-Allee 103, 79110 Freiburg, Germany
dDepartment of Materials Chemistry, National Institute of Chemistry, Hajdrihova ulica 19, 1000 Ljubljana, Slovenia
eReCatalyst d.o.o., Hajdrihova ulica 19, Ljubljana, 1000, Slovenia
fDepartment of Analytical Chemistry, National Institute of Chemistry, Hajdrihova ulica 19, 1000 Ljubljana, Slovenia
First published on 6th February 2023
Abstract
The design of catalysts with stable and finely dispersed platinum or platinum alloy nanoparticles on the carbon support is key in controlling the performance of proton exchange membrane (PEM) fuel cells. In the present work, an intermetallic PtCo/C catalyst is synthesized via double-passivation galvanic displacement. TEM and XRD confirm a significantly narrowed particle size distribution for the catalyst particles compared to commercial benchmark catalysts (Umicore PtCo/C). Only about 10% of the mass fraction of PtCo particles show a diameter larger than 8 nm, whereas this is up to or even more than 35% for the reference systems. This directly results in a considerable increase in electrochemically active surface area (96 m2 g−1 vs. >70 m2 g−1), which confirms the more efficient usage of precious catalyst metal in the novel catalyst. Single-cell tests validate this finding by improved PEM fuel cell performance. Reducing the cathode catalyst loading from 0.4 mg cm−2 to 0.25 mg cm−2 resulted in a power density drop at an application-relevant 0.7 V of only 4% for the novel catalyst, compared to the 10% and 20% for the commercial benchmarks reference catalysts.
1 Introduction
On the way to developing and implementing sustainable clean energy technologies to combat climate change and pollution, proton exchange membrane fuel cells (PEMFCs) present themselves as promising technology for power source applications in the automobile and energy industries due to their high power density, low operating temperature and fast refuelling times.1 A major barrier to large-scale commercialization is the high cost of PEMFCs, with the common platinum-based catalyst accounting for a substantial portion of the price. At high production volume (500 k systems/annually), the platinum-based catalyst represents >40% of the total system costs, which will not benefit from the economies of scale.2–4 Thus, while economies of scale in general will be a crucial cost reduction driver, it is also critical to reduce the required amount of noble metals in the PEMFC.Numerous studies showed that PtM/C alloy catalysts (M e.g. Co, Ni, Cu, Fe) exhibit an increased activity for the oxygen reduction reaction (ORR) and thus lead to a remarkable performance improvement of the corresponding membrane electrode assembly (MEA) in PEMFCs compared to pure Pt/C based systems.5–10 Advantageously, the dilution of the particle core with these 3d transition metals simultaneously leads to a reduction in the total amount of noble metal.11 However, the use of these alloying transition metals also comes with two major disadvantages: the higher complexity of the overall catalyst, e.g. regarding the more difficult synthetic access, and the intrinsic thermodynamic instability of the alloy catalysts under acidic conditions.12 One approach to the latter problem is to selectively deplete the transition metal concentration at the particle surface to practically prevent the dissolution while preserving the positive influences of alloy formation (ligand and crystal strain effects) as much as possible. For example, PtCo nanoparticles with Pt-rich shell of ∼3 atomic layers still exhibits significantly improved ORR activity.13,14 The process of Pt-rich shell formation (in other words, depletion of M from the PtM shell) can be accomplished by electrodissolution or by washing in acidified solutions.15–17
In addition to fundamental research focusing on improving the performance and stability of the catalysts, studies on optimization and scalable synthesis routes are as important.17–22 In the last years, some research has tackled this topic. For instance, as part of our recent work we reported a novel double passivation galvanic displacement method for Pt-alloy catalysts with high reproducibility and great flexibility allowing a highly targeted catalyst design, where the chemical composition and loading of the alloy on the carbon support can be tuned very precisely.23 This approach can potentially be applied to a wide range of sacrificial metals M and on a variety of carbon supports while allowing the production of the resulting catalyst on a multigram scale.24 In these studies, the reported synthesized catalysts showed promising electrochemical improvements in both rotating-disk electrode (RDE) and gas-diffusion electrode (GDE) tests compared to commercially available catalysts, including higher specific activities (SA), mass activities (MA) and electrochemical surface areas (ECSA).
In the present study, we take advantage of the promising catalytic activity for the ORR and the high ECSA of the Pt-alloy catalyst based on double passivation galvanic displacement.23–25 We confirm the significantly narrower particle size distribution of the novel PtCo/C catalysts compared to commercial PtCo/C catalysts via transmission scanning electron microscopy (TEM) image analysis and X-ray powder diffraction (XRD). In addition, the effect of the significantly narrowed particle size distribution obtained by the optimized synthesis route on the performance is demonstrated with thin-film rotating disk electrodes (TF-RDEs) and in single-cell MEA validation.
2 Experimental
2.1 Catalyst synthesis
The synthesis of the new experimental PtCo/C catalyst (hereafter referred to as ReCatalyst) was based on the previous works and can be conceptualized into four main steps (Scheme 1).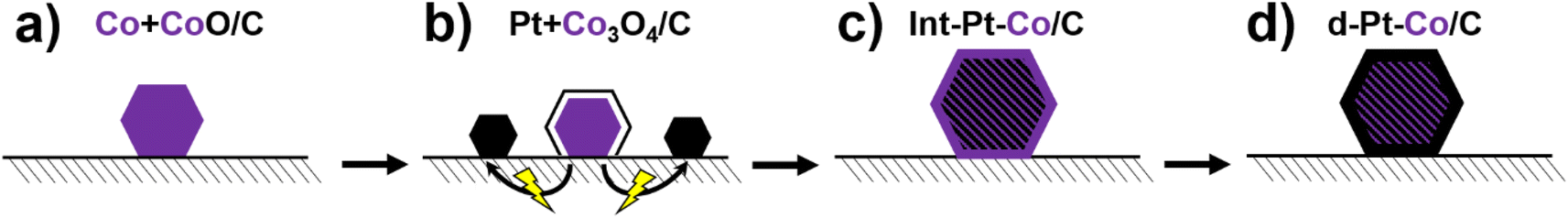 | ||
| Scheme 1 Alternative preparation method of de-alloyed intermetallic PtCo/C catalysts via the 4 step process of using (a) sacrificial Co-based precursor, (b) double passivation galvanic displacement to deposit Pt NPs, (c) thermal annealing to obtain the intermetallic crystal structure as well as (d) de-alloying to obtain the final catalyst material. | ||
2.2 Characterization
Aberration-corrected STEM (Cs-STEM) micrographs were acquired using a JEM-ARM200CF (JEOL Ltd, Cold FEG emitter) at 80 kV acceleration voltage with a convergence angle of 25 mrad.
As a reference electrode (RE), the reversible hydrogen electrode (RHE; HydroFlex, Gaskatel) was used, while the graphite rode electrode was used as a counter electrode. The working electrode (WE) was a 0.196 cm2 glassy carbon disc embedded in Teflon (Pine Instruments). The WE was prepared following the procedure, which was also reported in the previous work:28
- Polishing to a mirror finish with Al2O3 paste (particle size 0.05 μm, Buehler) on a polishing cloth (Buehler).
- Rinsing and ultrasonication (Ultrasound bath Iskra Sonis 4) in Milli-Q water for 5 min.
- Pipetting 20 μl of a freshly prepared water-based catalyst ink (1 mgcatalyst per 1 mlMilli-Q water) on the WE so that the WE is completely covered by the dispersion.
- After the drop had dried, 5 μl of Nafion solution (EelctroChem, 5% aqueous solution) diluted in isopropanol (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 v/v) was added to the WE with the catalyst ink to bind the catalyst thin-film on the glassy carbon electrode.
:50 v/v) was added to the WE with the catalyst ink to bind the catalyst thin-film on the glassy carbon electrode.
After preparing the WE, it was mounted on the rotator (Pine Instruments). The RDE measurements performed in 0.1 M HClO4 are as follows: ORR polarization curves were measured in an oxygen saturated electrolyte with rotation at 1600 rpm between 0.05 and 1.0 VRHE with a scan rate of 20 mV s−1. After the ORR polarization curve measurement, the electrolyte (0.1 M HClO4) was purged with carbon monoxide (CO) under potentiostatic mode (0.05 VRHE) for CO-stripping experiments, followed by Ar to saturate the electrolyte. CO-electrooxidation (“stripping”) was performed using the same potential window (0.05–1.0 VRHE) and scan rate (20 mV s−1) as in ORR polarization curves, but without rotation and in an Ar-saturated electrode.29
Kinetics parameters from RDE experiments were calculated at 0.9 VRHE with Koutecky–Levich equation, as described in literature30,31
| i−1 = ik−1 + id−1 |
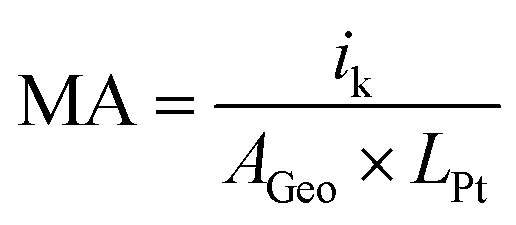
The ECSA was determined by integrating the charge in CO electrooxidation experiments between 0.4 VRHE and 1.0 VRHE, following the approach reported in literature.29,30
2.3 Fabrication of membrane electrode assemblies
The novel PtCo/C catalyst was prepared by ReCatalyst. The commercial PtCo/C catalysts were purchased from Umicore N.V. 3M PFSA ionomer was supplied by 3M and PFSA FS715RFS membranes were supplied by Fumatech BWT GmbH.Anode catalyst inks (2 wt% solids in 1:4 w/w IPA/water) were prepared using Pt/C (45 wt% Pt content, Elyst Pt50 0550, Umicore) and an ionomer-to-carbon ratio (I/C) of 0.7. Three cathode catalyst inks (2 wt% solids in 1:4 w/w IPA/water) were prepared using three different catalysts; PtCo/C (35 wt% Pt content provided by ReCatalyst). As references, two commercial PtCo/C from Umicore were chosen: Elyst Pt50 0690 (41 wt% Pt content) and Elyst Pt30 0690 (27 wt% Pt content), as these are two commercial PtCo/C catalysts reported in the literature with state-of-the-art performance32–34 and are available with comparable metal content to the ReCatalyst catalyst. The membrane electrode assemblies (MEA) with the PtCo/C from ReCatalyst are denoted ReCatalyst. The references MEAs with PtCo/C from Umicore are denoted UM50 for Elyst Pt50 0690 and UM30 for Elyst Pt30 0690. The ionomer-to-carbon (I/C) ratio was adjusted to 0.4 for all cathodes, based on previously published work from our group.35
The catalyst layers were applied onto pristine membranes using an automated ultrasonic spray-coating system (Sonaer Sono-Cell). Anode and cathode catalyst inks were applied onto commercial Fumapem® membrane (725 EW, mechanically reinforced, chemically stabilized, nominal thickness: 15 μm). The desired Pt-loading of all MEAs was 0.1 mg cm−2 for the anode and two different loadings for the cathode: 0.25 mg cm−2 and 0.4 mg cm−2. These loadings were chosen as 0.4 mg cm−2 is a standard Pt loading widely reported in literature and also present in typical commercial MEAs, while 0.25 mg cm−2 is a typical Pt loading envisioned for upcoming heavy-duty applications (e.g. summarized in the article of Cullen et al.).36–40 The loading was controlled by weighing a thin metal pad of 2 cm2 area before and after the spraying with a microbalance (ME36S, Satorius AG), as reported in a previous study.41 The resulting catalyst-coated membranes were sandwiched between two 4 cm2 gas diffusion layers (H14Cx653, Freudenberg). The performances of the 4 cm2 active area MEAs were evaluated using a fuel cell test station (Scribner 850e). All MEAs were tested with the same experimental protocol.
2.4 Characterization of membrane electrode assemblies
The protocol applied to all the MEAs in this work consisted of a break-in procedure followed by voltage recovery (VR). This procedure was shown to be a valuable step for removing sulfate, which improves the electrochemical performance.32,42 After the voltage recovery, polarization measurements were conducted in H2/O2, followed by cyclic voltammograms for the determination of electrochemical surface area (ECSA) and a hydrogen crossover measurement for a proper mass activity assessment. Lastly, polarization measurements in H2/air were performed.The break-in procedure was reported in our previous work.35 The voltage recovery protocol is based on the works of Zhang et al.43 and Kabir et al.32 During the VR, the cells were held under 55 °C, above saturation (198% RH) and ambient pressure. A series of potential cycles between 0.08 V and 0.12 V was applied for 20 s each on the cells. The voltage cycles were repeated 180 times.
The H2/O2 polarization curves (0.25 slpm/1 slpm) were measured under 80 °C, 96% RH and 150 kPaabs. The current density was scanned from 0 mA cm−2 to 125 mA cm−2 in 5 mA cm−2 steps for 5 minutes per point (average of last 5 seconds used). High-frequency resistances (HFRs) were measured at a frequency of 3200 Hz by the fuel cell test station's integrated Frequency Resistance Analyzer (FRA) throughout all polarization characterizations and used to correct for membrane, contact, and electronic resistances, as previously reported in our work.35
The Tafel plots were corrected for the high-frequency resistances (HFR) and the hydrogen crossover current densities following the approach by Neyerlin et al.44 The mass activity is obtained by dividing the current density corrected with hydrogen crossover (i + ix-over) at 0.9 VHFR-free with the cathode Pt-loading of the cell (0.25 mgPt cm−2).
The cyclic voltammograms (CVs) were performed under H2/N2, 35 °C, 95% RH and ambient pressure. The potential was swept from 0.05 to 1.0 V versus RHE at a scan rate of 50 mV s−1. The CVs were repeated 8 times to reach saturation. The cyclic voltammograms of the three samples are provided in the ESI.† We acknowledge that the hydrogen underpotential deposition (Hupd) method for Pt-alloy complicates the quantitative ECSA measurement due to the altered electronic properties of PtCo, as this change affects the adsorption behaviour of hydrogen.45,46 Still, the Hupd method is widely used as a standard in literature for both Pt47 and Pt-alloy catalysts48,49 and is therefore employed in this work. As reported in the literature, the factor used to calculate the ECSA from the Hupd charge was 210 μC cm−2.32,35 The more accurate values for the ECSA are obtained by CO-stripping (see section 2.2.5), as the adsorption of CO is less affected by the altered electronic properties of PtCo.46 However, differences in ECSAs will be discussed in section 3.2.1.
Hydrogen crossover currents were measured via linear sweep voltammetry (LSV) under H2/N2 and under the same conditions as the H2/O2 polarization curves, i.e. 80 °C, 96% RH and 150 kPaabs, to correct the current densities from the H2/O2 polarization curves.
The H2/air polarization curves (0.25 slpm/0.5 slpm) were measured at 80 °C, 96% RH and ambient pressure. This condition ensures no additional effects on the water management induced by the backpressure or the proton conductivity caused by the reduced gas humidity, providing a simple comparison between the catalysts on the MEA level.
The current density was scanned from zero to 250 mA cm−2 in 12.5 mA cm−2 increments with a 1 minute hold at each current step.35 Full polarization curves were obtained by scanning the current density from 375 mA cm−2 to the final current density in 125 mA cm−2 increments with a 3 minute hold per current step.35
3 Results and discussion
3.1 Particle composition and morphology
ICP-OES measurements revealed the Pt:Co compositions of the three catalysts to be Pt2.8Co for Umicore Elyst Pt30 0690 (UM30), Pt2.4Co for Elyst Pt50 0690 (UM50) and Pt2.9Co for PtCo/C synthesized via double-passivation galvanic displacement (ReCatalyst). XRD patterns of all three samples (Fig. 1) confirm the high-crystallinity and bimetallic alloy nature of all PtCo/C catalysts, whereby both are also evidenced by aberration-corrected STEM and STEM-EDX element mapping (Fig. S1 and S2†), respectively. The diffractograms suggest the close similarity of the bulk chemical composition as well as the crystal structure of the as-prepared ReCatalyst and PtCo/C references from Umicore. The diffraction peaks at 2θ = 24–26°, 33–34°, 40.9–41.3°, 47.6–47.9° and 54° correspond to the (001), (110), (111), (200) and (201) planes of intermetallic tetragonal P4/mmm PtCo. The peak at 2θ = 24–26° is superimposed by the (002) plane of the carbon support.50 Compared to standard face-centered cubic (fcc) platinum, the dominant (111) and (200) plane peaks (denoted by the dashed lines, PDF(Pt)#00-004-0802) are shifted to higher 2θ values, indicating a substantial lattice contraction due to the formation of the alloy. Employing Bragg's law, interplanar spacings of 2.25 Å (UM30), 2.20 Å (UM50), and 2.21 Å (ReCatalyst) for the (111) plane were obtained.51 These values are approximately in agreement with d-spacings of PtCo alloys reported in the literature and are slightly shortened compared to the d-spacings reported for pure platinum nanoparticles (∼2.3 Å).52–56 | ||
| Fig. 1 Top: X-ray diffraction patterns of the ReCatalyst (PtCo/C synthesized via galvanic displacement, red) and the two benchmark catalysts Umicore UM30 (blue) and UM50 (black). Bottom: Line patterns of reference materials and their commonly assigned crystal planes. | ||
The observed lattice distance decrease could result from the incorporation of the smaller cobalt atoms (atomic radius of 1.26 Å) in place of the larger platinum atoms (1.36 Å).57,58 The pronounced broadening of these reflections in all three samples suggests a small mean crystallite size. As the width of a diffraction peak is not only influenced by the crystallite size, but also by crystal lattice imperfections such as dislocations, chemical inhomogeneities and residual stress, the simple measurement of the full width at half maximum (FWHM) to calculate specific crystallite size values (e.g. by the often used Scherrer equation59) can be biased by the mentioned effects.60 Still, a decreasing FWHM was observed in order of UM50 (2.0°) < UM30 (2.4°) < ReCatalyst (2.7°) for the (111) plane, suggesting a slightly larger crystallite size for the Umicore references. To obtain more precise information about the particle morphology, additional TEM micrographs over a larger field of view were acquired (Fig. 2, more micrographs can be found in Fig. S3–S5†). The carbon support in all three samples consists of 30–80 nm primary carbon particles, which coalesce into aggregates. These primary carbon particles are characterized by core-filling amorphous and shell-like graphitic carbon (see more resolved HRTEM micrographs in Fig. S6†). The resemblance of the carbon support in all samples, as well as the similarity of the XRD patterns and former findings in their electrochemical behaviour confirms that the Umicore references also contain a high surface area carbon as support material.25 It can be seen that on all samples, the PtCo nanoparticles are finely distributed over the entire carbon surface. From 2D TEM micrographs it is not possible to determine the precise proportion of nanoparticles located inside or outside the carbon pores. As proposed by Harzer et al., however, some nanoparticles can be reliably assigned to be on the outside of the carbon support if the nanoparticle is clearly visible outside the projection of the carbon primary particle. In turn, it cannot be resolved whether a particle sits on the surface or in a carbon pore if that nanoparticle is completely enclosed by the projection of the primary carbon particle.61
 | ||
| Fig. 2 Representative TEM micrographs of the three catalysts UM50 (black), UM30 (blue) and ReCatalyst (red). | ||
With this counting method, it can be estimated that more nanoparticles are located on the carbon surface for the ReCatalyst compared to the Umicore catalysts, as notably more particles are located outside the carbon projections. This is supported by TEM tilt series (ESI movies 1–3†), where the rotation of the respective carbon particles over a wide tilt range (typically 140–144°) allows a better determination of the nanoparticle position.
One of the main advantages of the developed alternative synthesis route is the ability to obtain a narrower particle size distribution, which in general could also be considered as advantageous towards the reduction of excessive Ostwald ripening of the particles upon aging.62 Fig. 3 shows a violin plot with the particle size distribution obtained by measuring the diameters of the nanoparticles in the 2D TEM micrographs to enable a facile comparison of the PSDs of all three catalysts.
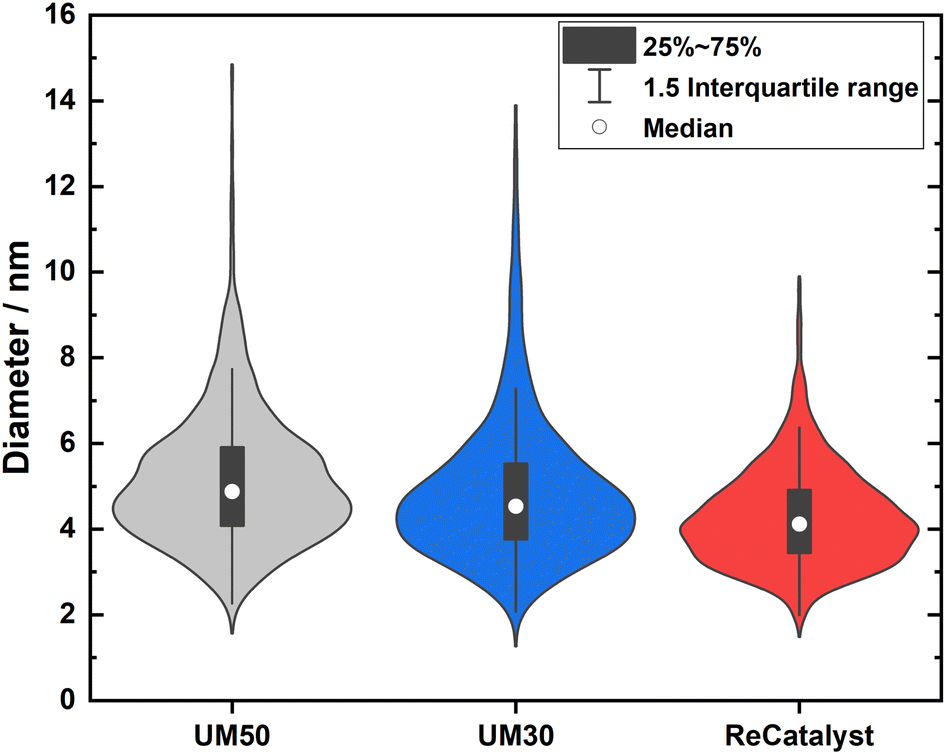 | ||
| Fig. 3 Violin plots of the particle size distribution for the three PtCo/C catalysts. | ||
The vast majority of the measured nanoparticles for all three samples feature a diameter of 3 to 6 nm. The number-weighted diameter was determined to be 4.8 nm for UM50, 4.5 nm for UM30 and 4.1 nm for ReCatalyst, while the surface normalized diameters were calculated to be 6.3 nm for UM50, 6.1 nm for UM30 and 5.1 nm for ReCatalyst, respectively.61 In comparison, a diameter of ∼4.4 nm was reported for the Umicore Elyst Pt 50 0670 variant.63,64 Following Schulenburgs' et al. approach for more realistic determinations of the surface area by TEM micrograph evaluation, we approximated the TEM derived surface areas of the samples to be 68 m2 gPt−1 for UM50, 75 m2 gPt−1 for UM30 and 86 m2 gPt−1 for ReCatalyst assuming spherical particles.45 It is important to note that although the majority of nanoparticles have comparable diameters, it is the significantly smaller number of particularly large (>8 nm) nanoparticles in the ReCatalyst sample that plays a crucial role in the utilization of the employed precious metal, which is directly quantified by the ECSA (see Table 1). While the surface area of the ReCatalyst catalyst from purely geometric considerations is already 26% and 17% higher compared to UM30 and UM50, respectively, these differences are substantially greater for the electrochemical surface area and are discussed in section 3.3. Both size and geometry are fundamentally determinating the active sites of the particles (besides the chemical composition) and thus influence the critical selectivity and stability of the catalyst.65 This is called the particle size effect where smaller particles exhibited lower specific ORR activity and lower stability.66–68 To not underutilize precious metal in the bulk of the particle and risk mass activity losses, it is essential to ensure large enough diameters that allow electrochemical activity and sufficient stability of the particles, but at the same time show a high surface-to-volume ratio for electrochemical surface maximization. These relationships have been extensively modelled and experimentally investigated for the simpler Pt/C system, where Pt nanoparticles with diameters of ∼2–4 nm have been determined to feature optimal mass activity.69–72 Even though particle size studies of PtCo nanoparticles are not similarly extensive and are more difficult to obtain due to the alloy nature of nanoparticles with varying intraparticle elemental (e.g. Pt/Co ratio) and structural (e.g. degree of ordering) compositions, similar trends as in pure Pt-particles seem to apply.19,73,74 E.g. according to Wang et al. the maximum mass activity for Pt3Co nanoparticles is found at particle sizes of ∼4.5 nm, although it must be pointed out that this activity also depends strongly on numerous other parameters (e.g. element composition, particle shape).75,76 In Fig. 4, the highest mass fraction for ReCatalyst is found at 4–5 nm, which lies in this optimal range for catalyst activity. For the commercial catalysts, the distribution is slightly shifted to 5–6 nm. This trend is in line with the improvement in RDE derived ECSA and mass activity of the ReCatalyst material compared to the UM references (see below, Fig. S7 and S8 and Table S1†).
| Sample | Pt loading in mg cm−2 | CL thickness in μm | ECSA in m2 g−1 Hupd | Roughness factor in cmPt2 cmMEA−2 (with ECSA from Hupd) | ECSA in m2 g−1 TEM | ECSA in m2 g−1 TF-RDE |
|---|---|---|---|---|---|---|
| UM50 | 0.4 | 8 | 31 | 124 | 68 | 59 |
| 0.25 | 5 | 23 | 58 | |||
| ReCatalyst | 0.4 | 17 | 38 | 152 | 86 | 96 |
| 0.25 | 10 | 45 | 113 | |||
| UM30 | 0.4 | 32 | 37 | 148 | 75 | 70 |
| 0.25 | 18 | 43 | 108 |
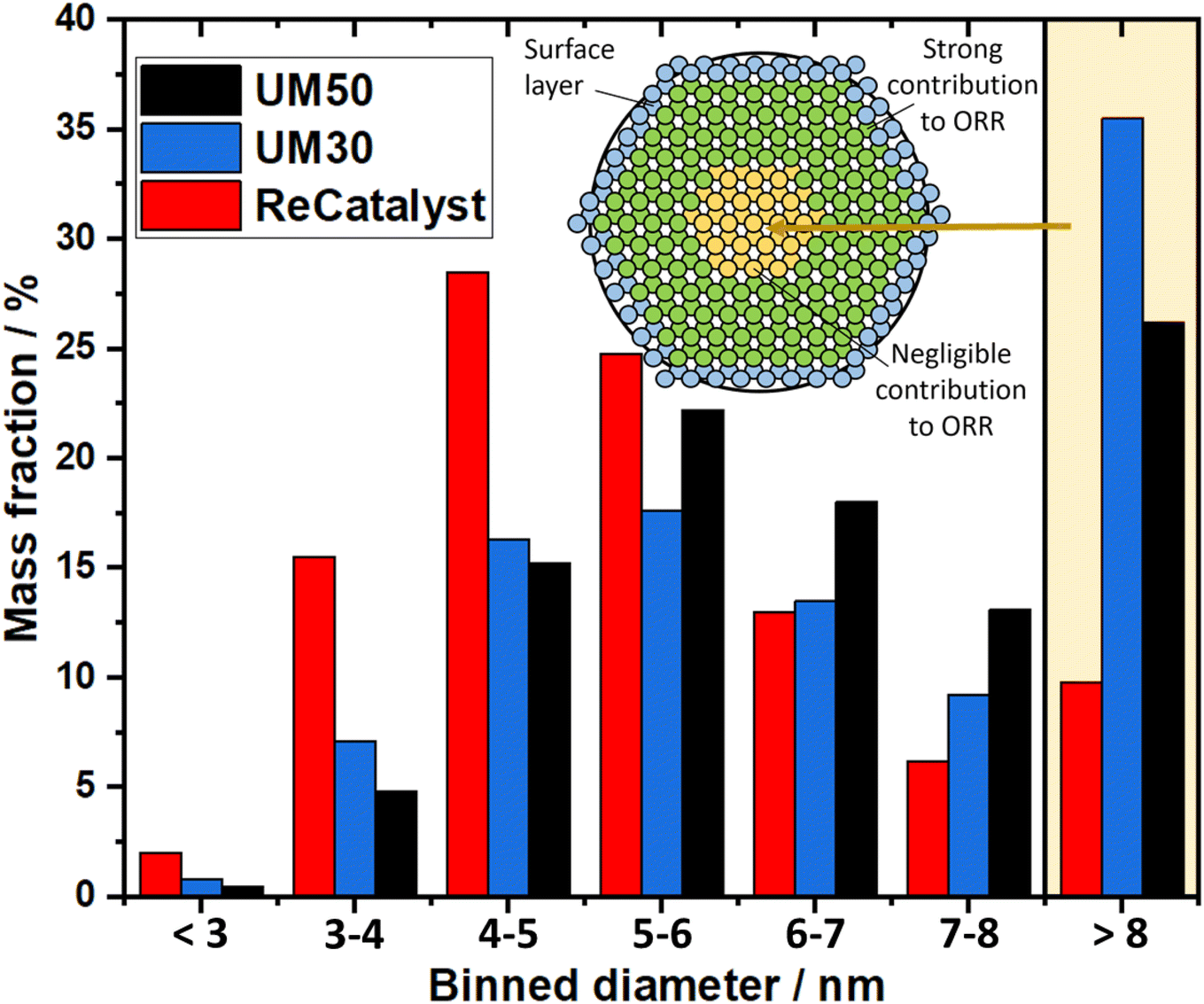 | ||
| Fig. 4 Mass distribution of the three PtCo/C catalysts. | ||
A second important factor influencing the catalyst performance is the loss of active catalyst material in large catalyst particles: For assumed solid spherical particles with a diameter of 6 nm or more, more material is found inside the particle than at the surface marking the lower limit above which the proportion of inside and outside material inflects, due to spheres having the lowest surface to volume ratio. In nature, nanoparticles are not perfectly spherical and not only the first atomic surface layer but also the subsequent atomic layers below have a crucial influence on the reaction, as shown with DFT calculations by Patrick et al., albeit with decreasing significance.14 It can be expected that above a particle size of ∼8 nm the majority of the core atoms play a negligible role in the oxygen reduction reaction and thus the contribution of the fractional mass activity of these particles to the total mass activity of the catalyst is almost zero. Applying this consideration to the analysis of our TEM investigation summarized in the binned histogram in Fig. 4 (binned values in Table S2†), we find a significantly reduced mass ratio of particles >8 nm for the ReCatalyst (∼10%) vs. the Umicore references (∼26% and 35% for Umicore 50 and 30, respectively), a consequence of the strong weighting of large particles due to the cubic relationship between the diameter and volume of spheres and, consequently, the mass of PtCo particles. This finding clearly confirms the advantage of the alternative fabrication procedure given the sharper particle size distribution for the ReCatalyst material.23–25 Nevertheless, one can assume considerable benefits are still accessible with further optimization of the particle size distribution if a complete elimination of particles with diameters >8 nm is achieved. The mass fraction of particles <3 nm is very low in all three samples (below ∼2.5% for all samples). Since very small particles feature a strong curvature, they are prone to faster decomposition due to increased surface energy and are therefore unfavourable for catalyst long-term durability.65,77 We also acknowledge, however, that the detection of nanoparticles in the sub-nm range is difficult due to the acquisition of TEM micrographs with an intermediate field of view (typically 50–200 nm) with associated reduced pixel resolution, which could be circumvented by improved imaging equipment in future work.
3.2 Electrochemical performance
Given the different size distributions of the three catalysts, the following sections show the concomitant ECSA (section 3.2.1) and the samples' electrochemical performance at the MEA level (section 3.2.2).At the same Pt loading on the cathode as both catalyst powder and as an MEA and independent from the measurement technique (MEA, TEM, RF-RDE), the ECSA of ReCatalyst is the highest compared to those of the reference samples, especially compared to that of UM50, i.e. ReCatalyst has the highest roughness factor, which is the product of the ECSA and the CL loading.78 The higher roughness factor of the ReCatalyst PtCo/C correlates well with its narrower particle size distribution, especially compared to UM50 (Fig. 3).
Theoretically, the ECSA should not be affected by the Pt loading of the cathode CL. However, the ECSA was practically affected by the loading, which is related to other properties, e.g. the CL thickness and utilization. For instance, the ECSA of the CL with UM50 increased significantly (by 35%) with higher loading (and thickness), while a reverse trend was observed for ReCatalyst and UM30: the ECSA decreased (by less than 20%) with higher loading. It is important to note that the thickness of the CL with UM50 is increased to less than 10 μm (from 0.25 mgPt cm−2 to 0.4 mgPt cm−2), while the CL thicknesses were greater than 17 μm for ReCatalyst with 0.4 mgPt cm−2 and UM30 with both loadings (Table 1 and Fig. S12†). Based on these observations, CL thicknesses higher than 10 μm could have detrimental impacts on the ECSA of the MEAs. Further information obtained from the in situ CVs is shown in Fig. S9.†
At similar thicknesses (8–10 μm), ReCatalyst PtCo/C has a higher mass activity (146 A g−1) than that of UM50 (138 A g−1). This trend aligns with the ECSA measurements and TF-RDE (ESI Table S1†) for the two catalysts. The higher mass activity can be attributed to the narrower particle size distribution of ReCatalyst especially compared to UM50 (Fig. 3). However, also at similar CL thicknesses (17–18 μm), ReCatalyst PtCo/C has a lower mass activity (105 A g−1) than that of UM30 PtCo/C (173 A g−1). The trend in mass activity is again in agreement with that of the ECSAHupd (ReCatalyst < UM30) (Table 2), however, it does not match the results obtained by TF-RDE (Table S1†), and direct comparisons between Hupd derived ECSAs and TF-RDE derived ECSAs should be made with caution for the reasons stated above.
| Sample | Pt loading in mg cm−2 | CL thickness in μm | Mass activity in A g−1 | ECSA (Hupd) in m2 g−1 |
|---|---|---|---|---|
| UM50 | 0.4 | 8 | 138 | 31 |
| 0.25 | 5 | 137 | 23 | |
| ReCatalyst | 0.4 | 17 | 105 | 38 |
| 0.25 | 10 | 146 | 45 | |
| UM30 | 0.4 | 32 | 121 | 37 |
| 0.25 | 18 | 173 | 43 |
The mass activity of ReCatalyst PtCo/C is slightly lower than that of UM30 in MEA configuration. This might be linked to the fact that ReCatalyst PtCo/C features a higher fraction of exterior PtCo on carbon than the references (as discussed earlier), which increases the chance for ionomer-induced poisoning for ReCatalyst catalyst vs. UM30 and was reported to lower mass activity.79–82 Further, the lower PtCo content in UM30 (with a lower fraction of exterior PtCo on carbon) might help to reduce the exposure of the catalyst to ionomer.83
Fig. 5 shows the polarization curves of the MEAs with the three catalysts under H2/air, 80 °C, 96% RH and ambient pressure. In particular, at high current densities (>750 mA cm−2), the MEA performance with ReCatalyst is superior at the same Pt loadings. This result is most likely linked to the significantly higher roughness factor of ReCatalyst (Table 1) and probably also to a higher fraction of exterior PtCo particles on carbon (as discussed earlier).
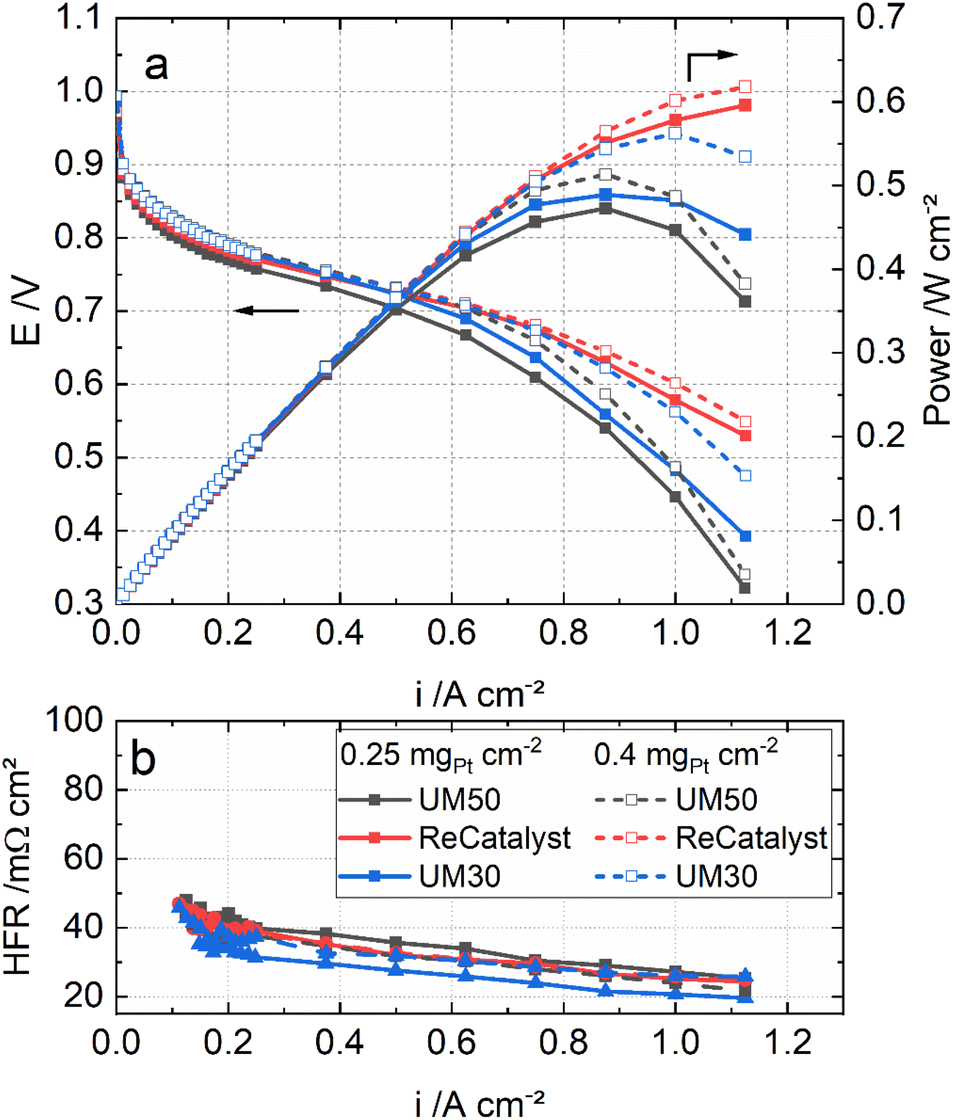 | ||
| Fig. 5 Polarization curves (a) and the high-frequency resistances (b) of the MEAs with UM50, ReCatalyst and UM30 at 0.25 mgPt cm−2 loading and 0.4 mgPt cm−2 loading under H2/air, 80 °C, 96% RH and ambient pressure. | ||
Fig. 5 also compares the MEAs with the three catalysts in two different cathode loadings: 0.4 mgPt cm−2 and 0.25 mgPt cm−2. It can be seen that the performance of the ReCatalyst MEA is not affected much by the reduced loading. The MEA with 0.25 mgPt cm−2 ReCatalyst PtCo/C still outperforms that with 0.4 mgPt cm−2 UM50 in both conditions. This result indicates that the higher peak performance of the ReCatalyst MEA (0.25 mgPt cm−2) is mainly attributed to the greater ECSA and the related roughness factor in combination with a possibly higher fraction of exterior PtCo on carbon than the references. These factors improve the oxygen assessment to the Pt surface at high current densities.4,78 Further, as the catalyst layer thicknesses of UM50 at 0.4 mgPt cm−2 and ReCatalyst at 0.25 mgPt cm−2 are both approx. 10 μm (Table 1), the performance improvement at high current densities can be considered independent from the catalyst layer thickness. This is reflected in the cell metrics: The performance of the ReCatalyst-MEA was reduced only by 4% at 0.7 V and peak power density (PPD) when the Pt-loading was reduced from 0.4 to 0.25 mgPt cm−2. The performance at 0.7 V was reduced by 20%, while the peak power density was reduced by 8% with UM50. The performance at 0.7 V was reduced by 10% and 14% at PPD with UM30.
4 Conclusion and outlook
Based on an alternative catalyst synthesis approach via the double passivation galvanic displacement, we have shown that a significant improvement in the particle size distribution can be obtained compared to commercial benchmark PtCo catalysts. TEM and XRD characterization confirmed that our optimized ReCatalyst PtCo/C catalyst features a considerably lower mass fraction of particles >8 nm vs. the commercial references. In addition, higher fractions of nanoparticles are located on the carbon surface for the ReCatalyst compared to the Umicore catalysts. These two features enable more effective usage of the available active catalyst material, which is reflected in superior single-cell performance in particular at reduced Pt-loading of 0.25 mg cm−2 at the cathode. Future works should further optimize this new catalyst synthesis and MEAs preparation, also in relation to an extended range of cathode loadings (e.g. low-loaded cathodes with <0.1 mg cm−2 Pt loading for light-duty applications). In particular, screening different carbon support pore sizes and closing the herein observed gap between RDE and MEA mass activity values to finally exploit the full potential of those novel catalysts. In further studies, the long-term stabilities of the electrocatalysts and its contribution to the overall MEA durability would also be of vital research importance.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors acknowledge funding from the Federal Ministry of Education and Research of Germany (BMBF) within the project FC-CAT (Grant 08SF0579B) and the Federal Ministry for Economic Affairs and Energy (BMWi) within the project DirectStack (Grant 03ETB024D). We would also like to thank the Slovenian research agency (ARRS) programs P2-0393, P1-0034; the projects NC-0007, NC-0016 and N2-0106; NATO Science for Peace and Security Program under Grant G5729; and European Research Council (ERC) Starting Grant 123STABLE (Grant agreement ID: 852208) and Proof of Concept Grant StableCat (Grant agreement ID: 966654) for funding the study. The authors would like to thank Ralf Thomann for valuable discussions concerning TEM imaging. The authors would like to acknowledge Mark Muggli from 3M for providing PFSA ionomer and Fumatech for providing PFSA membrane.References
- M. K. Debe, Nature, 2012, 486(7401), 43–51 CrossRef CAS PubMed.
- A. Wilson, G. Kleen and D. Papageorgopoulos, DOE Fuel Cell Technologies Office Record 17007: Fuel Cell System Cost – 2017, 2017 Search PubMed.
- C. S. Gittleman, A. Kongkanand, D. Masten and W. Gu, Curr. Opin. Electrochem., 2019, 18, 81–89 CrossRef CAS.
- J. Fan, M. Chen, Z. Zhao, Z. Zhang, S. Ye, S. Xu, H. Wang and H. Li, Nat. Energy, 2021, 6(5), 475–486 CrossRef CAS.
- D. Wang, H. L. Xin, R. Hovden, H. Wang, Y. Yu, D. A. Muller, F. J. DiSalvo and H. D. Abruña, Nat. Mater., 2013, 12(1), 81–87 CrossRef CAS PubMed.
- L. Bu, S. Guo, X. Zhang, X. Shen, D. Su, G. Lu, X. Zhu, J. Yao, J. Guo and X. Huang, Nat. Commun., 2016, 7, 11850 CrossRef CAS PubMed.
- D. S. Choi, A. W. Robertson, J. H. Warner, S. O. Kim and H. Kim, Adv. Mater., 2016, 28(33), 7115–7122 CrossRef CAS PubMed.
- S. B. Barim, S. E. Bozbag, B. Deljoo, M. Aindow and C. Erkey, Fuel Cells, 2020, 20(3), 285–299 CrossRef CAS.
- P. Strasser, S. Koh, T. Anniyev, J. Greeley, K. More, C. Yu, Z. Liu, S. Kaya, D. Nordlund, H. Ogasawara, M. F. Toney and A. Nilsson, Nat. Chem., 2010, 2(6), 454–460 CrossRef CAS PubMed.
- X. Cai, R. Lin, D. Shen and Y. Zhu, ACS Appl. Mater. Interfaces, 2019, 11(33), 29689–29697 CrossRef CAS PubMed.
- V. R. Stamenkovic, B. Fowler, B. S. Mun, G. Wang, P. N. Ross, C. A. Lucas and N. M. Marković, Science, 2007, 315(5811), 493–497 CrossRef CAS PubMed.
- M. Pourbaix, Atlas of Electrochemical Equilibria in Aqueous Solutions, NACE International, 1974 Search PubMed.
- C. E. Carlton, S. Chen, P. J. Ferreira, L. F. Allard and Y. Shao-Horn, J. Phys. Chem. Lett., 2012, 3(2), 161–166 CrossRef CAS.
- B. Patrick, H. C. Ham, Y. Shao-Horn, L. F. Allard, G. S. Hwang and P. J. Ferreira, Chem. Mater., 2013, 25(4), 530–535 CrossRef CAS.
- S. Koh and P. Strasser, J. Am. Chem. Soc., 2007, 129(42), 12624–12625 CrossRef CAS PubMed.
- R. Srivastava, P. Mani, N. Hahn and P. Strasser, Angew. Chem., Int. Ed. Engl., 2007, 46(47), 8988–8991 CrossRef PubMed.
- M. Gatalo, L. Moriau, U. Petek, F. Ruiz-Zepeda, M. Šala, M. Grom, T. Galun, P. Jovanovič, A. Pavlišič, M. Bele, N. Hodnik and M. Gaberšček, Electrochim. Acta, 2019, 306, 377–386 CrossRef CAS.
- M. Gatalo, P. Jovanovič, U. Petek, M. Šala, V. S. Šelih, F. Ruiz-Zepeda, M. Bele, N. Hodnik and M. Gaberšček, ACS Appl. Energy Mater., 2019, 2(5), 3131–3141 CrossRef CAS.
- C. Zalitis, A. Kucernak, X. Lin and J. Sharman, ACS Catal., 2020, 10(7), 4361–4376 CrossRef CAS.
- F. Xiao, X. Qin, M. Xu, S. Zhu, L. Zhang, Y. Hong, S.-I. Choi, Q. Chang, Y. Xu, X. Pan and M. Shao, ACS Catal., 2019, 9(12), 11189–11198 CrossRef CAS.
- M. Ko, E. Padgett, V. Yarlagadda, A. Kongkanand and D. A. Muller, J. Electrochem. Soc., 2021, 168(2), 24512 CrossRef CAS.
- Y. Xiong, Y. Yang, H. Joress, E. Padgett, U. Gupta, V. Yarlagadda, D. N. Agyeman-Budu, X. Huang, T. E. Moylan, R. Zeng, A. Kongkanand, F. A. Escobedo, J. D. Brock, F. J. DiSalvo, D. A. Muller and H. D. Abruña, Proc. Natl. Acad. Sci. U. S. A., 2019, 116(6), 1974–1983 CrossRef CAS PubMed.
- M. Gatalo, M. Bele, F. Ruiz-Zepeda, E. Šest, M. Šala, A. R. Kamšek, N. Maselj, T. Galun, P. Jovanovič, N. Hodnik and M. Gaberšček, Angew. Chem., Int. Ed. Engl., 2019, 58(38), 13266–13270 CrossRef CAS PubMed.
- L. Pavko, M. Gatalo, G. Križan, J. Križan, K. Ehelebe, F. Ruiz-Zepeda, M. Šala, G. Dražić, M. Geuß, P. Kaiser, M. Bele, M. Kostelec, T. Đukić, N. van de Velde, I. Jerman, S. Cherevko, N. Hodnik, B. Genorio and M. Gaberšček, ACS Appl. Energy Mater., 2021, 4(12), 13819–13829 CrossRef CAS PubMed.
- T. Đukić, L. J. Moriau, L. Pavko, M. Kostelec, M. Prokop, F. Ruiz-Zepeda, M. Šala, G. Dražić, M. Gatalo and N. Hodnik, ACS Catal., 2022, 12(1), 101–115 CrossRef PubMed.
- D. Myers, N. Kariuki, R. Ahluwalia, X. Wang and J.-K. Peng, Rationally Designed Catalyst Layers for PEMFC Performance Optimization, 2015 Search PubMed.
- A. Kongkanand, R. Leach, D. Papageorgopoulos, G. Kleen, P. Strasser, R. O'Malley, Y. Shao-Horn, D. E. Ramaker and S. Mukerjee, High-Activity Dealloyed Catalysts, 2014, DOI:10.2172/1262711.
- N. Maselj, M. Gatalo, F. Ruiz-Zepeda, A. Kregar, P. Jovanovič, N. Hodnik and M. Gaberšček, J. Electrochem. Soc., 2020, 167(11), 114506 CrossRef CAS.
- M. Gatalo, A. Martinez Bonastre, L.-J. Moriau, H. Burdett, F. Ruiz-Zepeda, E. Hughes, A. Hodgkinson, M. Šala, L. Pavko, M. Bele, N. Hodnik, J. Sharman and M. Gaberšček, ACS Appl. Energy Mater., 2022, 5, 8862–8877 CrossRef CAS PubMed.
- K. Mayrhofer, D. Strmcnik, B. B. Blizanac, V. Stamenkovic, M. Arenz and N. M. Markovic, Electrochim. Acta, 2008, 53(7), 3181–3188 CrossRef CAS.
- A. J. Bard and L. R. Faulkner, Electrochemical methods: fundamentals and applications, Wiley, Hoboken, NJ, 2001 Search PubMed.
- S. Kabir, D. J. Myers, N. Kariuki, J. Park, G. Wang, A. Baker, N. Macauley, R. Mukundan, K. L. More and K. C. Neyerlin, ACS Appl. Mater. Interfaces, 2019, 11(48), 45016–45030 CrossRef CAS PubMed.
- T. van Cleve, G. Wang, M. Mooney, C. F. Cetinbas, N. Kariuki, J. Park, A. Farghaly, D. Myers and K. C. Neyerlin, J. Power Sources, 2021, 482, 228889 CrossRef CAS.
- Y. Garsany, R. W. Atkinson, B. D. Gould and K. E. Swider-Lyons, J. Power Sources, 2018, 408(1), 38–45 CrossRef CAS.
- H. T. T. Nguyen, F. Lombeck, C. Schwarz, P. A. Heizmann, M. Adamski, H.-F. Lee, B. Britton, S. Holdcroft, S. Vierrath and M. Breitwieser, Sustainable Energy Fuels, 2021,(5), 3687–3699 RSC.
- D. A. Cullen, K. C. Neyerlin, R. K. Ahluwalia, R. Mukundan, K. L. More, R. L. Borup, A. Z. Weber, D. J. Myers and A. Kusoglu, Nat. Energy, 2021, 2, 1 Search PubMed.
- Z. Xie, X. Zhao, M. Adachi, Z. Shi, T. Mashio, A. Ohma, K. Shinohara, S. Holdcroft and T. Navessin, Energy Environ. Sci., 2008, 1(1), 184 RSC.
- T. Soboleva, K. Malek, Z. Xie, T. Navessin and S. Holdcroft, ACS Appl. Mater. Interfaces, 2011, 3(6), 1827–1837 CrossRef CAS PubMed.
- J. Peron, A. Mani, X. Zhao, D. Edwards, M. Adachi, T. Soboleva, Z. Shi, Z. Xie, T. Navessin and S. Holdcroft, J. Membr. Sci., 2010, 356(1–2), 44–51 CrossRef CAS.
- T. Soboleva, X. Zhao, K. Malek, Z. Xie, T. Navessin and S. Holdcroft, ACS Appl. Mater. Interfaces, 2010, 2(2), 375–384 CrossRef CAS PubMed.
- C. Klose, T. Saatkamp, A. Münchinger, L. Bohn, G. Titvinidze, M. Breitwieser, K.-D. Kreuer and S. Vierrath, Adv. Energy Mater., 2020, 10(14), 1903995 CrossRef CAS.
- J. Zhang, B. A. Litteer, F. D. Coms and R. Makharia, J. Electrochem. Soc., 2012, 159(7), F287–F293 CrossRef CAS.
- J. Zhang, L. Paine, A. Nayar and R. Makharia, Methods and processes to recover voltage loss of PEM fuel cell stack, US Pat.2011/0195324 A1, 2010 Search PubMed.
- K. C. Neyerlin, W. Gu, J. Jorne, A. Clark and H. A. Gasteiger, J. Electrochem. Soc., 2007, 154(2), B279 CrossRef CAS.
- H. Schulenburg, J. Durst, E. Müller, A. Wokaun and G. G. Scherer, J. Electroanal. Chem., 2010, 642(1), 52–60 CrossRef CAS.
- D. F. van der Vliet, C. Wang, D. Li, A. P. Paulikas, J. Greeley, R. B. Rankin, D. Strmcnik, D. Tripkovic, N. M. Markovic and V. R. Stamenkovic, Angew. Chem., 2012, 124(13), 3193–3196 CrossRef.
- H. A. Gasteiger, S. S. Kocha, B. Sompalli and F. T. Wagner, Appl. Catal., B, 2005, 56(1–2), 9–35 CrossRef CAS.
- T. Takahashi, T. Ikeda, K. Murata, O. Hotaka, S. Hasegawa, Y. Tachikawa, M. Nishihara, J. Matsuda, T. Kitahara, S. M. Lyth, A. Hayashi and K. Sasaki, J. Electrochem. Soc., 2022, 169(4), 44523 CrossRef.
- S. Rudi, C. Cui, L. Gan and P. Strasser, Electrocatalysis, 2014, 5(4), 408–418 CrossRef CAS.
- T. Kim and B. N. Popov, Int. J. Hydrogen Energy, 2016, 41(3), 1828–1836 CrossRef CAS.
- W. H. Bragg and W. L. Bragg, Proc. R. Soc. London, Ser. A, 1913,(88), 428–438 CAS.
- S. Dai, Y. You, S. Zhang, W. Cai, M. Xu, L. Xie, R. Wu, G. W. Graham and X. Pan, Nat. Commun., 2017, 8(1), 204 CrossRef PubMed.
- K. Miyazawa, M. Yoshitake and Y. Tanaka, J. Nanopart. Res., 2017, 19(6), F57 CrossRef.
- J. Y. Song, E.-Y. Kwon and B. S. Kim, Bioprocess Biosyst. Eng., 2010, 33(1), 159–164 CrossRef CAS PubMed.
- C. Zhang, R. Zhang, X. Gao, C. Cheng, L. Hou, X. Li and W. Chen, ACS Omega, 2018, 3(1), 96–105 CrossRef CAS PubMed.
- N. V. Long, M. Ohtaki, M. Uchida, R. Jalem, H. Hirata, N. D. Chien and M. Nogami, J. Colloid Interface Sci., 2011, 359(2), 339–350 CrossRef PubMed.
- S.-I. Choi, S.-U. Lee, W. Y. Kim, R. Choi, K. Hong, K. M. Nam, S. W. Han and J. T. Park, ACS Appl. Mater. Interfaces, 2012, 4(11), 6228–6234 CrossRef CAS PubMed.
- B. Cordero, V. Gómez, A. E. Platero-Prats, M. Revés, J. Echeverría, E. Cremades, F. Barragán and S. Alvarez, Dalton Trans., 2008,(21), 2832–2838 RSC.
- P. Scherrer, Nachr. Akad. Wiss. Goettingen, Math.-Phys. Kl., 2, 1918, 98–100 Search PubMed.
- J. I. Langford and A. Wilson, J. Appl. Crystallogr., 1978,(11), 102–113 CrossRef CAS.
- G. S. Harzer, A. Orfanidi, H. El-Sayed, P. Madkikar and H. A. Gasteiger, J. Electrochem. Soc., 2018, 165(10), F770–F779 CrossRef CAS.
- M. Watanabe, H. Yano, H. Uchida and D. A. Tryk, J. Electroanal. Chem., 2018, 819, 359–364 CrossRef CAS.
- R. K. Ahluwalia, D. D. Papadias, N. Kariuki, J.-K. Peng, X. Wang, Y. Tsai, D. G. Graczyk and D. J. Myers, J. Electrochem. Soc., 2018,(265), F3024–F3035 CrossRef CAS.
- D. D. Papadias, R. K. Ahluwalia, N. Kariuki, D. J. Myers, K. L. More, D. A. Cullen, B. Sneed, K. C. Neyerlin, R. Mukundan and R. L. Borup, J. Electrochem. Soc., 2018,(165), F3166–F3177 CrossRef CAS.
- J. C. Meier, C. Galeano, I. Katsounaros, J. Witte, H. J. Bongard, A. A. Topalov, C. Baldizzone, S. Mezzavilla, F. Schüth and K. J. J. Mayrhofer, Beilstein J. Nanotechnol., 2014, 5, 44–67 CrossRef PubMed.
- M. Nesselberger, S. Ashton, J. C. Meier, I. Katsounaros, K. J. J. Mayrhofer and M. Arenz, J. Am. Chem. Soc., 2011, 133(43), 17428–17433 CrossRef CAS PubMed.
- F. J. Perez-Alonso, D. N. McCarthy, A. Nierhoff, P. Hernandez-Fernandez, C. Strebel, I. E. L. Stephens, J. H. Nielsen and I. Chorkendorff, Angew. Chem., Int. Ed. Engl., 2012, 51(19), 4641–4643 CrossRef CAS PubMed.
- P. Jovanovič, U. Petek, N. Hodnik, F. Ruiz-Zepeda, M. Gatalo, M. Šala, V. S. Šelih, T. P. Fellinger and M. Gaberšček, Phys. Chem. Chem. Phys., 2017, 19(32), 21446–21452 RSC.
- Z. Xu, H. Zhang, H. Zhong, Q. Lu, Y. Wang and D. Su, Appl. Catal., B, 2012, 111–112, 264–270 CrossRef CAS.
- M. Shao, A. Peles and K. Shoemaker, Nano Lett., 2011, 11(9), 3714–3719 CrossRef CAS PubMed.
- K. Kinoshita, J. Electrochem. Soc., 1990,(137), 845 CrossRef CAS.
- G. A. Tritsaris, J. Greeley, J. Rossmeisl and J. K. Nørskov, Catal. Lett., 2011, 141(7), 909–913 CrossRef CAS.
- K. Matsutani, K. Hayakawa and T. Tada, Platinum Met. Rev., 2010, 54(4), 223–232 CrossRef CAS.
- M. Gummalla, S. Ball, D. Condit, S. Rasouli, K. Yu, P. Ferreira, D. Myers and Z. Yang, Catalysts, 2015, 5(2), 926–948 CrossRef CAS.
- C. Wang, D. van der Vliet, K.-C. Chang, H. You, D. Strmcnik, J. A. Schlueter, N. M. Markovic and V. R. Stamenkovic, J. Phys. Chem. C, 2009, 113(45), 19365–19368 CrossRef CAS.
- C. Wang, G. Wang, D. van der Vliet, K.-C. Chang, N. M. Markovic and V. R. Stamenkovic, Phys. Chem. Chem. Phys., 2010, 12(26), 6933–6939 RSC.
- W. Sheng, S. Chen, E. Vescovo and Y. Shao-Horn, J. Electrochem. Soc., 2011,(159), B59 Search PubMed.
- A. Kongkanand and M. F. Mathias, J. Phys. Chem. Lett., 2016, 7(7), 1127–1137 CrossRef CAS PubMed.
- K. Kodama, A. Shinohara, N. Hasegawa, K. Shinozaki, R. Jinnouchi, T. Suzuki, T. Hatanaka and Y. Morimoto, J. Electrochem. Soc., 2014, 161(5), F649–F652 CrossRef CAS.
- R. Jinnouchi, K. Kudo, K. Kodama, N. Kitano, T. Suzuki, S. Minami, K. Shinozaki, N. Hasegawa and A. Shinohara, Nat. Commun., 2021, 12(1), 4956 CrossRef CAS PubMed.
- K. Kodama, K. Motobayashi, A. Shinohara, N. Hasegawa, K. Kudo, R. Jinnouchi, M. Osawa and Y. Morimoto, ACS Catal., 2018, 8(1), 694–700 CrossRef CAS.
- K. Kodama, T. Nagai, A. Kuwaki, R. Jinnouchi and Y. Morimoto, Nat. Nanotechnol., 2021, 16(2), 140–147 CrossRef CAS PubMed.
- T. Lazaridis and H. A. Gasteiger, J. Electrochem. Soc., 2021, 168(11), 114517 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ra07780a |
| ‡ Equal contribution. |
| This journal is © The Royal Society of Chemistry 2023 |