DOI:
10.1039/D2RA07671C
(Paper)
RSC Adv., 2023,
13, 7293-7299
NiII and CuII complexes of a salen ligand bearing ferrocenes in its secondary coordination sphere†
Received
2nd December 2022
, Accepted 24th February 2023
First published on 6th March 2023
Abstract
Herein, we report the synthesis, spectroscopic characterization and electrochemical investigation of the NiII and CuII complexes of a novel Sal ligand bearing two ferrocene moieties attached at its diimine linker, M(Sal)Fc. The electronic spectra of M(Sal)Fc are near identical to its phenyl-substituted counterpart, M(Sal)Ph, indicating the ferrocene moieties exist in the secondary coordination sphere of M(Sal)Fc. The cyclic voltammograms of M(Sal)Fc exhibit an additional two-electron wave in comparison to M(Sal)Ph, which is assigned to the sequential oxidation of the two ferrocene moieties. The chemical oxidation of M(Sal)Fc, monitored by low temperature UV-vis spectroscopy, supports the formation of a mixed valent FeIIFeIII species followed by a bis(ferrocenium) species upon sequential addition of one and two equivalents of chemical oxidant. The addition of a third equivalent of oxidant to Ni(Sal)Fc yielded intense near-IR transitions that are indicative of the formation of a fully delocalized Sal-ligand radical (Sal˙), while the same addition to Cu(Sal)Fc yielded a species that is currently under further spectroscopic investigation. These results suggest the oxidation of the ferrocene moieties of M(Sal)Fc does not affect the electronic structure of the M(Sal) core, and these are thus in the secondary coordination sphere of the overall complex.
Introduction
Salen, a common abbreviation for the tetradentate N2O2 bis(iminophenoxide) ligand scaffold (referred to as Sal herein), is a highly popular ligand scaffold due to the ease of its synthesis and the ability to robustly coordinate transition metal ions in a variety of oxidation states.1–6 The Sal ligand scaffold is a redox active ligand: M(Sal) complexes can undergo metal- or ligand- oxidation depending on the relative energies of the metal and ligand frontier orbitals. The locus of oxidation is highly sensitive to experimental conditions such as solvent, presence of extraneous ligands and even temperature, illustrating the similarity in energy between the two oxidized states.7–13 For example, the oxidation of NiII(Sal) yields a ligand-oxidized species [NiII(Sal)˙]+ in non-coordinating solvents such as CH2Cl2,14,15 which is converted to a metal-oxidized species [NiIII(Sal)(L)2]+ in the presence of coordinating solvents or extraneous ligands, where the shift in locus of oxidation in accompanied by concomitant axial ligation.14–18 The locus of oxidation is also dependant on the substituents on the Sal scaffold: oxidation of M(Sal)R, where R is an electron donating substituent at the distal para-position of the phenoxide moiety, generally yields ligand oxidized products.14,15,19–25 On the other hand, metal oxidized products are observed if R is an electron withdrawing substituent at the same position.21,22,26 Similarly, the oxidation of CuII(Sal) yields metal- ([CuIII(Sal)]+) and ligand-oxidized ([CuII(Sal˙)]+) products that can exist in a temperature dependant spin equilibrium.19,20
The N2O2 donor atoms of the Sal ligand scaffold, which are directly ligated to metal ions upon coordination, constitute the primary coordination sphere of M(Sal). Beyond this coordination sphere is the secondary coordination sphere, consisting of groups that are not directly coordinated to the metal center. An archetypal example is the enzyme Ni–Fe carbon monoxide dehydrogenase, which contains non-coordinated, Brønsted basic histidine and lysine amino acid residues in its secondary coordination sphere that facilitates the reduction of a bound CO2 substrate via hydrogen bonding.27 Inspired by this, the incorporation of similar Brønsted acidic and basic groups in the secondary coordination spheres of inorganic complexes have received significant research interest in recent years.28–31
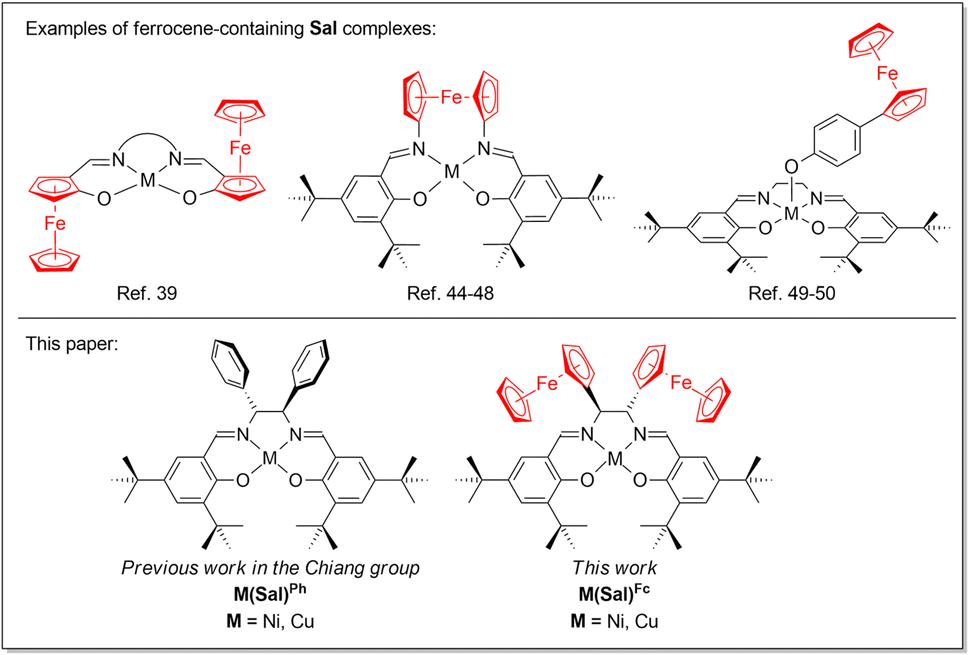
In general, the Sal ligand platform is capable of supplying two electron equivalents upon its oxidation, although further oxidizing equivalents are possible when electron-donating substituents are incorporated on the ligand scaffold. Our research group is specifically interested in incorporating redox active moieties, such as ferrocene, onto the already redox active Sal ligand scaffold. Ferrocene moieties have been introduced to redox-active ligand scaffolds, resulting in complexes that exhibit rich electrochemical behaviour.32–35 Ferrocene moieties have been incorporated into M(Sal) complexes, most commonly via the aromatic system of Sal,36–43 diamine linker,44–48 or axial coordination of a ferrocene-containing ligand (Chart 1).49,50 However their incorporation into the secondary coordination sphere of Sal via the sp3-carbon of the diamine linker have been far less explored.51 Furthermore, the electronic spectroscopy and electrochemical behaviors of these compounds have yet to be reported. To this end, this paper will describe the synthesis, spectroscopic characterization and electrochemical studies of a NiII and CuII complexes of a novel Sal ligand scaffold in which two ferrocene moieties are incorporated into its secondary coordination sphere via its diimine linker (Chart 1). The formation of their corresponding oxidized forms using a suitable chemical oxidant were investigated using low temperatures UV-vis spectroscopy.
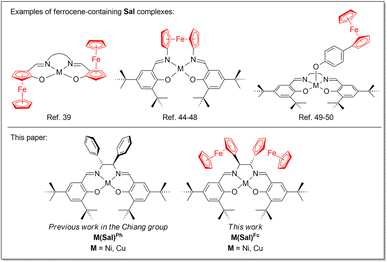 |
| | Chart 1 Ferrocene-containing Sal complexes. | |
Experimental
Materials and methods
All chemicals used were of the highest grade available and were used as received. Schiff base 1 was synthesized based on a previous report.51 The tris(2,4-dibromophenyl)aminium hexafluoroantimonate oxidant (N(C6H3Br2)3SbF6, E1/2 = 1.14 V vs. Fc/Fc+ in MeCN)52,53 was prepared from commercially available triphenylamine by reported procedures.17,54 1H and 13C NMR spectra were collected at Trinity Western University on a Bruker ASCEND III 400 MHz with Bruker AVANCE III 400 MHz instrument running the TopSpin 3.1.6 program. Mass spectrometry data (electrospray ionization (ESI) positive ion or ESI negative ion) were collected at Simon Fraser University on an Agilent 6210 TOF ESI-MS instrument. Elemental analyses (C, H, N) were performed at the University of British Columbia on a Thermo Flash 2000 elemental analyzer. Electronic spectra were collected on a Cary 50 spectrophotometer using a custom designed immersion fiber-optic probe with a path length of 1 mm (Hellma Inc.). Temperatures were maintained during data collection using a dry ice/acetone bath (−78 °C). Cyclic voltammetry (CV) data were collected on a CHI-630E potentiostat connected to a three electrode voltammetry cell (Ag/AgCl wire reference electrode, glassy carbon working electrode, and a Pt auxiliary electrode) with tetra-n-butylammnoiumperchlorate (nBu4NClO4, 0.1 M) as supporting electrolyte. Cobaltaceniumhexafluorophosphate (E1/2 = −1.34 V vs. Fc/Fc+ in 0.1 M nBu4NClO4 in CH2Cl2) were used as internal standards.
Oxidation protocol
Under an Ar atmosphere at 195 K, 1.0 mL of a CH2Cl2 solution of the metal complex (10.0 mM) was added to 10.0 mL of CH2Cl2. Monitored by UV-vis spectroscopy, a saturated solution of N(C6H3Br2)3SbF6 was added in 100 μL aliquots to yield the corresponding one-electron oxidized species.
Synthesis
H2(Sal)Fc. To a clear red solution of Schiff base 1 (1.9926 g, 3.131 mmol) in THF (30 mL) was added 37% aqueous HCl (6 mL, 37%), and the solution stirred at room temperature for 3 hours. The resulting dark yellow precipitate was collected by vacuum filtration, washed with THF, and dried under vacuum to afford a pale-yellow powder that is presumably the diammonium salt (1.347 g, yield: 88%). This material was used in the subsequent steps immediately without purification. A portion of this salt (0.1000 g, 0.200 mmol) was suspended in methanol (5 mL), where triethylamine (0.18 mL, 1.29 mmol) and 3,5-di-tert-butylsalicylaldehyde (0.0935 g, 0.399 mmol) were added sequentially. The resulting reaction mixture became orange and clear immediately, which was left stirring at room temperature overnight. The resulting bright yellow precipitate was collected via vacuum filtration and washed with cold EtOH to yield H2(Sal)Fc as a fluffy light yellow solid (0.1162 g, yield: 68%). 1H NMR (CDCl3, 400 MHz): δ = 13.82 (s, 2H, OH), 8.27 (s, 2H, imine-H), 7.34 (d, 2H, Ar–H), 6.95 (d, 2H, Ar–H), 4.28 (m, 2H, Fc-H), 4.13 (m, 2H, Fc-H), 4.09 (s, 2H, sp3-CH), 3.99 (m, 2H, Fc-H), 3.95 (s, 10H, Fc-H), 3.72 (m, 2H, Fc-H), 1.48 (s, 18H, tBu-H), 1.23 (s, 18H, tBu-H). 13C NMR (CDCl3, 100 MHz): δ = 166.4 (N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH), 158.2 (Ar–C), 140.1 (Ar–C), 136.6 (Ar–C), 127.2 (Ar–C), 126.2 (Ar–C), 117.7 (Ar–C), 88.8 (Fc-CC), 75.9 (N–CH-Fc), 69.4 (Fc-CH), 68.7 (Fc-CH), 68.1 (unsubstituted Fc-CH), 67.0 (Fc-CH), 66.9 (Fc-CH), 35.2 (C-(CH3)3), 34.2 (C-(CH3)3), 31.6 (C-(CH3)3), 29.6 (C-(CH3)3). MS (ESI): Found (m/z (%)): 861.3733 (100); calculated (m/z (%)): 861.3739 (100) [H2(Sal)Fc + H+]+. Elemental analysis calculated (%) for H2(Sal)Fc: C: 72.56, H: 7.49, N: 3.25; Found (%) C: 72.60, H: 7.37, N: 3.23.
CH), 158.2 (Ar–C), 140.1 (Ar–C), 136.6 (Ar–C), 127.2 (Ar–C), 126.2 (Ar–C), 117.7 (Ar–C), 88.8 (Fc-CC), 75.9 (N–CH-Fc), 69.4 (Fc-CH), 68.7 (Fc-CH), 68.1 (unsubstituted Fc-CH), 67.0 (Fc-CH), 66.9 (Fc-CH), 35.2 (C-(CH3)3), 34.2 (C-(CH3)3), 31.6 (C-(CH3)3), 29.6 (C-(CH3)3). MS (ESI): Found (m/z (%)): 861.3733 (100); calculated (m/z (%)): 861.3739 (100) [H2(Sal)Fc + H+]+. Elemental analysis calculated (%) for H2(Sal)Fc: C: 72.56, H: 7.49, N: 3.25; Found (%) C: 72.60, H: 7.37, N: 3.23.
Ni(Sal)Fc. To a solution of H2(Sal)Fc (0.1000 g, 0.12 mmol) Et2O (1 mL) was added a solution of Ni(OAc)2·4H2O (0.0289 g, 0.12 mmol) in MeOH (1 mL), which was left stirring overnight. A dark amber coloured solution with brown precipitate remained, where the solvent was evaporated via rotary evaporation to yield a yellow-brown powdered product. This crude material was purified by silica gel column chromatography using a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v pentane and dichloromethane as eluent (Rf = 0.70) to yield a dark brown powder of Ni(Sal)Fc (0.017 g, 16%). 1H NMR (400 MHz, CDCl3): δ 7.54 (s, 1H, imine H), 7.31 (d, 1H, aromatic H), 6.91 (d, 1H, aromatic H), 4.80 (s, 1H, CH), 4.39 (s, 1H, Fc H), 4.27 (s, 6H, Fc H), 1.40 (s, 9H, tBu H), 1.30 (s, 9H, tBu H). 13C NMR (CDCl3, 100 MHz): δ = 163.6 (NCH), 162.4 (Ar–C), 140.5 (Ar–C), 135.9 (Ar–C), 129.2 (Ar–C), 125.7 (Ar–C), 119.7 (Ar–C), 91.9 (Fc-CC), 75.4 (N–CH-Fc), 69.1 (Fc-CH), 68.4 (Fc-CH), 68.1 (Fc-CH), 66.5 (Fc-CH), 35.9 (C-(CH3)3), 33.9 (C-(CH3)3), 31.4 (C-(CH3)3), 29.7 (C-(CH3)3). MS (ESI): Found (m/z (%)): 917.2946 (100); calculated (m/z (%)): 917.2925(100) (917.2946 (66) [Ni(Sal)Fc + H+]+, 916.2858 (34) [Ni(Sal)Fc − e−]+). Elemental analysis calculated (%) for Ni(Sal)Fc: C: 68.08, H: 6.81, N: 3.05; Found (%) C: 68.51, H: 6.93, N: 2.84.
:1 v/v pentane and dichloromethane as eluent (Rf = 0.70) to yield a dark brown powder of Ni(Sal)Fc (0.017 g, 16%). 1H NMR (400 MHz, CDCl3): δ 7.54 (s, 1H, imine H), 7.31 (d, 1H, aromatic H), 6.91 (d, 1H, aromatic H), 4.80 (s, 1H, CH), 4.39 (s, 1H, Fc H), 4.27 (s, 6H, Fc H), 1.40 (s, 9H, tBu H), 1.30 (s, 9H, tBu H). 13C NMR (CDCl3, 100 MHz): δ = 163.6 (NCH), 162.4 (Ar–C), 140.5 (Ar–C), 135.9 (Ar–C), 129.2 (Ar–C), 125.7 (Ar–C), 119.7 (Ar–C), 91.9 (Fc-CC), 75.4 (N–CH-Fc), 69.1 (Fc-CH), 68.4 (Fc-CH), 68.1 (Fc-CH), 66.5 (Fc-CH), 35.9 (C-(CH3)3), 33.9 (C-(CH3)3), 31.4 (C-(CH3)3), 29.7 (C-(CH3)3). MS (ESI): Found (m/z (%)): 917.2946 (100); calculated (m/z (%)): 917.2925(100) (917.2946 (66) [Ni(Sal)Fc + H+]+, 916.2858 (34) [Ni(Sal)Fc − e−]+). Elemental analysis calculated (%) for Ni(Sal)Fc: C: 68.08, H: 6.81, N: 3.05; Found (%) C: 68.51, H: 6.93, N: 2.84.
Cu(Sal)Fc. A solution of H2(Sal)Fc (0.1000 g, 0.12 mmol) in Et2O (2 mL) was added a solution of Cu(OAc)2·H2O (0.0232 g, 0.12 mmol) in MeOH (2 mL), which was left stirring overnight. The murky green solution that remained was concentrated in vacuo to yield a green powder. This crude material was purified by silica gel column chromatography using 3:1 v/v pentane and dichloromethane as eluent (Rf = 0.71) to yield a dark green powder of Cu(Sal)Fc (0.079 g, 74%). MS (ESI): Found (m/z (%)): 922.2880 (100); calculated (m/z (%)): 922.2866 (100) (922.2879 (87) [Cu(Sal)Fc + H+]+, 921.2801 (13) [Cu(Sal)Fc − e−]+). Elemental analysis calculated (%) for Cu(Sal)Fc: C: 67.72, H: 6.78, N: 3.04; Found (%) C: 67.89, H: 6.68, N: 2.88.
Results and discussion
Synthesis of ligands and complexes
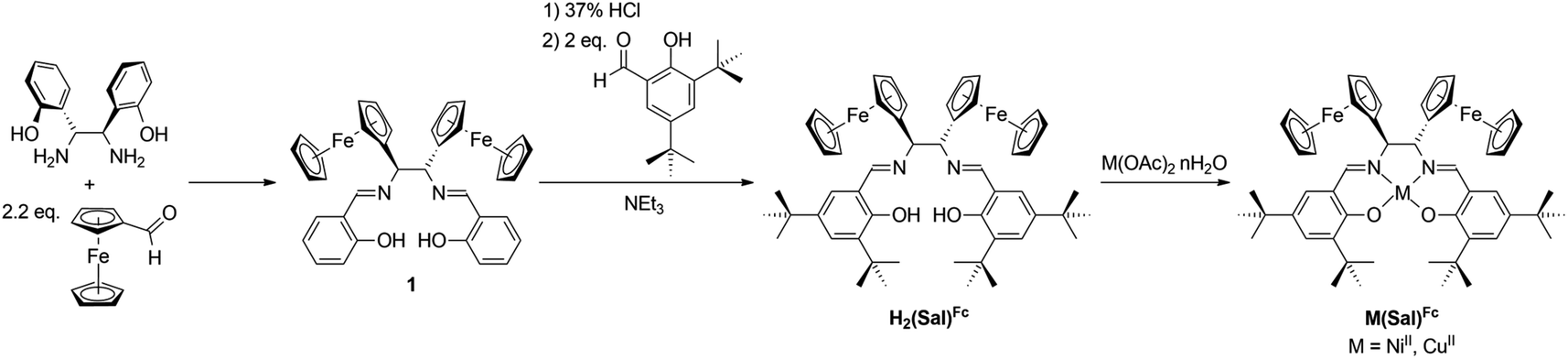
Ligand H2(Sal)Fc was synthesized in three steps from commercially available starting materials. First, a diaza-Cope rearrangement reaction between (1R,2R)-1,2-bis(2-hydroxyphenyl)-1,2-diaminoethane and 2.2 equivalents of ferrocenecarboxaldehyde yields Schiff-base 1.51 While 1 is a ferrocene-containing Sal ligand, the lack of ortho- and para-substituents relative to the phenol oxygen is envisaged to produce less stable oxidized products, as unsubstituted M(Sal) complexes are known to undergo decomposition via oxidative radical polymerization at these positions.55–59 As such, 1 was hydrolyzed under acidic conditions, then condensed with 2 equivalents of 3,5-di-tert-butylsalicylaldehyde in the presence of an excess of NEt3 to afford the target ligand H2(Sal)Fc. Equimolar portions of H2(Sal)Fc and the appropriate metal(II) acetate reagent were reacted to yield Ni(Sal)Fc and Cu(Sal)Fc (Scheme 1). All attempts to obtain single crystals of M(Sal)Fc for X-ray diffraction analysis using column purified material resulted in oils due to their high solubility in nearly all organic solvents.
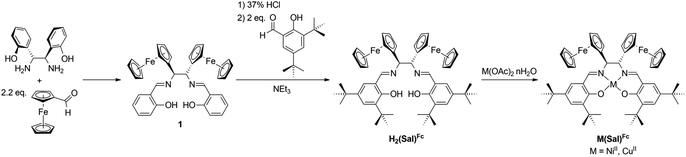 |
| | Scheme 1 Synthetic scheme of ligand H2(Sal)Fc and their corresponding complexes M(Sal)Fc. | |
Characterization of M(Sal)R
In lieu of a solid state structure for M(Sal)Fc, a combination of solution-based high resolution ESI mass spectrometry, NMR spectroscopy and theoretical DFT calculations were employed to establish their structures. H2(Sal)Fc exhibit ESI-MS signals at m/z = 861.3733, as expected for its corresponding mono-protonated [H2(Sal)Fc + H+]+ species (Fig. S1†). Similar mono-protonated species were observed in the mass spectra of M(Sal)Fc (m/z (%): [Ni(Sal)Fc + H+]+: 917.2946 (66); [Cu(Sal)Fc + H+]+: 922.2879 (87)). However, the observed isotope pattern can only be accurately modeled when a minor species at ca. 1 m/z unit less was considered both M(Sal)Fc, which can be attributed to a mono-oxidized species (m/z (%): [Ni(Sal)Fc − e−]:+: 916.2858 (34); [Cu(Sal)Fc − e−]+: 921.2801 (13); Fig. S2 and S3†). Taken together, these data suggest that both M(Sal)Fc are 1:1 metal to Sal complexes that forms with the concomitant deprotonation of H2(Sal)Fc.
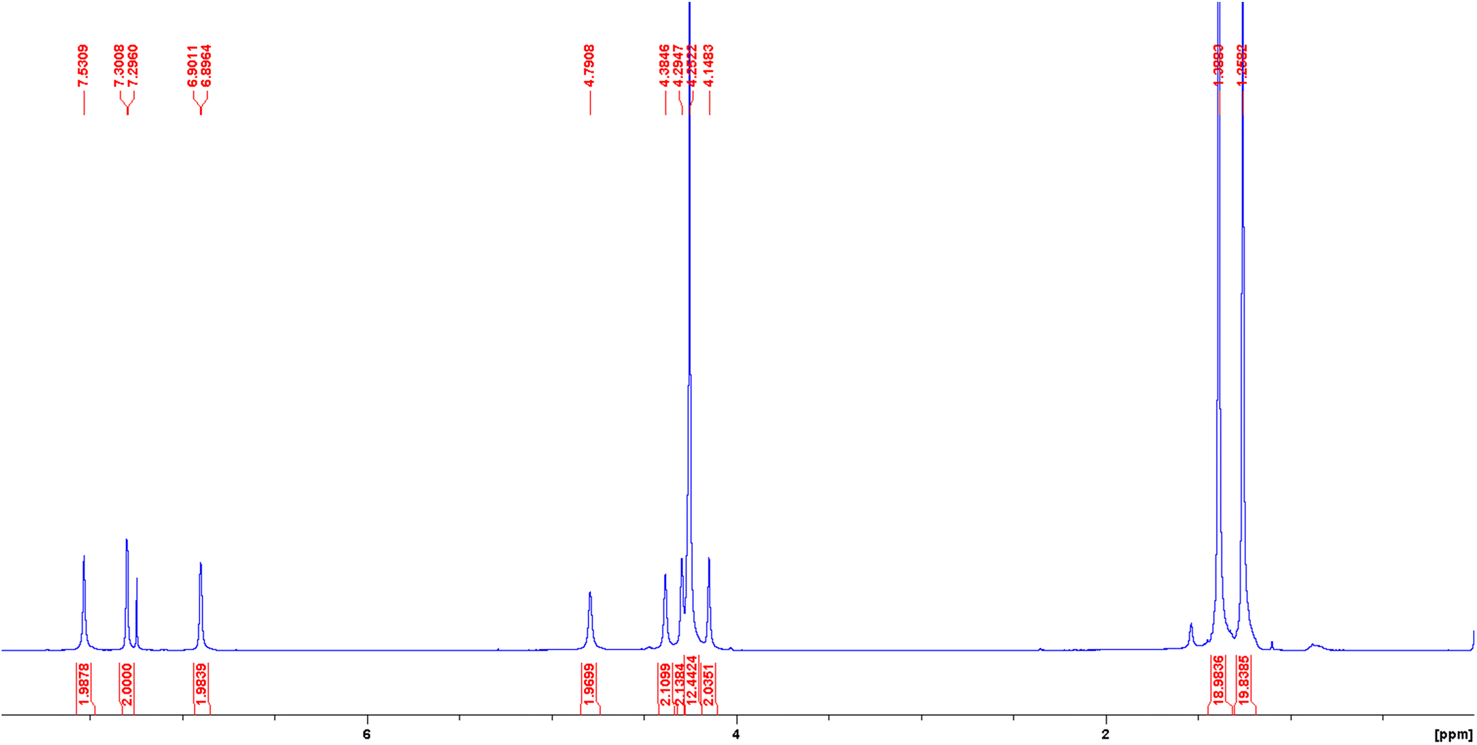
The 1H NMR spectra of both diamagnetic H2(Sal)Fc and Ni(Sal)Fc reveal their C2-symmetry on the NMR timescale (Fig. 1 and S4–S6†). The imine (δ = 7.5–8.5 ppm), phenol/phenolate (δ = 6.9–7.4 ppm) and tert-butyl (δ = 1.2–1.5 ppm) signals are consistent with those of H2(Sal)Ph and Ni(Sal)Ph.60 Four signals were observed in the δ = 4–5 ppm region, which can be assigned to the proton environments of the monosubstituted η5-C5H4 ring due to their diastereotopic nature as they are adjacent to the stereogenic center on the diimine linker.61 1H–1H COSY NMR experiments confirm this assignment by revealing the coupling between the protons on the monosubstituted η5-C5H4 ring, while 1H–13C HSQC NMR experiments demonstrate the coupling between these protons and the aromatic carbon signals (Fig. S7–S13†). The remaining signals that do not exhibit coupling to the protons of the monosubstituted η5-C5H4 ring are then assigned to the protons on the unsubstituted η5-C5H5 aromatic system and the proton on the diimine linker.
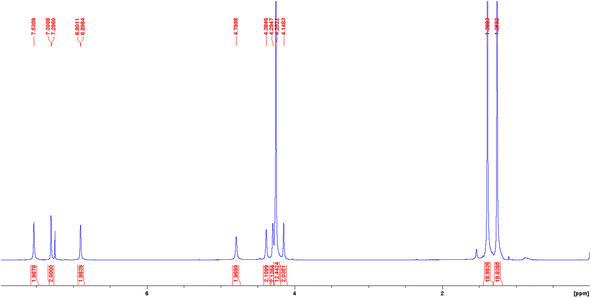 |
| | Fig. 1 1H NMR spectrum of Ni(Sal)Fc recorded in CDCl3 at 298 K. | |
The structure of M(Sal)Fc was examined via DFT calculations using the B3LYP/6–31g* functional/basis set combination and a PCM solvent model, parameters that have been used to investigate similar M(Sal) compounds. The optimized structures of M(Sal)Fc both exhibit two ferrocene moieties on the diimine linker separated by large dihedral angles that results from their occupation of trans, diaxial sites (Ni(Sal)Fc: 158.2°; Cu(Sal)Fc: 159.8°, Fig. S14 and S15†). This configuration has been observed previously via crystallography for structurally similar compounds in which two ferrocene moieties are also attached on a diimine linker, and is expected due to their steric bulk.51 Both M(Sal)Fc exhibit calculated coordinative bonds that are within 0.03 Å to their M(Sal)Ph counterpart under the same computational parameters (Table S1†). In addition, the singly occupied molecular orbitals (SOMO) of Cu(Sal)Fc consist predominantly of a dx2–y2 orbital with spin covalency with the coordinating atoms, which is expected for a square planar, d9, CuII complex (Fig. S16†). The predicted spin density on the CuII center of Cu(Sal)Fc is identical to Cu(Sal)Ph at ca. 57%.21 Overall, these results suggest that the two ferrocene moieties in the secondary coordination sphere of M(Sal)Fc does not significantly affect the tetradentate and square planar geometry around the NiII and CuII centers afforded by the primary coordination sphere in M(Sal)Fc.
Electronic spectroscopy of neutral M(Sal)R
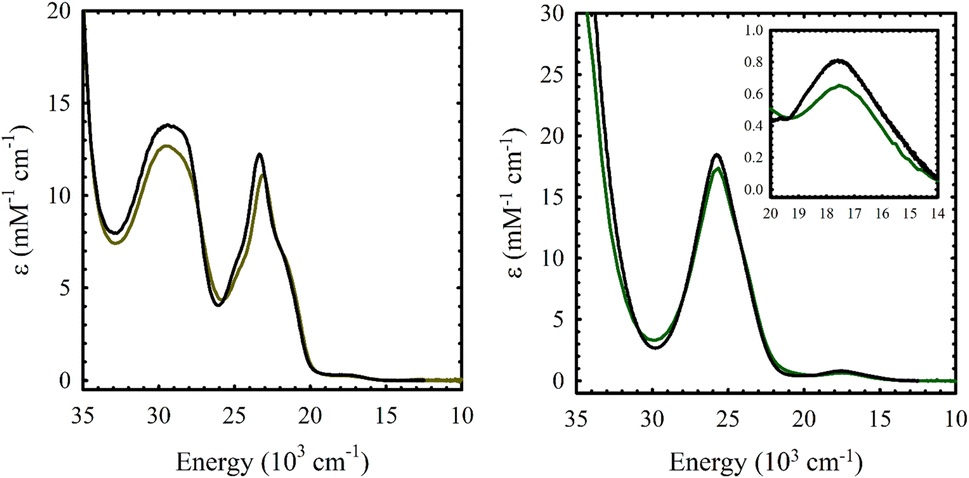
The electronic absorption spectrum of M(Sal)Fc is nearly identical to its M(Sal)Ph counterpart in CH2Cl2 (Fig. 2 and Table 1). A series of charge transfer transitions above 20000 cm−1 and weak d–d transitions below 18000 cm−1, typical of square-planar, d8 NiII bis-phenoxide salen complexes, were observed for Ni(Sal)Fc.15,19,26 On the other hand, an intense charge transfer transition above 25000 cm−1 and a d–d transition above 17300 cm−1 were observed for Cu(Sal)Fc, typical of square-planar, d9 CuII bis-phenoxide salen complexes.19,20,26 The d–d transitions of Cu(Sal)Fc, an indicator of ligand field strength, is observed at similar energies to its Cu(Sal)Ph counterpart (E(Cu(Sal)Fc): 17500 cm−1; E(Cu(Sal)Ph): 17600 cm−1). This suggests the two ferrocene moieties on the diimine linker only minimally affects the primary coordination sphere afforded by the N2O2 donor atoms of the Sal scaffold, and thus exists in the secondary coordination sphere of the complex.
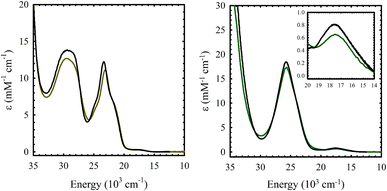 |
| | Fig. 2 UV-vis spectra of ca. 0.1 mM solutions of (a) Ni(Sal)Fc (left, brown) vs. Ni(Sal)Ph (left, black) and (b) Cu(Sal)Fc (right, green) vs. Cu(Sal)Ph (right, black) in CH2Cl2. (Inset) Magnified view of the d–d transitions. | |
Table 1 UV-vis spectroscopy data of M(Sal)R in CH2Cl2
| Compound |
UV-vis [E, 103 cm−1 (ε, mM−1 cm−1)] |
| Ni(Sal)Ph |
29.5 (13.8), 23.4 (12.2), 17.5 (0.3) |
| Ni(Sal)Fc |
29.5 (12.7), 23.1 (11.1), 17.5 (0.3) |
| Cu(Sal)Ph |
25.8 (18.3), 17.6 (0.8) |
| Cu(Sal)Fc |
25.7 (17.3), 17.5 (0.6) |
Electrochemistry
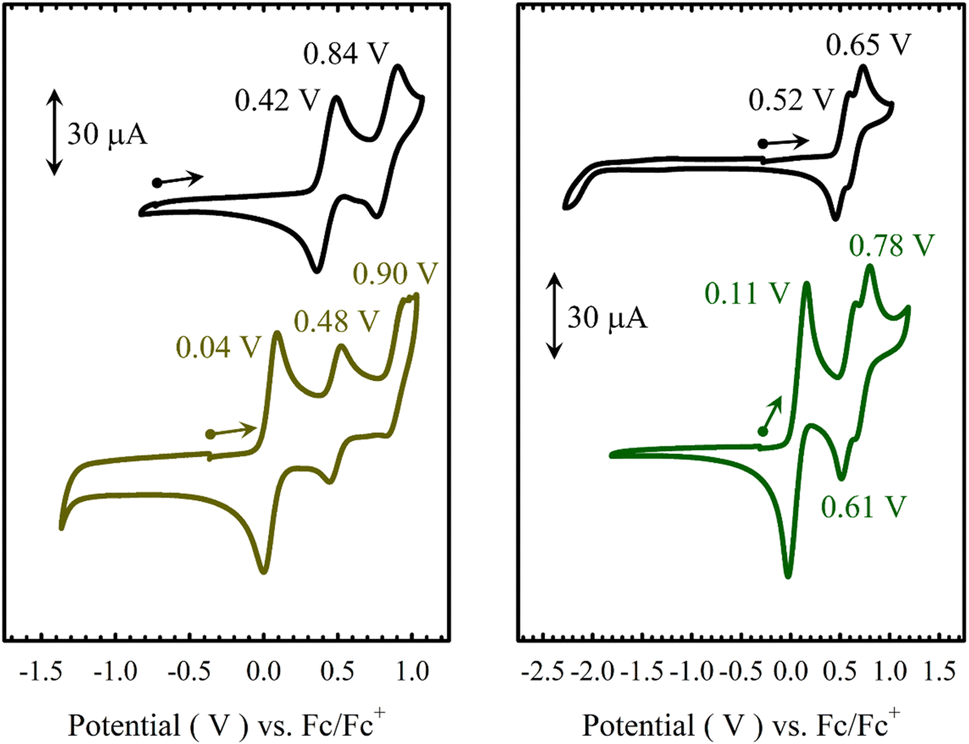
The electrochemical behaviours of M(Sal)Fc in CH2Cl2 were probed by cyclic voltammetry (CV) in the presence of tetra-n-butylammonium perchlorate (nBu4NClO4, 0.1 M) as supporting electrolyte (Fig. 3 and Table 2). Three quasi-reversible oxidative waves are observed for both M(Sal)Fc (Ni(Sal)Fc: 0.04 V, 0.48 and 0.90 V vs. Fc/Fc+; Cu(Sal)Fc: 0.11 V, 0.61 and 0.78 V vs. Fc/Fc+). The least positive redox couple is assigned to the oxidation of the two ferrocene moieties on the basis of its similarity in E1/2 to the Fc/Fc+ reference couple and its absence in the CV of the corresponding M(Sal)Ph complexes.60 The current response of this redox couple is nearly twice that of the remaining couples on CV. This suggests the successive oxidation of the two ferrocene moieties first affords a one-electron oxidized, charge localized, mixed valence FeIIFeIII species followed by a two-electron oxidized FeIIIFeIII species under these experimental conditions, in which the two ferrocenes are electronically isolated. This is in contrast to the electrochemical behaviour of biferrocenes that are connected such that the iron centers are closely associated or via a conjugated linker, both of which allow the two iron centers to interact electronically.62–64 The formation of a mixed valence species is expected for one-electron oxidized [M(Sal)Fc]+, as the two ferrocene moieties are connected via two unsaturated sp3-hybridized carbons,62,65 and are likely well separated due to the steric bulk afforded by the ferrocene moieties.51
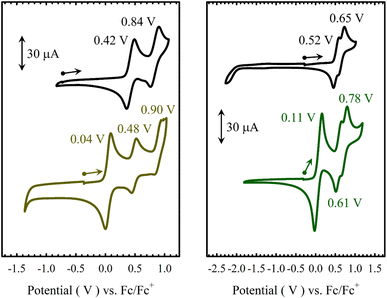 |
| | Fig. 3 Cyclic voltammograms of ca. 1.5 mM solutions in CH2Cl2 of (a) Ni(Sal)Ph (left, black) vs. Ni(Sal)Fc (left, brown), and (b) Cu(Sal)Ph (right, black) vs. Cu(Sal)Fc (right, green). | |
Table 2 Redox potentials (V) of M(Sal)R complexes vs. Fc+/Fc in CH2Cl2a,b
| Compound |
E1/21 |
E1/22 |
E1/23 |
| Peak-to-peak difference (Eap − Ecp) are provided in brackets (V). Peak-to-peak difference for the Cp2Co+/Cp2Co at 298 K is 0.08 V. |
| Ni(Sal)Ph |
0.42 (0.13) |
0.84 (0.14) |
— |
| Ni(Sal)Fc |
0.04 (0.09) |
0.48 (0.08) |
0.90 (0.12) |
| Cu(Sal)Ph |
0.52 (0.15) |
0.65 (0.16) |
— |
| Cu(Sal)Fc |
0.11 (0.12) |
0.61 (0.14) |
0.78 (0.15) |
The remaining two couples of M(Sal)Fc, E1/22 and E1/23, are thus assigned to the same oxidative events as those observed for M(Sal)Ph.21,60 The quasi-reversible nature of these couples suggests that oxidation of the ferrocene moieties does not affect the chemical stability of the electrogenerated species on the timescale of the electrochemistry experiment. The E1/22 and E1/23 of M(Sal)Fc are slightly anodically shifted in comparison to the E1/21 and E1/22 of M(Sal)Ph, which can be attributed to the dicationic nature of the overall complex before the next oxidation occurs (i.e. [M(Sal)Fc]2+ → [M(Sal)Fc]3+ vs. M(Sal)Ph → [M(Sal)Ph]+). In addition, the difference in potentials between E1/22 and E1/23 of Ni(Sal)Fc is identical to E1/21 and E1/22 of Ni(Sal)Ph (ΔE1/2 = 0.42 V). The magnitude of ΔE1/2 between the first two oxidative couples of Ni(Sal) compounds have been correlated to the degree of delocalization of ligand radical formed upon one-electron oxidation.15 This suggests that [Ni(Sal)Fc]3+ and [Ni(Sal)Ph]+ must exhibit similar Sal˙ electronic structures. Taken together, these data support the ferrocene moieties are indeed in the secondary coordination sphere of M(Sal)Fc as their oxidation does not affect the subsequent oxidation of M(Sal) significantly (Table 2).
Chemical oxidation of M(Sal)Fc
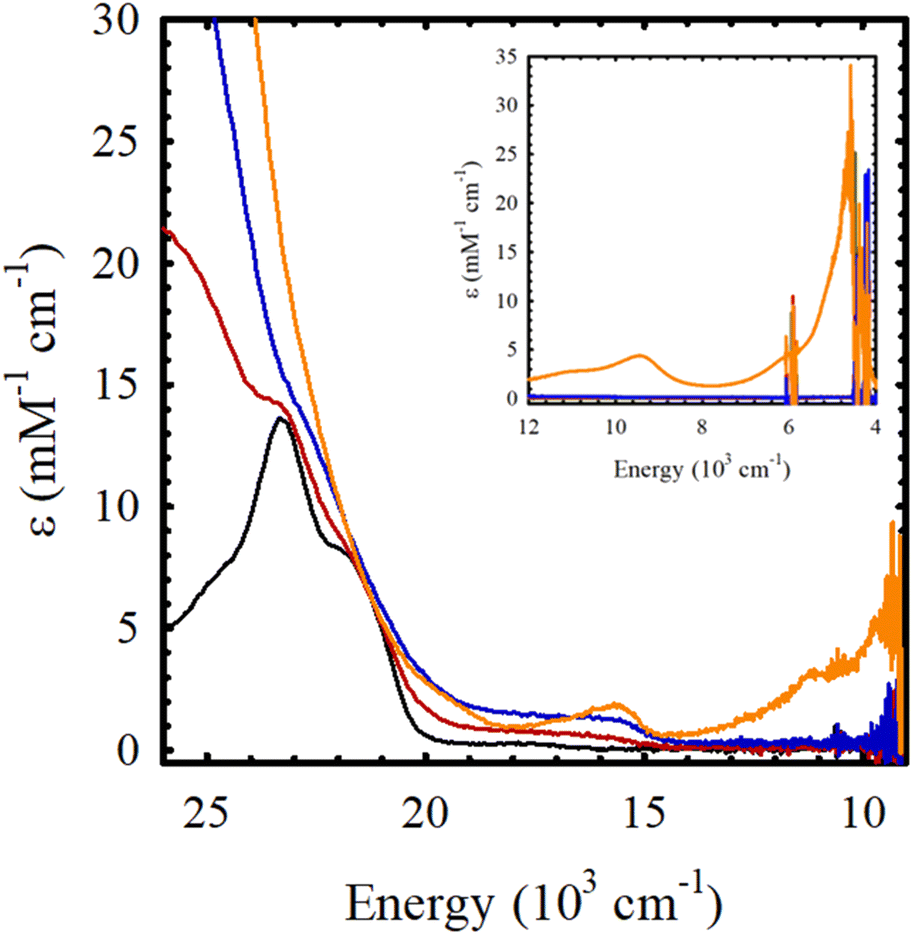
Chemical oxidation of Ni(Sal)Fc by incremental addition of two equivalents of the aminyl oxidant N(C6H3Br2)3SbF6 (E = 1.14 V vs. Fc/Fc+ in CH2Cl2) in CH2Cl2 at 195 K leads to the formation of a broad envelope of weak transitions between 15000–20000 cm−1 (ε = 1 – 2 mM−1 cm−1), that can be attributed to the formation of a ferrocenium-containing species (Fig. 4 and Table 3). The doubling in intensity of these transition, alongside the lack of near-IR transitions upon the addition of a second equivalent of oxidant strongly supports the formation of a charge-localized mono(ferrocenium) (FeIIFeIII) species, followed by a bis(ferrocenium) (FeIIIFeIII) species upon the addition of the first two equivalents of oxidant. The addition of a third equivalent of the aminyl oxidant to Ni(Sal)Fc yields NIR transitions at 9400 cm−1 (ε = 4.3 mM−1 cm−1) and 4600 cm−1 (ε = 23.5 mM−1 cm−1) that are demonstrative of the oxidation of Sal to a fully delocalized [Sal˙]+ ligand radical.15 These results are fully consistent with the electrochemical data of Ni(Sal)Fc, where the ferrocene-based oxidative couple at 0.04 V vs. Fc/Fc+ exhibits minimal separation, suggesting the sequential formation of a mixed-valent ferrocene-ferrocenium species followed by a bis(ferrocenium) species.66,67 The anodic wave observed at 0.48 V vs. Fc/Fc+, attributed to Sal oxidation, is separated from the next anodic wave by 0.42 V, indicates the formation of a fully delocalized Sal˙ ligand radical (vide supra).
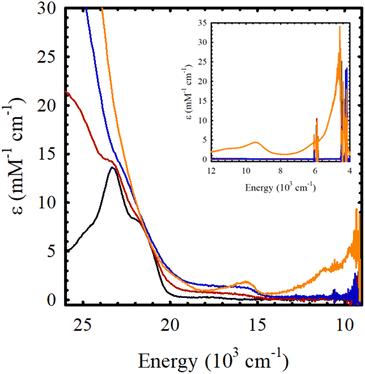 |
| | Fig. 4 Sequential chemical oxidation of Ni(Sal)Fc (black) to its one-electron oxidized (red), two-electron oxidized (blue), and three-electron oxidized (orange) species. (Inset) Near-IR region. Conditions: 0.4 mM Ni(Sal)Fc in CH2Cl2, 195 K. | |
Table 3 UV-vis spectroscopy data of the chemical oxidation M(Sal)Fc in CH2Cl2
| Compound |
UV-vis [E, 103 cm−1 (ε, mM−1, cm−1)] |
| Ni(Sal)Fc |
23.1 (9.9), 17.5 (0.3) |
| [Ni(Sal)Fc]+ |
23.0 (8.8), ca. 17.4 (br, 0.8) |
| [Ni(Sal)Fc]2+ |
22.6 (9.1), ca 17.4 (br, 1.6) |
| [Ni(Sal)Fc]3+ |
23.1 (10.6), 15.7 (1.8), 9.4 (4.3), 4.6 (23.5) |
Similarly, addition of two equivalents of aminyl oxidant to Cu(Sal)Fc yields weak transitions between 15000–20000 cm−1 (ε = 1 – 2 mM−1 cm−1) that can be attributed to the formation of a bis(ferrocenium) species (Fig. S17†). However, the addition of a third equivalent of the aminyl oxidant to Cu(Sal)Fc yields broad and weak transitions at 12000 cm−1, 17500 cm−1, and 22500 cm−1. The lack of an intense transition at ca. 18000 cm−1 (ε = ca. 15 mM−1 cm−1), which is diagnostic for the formation of a metal-oxidized [CuIIISalPh]+ species,21 supports the formation of a ligand-oxidized species for [Cu(Sal)Fc]3+ instead. Further spectroscopic investigation of the electronic structure of [Cu(Sal)Fc]3+ is currently underway.
Summary
In this paper, we report the synthesis, spectroscopic characterization and electrochemical studies of M(Sal)Fc, the NiII and CuII complexes of a novel Sal ligand bearing two ferrocene moieties in its secondary coordination sphere. The cyclic voltammograms of M(Sal)Fc both exhibit an additional two-electron wave in comparison to its M(Sal)Ph counterpart, which is assigned to the sequential oxidation of the two ferrocene moieties. The minimal separation between these two waves suggests the ferrocene moieties are incapable of electronic communication under these experimental conditions, consistent with their connection via a non-conjugated linker. This was further confirmed via the chemical oxidation of M(Sal)Fc monitored by low temperatures UV-vis spectroscopy, where no near IR transitions were observed upon the addition of one equivalent of oxidant. This supports the formation of a mixed valent FeIIFeIII intermediate species upon one-electron oxidation, while the addition of a second equivalent of oxidant yields a bis(ferrocenium) species. The subsequent anodic waves occur at similar potentials to the anodic waves of M(Sal)Ph, suggesting the ferrocene moieties are indeed in the secondary coordination sphere of M(Sal)Fc and does not affect the electronic structure of the M(Sal) core. Accordingly, the addition of a third equivalence of oxidant to Ni(Sal)Fc yielded intense near-IR transitions that are indicative of the formation of a fully delocalized Sal-ligand radical (Sal˙), while the same addition to Cu(Sal)Fc yielded a species that is currently under further spectroscopic investigation.
Author contributions
R. S., W. V., K. T.: investigation, formal analysis, methodology, visualization, writing – review & editing. L. C.: conceptualization, funding acquisition, project administration, resources, supervision, writing – original draft.
Conflicts of interest
There are no conflicts to declare.
References
- L. Canali and D. C. Sherrington, Chem. Soc. Rev., 1999, 28, 85–93 RSC.
- D. J. Darensbourg, Chem. Rev., 2007, 107, 2388–2410 CrossRef CAS PubMed.
- R. Irie, K. Noda, Y. Ito, N. Matsumoto and T. Katsuki, Tetrahedron Lett., 1990, 31, 7345–7348 CrossRef CAS.
- E. N. Jacobsen, W. Zhang, A. R. Muci, J. R. Ecker and L. Deng, J. Am. Chem. Soc., 1991, 113, 7063–7064 CrossRef CAS.
- E. M. McGarrigle and D. G. Gilheany, Chem. Rev., 2005, 105, 1563–1602 CrossRef CAS PubMed.
- R. M. Clarke and T. Storr, Dalton Trans., 2014, 43, 9380–9391 RSC.
- D. N. Hendrickson and C. G. Pierpont, in Spin Crossover in Transition Metal Compounds II, ed. P. Gütlich and H. A. Goodwin, Springer, Berlin, Heidelberg, 2004, pp. 63–95 Search PubMed.
- C. G. Pierpont, Coord. Chem. Rev., 2001, 219–221, 415–433 CrossRef CAS.
- C. G. Pierpont, Coord. Chem. Rev., 2001, 216–217, 99–125 CrossRef CAS.
- C. G. Pierpont and C. W. Lange, in Progress in Inorganic Chemistry, 1994, vol. 41, pp. 331–442 Search PubMed.
- D. Dolphin, T. Niem, R. Felton and I. Fujita, J. Am. Chem. Soc., 1975, 97, 5288–5290 CrossRef CAS PubMed.
- H. Ohtsu and K. Tanaka, Angew. Chem., Int. Ed., 2004, 43, 6301–6303 CrossRef CAS PubMed.
- F. F. Puschmann, J. Harmer, D. Stein, H. Rüegger, B. de Bruin and H. Grützmacher, Angew. Chem., Int. Ed., 2010, 49, 385–389 CrossRef CAS PubMed.
- T. Storr, E. C. Wasinger, R. C. Pratt and T. D. P. Stack, Angew. Chem., Int. Ed., 2007, 46, 5198–5201 CrossRef CAS PubMed.
- L. Chiang, A. Kochem, O. Jarjayes, T. J. Dunn, H. Vezin, M. Sakaguchi, T. Ogura, M. Orio, Y. Shimazaki, F. Thomas and T. Storr, Chem.–Eur. J., 2012, 18, 14117–14127 CrossRef CAS PubMed.
- Y. Shimazaki, F. Tani, K. Fukui, Y. Naruta and O. Yamauchi, J. Am. Chem. Soc., 2003, 125, 10512–10513 CrossRef CAS PubMed.
- T. J. Dunn, L. Chiang, C. F. Ramogida, K. Hazin, M. I. Webb, M. J. Katz and T. Storr, Chem.–Eur. J., 2013, 19, 9606–9618 CrossRef CAS PubMed.
- L. Benisvy, R. Kannappan, Y.-F. Song, S. Milikisyants, M. Huber, I. Mutikainen, U. Turpeinen, P. Gamez, L. Bernasconi, E. J. Baerends, F. Hartl and J. Reedijk, Eur. J. Inorg. Chem., 2007, 2007, 637–642 CrossRef.
- M. Orio, O. Jarjayes, H. Kanso, C. Philouze, F. Neese and F. Thomas, Angew. Chem., Int. Ed., 2010, 49, 4989–4992 CrossRef CAS PubMed.
- T. Storr, P. Verma, R. C. Pratt, E. C. Wasinger, Y. Shimazaki and T. D. P. Stack, J. Am. Chem. Soc., 2008, 130, 15448–15459 CrossRef CAS PubMed.
- A. Sharma, K. Mejia, H. Ueno, W. Zhou and L. Chiang, Inorg. Chim. Acta, 2022, 542, 121106 CrossRef CAS.
- R. M. Clarke and T. Storr, J. Am. Chem. Soc., 2016, 138, 15299–15302 CrossRef CAS PubMed.
- K. Herasymchuk, L. Chiang, C. E. Hayes, M. L. Brown, J. S. Ovens, B. O. Patrick, D. B. Leznoff and T. Storr, Dalton Trans., 2016, 45, 12576–12586 RSC.
- T. Kurahashi and H. Fujii, Inorg. Chem., 2013, 52, 3908–3919 CrossRef CAS PubMed.
- T. Kurahashi and H. Fujii, Bull. Chem. Soc. Jpn., 2012, 85, 940–947 CrossRef CAS.
- L. Chiang, K. Herasymchuk, F. Thomas and T. Storr, Inorg. Chem., 2015, 54, 5970–5980 CrossRef CAS PubMed.
- J.-H. Jeoung and H. Dobbek, Science, 2007, 318, 1461–1464 CrossRef CAS PubMed.
- A. W. Nichols and C. W. Machan, Front. Chem., 2019, 7, 397 CrossRef CAS PubMed.
- M. Zhao, H.-B. Wang, L.-N. Ji and Z.-W. Mao, Chem. Soc. Rev., 2013, 42, 8360–8375 RSC.
- J. Trouvé and R. Gramage-Doria, Chem. Soc. Rev., 2021, 50, 3565–3584 RSC.
- M. W. Drover, Chem. Soc. Rev., 2022, 51, 1861–1880 RSC.
- D. D. Swanson, K. M. Conner and S. N. Brown, Dalton Trans., 2017, 46, 9049–9057 RSC.
- Y. Shen, S. M. Shepard, C. J. Reed and P. L. Diaconescu, Chem. Commun., 2019, 55, 5587–5590 RSC.
- X. Wang, A. Thevenon, J. L. Brosmer, I. Yu, S. I. Khan, P. Mehrkhodavandi and P. L. Diaconescu, J. Am. Chem. Soc., 2014, 136, 11264–11267 CrossRef CAS PubMed.
- S. V. Baryshnikova, A. I. Poddel’sky, A. V. Cherkasov and I. V. Smolyaninov, Inorg. Chim. Acta, 2019, 495, 118963 CrossRef CAS.
- X.-X. Yan, C.-G. Liang, Y. Zhang, W. Hong, B.-X. Cao, L.-X. Dai and X.-L. Hou, Angew. Chem., Int. Ed., 2005, 44, 6544–6546 CrossRef CAS PubMed.
- J. M. Longmire, B. Wang and X. Zhang, J. Am. Chem. Soc., 2002, 124, 13400–13401 CrossRef CAS PubMed.
- S.-L. You, X.-L. Hou, L.-X. Dai, B.-X. Cao and J. Sun, Chem. Commun., 2000, 1933–1934 RSC.
- M. Sawamura, H. Sasaki, T. Nakata and Y. Ito, Bull. Chem. Soc. Jpn., 1993, 66, 2725–2729 CrossRef CAS.
- H. Wölfle, H. Kopacka, K. Wurst, K.-H. Ongania, H.-H. Görtz, P. Preishuber-Pflügl and B. Bildstein, J. Organomet. Chem., 2006, 691, 1197–1215 CrossRef.
- H.-C. Lin, C.-C. Huang, C.-H. Shi, Y.-H. Liao, C.-C. Chen, Y.-C. Lin and Y.-H. Liu, Dalton Trans., 2007, 781–791 RSC.
- Y. Ouennoughi, H. E. Karce, D. Aggoun, T. Lanez, R. Ruiz-Rosas, B. Bouzerafa, A. Ourari and E. Morallon, J. Organomet. Chem., 2017, 848, 344–351 CrossRef CAS.
- A. Patti, S. Pedotti, F. P. Ballistreri and G. T. Sfrazzetto, Molecules, 2009, 14, 4312–4325 CrossRef CAS PubMed.
- A. Shafir, D. Fiedler and J. Arnold, J. Chem. Soc., Dalton Trans., 2002, 555–560 RSC.
- E. M. Broderick and P. L. Diaconescu, Inorg. Chem., 2009, 48, 4701–4706 CrossRef CAS PubMed.
- S. M. Shepard and P. L. Diaconescu, Organometallics, 2016, 35, 2446–2453 CrossRef CAS.
- R. Dai, A. Lai, A. N. Alexandrova and P. L. Diaconescu, Organometallics, 2018, 37, 4040–4047 CrossRef CAS.
- S. M. Quan and P. L. Diaconescu, Chem. Commun., 2015, 51, 9643–9646 RSC.
- L. Mengozzi, M. El Garah, A. Gualandi, M. Iurlo, A. Fiorani, A. Ciesielski, M. Marcaccio, F. Paolucci, P. Samorì and P. G. Cozzi, Chem.–Eur. J., 2018, 24, 11954–11960 CrossRef CAS PubMed.
- G. Chen, W.-L. Man, S.-M. Yiu, T.-W. Wong, L. Szeto, W.-T. Wong and T.-C. Lau, Dalton Trans., 2011, 40, 1938–1944 RSC.
- K. Kiss, T. Holczbauer, M. Czugler, P. Sohár, A. Bodor and A. Csámpai, J. Organomet. Chem., 2012, 706–707, 46–51 CrossRef CAS.
- N. G. Connelly and W. E. Geiger, Chem. Rev., 1996, 96, 877–910 CrossRef CAS PubMed.
- E. Steckhan, in Electrochemistry I, Springer, 1987, pp. 1–69 Search PubMed.
- Y. Murata, F. Cheng, T. Kitagawa and K. Komatsu, J. Am. Chem. Soc., 2004, 126, 8874–8875 CrossRef CAS PubMed.
- K. A. Goldsby, J. K. Blaho and L. A. Hoferkamp, Polyhedron, 1989, 8, 113–115 CrossRef CAS.
- L. A. Hoferkamp and K. A. Goldsby, Chem. Mater., 1989, 1, 348–352 CrossRef CAS.
- P.-H. Aubert, P. Audebert, P. Capdevielle, M. Maumy and M. Roche, New J. Chem., 1999, 23, 297–301 RSC.
- P. Audebert, P. Hapiot, P. Capdevielle and M. Maumy, J. Electroanal. Chem., 1992, 338, 269–278 CrossRef CAS.
- C. E. Dahm, D. G. Peters and J. Simonet, J. Electroanal. Chem., 1996, 410, 163–171 CrossRef.
- K. Teindl, K. Mejia, W. Zhou and L. Chiang, 2022, Unpublished work.
- A. N. Nesmeyanov, G. B. Shul'pin, L. A. Fedorov, P. V. Petrovsky and M. I. Rybinskaya, J. Organomet. Chem., 1974, 69, 429–435 CrossRef CAS.
- N. Camire, U. T. Mueller-Westerhoff and W. E. Geiger, J. Organomet. Chem., 2001, 637–639, 823–826 CrossRef CAS.
- T. L. R. Bennett, L. A. Wilkinson, J. M. A. Lok, R. C. P. O'Toole and N. J. Long, Organometallics, 2021, 40, 1156–1162 CrossRef CAS.
- L. E. Wilson, C. Hassenrück, R. F. Winter, A. J. P. White, T. Albrecht and N. J. Long, Angew. Chem., Int. Ed., 2017, 56, 6838–6842 CrossRef CAS PubMed.
- W. H. Morrison Jr, S. Krogsrud and D. N. Hendrickson, Inorg. Chem., 1973, 12, 1998–2004 CrossRef.
- D. Gottwald, Q. Yuan, M. Speck, J. Mahrholdt, M. Korb, K. Schreiter, S. Spange and H. Lang, J. Organomet. Chem., 2020, 923, 121447 CrossRef CAS.
- L. E. Wilson, C. Hassenrück, R. F. Winter, A. J. P. White, T. Albrecht and N. J. Long, Eur. J. Inorg. Chem., 2017, 2017, 496–504 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ra07671c |
| ‡ Current address: The University of British Columbia, Vancouver, British Columbia V6T 1Z4, Canada. |
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  b,
Kaeden Teindl‡
a and
Linus Chiang
b,
Kaeden Teindl‡
a and
Linus Chiang