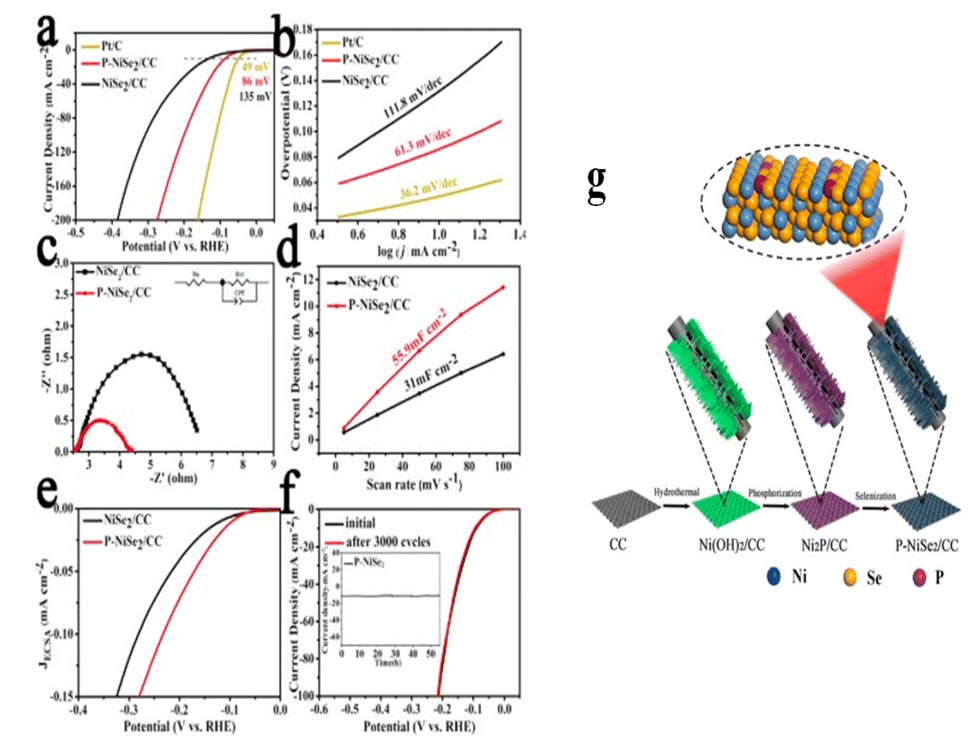
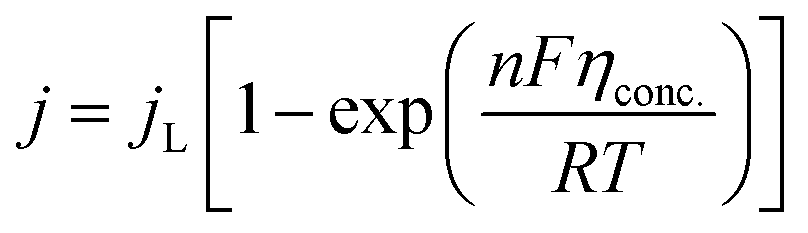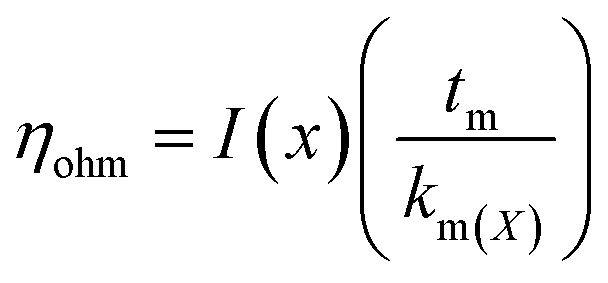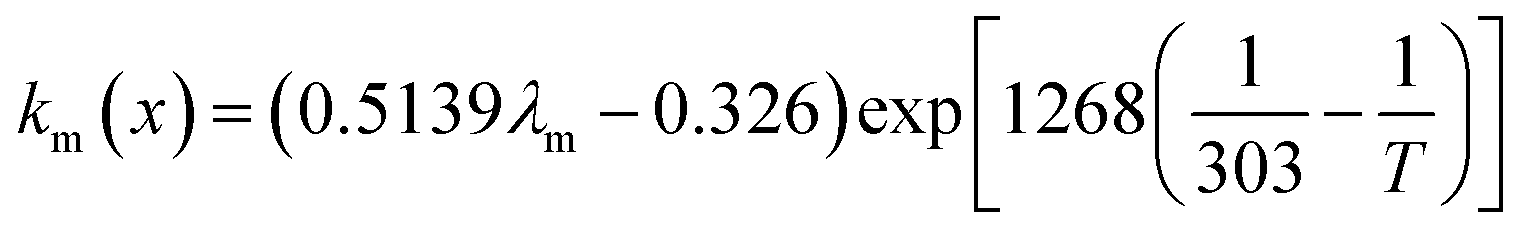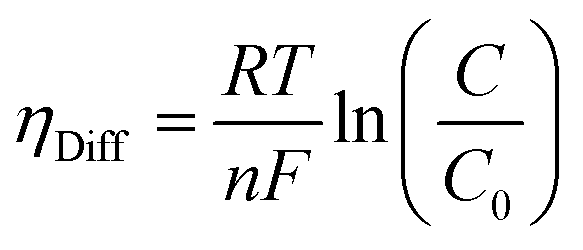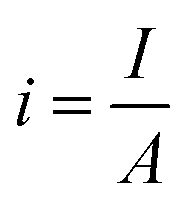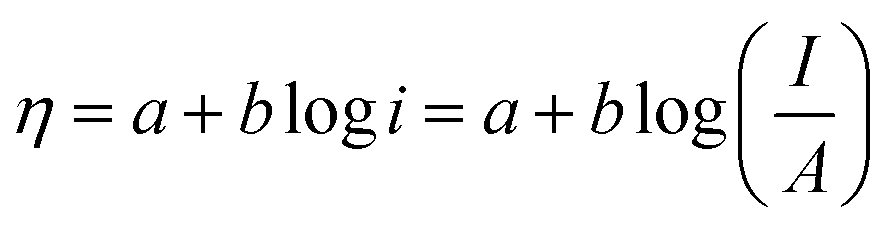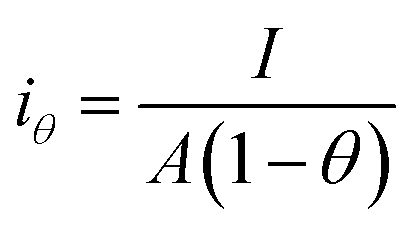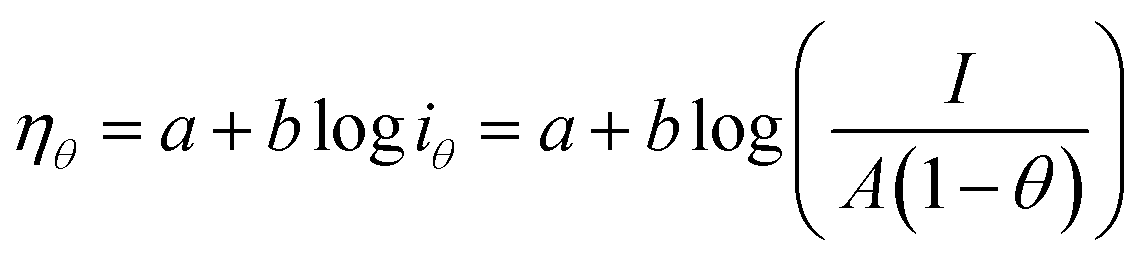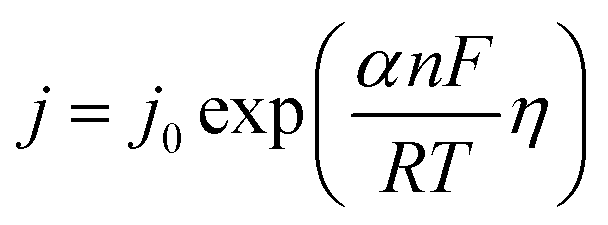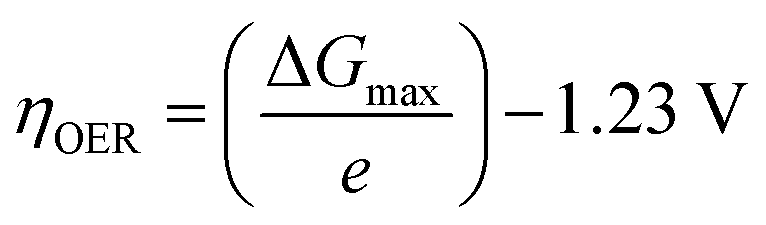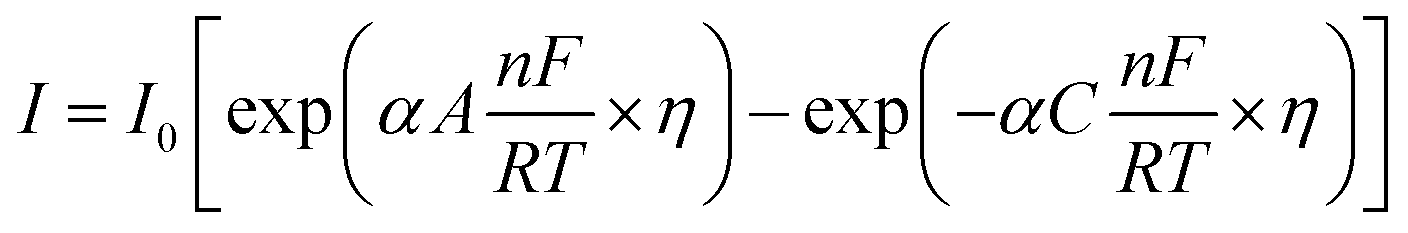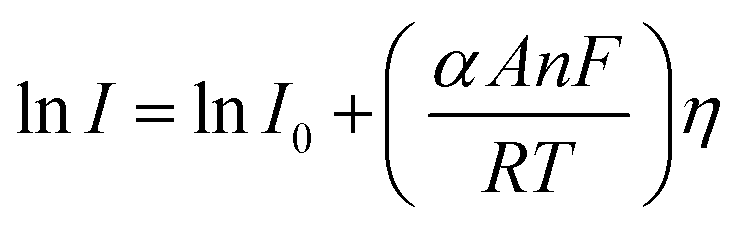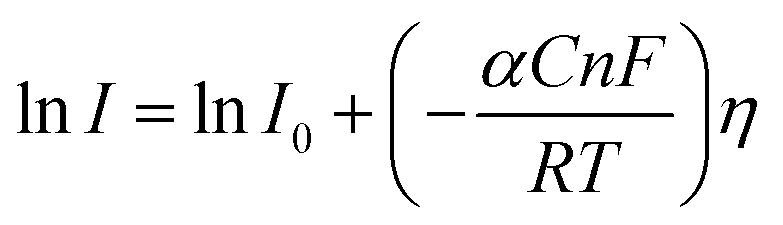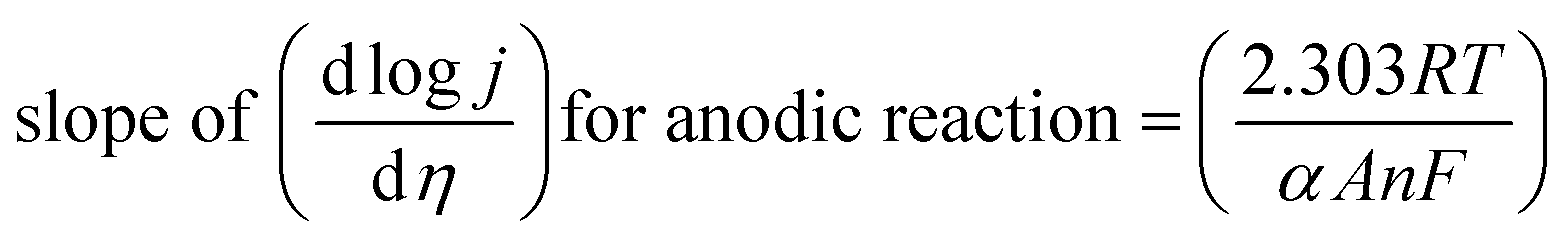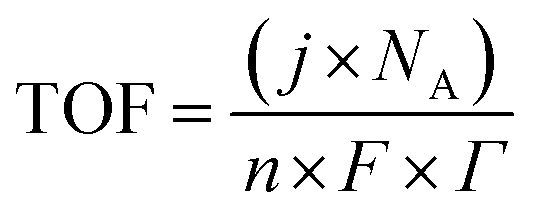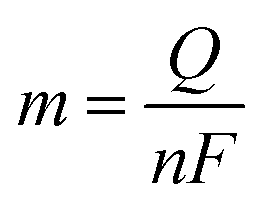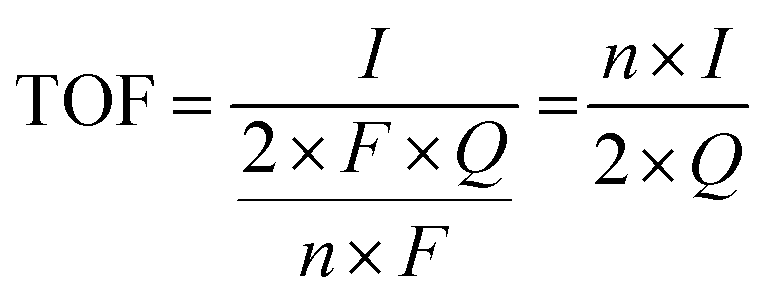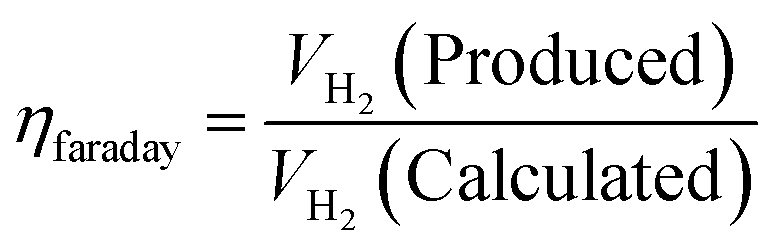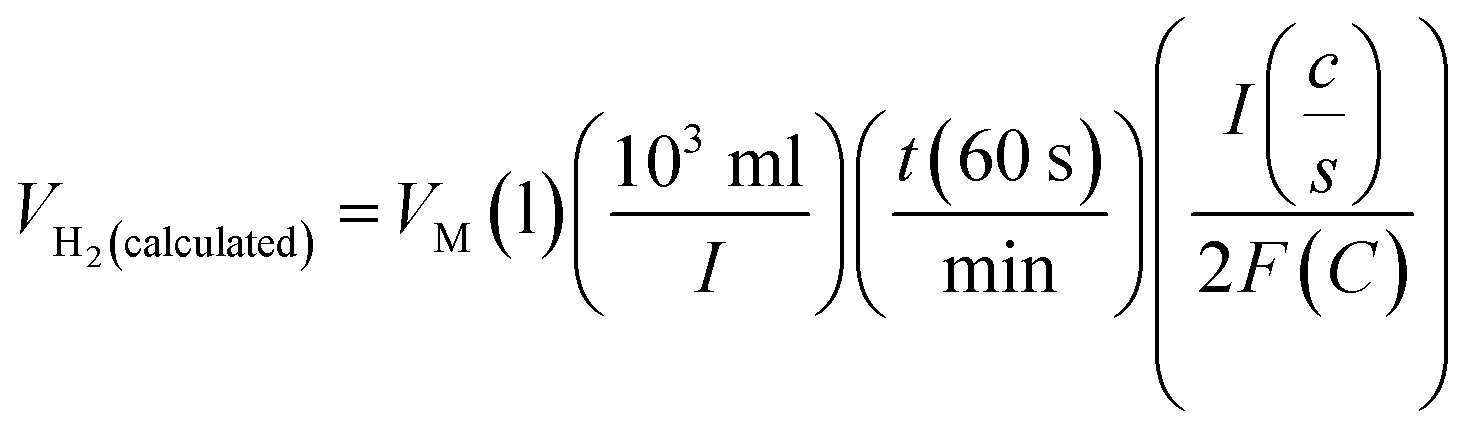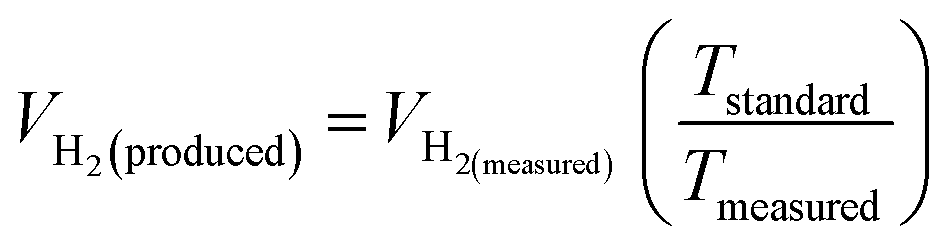DOI:
10.1039/D2RA07642J
(Review Article)
RSC Adv., 2023,
13, 3843-3876
A comprehensive review on the electrochemical parameters and recent material development of electrochemical water splitting electrocatalysts†
Received
1st December 2022
, Accepted 18th January 2023
First published on 26th January 2023
Abstract
Electrochemical splitting of water is an appealing solution for energy storage and conversion to overcome the reliance on depleting fossil fuel reserves and prevent severe deterioration of the global climate. Though there are several fuel cells, hydrogen (H2) and oxygen (O2) fuel cells have zero carbon emissions, and water is the only by-product. Countless researchers worldwide are working on the fundamentals, i.e. the parameters affecting the electrocatalysis of water splitting and electrocatalysts that could improve the performance of the hydrogen evolution reaction (HER) and oxygen evolution reaction (OER) and overall simplify the water electrolysis process. Noble metals like platinum for HER and ruthenium and iridium for OER were used earlier; however, being expensive, there are more feasible options than employing these metals for all commercialization. The review discusses the recent developments in metal and metalloid HER and OER electrocatalysts from the s, p and d block elements. The evaluation perspectives for electrocatalysts of electrochemical water splitting are also highlighted.
 Asha Raveendran | Asha Raveendran is pursuing her PhD degree in the Department of Chemistry, National Institute of Technology Puducherry, Karaikal, India. After obtaining Master's degree from Central University of Kerala, India, she joined Nano Electrochemistry Lab (NEL) under the guidance of Dr Ragupathy Dhanusuraman. Her research is on the design, synthesis, and fundamental electrochemical studies of 2D materials as well as conducting polymers based nanoelectrocatalysts for energy conversion studies like electrochemical water splitting, direct alcohol fuel cell applications and energy storage systems, such as supercapacitors. |
 Mijun Chandran | Mijun Chandran completed his undergraduate studies (BSc) in Chemistry from Kannur University in 2013. He received his MSc degree (2015) and PhD (2021) in Chemistry from Central University of Kerala, India, under the supervision of Dr M. Bhagiyalakshmi. His research focuses on the development and characterization of 2-dimensional MXene-based electrocatalysts for supercapacitors, direct alcohol fuel cell and biosensor applications. He has also worked as a DST-SERB Junior Research fellow entitled “Exploration on the bio-electrocatalytic system for converting CO2 to formic acid/methanol in CO2 capture and utilisation processes”. He is currently working as Technical Assistant at Department of Chemistry, Central University of Tamil Nadu, India. |
 Ragupathy Dhanusuraman | Ragupathy Dhanusuraman is Associate Professor at National Institute of Technology Puducherry, Karaikal, India. Previously, he worked as an Assistant Professor at Department of Chemical System Engineering, Keimyung University, Daegu, South Korea (2012). Between 2010 and 2012, he pursued as Postdoctoral Fellow and Post-Doctoral Researcher at Kyungpook National University & Pusan National University. He has 91 Peer-reviewed research articles, 4 Patents, and 5 book chapters and has received “Young Scientist Award – 2014” from Department of Science & Technology, Government of India, New Delhi. His recent research includes Nanotechnology for Energy, Catalytic and Biomedical Applications. |
1. Introduction
One of the significant challenges we face, inevitable in the 21st century, is the intensifying energy demand, depletion of fossil fuels and detrimental effects of the utilization of non-renewable resources on our environment.1–3 Renewable energy resources like solar, tidal and wave power are alternative energy sources for meeting energy demands, but they are erratic, owing to inconsistencies due to seasonal and regional variability.4,5 Electrochemical water splitting is considered friendly to the environment and a promising route to address the intermittency problem by converting electrical energy from renewable resources into chemical energy resulting in the electrocatalytic generation of H2.6 Hydrogen is termed as the future fuel7 because of its high gravimetric energy density (120 MJ kg−1) compared to gasoline (44 MJ kg−1), excellent energy conversion efficiency, environmental compatibility and zero carbon dioxide emission with a byproduct of just water.8–12 Moreover, Hydrogen has been applied in the synthesis of ammonia (Haber's Process), methanol synthesis, hydrocracking of crude oil, production of hydrochloric acid, and hydrogenation processes of oils and fats.13,14 As no natural hydrogen exists on the earth, hydrogen production is currently via steam reforming hydrocarbons at high temperatures and pressure, which inevitably leads to consumption of limited fossil fuels and carbon dioxide emission.15 Moreover, the H2 obtained by this method is accompanied by the oxides of C, N and S, which would poison the surface of the catalyst, reducing their cycle life.16,17 Other methods include photoelectrochemical water splitting, which utilizes photons to produce H2.18,19 Though greener and evolve pure H2, they make insufficient quantity per unit time attributable to their low solar to hydrogen (STH) conversion efficiencies and, as a result, cannot be replaced for bulk and immediate generation.20,21 Hydrolysis of metal hydrides and reactive metal can be utilized to produce large quantities of H2 rapidly.22,23 Nonetheless, their precursors are frequently toxic metals and are synthesized through fine chemical industries polluting the environment and cannot be opted as a greener method of production.24–27 Thus, water electrolysis is the only environmental friendly method to generate the immediate and large scale H2, and studies on improving the water electrolyzers performance by developing cost-effective electrocatalysts with excellent efficiency for the decomposition of water is a raging hotspot among researchers.28–30
2. Electrochemistry involved in water electrolysis
The electrolytic cell was first proposed in 1789 and composed of three parts: cathode, anode and an electrolyte.31 The reaction involved in electrochemical water splitting;| |
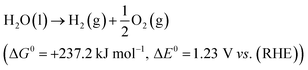 | (1) |
One mole of hydrogen and half a mole of oxygen gas are produced during the electrolysis of one mole of water. The process utilizes the thermodynamic potentials and the first law of thermodynamics. The pertinent values are taken from a table of thermodynamic properties, assuming the process at 298 K and one pressure atmosphere (Table 1).32
Table 1 Thermodynamic quantites of H2, O2 and H2O
| Quantity |
H2O |
H2 |
0.5O2 |
Change |
| Enthalpy |
−285.85 kJ |
0 |
0 |
ΔH = 285.83 kJ |
| Entropy |
69.91 J k−1 |
130.68 J k−1 |
0.5 × 205.14 J k−1 |
TΔS = 48.7 kJ |
Energy for the dissociation and the expansion of the gases that are produced must be supplied by the process, and they are accounted for in the change in enthalpy shown in the table above. Considering the system works at 298 K and one-atmosphere pressure;
| | |
W = PΔV = (101.3 × 103 Pa)(1.5 moles)(22.4 × 10−3 m3 mol−1)(298 K/273 K) = 3715 J
| (2) |
As the enthalpy H= U + PV, the change in internal energy U is then
| | |
ΔU = ΔH − PΔV = 285.83 kJ − 3.72 kJ = 282.1 kJ
| (3) |
The expansion of the gases produced occurs along with this change in internal energy; hence change in enthalpy represents the energy required to complete the electrolysis. Therefore
| | |
ΔG0 = ΔH − TΔS = 285.83 kJ − 48.7 kJ = 237.1 kJ
| (4) |
Water splitting is processed via two half-cell reaction; water reduction & water oxidation. Hydrogen evolution reaction (HER) is the reduction of protons at the cathode;
| | |
2H+(aq) + 2e− → H2(g)
| (5) |
and oxygen evolution reaction (OER) is oxidation of water.
33| | |
2H2O(l) → O2(g) + 4H+(aq) + 4e−
| (6) |
Subsequently water is highly stable and weakly conductive, its conductivity is enhanced by electrolytes like sulphuric acid, potassium hydroxide and potassium phosphate buffer making the medium acidic, basic and neutral.34
In acidic medium;35
| | |
Cathode: 4H+ + 4e− → 2H2, E° = 0.0 V
| (7) |
| | |
Anode: 2H2O → O2 + 4H+ + 4e−, E° = +1.23 V
| (8) |
In basic medium;
| | |
Cathode: 4H2O + 4e− → 2H2 + 4OH−, E° = −0.828 V
| (9) |
| | |
Anode: 4OH− → O2 + 2H2O + 4e−, E° = +0.401 V
| (10) |
In neutral medium
| | |
Cathode: 4H2O + 4e− → 2H2 + 4OH−, E° = +0.413 V
| (11) |
| | |
Anode: 2H2O → O2 + 4H+ + 4e−, E° = +0.817 V
| (12) |
Water electrolysis conducted in utmost pH conditions has its pros and cons. As plenty of protons are accessible for immediate charge and discharge in acidic medium, the efficiency of HER is better in acidic medium.36,37 Nonetheless, OER can be catalysed only by oxides of precious and expensive iridium (Ir) or ruthenium (Ru) alloys at minimum overpotential at this medium. It is crucial to note that only a small number of electrocatalytically active oxides exhibit metallic-type conductivity. With a relatively high electrical conductivity of 104 cm−1, both IrO2 and RuO2 exhibit metallic behaviour. This is due to the lack of a band gap caused by the relatively small electronegativity difference between oxygen and metal (less than 1.3 units). It is owing to weak oxygen binding and comparatively stronger hydroxyl (HO*) binding on the surface, both oxides exhibit lower over-potentials of O2-evolution than other metal anodes. Additionally, the –O, –OH and –OOH binding energies on the (1 1 0) surfaces satisfy universal linear relations comparable to those observed on metallic surface, such as RuO2 and IrO2.38,39 Thus OER in acidic medium remains a bottleneck for electrochemical researchers. The efficiency of OER is observed to increase drastically in alkaline medium utilizing oxides/hydroxides of less expensive 3d transition metals; the H2 production is, however, low as there is no instantaneous supply of H+ to cathode at high pH.40,41
Moreover, water splitting occurs at considerable cell potential (1.8–2.0 V) for commercial electrolysers, about 0.55 to 0.77 V higher than the theoretical value (1.23 V).42 This excess potential is obligatory to overcome the energy barrier of the reaction system, and it is referred to as overpotential. Overpotential may be due to the contact and solution resistance by the electrode, electrolyte, their distance, and circuit and by the activation hindrance from the kinetic steps in both cathode and anode. Thus to practically carry out the process of water splitting, potential higher than thermodynamic potential is required. A decrease in overpotential can be achieved by utilizing highly efficient, sustainable and eco-friendly HER and OER electrocatalysts & other resistances can be minimalized by augmenting the electrolyzer design, thus making electrochemical water splitting greener and affordable.43,44
3. Hydrogen evolution reaction (HER)
Despite the pH of the medium, the primary step in the multi-step HER is the adsorption of hydrogen on the surface of the electrode by the discharged protons. The reaction is stated as the Volmer reaction. The second step involves the desorption of H2 from the cathode either via electrochemical (Heyrovsky step) or via the chemical route (Tafel step), as observed in (Fig. 1).45 Between the two, the Tafel mechanism is more facile.
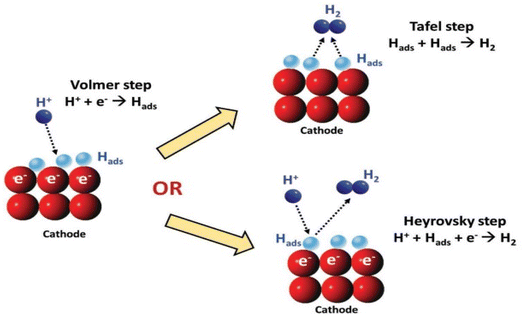 |
| | Fig. 1 A schematic illustration of a potential HER reaction pathway on cathode.48 | |
HER in acidic media (* stands for an active center on the electrocatalyst):46,47
| | |
H+ + e− → H*(Volmer)
| (13) |
| | |
H+ + e− + H* → H2(Heyrovsky)
| (14) |
| | |
(or) 2H* → H2(Tafel)
| (15) |
HER in alkaline media:
| | |
H2O + e− → H* + OH−(Volmer)
| (16) |
| | |
H2O + e− → H2 + OH−(Heyrovsky)
| (17) |
| | |
(or) 2H* → H2(Tafel)
| (18) |
The mechanism by which HER proceed is studied by the Tafel slope value is based on the coverage of the Hads. When the Hads surface coverage is low, Hads react with a proton and electron simultaneously since there are active sites adjacent to the adsorbed hydrogen on the cathode surface leading to Heyrovsky reaction. If the surface coverage is high, the adjacent two Hads combine, and H2 is generated.48,49 Tafel slope values are expressed as follows:
| |
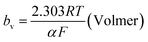 | (19) |
| |
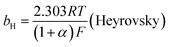 | (20) |
| |
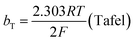 | (21) |
where
b stands for the Tafel slope (
bv: Volmer,
bH: Heyrovsky,
bT: Tafel)
α is the symmetry value 0.5,
R is the ideal gas constant,
T is the temperature, and
F is the Faraday constant. The quicker the kinetics of the reaction, the lower would the Tafel slope value.
50 The observed slope values are 11 mV dec
−1 for the Volmer step, 25–35 mV dec
−1 for the Tafel reaction and slope values greater than 39 mV dec
−1 are observed in the slower Heyrovsky reaction. Ultimately, the free energy for the adsorption of hydrogen (Δ
GH*) can be a favourable indicator for HER performance of an electrocatalyst. For instance, electrocatalysts with Δ
GH* value approx. zero, Pt-based exhibits the most efficient activity for HER.
If ΔGH* is too negative, the adsorbed H have difficulty desorbing from the electrocatalyst surface leading to slow Tafel or Heyrovsky steps. If ΔGH* is too positive due to the poor interaction of Hydrogen and electrocatalyst, on contrary is the Volmer step. Optimizing the design of the HER electrocatalyst depends on adjusting the value of ΔGH*.51
The HER activities of different metals are reflected in the Volcano relationship, which is experimentally plotted ΔGH* calculated from density functional theory (DFT) versus the logarithm of their corresponding exchange current densities (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) j0) of a wide range of catalysts. The so-called volcano diagram in (Fig. 2) is a simple approach to illustrate and compare and optimize the activity and design of different metals. Thus a good HER catalyst should have numerous active sites with optimal electron density for moderate bonding strength with adsorbed H(Sabatier principle), minimal charge transfer resistance between electrode–electrolyte interfaces, and should be unwavering in electrolyte medium.52,53
j0) of a wide range of catalysts. The so-called volcano diagram in (Fig. 2) is a simple approach to illustrate and compare and optimize the activity and design of different metals. Thus a good HER catalyst should have numerous active sites with optimal electron density for moderate bonding strength with adsorbed H(Sabatier principle), minimal charge transfer resistance between electrode–electrolyte interfaces, and should be unwavering in electrolyte medium.52,53
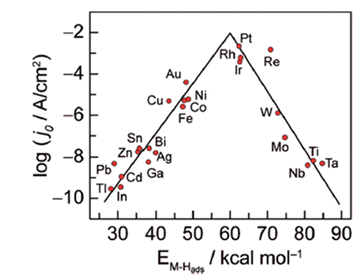 |
| | Fig. 2 Hydrogen evolution reaction (HER) volcano plot on metal electrodes under low pH environment. For each metal surface, the logarithm of the exchange current density j0 is plotted against M–H bond energy. Reproduced with permission2 Copyright 2010, American Chemical Society. | |
4. Oxygen evolution reaction (OER)
In comparison to HER, Volcano relationship reflects a sluggish reaction involving a multistep four-electron transfer process at the anode. In an acidic electrolyte, H2O is oxidized into hydrogen and oxygen, while in alkaline or neutral electrolyte, the hydroxyl ions are oxidized as oxygen and water. The kinetics with which the reaction proceeds are entirely dependent on the material by which the reaction is catalysed. Ir and Ru based catalysts show better efficiency in acidic than in alkaline medium. Transition metals like iron (Fe), nickel (Ni) and cobalt (Co) based catalysts catalyze OER better in alkaline medium.54,55 OER mechanisms in both acidic and basic mediums were reviewed in detail in 1986 by Matsumoto and Sato56 including the Yeager and wade, Krasil shchkov, Bockris57 and Hackerman58 pathway along with the renowned electrochemical oxide and oxide pathway. In basic medium, all the listed mechanisms commence with the coordination of hydroxide to the active site as their elementary step and would proceed through different steps. The overpotential is higher due to kinetic barriers in each steps of the reaction mechanism.58,59
OER in acidic media;
(i) Electrochemical oxide path
| | |
* +H2O → *OH + H+ + e−
| (22) |
| | |
*O + H2O → OOH* + H+ + e−
| (24) |
(ii) Oxide path
| | |
* + H2O → *OH + H+(aq) + e−
| (26) |
(iii) Krasil' shchkov path
| | |
* + H2O → *OH + H+(aq) + e−
| (29) |
(iv) Wade and Hackerman's path
| | |
2* + 2H2O → *O + *H2O + 2H+(aq) + 2e−
| (33) |
| | |
*O + 2*OH− → *H2O + 2* + O2(g) + 2e−
| (34) |
OER in basic media;
(i) Electrochemical oxide path
| | |
*OH + OH−(aq) → *O + H2O + e−
| (36) |
(ii) Oxide path
| | |
* + OH−(aq) → *OH + e−
| (38) |
(iii) Krasil' shchkov path
| | |
* + OH−(aq) → *OH + e−
| (41) |
| | |
*OH + OH−(aq) → *O− + H2O
| (42) |
| | |
*O + *O → 2* + O2(g)
| (44) |
(iv) Yeager's path
| | |
* + OH−(aq) → *OH + e−
| (45) |
| | |
*zOH → *z+1OH + * + e−
| (46) |
| | |
2*z+1OH + 2OH−(aq) → 2* + 2H2O + O2(g)
| (47) |
(v) Bockris path
| | |
* + OH−(aq) → *OH + e−
| (48) |
| | |
*OH + OH−(aq) → *H2O2 + e−
| (49) |
| | |
*H2O2 + OH−(aq) → *OOH− + H2O
| (50) |
| | |
*H2O2 + *OOH− → 2* + OH−(aq) + O2(g)
| (51) |
The mechanisms mentioned above match the experimental results of both hydroxide and oxide catalysts but shows inconsistencies in perovskite materials.60 18O isotope labelling and DFT studies are conducted to address these inconsistencies, and it was observed that some material prefer adsorbate evolution mechanism & some others prefer Lattice oxygen participation mechanism (LOM).61,62
Steps involved in AEM;
| | |
* + H2O → OH* + H* + e−
| (52) |
| | |
O* + H2O → OOH* + H* + e−
| (54) |
| | |
OOH* → O2(g) + H* + e−
| (55) |
Steps involved in LOM (Vo: oxygen vacancy due to lattice oxygen atom participation in OER)
| | |
OH* → (Vo + OO*) + H* + e−
| (56) |
| | |
(Vo + OO*) + H2O(l) → O2(g) + (Vo + OH*) + H+ + e−
| (57) |
| |
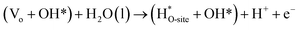 | (58) |
| |
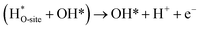 | (59) |
The usage of parenthesis is to indicate adsorbate of the same supercell. AEM and LOM pathways compete with each other and are based on the material used to synthesise of the electrocatalyst. In all the mechanisms, the active sites undergo cycles of oxidation & reduction in both acidic and basic mediums of OER, as shown in (Fig. 3). Thus metals with stable & variable oxidation states can behave as electrocatalysts for OER. Nonetheless, for all the possible OER mechanisms, the formation of intermediates O*, OH* and OOH* on the surface of the electrocatalyst is a prerequisite and behaves as the rate-determining step.63
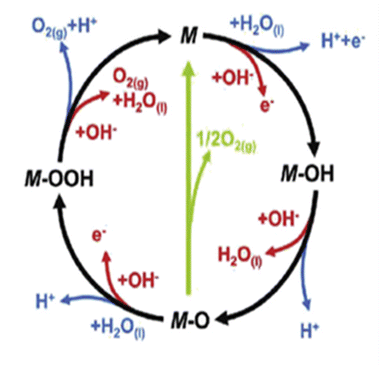 |
| | Fig. 3 The mechanisms governing the oxygen evolution reaction in acidic (blue line) and alkaline electrolytes, respectively. The corresponding black and green lines represent the two likely intermediates, M–OOH and M–O. Reproduced with permission64 Copyright 2020, Elsevier. | |
The free energy for the adsorption value of ΔG*OOH − ΔG*OH of an electrocatalyst has an impact on the OER activity as they provide the thermodynamic information between the intermediates as well as the electrocatalyst (too strong & weak bonds cause sluggish reactions), while the Tafel slope gives the kinetic information of the catalyst.64,65 Evolution of oxygen gas is generally from the oxide surface of the catalyst and not the bare surface, and thus the OER mechanism varies for different oxides having different surface morphology. Oxides with similar compositions portray different kinetics due to differences in preparation methods leading to different structures and thickness of their oxide layers.
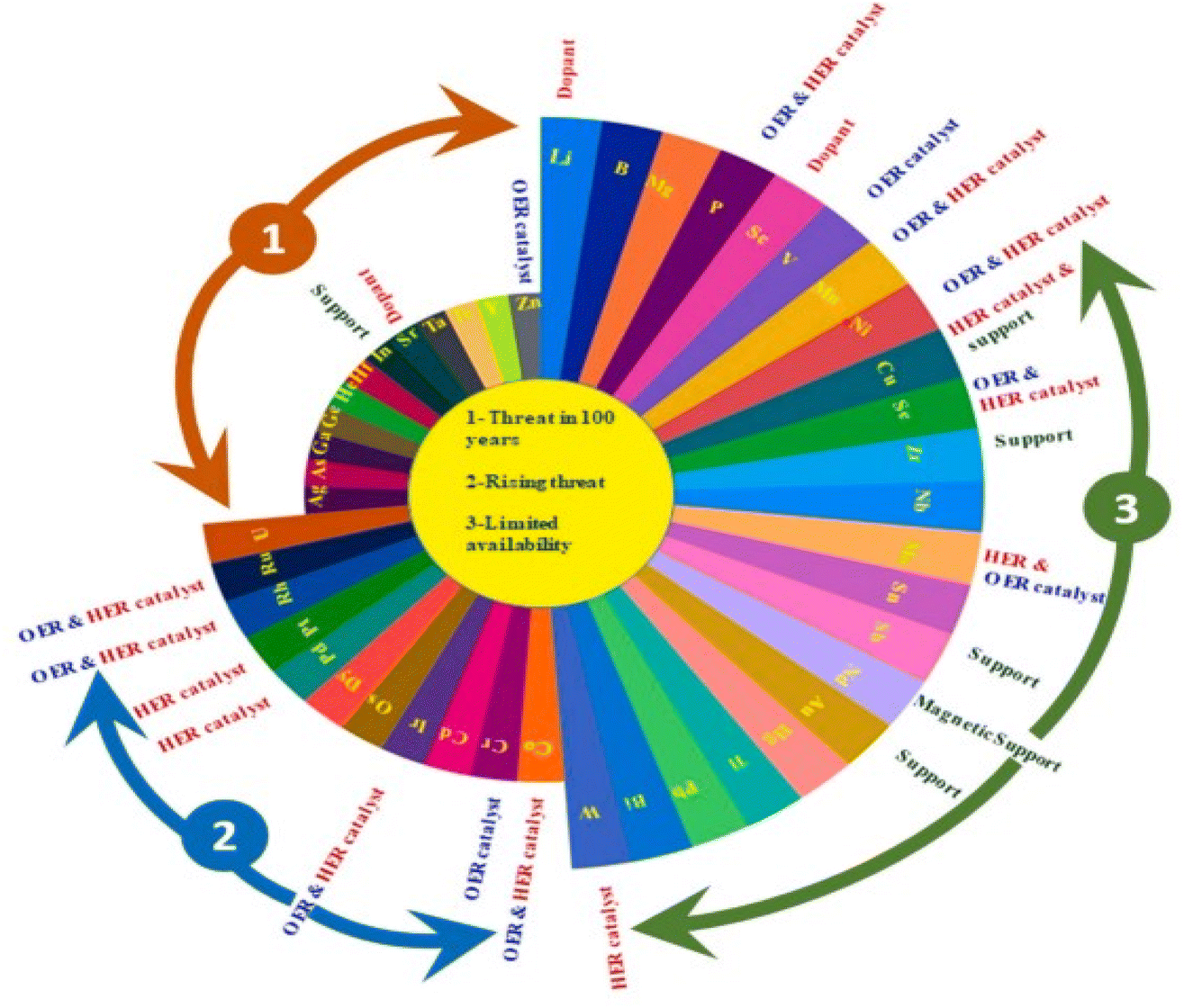
From the volcano plot portraying the activity O2 production from the surface of first row transition metal oxides in (Fig. 4), due to excellent electrical conductivity and low redox potential, metal oxides of noble Ru and Ir are on top of the plot. The drawback, however is their limited HER activity and being cost ineffective.56,66 Researchers are looking into affordable and sustainable metals electrocatalysts that demonstrate extraordinary activity for both HER and OER accompanied by stability and the list of elements suitable for water splitting are portrayed in (Fig. 5).67
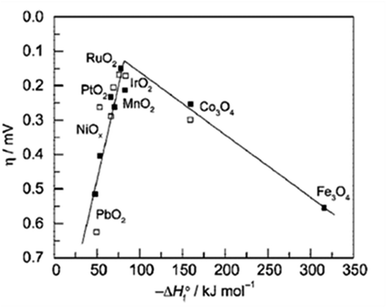 |
| | Fig. 4 Volcano plot demonstrates the relationship between the rate of O2 oxidation on transition metal oxide surfaces and change in enthalpy in acidic ( ) and basic ( ) and basic ( ) solutions. Measurement of overpotential in relation to 0.1 mA cm2 current density. Reproduced with permission2 Copyright 2010, American Chemical Society. ) solutions. Measurement of overpotential in relation to 0.1 mA cm2 current density. Reproduced with permission2 Copyright 2010, American Chemical Society. | |
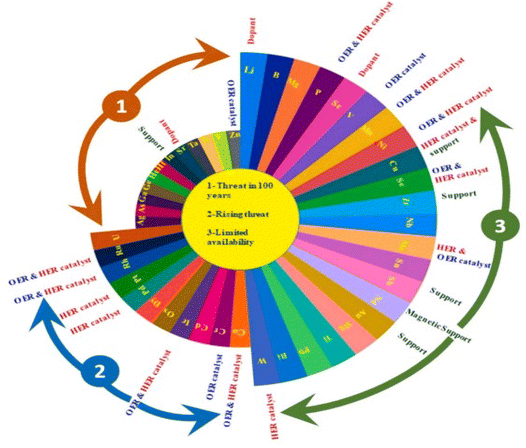 |
| | Fig. 5 Endangered species of elements and their potential application as electrocatalysts for water splitting, according to the aster diagram. Reproduced with permission67 Copyright 2022, Elsevier. | |
5. Parameters for the evaluation of electrochemical water splitting
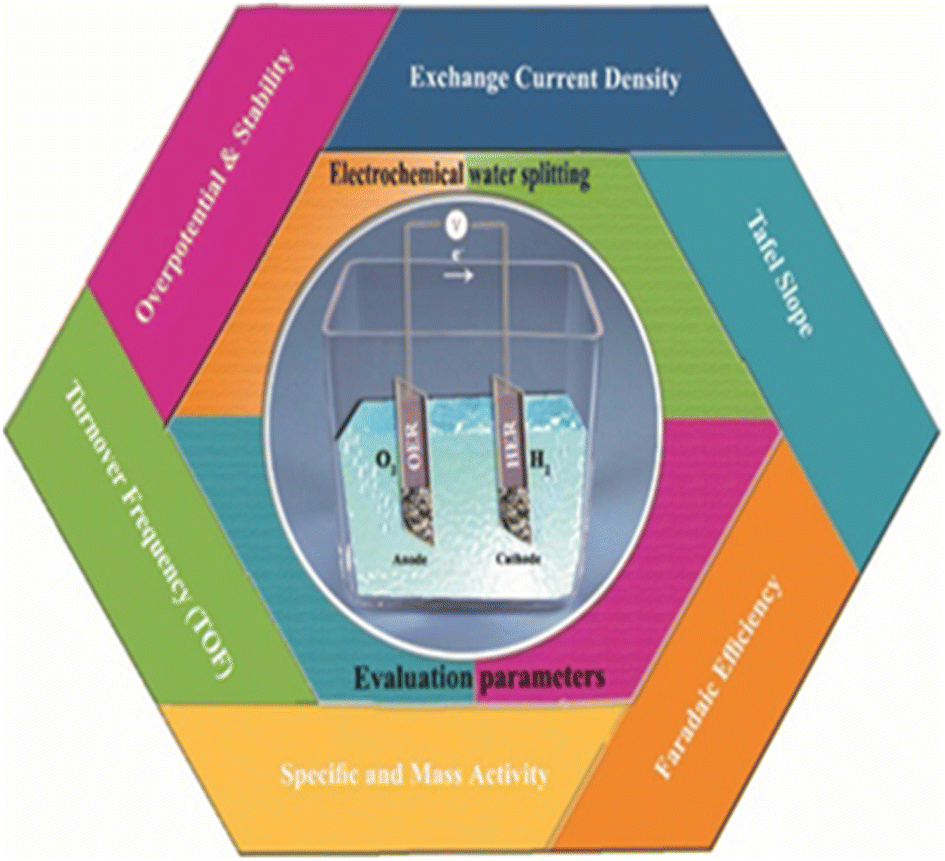
The main evaluation parameters for electrochemical splitting in (Fig. 6) include overpotential at a defined current density, electrochemical surface area (ECSA), Tafel slope, exchange current density, turnover frequency (TOF), faradaic efficiency, mass and specific activities. Tafel slope, exchange current density and TOF gives the kinetic activity i.e. how fast the reaction proceeds. All parameters however have both their merits and demerits.68,69
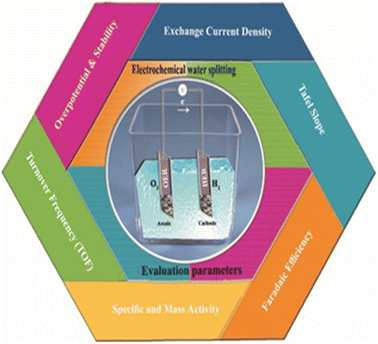 |
| | Fig. 6 The evaluation parameters of electrochemical water splitting. | |
5.1. Overpotential
As mentioned earlier, an extra potential is required to overcome the intrinsic kinetic hindrance in electrochemical water splitting (in both HER and OER) known as overpotential and is symbolized as η.58 The reversible thermodynamic potential for HER and OER are 0 and 1.23 V vs. the overpotential at desired current density without iR compensation are given by the equations; ηHER = ERHE − 0 V and ηOER = ERHE − 1.23 V.70–72 In commercial water electrolysis, the electrochemical setup works in a constant cell voltage which varies from 1.6 V to 2.0 V based on the electrocatalyst of HER and OER as well as the electrolyte pH. The accepted benchmark for current density is 10 mA cm−2 for comparing HER and OER electrocatalyst in mediums of different pH. The numerical figure of merit is the current density predicted for 10% competent solar to fuel conversion device, under 1 sun illumination.41,73 This benchmark as well as detailed convention for measuring the ECSA via CV as well as electrochemical impedance spectroscopy (EIS), faradaic efficiency using rotating ring disc voltammetry, catalytic activity using rotating disc voltammetry along with the long term cyclic stability of the catalysts in controlled electrolysis conditions was proposed by Jaramillo and co-workers.74
There are different types of overpotential in water splitting i.e., activation overpotential, ohmic overpotential, concentration overpotential, bubble overpotential and Kinetic overpotential of HER and OER.2
5.1.1. Activation overpotential. The kinetics of electrode at the reaction site determine the overpotential caused by the activation energy required for charge transfer, which is known as the activation overpotential. Thus it accounts for the electrochemical kinetics, electron migration, and proton migration. The Tafel equation can be used to calculate the anode activation overpotential (ηa) and cathode activation overpotential (ηC).| | |
ηa,a = aa + balnj
| (60) |
| | |
ηc,a = ac + bclnj
| (61) |
where ac and aa are the constants of the Tafel equation at the cathode and anode respectively, j is the current density (A m−2), and ba and bc are the Tafel slopes of the Tafel equation at the anode and cathode, respectively. By choosing an appropriate electrocatalyst, one could decrease the activation overpotential which is the inherent property of the electrocatalyst.42,75
5.1.2. Concentration overpotential. Concentration overpotential is caused by a rapid reduction in concentration at the interfaces due to concentration variance between ions in the bulk of the solution and on the electrode surface.The expression for j under pure mass transfer control can be written as
| |
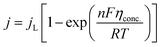 | (62) |
where
jL is the limiting current density and conc. is the concentration overpotential, the current at which the reactant's surface concentration falls to the limiting case of zero.
76 The anode and cathode concentration overpotentials make up this phenomenon, which is caused by mass transfer restrictions at higher current densities:
| | |
ηcon = ηacon + ηccon
| (63) |
where
ηacon is the anode concentration overpotential, and
ηccon is the cathode concentration overpotential, respectively. Due to the presence of a diffusion layer, it can be considerably decreased by agitating the solution.
5.1.3. Ohmic overpotential. The resistance between the surfaces and interfaces of the measuring system causes an additional voltage drop, resulting in the junction overpotential or resistance overpotential. Ohmic overpotential adheres to Ohm's law ηohmic = iR, where I is the applied current. According to this relationship, the ohmic drop changes linearly with the applied current. The ohmic overpotential can be expressed as| | |
ηohmic = ηabp + ηcbp + ηae + ηce + ηm
| (64) |
where ηabp is the ohmic losses of the anode bipolar plates and ηcbp, the ohmic losses of the cathode bipolar plates, respectively. The ohmic losses of the anode backing layers and the ohmic losses of the cathode electrode backing layers are denoted by ηae and ηce. The overpotential due to the polymer electrolyte membrane is ηm.Resistance overpotential could be excluded by performing ohmic drop compensation, also known as iR compensation which is available now in many electrochemical workstation.35 It could also be done manually by multiplying the resultant current density with uncompensated resistance (Ru) which gives a potential E. Thus iR drop should be subtracted from the experimental potential. Commercial water electrolyzers doesnot operate to the reported iR corrected overpotential in literature, thus its mandatory to report both iR uncompensated as well as compensated overpotential at the same current density.51,77,78
5.1.3.1. Proton transport and mass transport losses. The main losses occur in the middle of the operating range of PEM cell and are caused by the cell resistance of electrolyzer (Rcell) produced by electronic and ionic conduction. Application of Ohm's law is used to calculate the overall ohmic losses as mentioned above. Due to the high conductivity of frequently used materials, it is customarily believed that electron transport is significantly faster than protonic transport and that only ohmic losses resulting from proton transport through the PEM (the protons produced at the anode passes through the membrane and are transported to the cathode where they are reduced by electron) are taken into consideration.79 The ohmic overpotential with regard to proton transport across the membrane is given by;| |
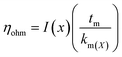 | (65) |
where x is the location in the membrane measured from the electrode-membrane interface, tm is the thickness of the membrane and the conductivity of the membrane is given as km which is further evaluated as| |
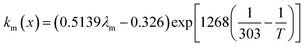 | (66) |
where λm is the water content in the membrane and the temperature of the electrolyzer is given as T.Since electrolyzers typically don't operate at high current densities, many authors neglect to take these mass transport limitations into account when calculating the major losses (such as the concentration losses), which usually occur at very high current densities.80 The mass transport losses are governed by the Nernst equation, which states that as the concentration of the product species at the reaction interface increases, the overpotential caused by mass transport limitation also increases. When the current density is high enough to prevent reactants from reaching active sites due to an excess of reacting molecules, mass transport losses take place, slowing down the rate of reaction. The Nernst equation can be used to calculate the mass transport overpotential (also known as the diffusion overpotential ηDiff).
| |
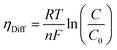 | (67) |
where
C0 is taking working concentration as reference concentration whereas
C is the hydrogen or oxygen concentration at the electrode-membrane interface. Until operating current densities are moderate, the mass transport losses are not significant. These losses can be neglected with no appreciable errors in voltage prediction at current densities of 1.6 A cm
−2.
81
5.1.4. Bubble overpotential. The bubbles (H2/O2) generated on the electrode's surface should not be avoided. While lot of the bubbles get away from the surface of the electrode & some cannot, as a result of which direct loss of effective active area, i.e. water is electrolyzed, and bubbles cannot be removed from the electrolytic system quickly; instead, they accumulate on the electrode surface or disperse in the electrolyte. The phenomenon, known as the “bubble effect,” would result in a high overpotential and a significant ohmic voltage drop.82Current density for gas evolution is given as
| |
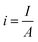 | (68) |
where
A is the geometric surface area and
I is current intensity. According to the Tafel equation, the reaction overpotential is
| |
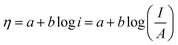 | (69) |
Since the active surface area is reduced by the bubble coverage, the overpotential (ηθ) and the real current density iθ
| |
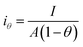 | (70) |
| |
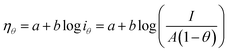 | (71) |
where
θ is the bubble coverage ratio on the electrode surface, 0 <
θ < 1. Severe bubble coverage decreases the electrode's effective active area. As a result, according to the Tafel equation,
iθ grows to be larger than
i, and the reaction overpotential also increases.
42
5.1.5. Kinetic overpotentials of HER and OER. An exponential relationship between the overpotential and the rate, or current density, j, of the desired reaction results from assuming an Arrhenius-type relationship for the interfacial charge-transfer kinetics.| |
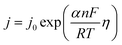 | (72) |
The exchange current density which is the current density that flows equally in both forward and backward direction at equilibrium is represented as j0, gas constant, Faraday constant and absolute temperature is represented as F, R and T respectively. α signifies the portion of the overpotential that lowers the reaction's kinetic barrier at the electrode/electrolyte interface.83 For every electrode and chemical reaction of interest, j0 and α are different because they depend on the materials. As a result, the overpotentials at all of the electrodes in an electrolyzer's electrolysis cell add up to form the device's overall kinetic overpotential. In comparison to electrocatalysts in contact with neutral electrolytes, the kinetic overpotentials for the water-splitting half-reactions are typically lower for electrocatalysts in contact with strongly acidic or strongly alkaline electrolytes. Electrocatalysts for the HER exhibit much lower overpotentials in acid than at neutral pH because it is simpler to reduce a positively charged proton than to reduce a neutral species, such as water. Electrocatalysts for the OER, however, display much lower overpotentials in alkaline media than at neutral pH because oxidation of a negatively charged hydroxide ion is easier than oxidation of a neutral species, water.84 The polarization curve obtained by plotting current density vs. overpotential is used to calculate the overpotential of a given current density, if value of the overpotential is lower, higher is the performance of the electrocatalyst. As the kinetics of HER are faster than that of OER, the activation overpotential has priority over the other overpotential as is referred to as onset overpotential. In the case of OER, other overpotentials are also important. Since, the proposed mechanisms of OER in both acidic and basic medium are multistep, Man et al. along with others proposed an expression for calculating the theoretical overpotential at U = 0 vs. SHE by studying the thermodynamics of the OER mechanism.59
| |
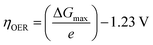 | (73) |
This theoretical value had huge difference compared to the experimental values of the standard free energy change related with the elementary step of conversion of peroxide form oxide. Thus, it's clear the kinetic constraints are not taken into consideration while studying thermodynamics of the overpotential. Due to the reason stated above, instead of an onset overpotential, overpotential (η10) at a fixed current density like 10 mA cm−2 is considered a quantitative parameter to evaluate an electrocatalyst for both HER and OER.85,86 Materials having intense redox peaks i.e. current density greater than 10 mA cm−2 and catalysts like layered double hydroxides which exhibit high performance, i.e. current density more than 500 mA cm−2, overpotentials at higher current densities such as 50 mA cm−2 and 100 mA cm−2 are used as alternative parameters.87–89 Overpotential of a catalyst is mass dependent i.e. it varies with different mass loading.90–92
5.2. Tafel slope and exchange current density
Tafel slope and exchange current density are the two kinetic parameters attained from Tafel plot. Tafel plot is retrieved by replotting the polarization curves i.e. LSV (linear sweep voltammogram) as log current density (j) versus overpotential. It is defined as the dependence of the current density with the iR compensated overpotential and is derived from the Butler–Volmer equation which gives the relationship between the overpotential and current at the electrical interface.93 It is represented as follows:| |
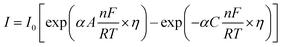 | (74) |
Here, I stands for the current density, I0 is the exchange current density i.e. the current at equilibrium potential, n is the number of electrons involved in the reaction (2 in case of HER and 4 in case of OER), F is the Faraday constant (96485C), T is the absolute temperature in Kelvin, η is the overpotential, αA and αC are the charge transfer coefficients for anodic reaction (OER) and cathodic reaction (HER) and R is the ideal constant. Tafel equations of anodic and cathodic polarization curves are the result of the high overpotential approximation of the Butler–Volmer equation.94 They are stated as follows:| |
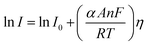 | (75) |
| |
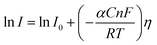 | (76) |
Both equations are in the form of y = b + mx and would give linear line on plotting logI vs. η and their slope for anodic and cathodic polarizations are
| |
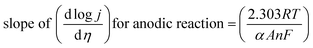 | (77) |
| |
 | (78) |
Tafel slope is related inversely to charge transfer coefficient (α) and number of electrons transferred, information about the charge rate kinetics and the reaction mechanism can be deduced. The smaller Tafel slope value indicates that increasing the same current density would need smaller overpotential suggesting a faster reaction rate.53 Usually the LSV procured at lowest possible scan rate gives a Tafel slope value with negligible experimental inaccuracies. The scan rate used for acquiring a Tafel plot would cause an issue only if the catalyst exhibit very high capacitive behaviour. This ultimately causes large error in the determination of the exchange current density. Exchange current density is obtained via the extrapolation of the linear fit which intersects at a point where equilibrium potential (zero potential) of the electrocatalytic process meet the respective current density in logarithmic scale. This current density is referred to as exchange current density. Higher the exchange current density, better is the electrocatalyst, it gives an insight on the intrinsic rate of reaction transfer between electrode and electrolyte under equilibrium conditions and is dependent on the electrocatalyst material, analyte in solution as well as the temperature.95,96 The electron transfer efficiency across the catalytic interface will be facile when exchange current density is high, which means very small activation energy is required. Hence the overpotential would be low. In case of electrochemical water splitting, at the potentials of interceptions of 0 and 1.23 V vs. RHE, the exchange current densities for HER and OER are calculated. Thus for electrocatalyst to be ideal for water splitting, they should have low overpotential, small Tafel slope value and a large exchange current density.97
As mentioned earlier in this review, Tafel slope is also correlated to the mechanism of HER, Tafel mechanism have lower Tafel slope value compared to Heyrovsky.98 For Tafel mechanism, the surface active sites separation should not be farther than van der Waals radius of the two adsorbed hydrogen atoms; i.e. smaller the space between them, greater would be the chance of chemical desorption. Comparatively, the Volmer–Heyrovsky mechanism takes place when the electroactive sites are relatively less available and the distance between adjacent sites is greater than the van der Waals radius of the adsorbed two hydrogen atoms. Due to this reason it unlikely for Tafel mechanism to occur. In case of OER, there are a number of mechanisms having different possible elementary steps with any of them being the rate determining step that changes as the pH of the medium changes. Though it is difficult to conclude the mechanism undertaken based on the Tafel slope value, it would be possible to predict the number of electrons transferred. This can be clearly understood from the Tafel slope values of IrO2 and RuO2 that are in between 30–50 mV dec−1 (acidic medium) and 60–120 mV dec−1 in case of the oxides/hydroxides of first row 3d transition metals like Fe, Co and Ni (basic medium). This is because the oxidation state of IrO2 and RuO2 changes from 4+ to 6+ by releasing two electrons per site during OER whereas the oxidation state of M(OH)2 (M = Fe, Co and Ni) changes from +2 to +3 by releasing one electron per site.71
There are different ways in extracting Tafel plots for both HER and OER. The electrochemical techniques used are voltammetry, chronopotentiometry, chronoamperometry and EIS. Tafel plots from voltammograms are in fact not the real steady state polarization curve as the overpotential and current obtained are not retrieved at steady state, instead they were changing as the time goes on increasing. These plots also retained the current contribution from the interface capacitance to the evolution of gas. Due to these demerits, researchers are giving importance to study steady-state polarization curves utilizing static electrochemical techniques like potentiometry and amperometry. After allowing sufficient time for the electrocatalytic interface to reach steady state, the current density and potential are measured for plotting the Tafel curves. The Tafel plot extracted from the steady state methods didn't alter the Tafel slope values, but a significant change in the exchange current density was observed due to the exclusion of the capacitance current in the chronoamperometric measurements.71
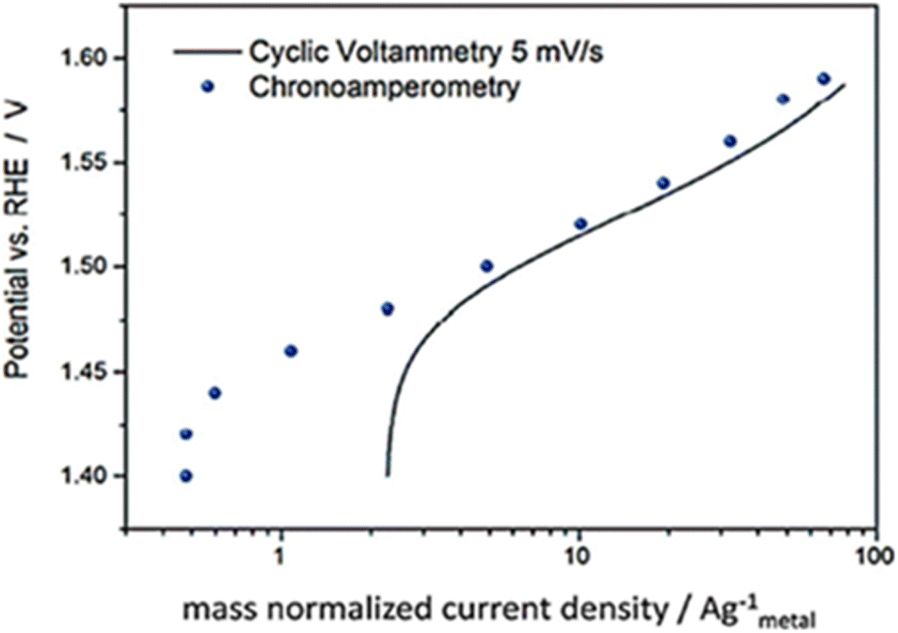
Based on the review by Fabbri et al., Tafel plots of IrTiO2 electrode obtained by voltammogram recorded at 5 mV s−1 in (Fig. 7) showed to be in agreement with amperometric measurements.58 Though these steady state electrochemical technique seem accurate in comparison to voltammetry, they too have issues. The current may be free of capacitance current and is obtained in steady state of potential, yet the different types of resistances like intrinsic resistance, solution resistance, electrode–electrolyte resistance and other resistances that occur due to the hetero-junction in the series were not eliminated. Hence the intrinsic and the real activity, the kinetics of the electrocatalyst is not correctly studied from the Tafel parameters gained via this method.
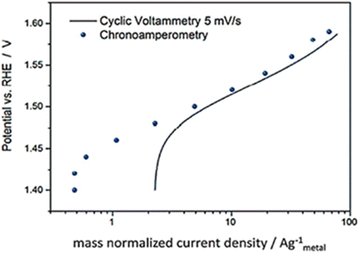 |
| | Fig. 7 IrTiO2 electrode's Tafel curves were obtained using both chronoamperometry in 0.1 M perchloric acid and CV at 5 mV s−1. Reproduced with permission from ref. 58 Copyright 2014, Royal Society of Chemistry. | |
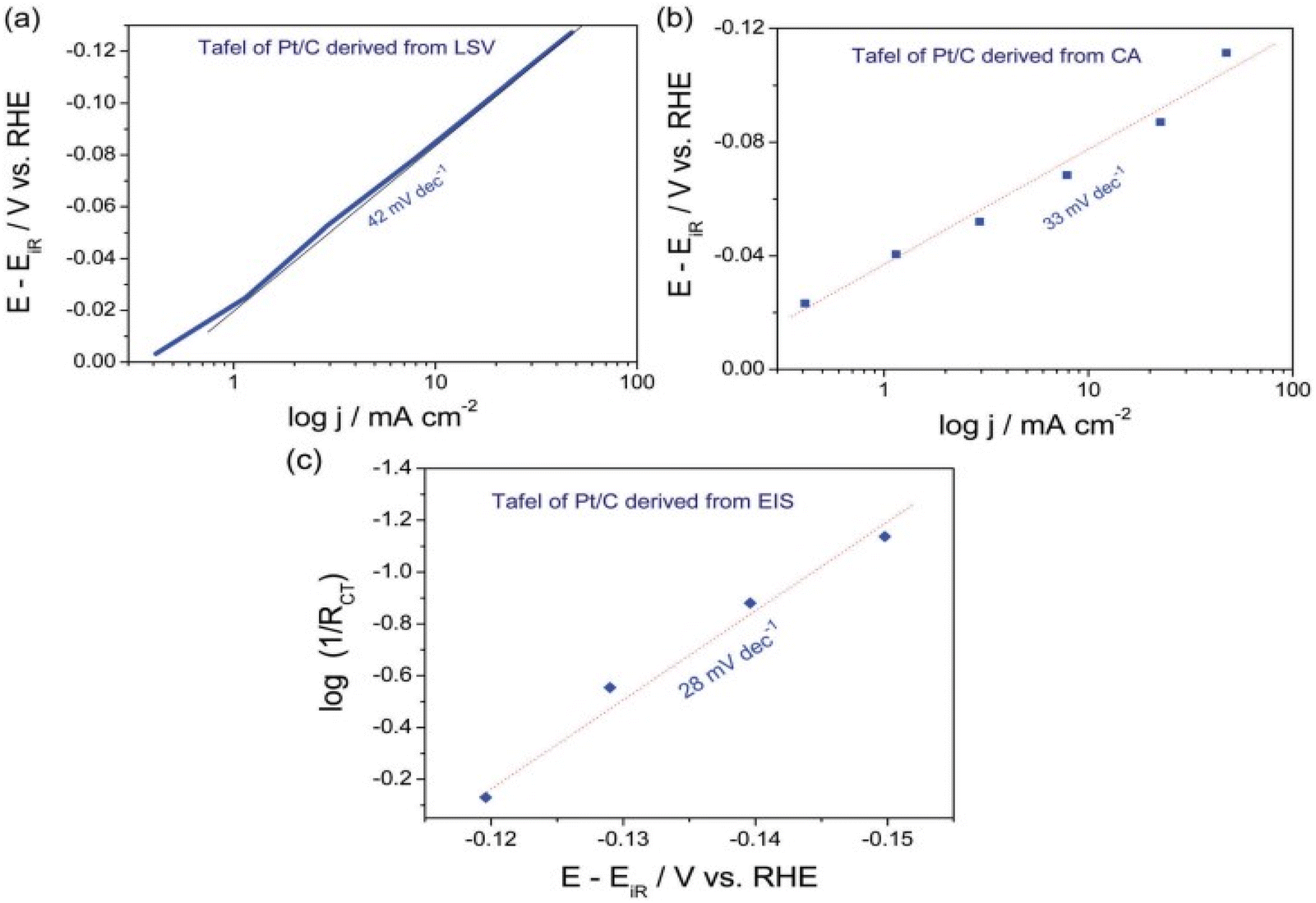
Tafel parameters from EIS technique was studied extensively by Hu and co-workers in 2012 who proposed a method to neglect the series resistance (Ru) and catalysts intrinsic resistance.99 EIS measurements were performed at different overpotential in intervals varying with catalyst activity range with an AC sine wave of amplitude ranging 10 to 50 mV from the operational overpotential. The Nyquist plot of log charge transfer resistance (Rct) vs. the overpotential gives the Tafel slope reflecting the kinetics of the catalyst excluding the influence of series resistance as it depends only on the charge transfer ability of the catalyst. S. Anantharaj et al. plotted the Tafel curves of commercial Pt/C 20 wt% in all three methods as seen in (Fig. 8). The Tafel slope observed in LSV recorded in 0.5 M H2SO4 at scan rate of 5 mV s−1 is 43 mV dec−1 and that derived chronoamperometry & EIS under the similar experimental circumstances are 33 and 28 mV dec−1. The theoretical Tafel slope value of Pt/C is 30 mV dec−1 and among the three, EIS derived Tafel curve exhibited the closest value. Thus EIS method gives the exact information of the electrocatalytic activity in terms of Tafel slope and is superior among the three. The precise exchange current density of the catalyst is established by the 1/Rct obtained at the onset overpotential. Based on the necessities and conditions, any one of these approaches can be opted for finding the Tafel slope of an electrocatalyst. The important steps to be followed regardless the method opted for Tafel analysis are; the potential of the Tafel region should be greater than at least 0.12 V from the onset potential of HER and OER, minimum 2 to 3 decades should be present within the linear portion of the slope being measured. The third condition applicable for reversible/quasi reversible redox reactions i.e. attaining a minimum of 1/10 of the limiting current isn't relevant in the case of HER and OER as they are irreversible redox reactions governed by the charge transfer.68
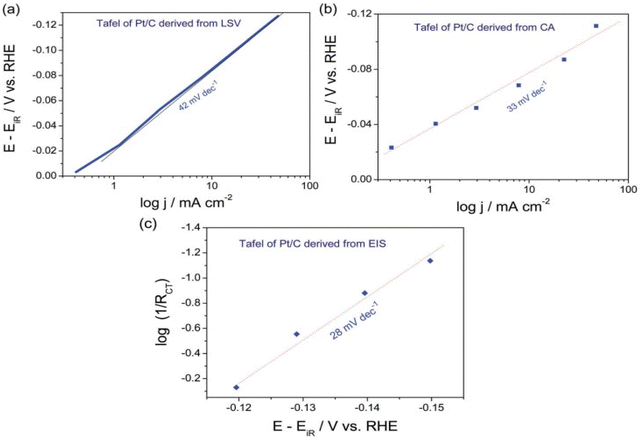 |
| | Fig. 8 (a) Linear sweep voltammograms recorded at 5 mV s−1 in 0.5 M H2SO4 exhibits the Tafel plot of Pt/C 20 wt% catalyst (b) & (c) are Tafel plots of the same obtained using Chronoamperometry & EIS methods, respectively,under similar experimental circumstances. Reproduced with permission from ref. 68 Copyright 2018, Royal Society of Chemistry. | |
5.3. Stability
Stability of an electrocatalyst is a crucial parameter determining its promotability to commercial level large scale operations by assessing their activity with a long term operation period. The stability of HER and OER is studied by the cycling of the catalyst within the potential window via two techniques: cyclic voltammetry (CV) or linear sweep voltammogram (LSV) at higher scan rate, also known as accelerated degradation test & prolonged potentiostatic or galvanostatic electrolysis measurements.34 The number of cycles determining the stability of the catalytic material is different in case of OER and HER. In HER, over thousands of cycles can be carried out as the polarization curve starts from 0 V vs. NHE. The accelerated degradation test beyond 1000 is unlikely to be observed in case of OER, as reports on OER catalysts having extreme stability (beyond 250–1000 cycles) are rare. The shift in both onset potential as well as overpotential at a defined current density of 10 mA cm−2 are indicative parameter of electro catalyst. It is considered apt if the overpotential does not increase more than 30 mV and if its final activity degradation after running the stability tests does not exceed 5%. A stable current density (e.g.: 10 mA cm−2 and 50 mA cm−2 or 100 mA cm−2 for high performance/catalysts with strong redox peaks) for over 12 h in chronoamperometric measurements or negligible increase in the overpotential at a current density of 10 mA cm−2 for more than 12 h in chronoamperometric measurements for an electrocatalyst is also another widely accepted parameter for analyzing the stability. The substrate, electrode being used, the catalyst fixation method all contribute to the stabilization of the electrocatalyst being studied.98
5.4. Turnover frequency (TOF)
TOF is another kinetic parameter that tells how fast the electrocatalyst can catalyze the electrochemical reaction at a defined overpotential. It is defined as the number of moles of O2 or H2 gas evolved at each available catalytic site. Both HER and OER undergo pseudo first order kinetics, thus both of them have TOF value in terms of time. TOF is independent of mass loading but is highly dependent on the high coverage, i.e. shows linear relationship only if the coverage is below 100%.99–101 Higher the TOF value better the catalyst. | |
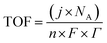 | (79) |
Here, j stands for current density (A cm−2), n is the number of electrons transferred (2 for HER and 4 for OER), NA is Avogadro number, F is the Faraday constant, Γ is the total/surface concentration of active sites of the catalysts or the number of participating atoms in electrocatalyst material. The challenges of estimating surface concentration render down the usage of this method as kinetic parameter. There are different methods in determining the total/surface concentration of catalysts. In the first, the surface concentration can be calculated from the redox peaks in the cyclic voltammogram after the activation of the catalyst by CV cycling. The TOF values of 3d transition metals in OER are determined from the redox peak in the CV. The surface active concentration of the metal sites is calculated from the region beneath the redox peak for the formation of oxyhydroxide from oxide/hydroxide. Prior to the onset of OER, metal sites that are undergoing in situ oxidation and reduction are thought to be involved in the catalysis.102,103 The total concentration of atoms can also be calculated using Avogadro number method utilizing the average particle diameter of the catalyst. When the catalyst surface is even and smooth or have sheet like morphology, one can use another method where it's assumed to have monolayer.36 All the three listed method have their demerits. Calculating TOF value from CV could lead to potential error if the entire catalyst is not activated, if the total current is not only contributed by the faradaic reaction but also have non-faradaic component. The problem can be rectified by using the ORR current measured during RRDE (rotating ring disk electrode) experimental in correspondence to OER at the disk and concurrent ORR at the ring is used. The current measured at ORR at ring electrode is solely due to O2 generated from the disk electrode giving more accurate TOF value. In case of HER, techniques like CO stripping, hydrogen under potential deposition (H-UPD), peroxide reactions are used. In the second method the atoms in the core are also taken into consideration though they donot contribute to the catalytic activity thus the exact catalytic property is not reflected and the TOF calculated differs by the order 102 to 103. The third method would lead to error when the material is uneven or may degrade under severe electrochemical conditions. Samar K et al. utilised the galvanostatic Tafel plots to calculate the relevant turnover frequency (TOF) values,104 using the formulawhere current value was acquired from the chronoamperometry electrolysis was represented as I, n is the number of active sites (mol), F is the Faraday constant (C mol−1), 2 represents the number of electrons needed to create one mole of hydrogen; the number of active sites m is calculated as follows;| |
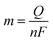 | (81) |
Q is derived by integrating the current vs. time plot produced from the trace of the cyclic voltammogram| |
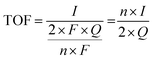 | (82) |
The method adopted for TOF calculation should be appropriate for the nature of the catalyst.105
5.5. Faradaic efficiency
It is the efficiency of electrocatalyst for electron transfer provided by the external circuit across the electrode-electrolyte surface which promote the electrochemical reaction i.e. HER and OER. There are two different ways in determining it, either via gas chromatography (GC) for both HER and OER and rotating ring disk electrode (RRDE) for OER to measure the amount of O2. If the reaction course is studied using GC, one could gather information on the how the reaction rate of gas evolution reaction is affected with the change in applied potential by associating the results with the calculated theoretical amount of the evolved gas at certain time interval.
To analysis the FE of an electrocatalyst of OER in both acidic and basic medium, RRDE method is used. In this technique, the O2 molecule evolved at the disk electrode are trapped and reduced at which the potential is swept at a very low scan rate of 5 mV s−1 by keeping potential of the ring electrode constant where complete reduction of O2 to water is possible. The potential of Pt ring is set at constant potential of ORR which varies depending on the pH of the solution. The setup includes glassy carbon disk and Pt ring/Au disk and Pt ring electrode. Faradaic efficiency of an OER electrocatalyst using RRDE is determined using the equation:
| | |
FE = jR × nD/(jD × nR × NCL)
| (83) |
In the above equation, jR is ring current density, nD is number of electron transferred at the disk electrode (4), jD is the current density of disk electrode and nR is number of electrons transferred for reduction of O2 (4, since the ring electrode is Pt and thus electron pathway probable for ORR on Pt surface) and NCL is collection efficiency of ring electrode. FE along with other parameters are mandatory for the screening of HER/OER/water splitting electrocatalyst as it reflects the selectivity of the electrocatalyst. FE is exempted from experimental inaccuracies as they are unaffected by the shape, size, and morphology of the catalyst.105 Quite recently, researchers have tried fluorescence spectroscopy for the measurement of gas evolved, at periodic intervals during galvanostatic or potentiostatic electrolysis instead of GC for both HER and OER to determine FE of their electrocatalysts. Here the oxygen evolved is excited from doublet to singlet state and relaxed back by fluorescence. The intensity of the fluorescent light is the direct measure of the amount of O2 evolved. The majority of faradaic loss are contributed by the formation of by products or due to heat loss.35,53 faradaic efficiency can be defined as the ratio of experimentally evolved volume of gas value (hydrogen or oxygen) to theoretically calculated volume of gas value.
| |
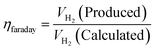 | (84) |
Assuming a 100% faradaic efficiency, the theoretical volume of gas can be calculated using the Faraday's second law, based on the current density, electrolysis time, and electrode area, as shown in eqn (85). when the water–gas displacement method or a gas chromatography analysis can be used to determine the practical amount that the experiment produced.
| |
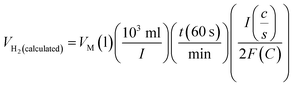 | (85) |
The theoretical hydrogen yield VH2, the ideal gas expression (VM = R(273 + T)/P) is represented as VM, ideal gas constant (0.0821 atm K−1 mol−1), pressure (atm), temperature is denoted as R, P, t respectively; Faraday's constant (96485C mol−1) and applied current (A) is represented F and I. By computing the electrolyte volume reduction during the electrolysis process at the operating temperature and pressure, the volume of hydrogen can be determined. Due to the hydrogen gas being assumed to be a perfect gas to facilitate calculations, the real volume (VH2 (produced)) will be as described in the following equation under normal circumstances.
| |
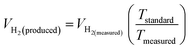 | (86) |
where
VH2(measured) is the volume obtained
via alkaline water displacement, the operating temperature and the standard temperatures in Kelvin is depicted as
Tstandard and
Tmeasured respectively.
106,107 The methods utilized for the calculation of FE depends on the demands of the catalysts. An electrocatalyst would become suitable for commercialization for water splitting if the FE is atleast 90%.
5.6. Mass and specific activities
The catalytic activity of the electro catalyst are also given by two other parameters; mass and specific activity.58,108 Similar to TOF, both are acquired at a particular overpotential. Mass activity is the overpotential at the current density normalized by mass and is expressed as A g−1 while the overpotential at ECSA or BET normalized current density is specific activity. At present, current density normalized by geometrical area are being used but they reflect the area of electrodes with planar and smooth surfaces only. This does not become applicable for electro catalysts with uneven or roughened surface area such as in situ grown catalysts or electrodes modified with nano powders. N2 gas adsorption and desorption measurements are used in BET (Brunauer–Emmett–Teller) adsorption isotherm studies to determine the real surface area. The measurements were made such that N2 are adsorbed and desorbed on all the sites, yet all these sites cannot be electrochemically active. The results acquired by this method have high chances of having inaccuracies, thus ECSA normalized activity is gaining attention over BET. ECSA compared to geometrical surface area is more accurate as they give an insight about the inherent catalytic property of the material of the catalyst. Their disadvantage is that there are number of methods for ECSA determination and the measured values varies from method to method. Thus ECSA normalized activity alone cannot be considered as an ideal parameter, but it along with other essential parameters can be used for the evaluation of the catalyst for water splitting.
6. Periodic classification of metal electrocatalysts of electrochemical water splitting
In this section of the review, elements acting as dopants, electrocatalysts exhibiting electrocatalytic activity for HER, OER and overall water splitting are discussed as per their periodic classifications below.
6.1. s- Block elements
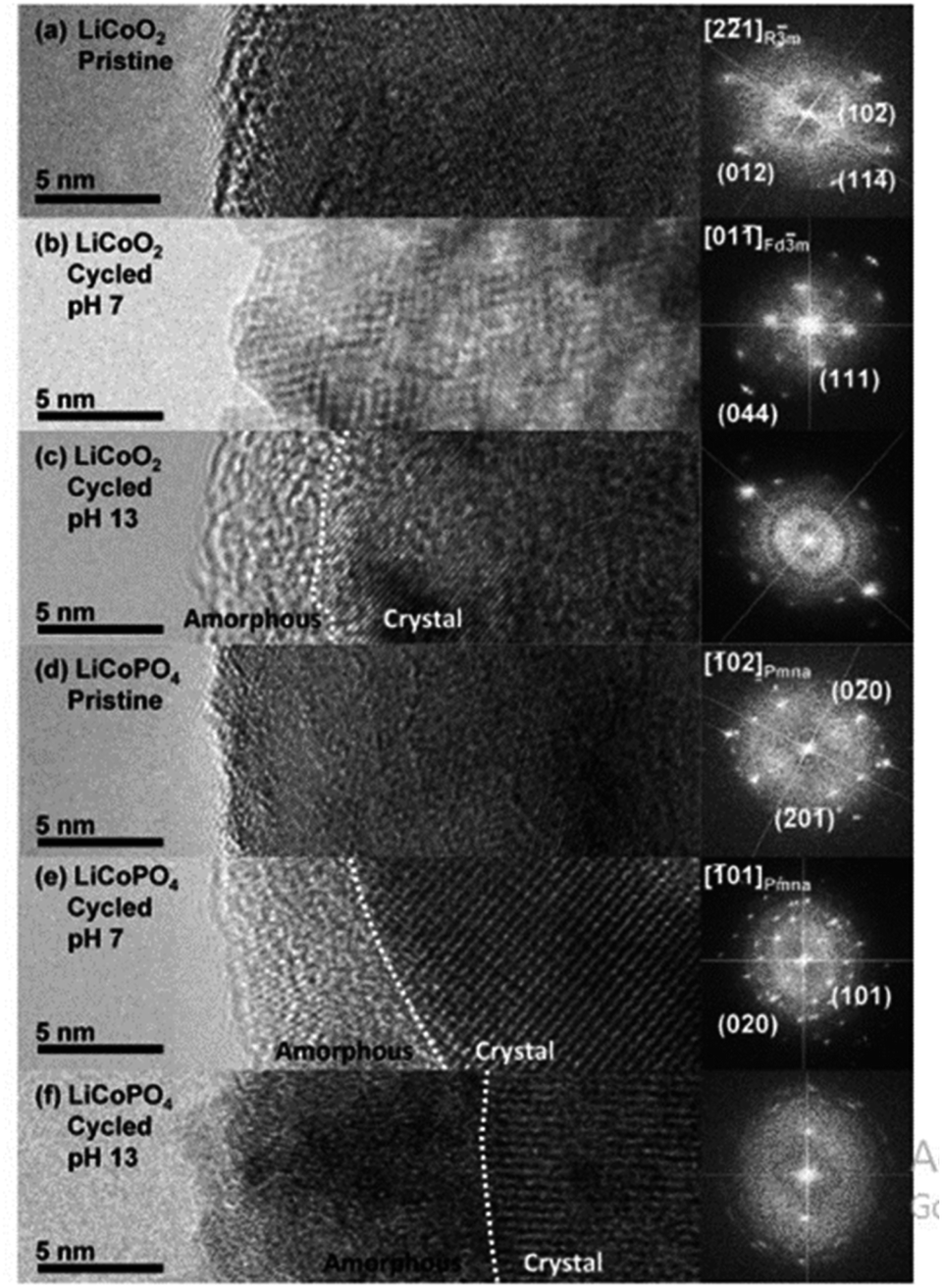
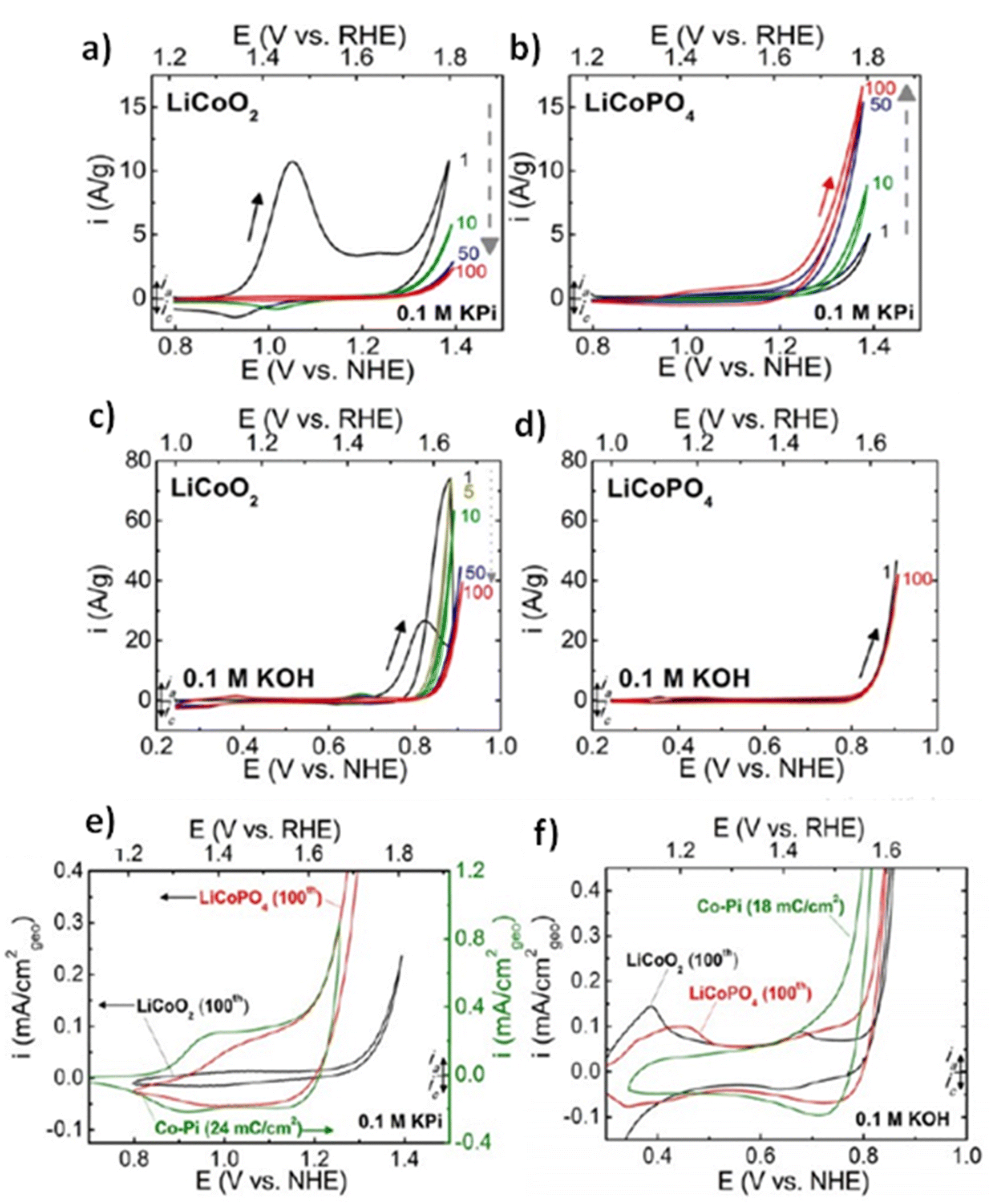
All elements of s block are metals, have low ionization enthalpies, are highly reactive and readily form ionic compounds. They have the ability to be easily oxidized in the presence of air and form oxides and have highly negative standard potential, thus acting as strong reducing agents. Owing to these properties, alkali metals and alkali earth metals have been used in many energy storage and conversion applications. Lithium plays essential and integral part in today's technology due to their small ionic size and negative electrode potential, increasing their ability to store more energy and hence act as ionic mediator in LIBs, Li–S etc.109 For electrochemical water splitting, Li incorporation of lithium as a dopant is seen to boost electrocatalysis. The super Lewis acidity of Li+ accelerated the hydrogen evolution activity in case of Li+–Ni(OH)2–Pt.110 Approximately a 10 fold of total increase in activity was seen due to the generation of hydrogen intermediates which increased due to the Li+- induced destabilization of HO–H bond. The high HER activity of LixMoS2 is because of key role played by Li adsorption. Upon the intercalation of Li, a phase transition from 2H- to 1T′-MoS2 occurs; altering conducting properties. The adsorption of Li direct ΔGH* in a favorable direction, promoting HER active edges sites.111 LiCoO2 is a non-precious metal-based catalyst and a prototype cation-exchanged layered metal oxide which is being used as a well-known cathode material for Li-ion batteries, which has its application in OER.112 Study conducted by Shao-Horn et al. on lithium cobalt oxide and phosphate (LiCoO3 and LiCoPO4) water splitting activity in neutral medium, further proved the importance of Li in the electrocatalysis of water splitting, their structure as well as their electrocatalytic activities are observed in (Fig. 9) and (Fig. 10) respectively. Upon cycling in neutral pH LiCoO2 and LiCoPO4 underwent deactivation and activation respectively coincidental with the morphological distortion leading to spinel like surface for LiCoO2 and amorphous surface for LiCoPO4.113
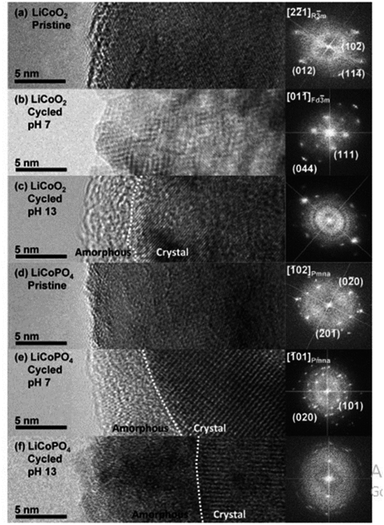 |
| | Fig. 9 The surface regions of LiCoO2 and LiCoPO4 are shown in HRTEM images and (right) FFTs in the following order: (a) pristine LiCoO2; (b) cycled LiCoO2 in 0.1 M KPi (pH 7) (c) cycled LiCoO2 in 0.1 M KOH (pH 13); (d) pristine LiCoPO4; (e) cycled LiCoPO4 in 0.1 M KPi (pH 7); (f) cycled LiCoPO4 in 0.1 M KOH (pH 13). The interface between amorphous and crystalline areas is denoted by white dashed lines. The electrodes underwent 100 uninterrupted cycles at a scan rate of 10 mV s−1 at 900 rpm between 1.2 and 1.8 V vs. RHE in 0.1 M KPi at pH 7 and between 1.0 and 1.7 V vs. RHE in 0.1 M KOH at pH 13. Reproduced with permission from ref. 113 Copyright 2018, American Chemical Society. | |
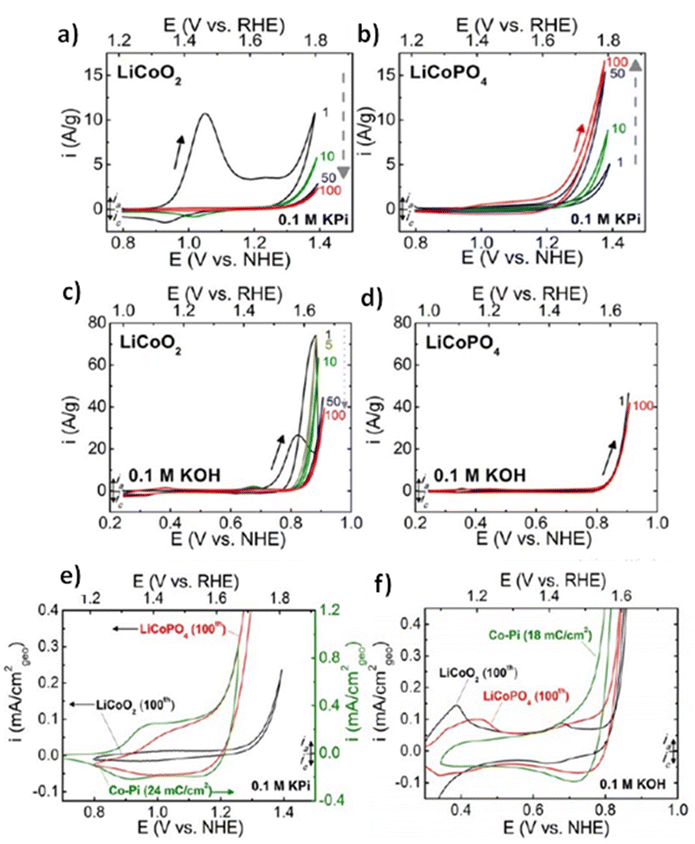 |
| | Fig. 10 (a) LiCoO2 and (b) LiCoPO4 electrodes were subjected to 10 mV s−1, 900 rpm CV scans in 0.1 M KPi electrolyte with a pH of 7.0. Dashed arrows indicate the progression of consecutive cycling, and the number labels indicate the number of cycles 0.1 M KOH at pH 13 was used for the CV scans of electrodes (c) LiCoO2 and (d) LiCoPO4 at a scan rate of 10 mV s−1 at 900 rpm. The results of 100 uninterrupted cycles are displayed CV scans in (e) 0.1 M KPi at pH 7 and (f) 0.1 M KOH at pH 13 for LiCoO2 (100th cycle), LiCoPO4 (100th cycle), and electrodeposited Co-Pi. Reproduced with permission from ref. 113. Copyright 2018, American Chemical Society. | |
Synthesis temperature of spinel-type lithium cobalt oxide also attribute to their activity towards OER. LT-LiCoO2, synthesized at 400 °C has higher activity than of layered lithium cobalt oxide synthesized at 800 °C (designated as HT-LiCoO2) for the oxygen evolution reaction.114 Zhiyi Lu et al.115 reported that electrochemical tuning (delithiation) of LiCoO2 improved oxygen evolution reaction. The monoclinic phase formation of Li0.5CoO2, improved the electronic structure altering the catalytic reaction. Compared to commercial Ir/C catalyst, De-LiCo0.33Ni0.33Fe0.33O2 showed optimum OER activity.116 Jianghao Wang et al. studies showcased ultrathin LiCoO2 nanosheets which displayed excellent activity due to high electron conduction and superior electrophilicity to the adsorbed oxygen. The mass activity of ultrathin LiCoO2 was 95.6 A g−1 at an over-potential of 410 mV, relatively greater than bulk LiCoO2.117 On the contrary, OER activity is quite low for Li1−xNiO2 since the intermixing of cations hinder the preparation of stoichiometric LiNiO2 with well-ordered Li+and Ni III into alternate (111) planes of the cubic close-packed oxygen sub lattices leading to the production of non-stoichiometric Li1−xNi1+xO2 which also include Ni II. Asha Gupta et al. reported that Al III doping helped stabilize the Ni III and via both conventional solid-state synthesis & solution-combustion synthesis, LiNi1−xAlxO2 with 0 ≤ x ≤ 0.4 was synthesized with/without Li2O2 (strong oxidizer) in the precursor which curbed the intermixing of Ni II in the Li+layer, improving the OER activity.114 The activity of LiNiO2 can be improved by electrochemical delithiation which assists the formation of superoxo/peroxo-like (O2)n− species like NiOO*.The adaptive heterojunction formed by OER-induced surface reconstruction is growth of NiOOH on delithiated LiNiO2.112 The lithium vacancies in the delithiated LiNiO2 augment the electronic structure of the surface NiOOH producing stable NiOO* species at this point, allowing for improved OER activity. Due to its inferior activity in terms of electrochemical water splitting, Mg as catalyst is least understood. Recently, Zheng Li et al.118 investigated how Mg doping increased the performance of a Ru-based electrocatalyst for acidic oxygen evolution process. It was observed that at different annealing temperatures, Mg-MOF-74 was able to generate metal oxide particles. Thus they were considered a good precursor for the introduction of Mg into RuO2 to decrease the Ru loading. Mg–RuO2 facilitates better oxygen evolution reaction (OER) in 0.5 M H2SO4 solution, with a low overpotential of 228 mV at 10 mA cm−2 and excellent stability in durability tests of 10000 cyclic voltammograms (CV) cycles as well as exhibited good lifetime for 30 h.
Introduction of beryllium, accelerated the hydrogen generation performance of Ru in both neutral and alkaline medium with the durability up to 50 h.119 However, they had poor activity in acidic medium. From density functional theory (DFT) calculations, it was observed charge on Be incorporation, charge transfer for electrocatalysis as well as improved water adsorption and lowered the energy barrier for dissociation of water, which are advantageous for better hydrogen production from water.
Various calcium based electrocatalysts have also been reported in water splitting especially in oxygen evolution reaction such as calcium copper titanate surface induced graphene oxide, reduced graphene oxide supported cobalt–calcium phosphate composite, F.
Fe-doped calcium cobaltites, layered calcium cobalt oxide, Cu-doping on the activity of calcium cobaltite, iron phosphate modified calcium iron oxide, Fe substitution in lanthanum calcium cobalt oxide are few among those reported.
Strontium is one of the most commonly used s block elements in electronic devices like in light emitting diodes (LED). This metal has shown their capability to act water splitting electrocatalyst while being part of perovskite material. Perovskites are mixed oxides with the general formula ABO3 and are effectively being used as OER electrocatalysts in both acidic and basic medium. Some of the low cost and effectual OER catalysts are Ba0.5Sr0.5Co0.8Fe0.2O3−δ and La1−xSrxCoO0.2Fe0.8O3−δ.120–122 Strontium cobaltite SrCoO3 due to their unique electronic structure exhibit excellent electrochemical property and is on the top of OER volcano chart.123 SrSc0.025Nb0.025Co0.95O3d (SSNC1) and SrSc0.175Nb0.025Co0.8O3d (SSNC2) as a series of extremely active and durable electrocatalysts for the OER in alkaline solution were also produced by Zhou et al. using scandium and niobium cation (Sc3+ and Nb5+) doped A-site strontium cobalt.124 In comparison to gold-standard of OER electrocatalysts, these A-site doped perovskites not only demonstrated up to a 50-fold increase in intrinsic activity, but also outstanding durability. Tafel slope of these catalysts rose to 70 mV dec−1 at lower voltage ranges (1.6 V), while the slopes of SrCo0.8Fe0.2O3d and RuO2 grew to 145 and 150 mV dec−1 correspondingly. Strontium palladium perovskite Sr2PdO3 when synthesized via citrate-combustion methodology portrayed excellent operation stability with the self-activation property and thus is employed as efficient electrocatalyst for OER as well as HER thus encouraging its use in fuel cells with water splitting application, where the catalyst remained stable for 3 h for OER activity.125 Mengran Li et al.126 reported that doping of Sr+ into A-site of the La(Ni,Fe)O3−δ optimizes the electronic structure, tunes the crystalline structure and enhance the number of OER active sites on the surfaces such as hydroxylation, oxygen vacancy defects, or transition metal ions with higher valences for Co or Fe-based perovskite OER catalysts. Excellent OER is achieved on the substitution of La3+ with Sr2+ by improving number of Ni III OER active species, elevating Ni/Fe surface ratio and increasing O22−/O− oxygen species.
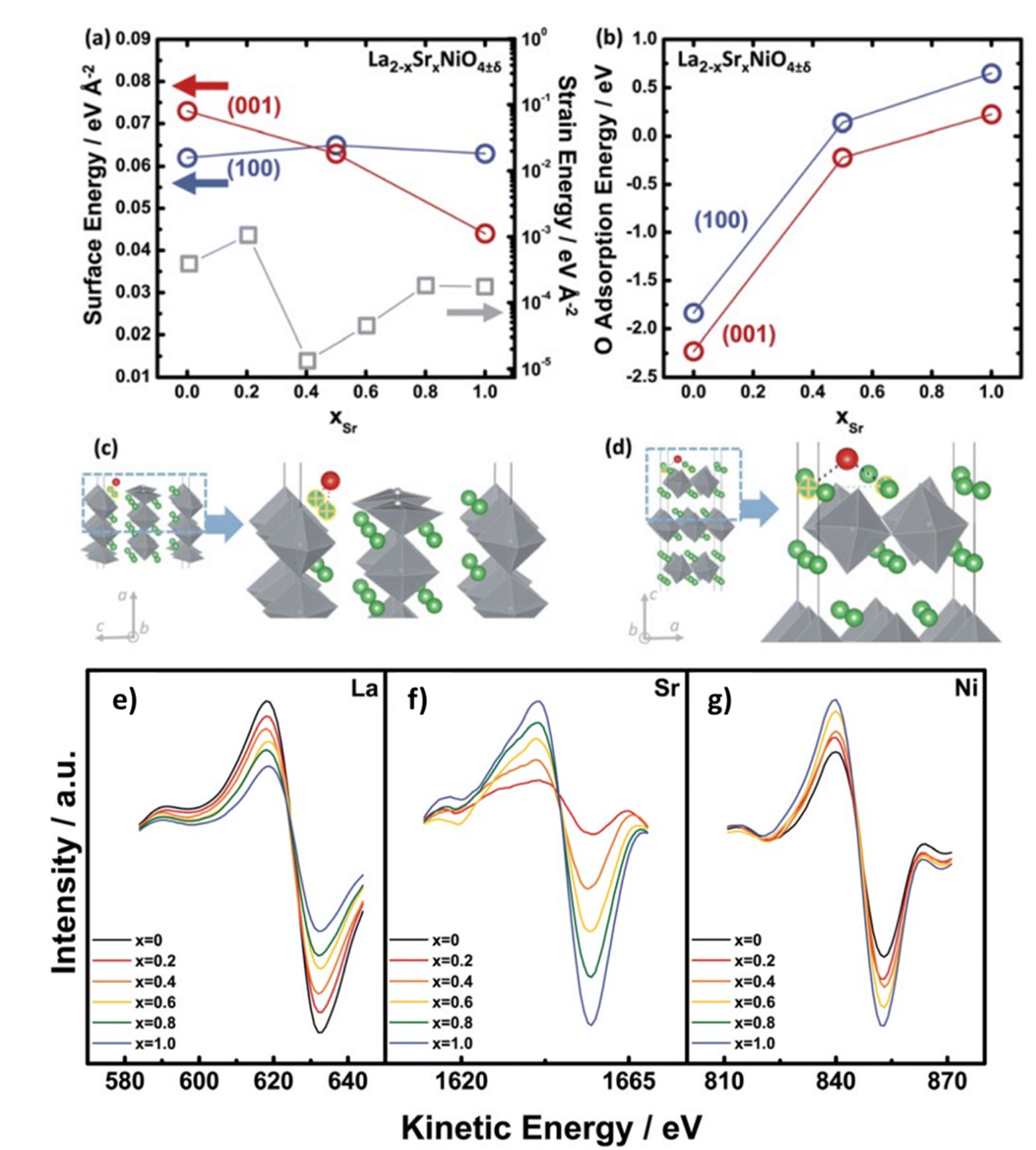
From AES and DFT studies of La2−xSrxNiO4−δ (LSNO), Dongkyu Lee et al. concluded that Sr substitution leads to reorientation to (001) tetra. (out-of-plane) from the (100) tetra. (in-plane) orientation as a function of the Sr content in (Fig. 11).127 DFT modelling also indicates a trend in adsorption energies of the LSNO system, the oxygen surface binding decreases with the increase in content of Sr.
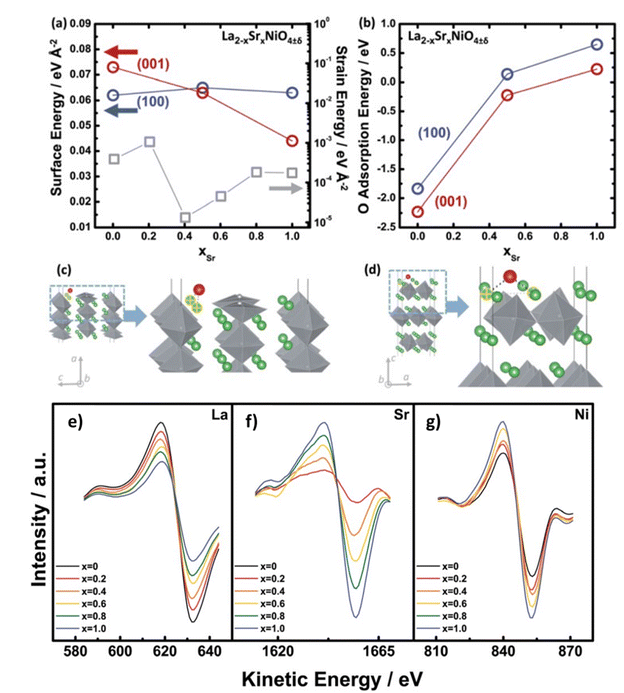 |
| | Fig. 11 Dependency on content of Sr (a) strain energies (gray) of the La2−xSrxNiO4±δ (LSNO) thin films with 0.0 ≤ xSr ≤ 1.0 derived using the surface energies and strain energy density equation, and (b) oxygen molecule adsorption energies on the (100)tetra. (blue) and (001)tetra. (red) surfaces of the LSNO thin films were acquired from density functional theory (DFT). Adsorption sites of surface oxygen on (c) the (100) tetra. slabs and (d) the (001)tetra. slabs. Anisotropy in LSNO thin films is represented by both surface and adsorption energies. AES data of the La2−xSrxNiO4±δ (LSNO) thin films with 0.0 ≤ xSr ≤ 1.0 annealed at 550 °C in an 1 atm partial pressure of oxygen. (e) LaMNN cation variation (RSF: 0.059), (f) SrLMM cation variation (RSF: 0.027), and (g) NiLMM cation variation (RSF: 0.277) as a function of Sr contents. The shift in the spectra of La and Sr cations scales with the concentration of La and Sr, indicating that the surface chemistry is characteristic of the surface cationic ratio desired. The increase in NiO6 octahedra population (normalised by surface area) with increasing Sr content is represented by a change in the Ni cation spectra which indicate a change in the thin film orientation. Reproduced with permission from ref. 127 Copyright 2014, Royal Society of Chemistry. | |
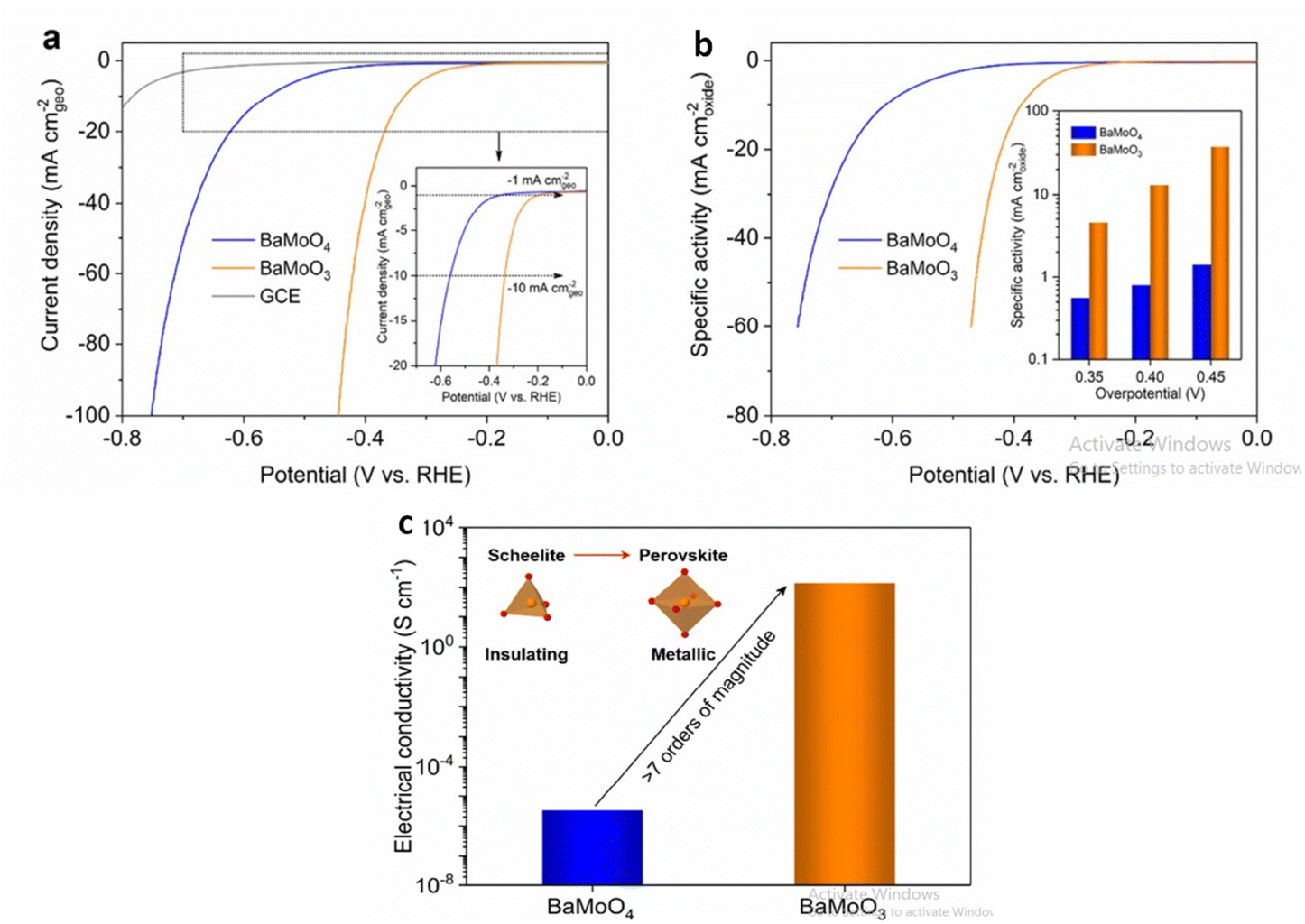
Barium based metal oxides are currently being studied for water splitting application. Through thermal reduction induced phase transformation, BaMoO3 perovskite is derived from Scheelite BaMoO4 which promotes alkaline HER activity.128 Compared to many bulk perovskite composed of Ni, Co, Fe; perovskite BaMoO3 have lower overpotential at current density of 10 mA cm−2. The higher electrical conductivity is due to interconnected MoO6 octahedra in the cubic-structured BaMoO3 perovskite compared to disconnected MoO4 tetrahedra in the tetragonal-structured BaMoO4 scheelite increasing the number of oxygen surface vacancies favoring the ensuing of the Volmer step of HER as shown in (Fig. 12). The precursor Barium salicylate is employed in the synthesis of BaMnO3 which enhanced the O2 evolution reaction.129 The nanoparticle size, shape as well as uniformity is affected on changing the precursor. Maximum TON and TOF accompanied with the high amount of O2 evolution is related to the nanoplate disc utilizing barium salicylate. Owing to the high charge transferability, May et al. reported Barium-based strontium-doped cobalt iron perovskite oxide Ba0.5Sr0.5Co0.8Fe 0.2O3−δ (BSCF82) to be an excellent catalyst for oxidation of water. The amorphous nature of the catalyst lead to high current for oxygen evolution as well as in double layer capacitance due to the lower O–P band centres w.r.t the Fermi level. On continuing the cyclic voltammetry, changes occurred in the surface morphology of BSCF82 which increased the OER activity.130 Barium based oxygen deficient bi-functional catalyst BaTiO3−x reported by Chen et al.131 was synthesised via sol–gel method executed better performance than noble and other precious metals synthesised through the same procedure. BaTiO3−x exhibited lower onset potential and greater current densities at low potentials <1.6 V than IrO2.
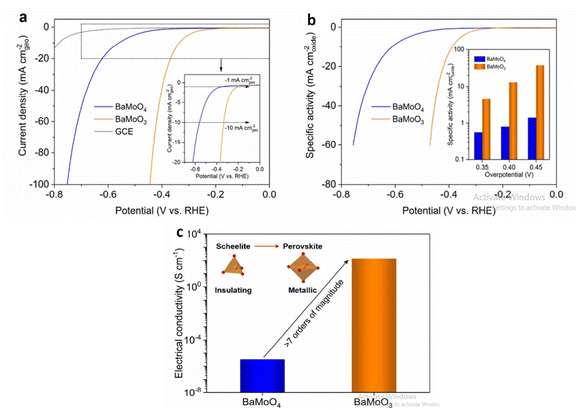 |
| | Fig. 12 In 1.0 M KOH, the electrocatalytic behaviour of BaMoO3 perovskite toward the HER was investigated. (a) BaMoO4 and BaMoO3 LSV curves, where currents are normalised to the geometric area of GCE (in mA cmgeo−2) and for reference, the GCE's LSV curve is shown in (a). The inset shows an enlarged version of the low current density region (b) LSV curves of BaMoO4 and BaMoO3, where currents are normalised to the BET surface area to calculate specific activity (in mA cmoxide−2). (c)BaMoO4 and BaMoO3's electrical conductivity was measured in ambient conditions. Schematic representations of the scheelite-to-perovskite phase transition and the shift from insulating to metallic conduction are shown in the inset. Reproduced with permission from ref. 128 Copyright 2020, Elsevier. | |
6.2. p- Block elements
p- Block comprises of a wide range of elements including metals, metalloids and nonmetal. They possess a relatively higher ionization energy and besides the group oxidation state, they show a number of other oxidation states. In this part of review, we would be discussing the recent development of most of the electrocatalysts based on metal as well as metalloid p block elements for OER, HER and electrochemical water splitting. The metal p block elements discussed here are Al, Ga, In, Sn and Bi.
Aluminium due to their nontoxic nature are considered as a good candidate in both acid and alkaline medium. They are inexpensive and are found abundant in the crust and are also observed to produce upto 11 wt% in the electrolysis of water. Aluminium doping onto transition metals like Co and Ni enable them to attain extra electrons from Al promoting interaction between reactant as well as transition metal sites. Al doped CoP nanoarray on carbon cloth (AlCoP/CC)132 exhibiting Pt like super high activity and good stability in acidic medium. From DFT calculations it was concluded that doping of Al lead to weakening of interaction between Co and H causing more thermo-neutral hydrogen adsorption free energy (ΔGH*). AlCoP/CC showed excellent activity in basic medium too as only 1.56 V was required to reach 10 mA cm−2 and the lifetime as HER electrocatalyst was studied up to 40 h. Gerken et al. and Chen et al. reported amorphous ternary metal oxide containing aluminium Al0.2Fe0.2Ni0.6Ox and Al0.4Fe0.2Ni0.4Ox had strong OER activity among various ternary metal-oxide combinations and the durability of Al0.4Fe0.2Ni0.4Ox was reported for 24 h.133 Phosphorus and aluminum co-doped porous nio nanosheets studied by Zhao Li134 identified that varying Al concentrations and comparing by LSV measurements, Al doping had significant effect and improve electrocatalytic activity. The elemental ratio of Ni:Al = 2:1 of PA-NiO had the highest catalytic activity OER and HER in series samples. For both HER/OER aluminum doped nickel-molybdenum oxide,135 the XPS results demonstrated Al acts as electron modulator by manipulating the electron density around Mo, inducing unsaturated coordination electron pair and causing electronic structural change. Al–NiMoO4-rods had overpotential of 259 mV to reach 10 mA cm−2 and low Tafel slope of 117 mV dec−1 for OER and overpotential of 131 mV to reach 10 mA cm−2 with low Tafel slope of 82 mV dec−1 for HER and the lifetime of catalyst was aquired up to 30 h. Hang Zhang et al. showed that the modification of electronic states of Ru nanosheets with Al in (Fig. 13) induced delocalize electrons, exposed more active sites due to formed holey structure.136 Metallic Al decoration on MoS2 ultrathin nanosheets have overpotential at 248 mV cm−2 in acid media and 198 mV cm−2 in basic media for HER as presence of Al reduces the band gap of MoS2 nanosheets, improving intrinsic conductivity and the improved is active surface area probability due to the activation of the inert surface of MoS2 by Al decoration.132 Inverse spinel NiFeAlO4 behaving as oxygen evolution electrocatalyst was reported by Jamie Y. C et al.137 revealed the mechanistic role of Al III is modulation of the redox properties of Ni and its ability to activate a water molecule for O–O bond formation owing to low overpotential and durability up to 15 h. DFT studied Co3−xAlxO4 Nanosheets for Water Oxidation rationalized Al-introduction lead occupancy of Co2+ Td sites by Al3+ atoms, decreasing the reaction barriers of intermediates and catalyst surface during the OER processes thereby enhancing the intrinsic activity.138 Co1.75Al1.25O4 is considered one of the best Co-based OER electrocatalysts to date with low overpotential of 248 mV to deliver a current density of 10 mA cm−2 and the authors studied the stability of the catalyst stability up to 30 h. Thus synthesis of Al incorporated transition metals, metal oxides is facial for obtaining high performance functional materials.
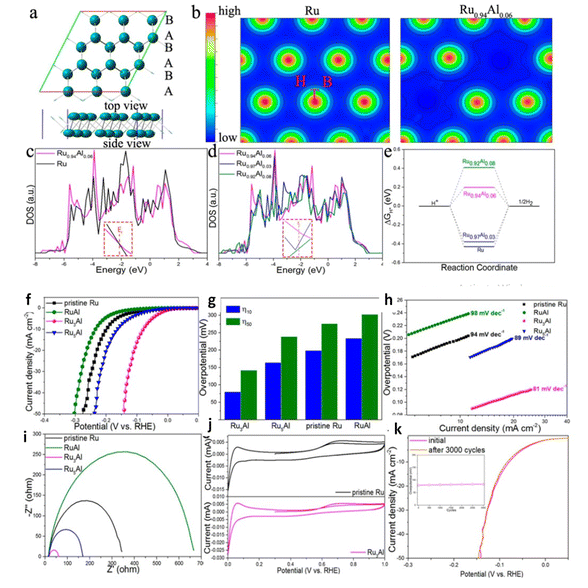 |
| | Fig. 13 Ru nanosheets with and without Al doping, calculated using DFT: optimised 2D Ru structure in (a); charge-density distribution on the surface with hydrogen adsorption sites indicated in (b); DOSs in (c and d) (inset: enlarged images for states near the Fermi level); and (e) HER free-energy diagrams. Electrochemical characteristics (f) LSV curves; (g) the necessary overpotential for 10 and 50 mA cm−2; (h) Tafel slopes; (i) the EIS at a 150 mV overpotential; (j) the CO stripping measurement; and (k) the LSV curves for the durability assessment. Inset: overpotential for 10 A cm−2 measured every 500 cycles. Reproduced with permission from ref. 136 Copyright 2019, American Chemical Society. | |
Gallium has attracted a lot of interest due to their low melting point and toxicity. Upon exposure of air, gallium can be self passivated by formation of ultrathin Ga oxide layer, this behaviour is similar to Al. Gallium oxide compared to iron oxide has better electrical conductivity, and α-Ga2O3 exhibited good water splitting ability. Gallium oxide nano fibers due to abundances their Lewis acid sites exhibit electrocatalytic behaviour.139 Their low onset potential of −0.34 V (vs. RHE), Tafel slope of this catalyst was 70 mV dec−1 and current density increment better than Pt/C, this catalyst in the absence of carbon additive and heteroatom doping show high performance. Ga intermetallic compounds like AuGa2 and AgGa2 obtained by the ultrasonic irradiation of Ga particles into aqueous solution or organic liquids possess positive reduction potential compared to metallic Ga. Suh-Ciuan Lim et al. reported popcorn-shaped GaPt3 as excellent HER catalyst synthesised via hot-solvent synthesis method.140 They exhibited exceptional durability and an overwhelmingly low overpotential (<80 mV). The same group also reported another Ga based HER electrocatalyst Gallium–Palladium (GaPd2)141 with high surface area and catalytic activity due to synergistic effect by the alloying of Pd–Ga with durability up to 24 h. Ni Wang et al. has stated the hydrogen evolution activity of gallium hydride and ligand-centered reduction.142 Their group was able to study the catalytic proton reduction cycle via mechanistic studies, identified the key intermediates and concluded that Ga III behave as the hydride binding site, whereas the porphyrin ligand behave as the electron transfer site enabling the redox inert metal ions to be catalytically active in the redox reactions. Tao Jing and group studied the Ga based electrocatalyst for oxygen evolution reaction via density functional theory studies.143
Using epitaxial graphene, graphene-like 2D gallium nitride (g-GaN) was prepared by migration-enhanced encapsulated growth method. All the three electrocatalysts, Fe/g-GaN, Ni/g-GaN, and Au/g-GaN are promising electrocatalysts for the OER. However among them Ni/g-GaN showed the least overpotential at 0.26 V. The g-GaN monolayer behave as excellent substrates and improved catalytic activity is related to the d-band center of active atoms. Introduction of Ga to the FeCoOx by Qiaoqiao Zhang via facile sol gel strategy produced amorphous iron–cobalt–gallium oxide (FeCoGaOx) as OER catalyst. The charge transfer kinetics improved due to the inclusion of third metal Ga, thus accelerating the water oxidation process. In 1.0 M KOH, FeCoGaOx on glassy carbon, platinum, and nickel foam had overpotential of 245, 275, and 215 mV respectively.144 Ga doped selenides are rare and recently shun Zhang reported Ga doped CoSe2 nanosheets for enhancing OER activity.145 Ga-doped CoSe2 was synthesised via magnetron sputtering and alloying on stainless steel mesh (SSM) and from XPS data it was revealed that electron drawing from Co to Ga, modulating the electronic structure of cobalt inducing electron deficiency in Co. This deficiency facilitated the transition of Co2+ in CoSe2 to Co3+/Co4+ during the OER process improving the intrinsic OER activity. Recently indium doped transition metal compounds were detected have improved catalytic performance due to adjustment in the strength of adsorption with the reactant, indium based spinels are quite rare and Jiajun Wang recently prepared cubic spinel CoIn2Se4 based on the polyol solution process in an environmental friendly manner where in the crystal site Co2+ occupied the tetrahedral and In3+ occupy the octahedral site as in (Fig. 14).146
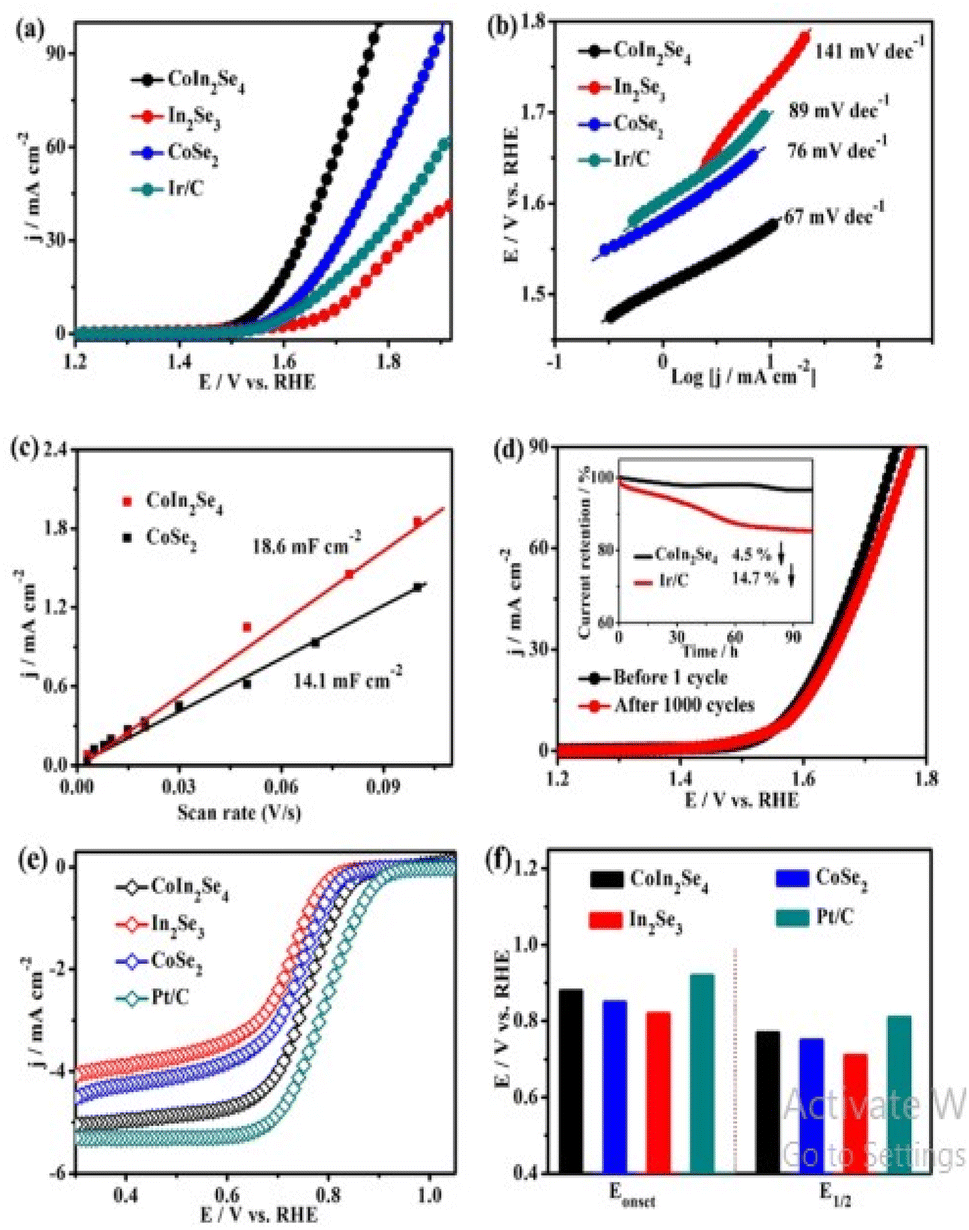
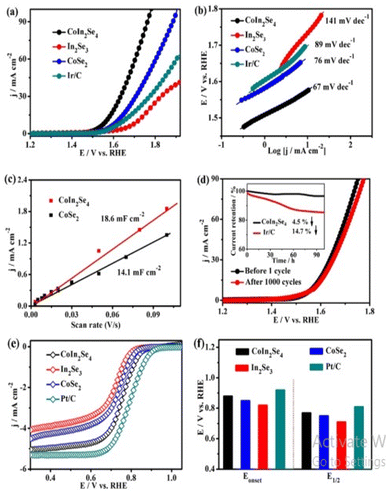 |
| | Fig. 14 (a) and (b) Represents the CoIn2Se4, In2Se3, CoSe2 and Ir/C electrocatalysts respectively (c) the determined capacitive currents plotted as a function of scan rate for CoIn2Se4, and CoSe2, (d) LSV curves of CoIn2Se4 before and after 1000 CV cycles (inset is the time-dependent current density curves on CoIn2Se4 and Ir/C), (e) ORR polarization curves and (f) the Eonset and E1/2 of CoIn2Se4, In2Se3, CoSe2 and Pt/C. Reproduced with permission from ref. 146 Copyright 2020, American Chemical Society. | |
Their boosted activity was contributed by charge transfer ability and numerous active site in alkaline electrolyte. Gd2O3In2O3–ZnO147 based ternary oxides in short GIZO was studied for overll water splitting activity i.e. both HER and OER. Firstly, the water molecule is reduced on the GIZO surface and liberated Hads, which proceeds to generate H2. For OER and HER; the Tafel slopes values were 121 and 64 mV dec−1 and the overpotential values at ±10 mA cm−2 are 282 and 271 mV respectively with the lifetime of catalyst of 10 h. One step solvent mediated process utilizing different solvents such as pure ethylene glycol, ethylene glycol/water, and ethylene glycol/ethanol mixtures was used to synthesis hollow and porous In2S3 spheres for overall electrocatalytic water splitting. The synthesised In2S3 exhibited overpotential at 230 mV for oxygen evolution reaction and 239 mV for hydrogen evolution reaction. Family of III–VI layered compounds, MX (M = Ga, In; X = S, Se, Te), are promising candidates for hydrogen production are held together via van der Waals interaction among stacked quaternary layers. InSe is seen to exhibit electrocatalytic property in both acidic and basic medium with overpotential of 549 and 471 mV, respectively.148 Jinjie Qian et al. reported (InOF-16, [(Me2NH2)2][In3(BTC)5Co2(DMF)6]·solvent) calcinated heterometallic oxides and/or carbides from a new bimetallic metal organic framework for increased electrocatalytic oxygen evolution activity. The MOF was studied in different thermal treatment i.e. InOF-16-Ar/O2-T (T = 550, 700 and 1000 °C).149 The composition was studied in different environment too.
In pure oxygen environment, there was low OER activity due to electrochemically inactive indium oxide and only binary oxides In(III)2O3 and Co(II,III)3O4 was be obtained. Superior performance was observed in argon atmosphere indicating In(III)2O3 and Co(I)3 In(0)C0.75 nanoparticles (NPs) are well embedded within these graphitic carbon material. The catalyst procured at 1000 °C, InOF-16-Ar/O2-1000 °C present best OER activities at 10 mA cm−2 with much lower overpotential compared to the non-precious OER catalysts. Indium tin oxide (ITO) electrodes are among the important electrodes for electrochemical measurements and is used as support for apprehending the NPs electrocatalytic activities such as HER and OER. Paolo Ciocci150 utilized an in situ optical microscopy approach to differentiate electrochemically active regions of ITO electrodes for hydrogen evolution. The electrochemical behaviour was studied by opto-electrochemical monitoring in a scanning electrochemical cell microscopy, SECCM in 5 mM H2SO4. The optical responses and the electrochemical behavior altered with the sample resistance however the onset potential of the HER remains mainly the same.
Doping of cost effective metal Tin (Sn) onto various electrocatalysts has also caught the attention of researchers in water splitting. Juan Jian reported the effects of doping tin onto Ni(OH)2 on water splitting performance.151 Compared to the undoped Ni(OH)2, Sn4+-doped Ni(OH)2, Sn–Ni(OH)2 processed via one step hydrothermal process had hugely enhanced catalytic activity for both HER and OER. Based on both computational as well as experimental data, Sn4+ was observed to act as active site for Ni sites for OER, while in HER Sn itself act active reaction site. Though Sn4+ has different effect for both hydrogen and oxygen evolution reaction, overall the catalyst act as overall water splitting. The overpotentials of Sn–Ni(OH)2 are 312 mV (OER) and 298 mV (HER), which are lower than the corresponding 396 and 427 mV of Ni(OH)2, respectively at 100 mA cm−2. Jing Y carried out in situ growth of Sn-doped Ni3S2 ultrathin nanosheets on Ni foam and studied their electrocatalytic activity. Its activity was studied in both acidic and basic media.152 Comparing the Nyquist plots of both doped and undoped Ni3S2, the electrochemical impedance was reduced in the case of doped catalyst. Tin due to its weak hydrogen binding and ability to maintain a good balance between adsorption and desorption of hydrogen atoms, considered a good choice to alloy with Ni. Sn doped Ni3S2 had lower Tafel slope value indicating accelerated kinetics and lower overpotential demonstrating they possess superior HER activity. Ni–Sn@C core/shell, though had lower stability in acid medium, their large active surface area and high conductivity led to good electrocatalytic activity toward HER.153 Ternary alloy including Sn as one of the metals for electrochemical water splitting application are also studied. Yuchan Liu successfully synthesised Ni–Co–Sn alloy electrodes154 via galvanostatic deposition onto copper foil and the synthesised catalyst exhibited both HER and OER activities as shown in (Fig. 15). The good performance is due to the synergistic effect of tin incorporation onto Ni–Co as well as the numerous active sites. The Tafel slope values are 63 mV dec−1 and 62 mV dec−1, for HER and OER activities and from chronoamperometric measurements, current density was maintained up to 92% even after 10 h. The bifunctional catalyst better activity with a small cell voltage of 1.76 V at 50 mA cm−2 for overall water splitting. Fang Liu et al. studied an efficient way to optimize the hydrogen evolution reaction activity by doping a main group metal like Sn onto CoS2 causing the activation of their sulphur sites.155 DFT studies evidences improved performance with long term durability which is attributed to increased intrinsic activity and the optimized ΔGH* by activating the inert S sites. Ravula et al. reported SnO2 enhanced the HER activity of MoS2 in the efficient HER electrocatalyst MoS2/SnO2/reduced graphene oxide (rGO) multilayer hybrid sheets.156 Yuksel Akinay et al. have reported 3D Sn doped Sb2O3 (ref. 157) as electrocatalysts for hydrogen evolution reaction in acidic medium & demonstrated they show excellent activity & are also stable electrocatalyst.
 |
| | Fig. 15 (a) Polarization curves of Cu substrate, commercial Pt/C, and Ni–Co–Sn catalysts with a scan rate of 2 mV s−1 in 1 M NaOH for HER (b) Ni–Co–Sn catalyst, Cu substrate, and commercial Ir/C polarisation curves in 1.0 M NaOH for OER at a scan rate of 2 mV s−1. (c) The double-layer capacitances for OER catalysts with pH values of 5, 6, and 7. Reproduced with permission from ref. 154 Copyright 2019, Elsevier. | |
Bismuth based catalyst owing to their cost effectiveness, high electrochemical active sites, environmental friendliness and good electrocatalytic activity towards water splitting has been investigated for numerous applications. Though Bi based catalysts are considered to showcase poor HER activity, the activity can be improved by the synergistic effect with oxy/hydroxide phases of metals. Rahmad Syah et al. incorporated metallic Bi with Bi2O3 (ref. 158) residuals on Ni and observed high catalytic performance. Evaluation of the EIS techniques indicated low Tafel slope value 55 mV dec−1 indicating rapid kinetics with low overpotential of 250 mV dec to the synergetic effect of metallic Bismuth and oxidized Bismuth in the crystal structure. The ion diffusion was readily reduced due to the 3D porous structure leading to high electron transport. Shuai Wang et al. reported succulent-like binary metal sulphide heterostructure activated by bismuth, Bi2S3/Ni3S2/NF prepared by mild solvothermal method as OER electrocatalyst in alkaline medium. Bi2S3/Ni3S2/NF, as a low overpotential of 268 mV with low Tafel slope (82 mV dec−1) and good stability for over 12 h at high current density.159 Various perovskite-based bifunctional catalysts are synthesised utilizing alkaline earth metals (Ba, Sr, Ca, and Mg)-doped bismuth iron oxides Bi0.6M0.4FeO3 (BFO).160 Among them, Ca(Bi0.6Ca0.4FeO3 (BCFO)) exhibited good OER performance in alkaline media, as BCFO160 have current density 6.93 mA cm−2 at a fixed overpotential of 0.42 V which is double compared to other catalysts having current density 3.06 to 4.04 mA cm−2. Bismuth molybedate, Bi2MoO6, have layered structure consisting perovskite layer (An−1BnO3n+1)2−, A = Ba, Bi, Pb, etc.; B = Ti, Nb, W, Mo, etc. and bismuth oxide layers (Bi2O2)2+ and is considered an advanced material. Sakila Khatun et al. reported the development of Bi2MoO6 along with a series of bismuth iron molybedate comprising of major component Bi2MoO6 and minor component monoclinic Bi3(FeO4)(MoO4)2 existing as solid solution and studied their performance in both OER and HER which exhibited overall ater splitting at 1.67 V @ 10 mA cm−2.161 The optimum blending of these components in solid solution exhibited overpotential at current density 10 mA cm−2 and 50 mA cm−2 at 266 and 288 mV respectively for OER and overpotential at 10 mA cm−2 at 207 mV for HER portraying the remarkable activity. Chenguang Duan et al. reported high catalytic activity of 2D Bi for hydrogen evolution reaction.162 The 2D Bi nanosheets are synthesised via sonication assisted liquid phase exfoliation method and their 2D structure has led to Bi nanosheets having shorter charge diffusion distance and more active sites compared to bulk counterpart. Pillai et al. calculated that adsorption energy of the hydrogen atom and oxygen atom on the surface of Bi were −1.418 and −3.963 eV respectively. The small atomic hydrogen adsorption value indicate that HER occur on the 2D Bi surface.
The metalloids of p Block elements being discussed here are B, Si, Ge, As, Sb, Te.
Transition metal borides in (Fig. 16) for electrochemical water splitting was studied as early as 2009 by Daniel Nocera and group by investigating the electrocatalytic properties of Co and Ni borates in near neutral medium. Followed by Hu and Patel for development of Mo and Co borides.163–167 Borates are referred to electrocatalysts prepared via electrodepostion and borides are electrocatalysts prepared by other techniques. Over the years many methods to improve the performance by usage of substrate like metal foams and plates, carbon and graphene based structures, nano structuring i.e. metal core-oxide shell, ternary alloys: Ni–Fe-b, Co–Mo–B, Co–Fe–B, Co–Ni–B, Co–W-b etc., quaternary alloy; Fe–Co–Ni–B, V–Co–Ni–B, alloying with P and different synthesis strategies; electrodeposition: amorphous coating of metal borates, solid state: molybdenum based borides, CVD/boronisation: thin films, boronizing metal surfaces, electro less deposition; nanostructured coating of Co and Ni borides. Chemical reduction: amorphous nanoparticles of Co and Ni borides have been studied. Though a number of reports and dedicated reviews are there for metal borides and borates, in this review, the most recent works will be discussed.
 |
| | Fig. 16 Performance of various metal boride/borate electrocatalysts was compared at pH 9.2. The graph also displays the most efficient catalysts from different nonprecious metal catalyst families and precious metal-based catalysts. (Acronyms used—C: Vulcan carbon; CC: carbon cloth; CP: carbon paper; NF: Ni foam; Ni: Ni foil/plate; Ti: Ti mesh/foil.)48 | |
Xuetao, Liu et al. successfully synthesised boron-doped cobalt–iron bimetal phosphides nanosheets by hydrothermal boronation of bimetallic metal–organic frameworks precursors with the subsequent low-temperature phosphidation for enhanced oxygen evolution.168 The cooperative effect between Co and Fe with B and P influenced the electronic configuration surrounding the metal centres. The catalyst exhibited excellent durability with low Tafel slope value of 49.5 mV dec−1 and overpotential of 294 mV at 10 mA cm−2 in 1.0 M KOH. Tungsten and phosphorus co-doped amorphous FeB in alkaline medium169 is considered to be among the efficient boride based OER catalysts due to low overpotential 209 mV of 10 mA cm−2 and durability for over 25 h. This study led by Zhijie Chen also offers an approach optimize metal borides such as NiBx, CoBx, and MoBx as precatalysts for OER. B doping onto 2D MXene nanosheets was seen to improve the adsorption kinetics of intermediate H* with reduction of charge transfer resistance towards HER based on quantum mechanical first-principles calculations.170 Munkhjargal Bat-Erdene incorporated Ru nanoparticles onto Boron doped Ti3C2Tx MXene, Ru@B-Ti3C2Tx as HER catalysts exhibited low overpotential of 62.9 and 276.9 mV at the current density 10 and 100 mA cm−2 with over 1000 cycles for stability. Dong et al. reported boron carbonitrides with tunable B/N Lewis acid/base sites for overall water splitting. B-BCN had onset potential at 1.4 V for OER with Tafel slope 65.2 mV dec−1.171 N-BCN had low onset potential of −50 mV for HER with a small Tafel slope of 61.3 mV dec−1 with the lifetime of 48 h and the overall water splitting occurs at 1.62 @ 10 mA cm−2 & 1.79 @ 100 mA cm−2. B-BCN//N-BCN exhibited onset potential of 1.52 V for overall water splitting. Thus BCN with different B/N contents exhibit electrocatalytic activity towards water electrolysis. B doped Ni (B-Ni) in basic medium exhibited bifunctional electro activity for both HER and OER and showcased excellent water splitting performance with 1.62 and 1.74 V (vs. RHE) to attain 10 and 100 mA cm−2, respectively.172 Due to electronegativity difference, doped B extract electron from Ni sites increasing their charge density and resulting in loss of charge density on the Ni atoms, this charge distribution favours HER processes. In-Kyoung Ahn et al. reported boron-doped nickel-iron layered double hydroxide to facilitate charge transfer in oxygen evolution electrocatalysts.173 B:NiFe LDH had 281 mV at 10 mA cm−2 which was further reduced to 229 mV through the electrochemical oxidation, maximizing the doing effect and activating the catalyst. EIS measurements confirmed that doping as well as activation reduced the charge transfer resistance. Boron doped FeNi solid solution grafted onto activated hydrophilic sponge (B-FeNi@HS) behave as a as a water splitting bifunctional catalyst.174 B-FeNi@HS exhibit good activity with overpotential at 10 mA cm−2 for HER and OER being 54 and 171 mV respectively. Hong ling Li fabricated and studied fabricate hybrid boron carbon oxynitride nano fibrous ((BCNONF)175 and they exhibit high performance and greater durability in alkaline electrolyte up to 10 h and overall water splitting 1.79 @ 10 mA cm−2.
Silicon is among the most abundant element in the Earth's crust. Silicon though compared to carbon has the same electronic configuration, carbides are widely explored in water splitting applications where as silicides are not much investigated. Mono silicide,176 RuSi which is recently reported showcased good HER activity, as interstitial silicon activated the recessive active Ru site on the catalyst surface. In the detailed study of transition metal monosilicides including silicides of Ti, Mn, Ru, Ni, Co, Pd, Fe and Rh as promising new HER catalysts by Yuan He, almost all of them exhibited excellent activity due to their large ECSA i.e. up to 211.5 cm2 and turn over frequency up to 118 s−1 at 100 mV. Yujia Huang176 reported for the first time reduced graphene oxide (rGO)/SiO2 ceramic composite as HER catalyst. The incorporation of SiO2 into the rGO layers avoided the aggregation of the rGO layers, prevent electrolyte diffusion and blockage of gas. The close contact between SiO2 and rGO improved the durability as well as electrical conductivity. Si based intermetallics are also investigated for OER activity. Porous Fe5Si3 has superior OER performance in extremely acidic condition showcasing their superior corrosion resistance.177 Amar A. Bhardwaj and group showed that Chlorine Evolution reaction can be suppressed greatly while still allowing OER to take place in acidic and pH neutral seawater, by the deposition of catalytically inert SiOx over layer planar Pt electrocatalyst, SiOx encapsulated electrocatalyst could be improved by enhancing the adhesion properties of the interface thus regulating the H2O/Cl− transport selectivity. Mesoporous silica (SBA-15) are also considered for evaluation for HER activity. Elham Chiani178 demonstrated that PdCu bimetallic nanoparticles decorated on ordered mesoporous silica (SBA-15)/MWCNTs showed excellent HER activity with high exchange current density of 165.24 mA cm−2 at −360 mV and low Tafel slope of 45 mV dec−1. The presence of mesoporous SBA-15 contributes to the improvement of the activity by ensuring abundant active sites for Pd and Cu NPs dispersion.
Another new candidate in the field of electrocatalysis is graphene like germanium carbide (g-GeC) that are being considered due to their adjustable electronic properties and large surface. Xin Chen et al. studied OER and HER activity of TM doped g-GeC (M-GeC). For OER, among the 3d, 4d, 5d transition metals, Pd-GeC and Ni-GeC exhibited highest activity with low overpotential values of 0.41 and 0.55 V, respectively.179 While for HER, Mn-GeC with ΔGH* equal to −0.04 eV possesses excellent electrocatalytic activity. Meihong Fan et al. based on DFT calculations and experimental study, explained how adsorption of germanium on ruthenium catalyst promote Hydrogen evolution activity.180 Preferential adsorption sites are altered from the hollow to top of Ru catalyst due to germanium. J. Niklas Hausmann reported intermetallic Fe6Ge5 synthesised via high temperature method, which consists of polyhedral of five different Fe atoms forming square pyramids whereas Ge atoms form pentagonal prisms with distorted octahedra.181 Fe6Ge5 behaves as a novel OER precatalyst in alkaline media with overpotential of 272 mV at 100 mA cm−2 and Tafel slope value of 32 mV dec−1.
The unique morphology and electronic structure of the natural mineral orthorhombic arsenic has attracted attention. Owing to their black colour they are represented as b-As.182 They have puckered honeycomb structure with semiconductor properties whose electronic structures are thickness related. Single and few layered b-As promote electron mobility in HER due to high carrier transport ability. Though pristine b-As has poor hydrogen atom binding and show weak HER properties, doping of atoms like N, S, C, O and P enhance their performance. Among them O is easily embedded into b-As lattice and reduce |ΔGH*| to as low as 0.044 eV close to Pt with |ΔGH*| = −0.090 eV. Joseph A. Gauthier et al. studied various transition metal arsendies and concluded CoAs and MoAs to be active arsenide materials for HER.183 MoAs exhibited weak hydrogen binding whereas CoAs bonds to hydrogen too strongly, both the catalyst exhibit moderate overpotential on the basis of normalized electrochemical surface area basis. Doping of grey arsenene (g-As) along with pristine black (b-As) with heteroatoms184 was studied for both HER and OER activities by Sengpajan Santisouk. Among them single O-doped g-AsO1 as well as bAsO2 exhibited OER activity whereas double C-doped g-AsC2 exhibited HER activity.
Elemental antimony nanoparticles as well as conductive antimony tin oxide (ATO) are used in electrochemistry Li ion and Na ion batteries. Raspberry like structures of SbPt synthesised via arc melting and sintering was observed to have ECSA 1.25 times greater than Pt black in (Fig. 17). Existence of Sb have synergistic effects from the hybridization of 3d orbital prevents the adsorption of hydrogen improving the catalytic performance.185 Song Lu et al. studied the DFT calculations to analyze the efficiency of single transition metal embedded antimonene monolayers as HER and OER catalyst. Transition metals like Sc, Ti, V, Cr, Mn, Fe, Co, Ni, Cu, Zn, Ru, Rh, Pd, Ag, Cd, Ir, Pt and Au are embedded onto the single Sb vacancy of the antimonene monolayer to form SACs (Single Atom Catalyst). Based on calculations, Ir@Sb has best electrocatalytic activity toward the HER as ΔGH* value is 0.009 eV and, Pt@Sb had best OER performance having the least overpotential of 0.48 V which lower than state of the art catalyst IrO2 with overpotential 0.56 V.186
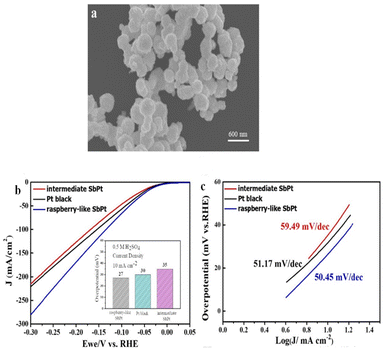 |
| | Fig. 17 At pH 9.2, the effectiveness of various metal boride/borate electrocatalysts was evaluated. Additionally, the most effective catalysts from various non-precious metal catalyst families and precious metal-based catalysts are shown in the graph. Reproduced with permission from ref. 185 Copyright 2021, Elsevier. | |
NiTe and CoTe187 are metal tellurides studied for bifunctional catalysts, OER and HER in metal chalcogenides due to their conductivity, specific area and chemical stability. It was explored that transition metal based catalysts when tellurized improves the intrinsic water splitting activity of the material by increasing the covalent character of the metal anion bond. Among all chalcogenides; tellurides are less toxic and has less electronegativity thus increasing their covalent character when bonded to metal, lowering redox potentials of metals generating active sites for catalysis. For HER, Nickel tellurides has overpotential of 679 mV and a Tafel slope of 151 mV dec−1 whereas CoTe nanostructures over carbon fiber paper show a low overpotential of just 230 mV at 100 mA cm−2 and CoTe2 with low overpotential of 246 mV at 10 mA cm−2.
CuTe-NS are deposited over conductive NiF substrate188 synthesised via hydrothermal approach demonstrate decent HER activity with low Tafel slope of 36 mV dec−1. Hagyeong Kwon recently reported nanoporous silver telluride on the basal plane of layered metallic support, WTe2.189 AgTe compared to other Ag related catalyst which have poor catalytic activity, reveal higher HER performance and better durability overcoming the problems faced by many state of the art catalysts.
6.3. d- Block elements
d- Block elements consist of first transition series (from Sc to Cu), the second transition series (from Y to Ag), and the third transition series (the element La and the elements from Hf to Au). They form stable complexes, have high melting and boiling point, exhibit variable doxidation states and form compounds with profound catalytic activity. As there are number of elements in d block, in this part of the review, the elements partaking in water electrolysis, HER and OER are divided into groups based on their properties. The groups are transition metal carbides/nitrides, transition metal phosphides, transition metal sulfides, and selenides and noble metal-based electrocatalysts that are recently reported will be discussed.
6.3.1. Transition metal carbides and nitrides. Transition metals carbides and nitrides are kind of interstitial compounds which, due to their noble metal like behaviour and electronic structure, are widely studied in water splitting. The interstitial sites of the parent metal are occupied by the carbon and nitrogen atoms. Their properties are a combination of covalent, ionic and metallic. Covalent character ensures tolerance to stress, and metallic character displays high electrical conductivity, whereas electronic structures like noble metals are due to the ionic character of bonding. The properties of monometallic carbides or nitrides are not up to that of state of the art catalysts. Thus further improvement can be achieved by modifying size, doping of heteroatom, usage of substrate etc.190,191 Recently, Mo2C was doped with different transition metals such as Cr, Fe, Co and Ni utilizing precursors, Anderson-type polyoxometalates (POMs) and their HER activities were studied. It was observed among the prepared four catalysts, Ni–Mo2C exhibited the highest activity followed by Co–Mo2C and Fe–Mo2C and the least activity was shown by Cr–Mo2C.192 POMs are a distinct class of nanosized clusters that may be used as a preliminary step for the synthesis of nanoscale material. Additionally, high-negative-charge polyoxoanions can readily combine with nearly all transition-metal elements in a single molecule, suggesting that they can be used as an ideal molecular platform for the design and preparation of multi-metallic composited electrocatalysts. In order to achieve 50 mA cm−2 current for HER and OER, respectively, Zhang et al.193 used a low-cost and effective catalyst of Co2P@Co2P/Co-POM/NF that delivered a minor overpotential of 130 and 336 mV with Tafel slope of 135 mV dec−1 and 57 mV dec−1 at a low cell voltage of 1.6 V @ 10 mA cm−2. Using the Anderson-type (NH4)4[Co(II)Mo6O24H6]·6H2O polyoxometalate as a bimetal precursor, Hou et al. recently demonstrated the synthesis of oxygenated CoMoS with low overpotentials of 97 mV and 272 mV for HER and OER at 10 mA cm−2 with Tafel slope of 70 and 45 mV in alkaline solution at low cell voltage of 1.6 V with the durability of 10 h.194 Samar K et al. K2[Ni(H2O)6]2[V10O28]·4H2O (1) synthesised is the first homogeneous POM catalyst with nickel that is used for electrochemical hydrogen evolution in acidic medium195 with overpotential of 127 mV, turn over frequency of 2.1 s−1 with durability of 5 h at 1 mA cm−2. For heterogenous water oxidation, surface-modifying agent APTS was used to immobilize the high nuclearity 16-Co(II)-containing POM over commercial Ni foam, and the synthesised electrode Co16-GeW9@NiF195,196 exhibited low overpotential of 370 mV at 10 mA cm−2 and a Tafel slope of 84 mV dec−1, in basic pH with stability of 8 h. K5[Bi(H2O)2SiW11O39]·13H2O (K51·13H2O). [Bi(OH2)2]3± functionalized Keggin polyoxometalate (POM) compound was fabricated with ZIF-8, a metal–organic framework containing zinc imidazole; H51@ZIF8 exhibited OER activity with 375 mV, the overpotential and Tafel slope is 188 mV dec−1 in 0.1 M KOH with stability studies carried up to 3 h.197 The overpotential value @ 10 mA cm−2 and Tafel slopes are found to be 726.04 mV and 173.80 mV dec−1 respectively for the anti-Lipscomb (Krebs type) compound incorporated with cobalt; Na6[{CoII(H2O)3}2{WVI(OH)2}2{(BiIIIWVI9 O33)2}]·8H2O suitable for HER activity to support up to 10 hours of chronoamperometry.198 Core shell-type metal–organic framework (MOF) derived Co-NC (Co-NC-POM) coated Keggin-type polyoxometalate (POM) nanoscale particles were prepared using a simplistic coprecipitation method by Huimin et al.199 where the as-synthesised Co-NC-POM hybrids exhibited excellent electrocatalytic performance for both HER and OER due to their active sites, distinctive structure, and defects as well as synergy effect. Co-NC-POM has lower overpotential and Tafel value of 133 mV at 10 mA cm−2 and 124 mV dec−1 for HER activity while for OER the catalyst also provides low overpotential of 400 mV with Tafel slope of 94 mV dec−1 with excellent stability up to 24 h and low voltage for overall water splitting 1.60 V. Due to their numerous active sites, atomic-level thickness and substantial surface area, the 2D nanosheets of metal–organic frameworks (MOFs) have recently become recognized as a promising material. Investigated in 1 M KOH, the OER performance of Te, Cl–NiFe MOF on nickel foam with overpotential of 224 mV at 30 mA cm−2 and Tafel slope value of 37.6 mV dec−1.200 The incorporation of Te in NiFe MOF improved charge transfer and allow for the adsorption of oxygen on the surface where as Cl helped to increase the number of active sites on the NiFe MOF surface. G/THTA-Co G/H3 [Co3(tht)(tha)], pristine MOF based electrocatalyst in acidic medium exhibited HER activity provided low overpotential of 230 mV with Tafel slope value of 70 mV dec−1 with stability of up to 4 h.201 MOF based electrocatalyst CoP/Co-MOF on carbon cloth exhibited overpotential of 49 mV and Tafel slope of 63 mV dec−1 in 1 M PBS, overpotential of 34 mV and Tafel slope of 56 mV dec−1 in 1 M KOH, overpotential 27 mV and Tafel slope 43 mV dec−1 in 0.5 M H2SO4 while MOF-derived electrocatalysts CoP/Mo2C-NCG with overpotential of 55.7 mV and Tafel slope of 49 mV dec−1 in 0.5 M H2SO4 and overpotential of 67.2 mV and 66 mV dec−1 in 1 M KOH. Co-Zn/PNC shows both HER and OER with overpotentials 180 and 348 mV and Tafel slopes 100 mV and 84 mV respectively with overall water splitting at 1.63 V @ 10 mA cm−2.202 Li et al. used the semi-MOF precursor NiCoFe-MOF-74 to create NiCo/Fe3O4 heteroparticles encased in MOF-74 (NiCo/Fe3O4/MOF-74) using a partial pyrolysis technique. Strong evidence of an interaction between NiCo and Fe3O4 was revealed by DFT calculations. A high activity with an overpotential of 238 mV at 10 mA cm2 and Tafel slope 29 mV dec−1 and exceptional long-term stability of 36 h with overall water splitting 1.47 V were observed when the NiCo/Fe3O4/MOF-74 was tested for the OER.203Porous molybdenum carbide nano-octahedrons was synthesised by Lou et al. using MOF based material (NENU-5; [Cu2(BTC)4/3(H2O)2]6[H3PMo12O40]) which shows excellent activity in both acidic and basic medium. They have low Tafel slope value 59 mV dec−1 with overpotential of mV at 10 mA cm−2.204 Both HER and OER activity is observed in Co doped Mo2C nanosheet (Co SAs/Mo2C), the Co-anchoring is carried out by sacrificial Zn strategy.205 α-Mo2C synthesised via simple urea-glass route of molybdenum carbide, due to small particle size (2–17 nm) exhibit enhanced HER performance in basic medium than in acidic medium.206 Wang et al. developed 3D molybdenum carbide (Mo2CTx) microflowers207 in (Fig. 18) with outstanding OER activity having an overpotential 180 mV to reach 10 mA cm−2.208
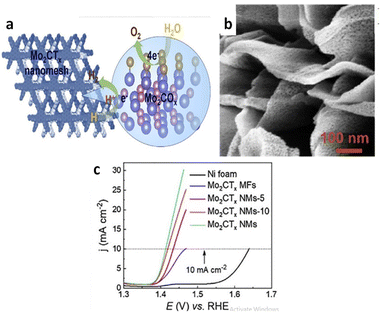 |
| | Fig. 18 (a) Mo2CTx electrocatalytic OER schematic illustration. (b) Image of the Mo2CTx MFs obtained using scanning electron microscopy. (c) A comparison of different Mo2CTx samples' Linear Sweep Voltammogram curves Reproduced with permission208 Copyright 2021, Elsevier. | |
Streb et al. using Mn/V oxide as precursor for metal oxides/carbides-CNT hybrid that it demonstrates higher activity and durability for OER activity.209 Titanium carbide (MXene) owing to their electronegative surfaces as well as large surface area has ability to be potential carrier material of MOF and FeNi-LDH. MOF/Ti3C2Tx and FeNi-LDH/Ti3C2-MXene exhibit good activity OER. Yang et al. introduced Mo into Nickel carbide to form Mo6Ni6C catalyst which shows good OER activity as at the atomic level, Mo6+ has integrated with the Ni species and the carbon element has been insert into MoNi array.210 Doping of Fe led to tuning of electronic properties of Ni3C enhancing its catalytic reactivity in both HER and OER. Among transition metal nitrides, molybdenum and tungsten based nitrides are popular than other TMNs. 2D nitrogen-rich hexagonal W2N3 (h-W2N3)211 developed and studied by Zhou and group showed HER activity with overpotential −98.2 mV at 10 mA cm−2 in acidic medium in seen (Fig. 19).212
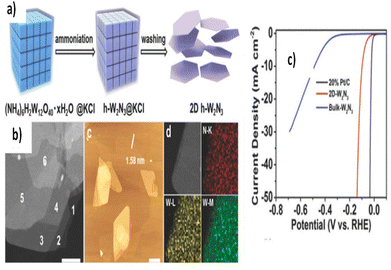 |
| | Fig. 19 (a) Schematic diagram depicts the preparation of 2D h-W2N3. (b) Images of 2D h-W2N3 nanosheets from STEM, AFM, and EDS. 2D h-W2N3, bulk-W2N3, and 20% Pt/C polarisation curves are shown in (c). Reproduced with permission212 Copyright 2021, Elsevier. | |
NiMoN nanowires reported by Hao et al.213 had remarkable HER and OER activity, with overpotential of 38 mV at 10 mA cm−2, lower compared Pt/C in HER and 290 mV at 50 mA cm−2, lower than Ru2O for OER, durability was studied up to 5 h and 1.498 @ 20 mA cm−2, 1.532 @ 50 mA cm−2, 1.559 @ 100 mA cm−2. Ni2Mo3N synthesised via urea glass route shows OER activity in alkaline medium with overpotential at 270 mV. Treating Co3Mo3N in ammonia at low temperature, close packed Co0.6Mo0.14N2 was procured by Khalifah and group214 with small Tafel slope and low overpotential, showing HER activity. By facile pyrolysis, among the series of bimetal embedded nitrogen-doped carbon framework synthesised, Co0.75Fe0.25-NC displayed low value of Tafel slope and small onset for both HER and OER. Ni3FeN supported Fe3Pt fabricated by Good enough et al. has more activity than OER catalyst (Ir/C).215 Zeng et al. synthesised plasmonic material TiOxNy nanoshells supported on Fe2N that have holes due to movement of hot electrons upon laser radiation which capture electrons from the reaction intermediate and elevate the OER activity.216 A series of VN support was synthesised by simple ammonolysis and Wang & group concluded that NiCo2O4 supported on VN due to the facile electron transfer from VN to NiCo2O4 surface shows more durability and catalytic activity than NiCo2O4 supported on carbon black.217 The same group studied MoS2 supported on VN which show superior HER performance.218
6.3.2. Transition metal phosphides. Transition metal phosphides are other promising class of electrocatalysts for electrochemical water splitting. They are being used extensively catalysts in hydro processing reactions like hydrodesulphurization, that involves hydrogen adsorption and dissociation steps, hence embarking potential adequacy as electrocatalyst for HER. As protons are not easily available in alkaline medium, the electrocatalytic activity of TMP was found to be unsatisfactory. Attempts to boost the catalytic activity is by doping via heteroatom, by bimetallic alloys enhancing surface area by architecture engineering or by usage of substrates. Among non-metal dopants sulphur is the most common, its notices that even the amount sulphur being doped has a drastic impact on the performance of the electrocatalyst. Varying amount of sulphur doped in nickel phosphide nanoplate arrays on carbon paper was evaluated for HER activity by Xing and group,219 it was observed 6 wt% of sulphur gave the maximum catalyst reactivity which have overpotential of 56 mV at 10 mA cm−2 and 104 mV at 100 mA cm−2. Doping of Zn along with sulphur has also improved overall water splitting activity both n neutral and alkaline medium and was extensively studied by Li et al. To drive HER process, the catalyst Zn0.075S–Co0.925P NRCs/CP in neutral medium (1.0 M PBS) has overpotential of 67 mV at 10 mA cm−2 & 37 mV at 10 mA cm−2 in 1.0 M KOH with durability of 20 h and overall water splitting of 1.70 V.220 The structural analysis accompanied the DFT calculation gave an insight on how doping of both Zn and S, optimized the electronic structure and increased the active sites. Boron doped TMP recently gained attention as HER electro activity as Boron being less electronegative than P The XPS studies of B-CoP anchored on MWCNT221 show that the electron density of the bonded Co is only weakly withdrawn by B than P, confirmed by the shift of the binding energies by 0.3 eV of both Co 2p3/2 and Co 2p1/2 making the oxidation state of B-CoP. Compared to undoped NiMoP2/Ni their oxygen doped counterpart had lower overpotential as ΔGH* value (DFT calculations) approached zero on doping making the HER kinetic facile.221,222 Ni2P when doped with oxygen and iron exhibited excellent performance for HER and OER at high pH.223 The electrochemical surface oxidation of Ni2P in 0.1 M potassium hydroxide led to the formation of a interface between Ni2P and amorphous nihydr(oxy)oxides (sc-Ni2Pδ−/NiHO) forming negatively charged Ni2P, confirmed by XANES and XPS studies. sc-Ni2Pδ−/NiHO compared to Ni2P exhibited higher HER activity and lifetime of 100 h.224 Fe doped CoP holey nanosheets in alkaline medium behave as bifunctional catalysts for both OER and HER as seen in (Fig. 20) with faradaic efficiency of 96.4% and 95.4% and overpotential 79 and 220 mV to achieve 10 mA cm−2, for hydrogen evolution reaction and oxygen evolution reaction respectively along with the overall water splitting at 1.55 V @ 10 mA cm−2 and lifetime of 20 h.225 Moreover there was no attenuation after running chronopotentiometric tests for one day which is viable quality for OER catalysts.
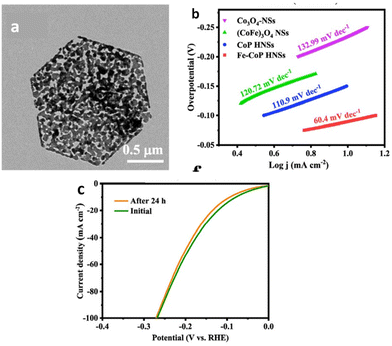 |
| | Fig. 20 Fe doped CoP electrocatalyst TEM (a), electrocatalyst Tafel plots (b), and LSV curves before and after a 24 hours test for Fe CoP HNSs (c). Reproduced with permission225 Copyright 2021, Elsevier. | |
Da Huo226 recently published their work on carbon encapsulated nickel phosphide as OER catalyst. A safe and simple strategy was considered, i.e. by pyrolysis of the precursor nickel triphenylphosphine which generate current density of 10 mA cm−2 at overpotential of 0.26 V and stability was ensured as there was no current change even after polarization for over 25 h. Core shell Fe2O3@C@CoP nanocomposite227 synthesised by Xia Zhong proposed that phosphorus vacancies promote Co O C bonds i.e. direct coupling of cobalt of CoP with the functional group oxygen from the carbon layer thereby enhancing OER performance having a small Tafel slope of 55 mV dec−1 with low overpotential of 230 mV and durability of 25 h. The electro activity is seen to increase even by doping with only trace amount of transition metal like Co and Ni to TMP which was evaluated by Zhang et al.228 Yihao Liu and group studied the pre and post electrolysis characterisation of Mn doped CoP and noticed that during OER Mn–CoP is converted to Mn–CoOOH hexagonal nanosheets which serve as active species.229 By doping Mn gap the gap states of the Fermi level of active O sites allowing the deprotonation of OH* thereby decreasing the RDS energy barrier. Doping of around 0.02% Ni or Co showed significant improvement in MoP as HER electrocatalyst in acidic medium with overpotential 102 mV at −10 mA cm−2 whereas MoP had around twice the overpotential i.e. 210 mV.228 3D core–shell CoP@CoFe-LDH/NF heterostructures exhibited enhanced the OER performance with small Tafel slope value of 69.2 mV dec−1 and high TOF value of 0.093 s−1 at 300 mV and durability of 20 h.230 The nanosheets are interconnected improving the number of active sites doping of TMP with noble metals have significantly bettered the activity and reduced the entire reliance on precious metals. Only 3.85 wt% ruthenium doped Ni5P4 synthesised via impregnation of Ru3+ into Ni(OH)2 followed by phosphidation as HER electrocatalyst with Tafel slope of 52 mV dec−1 and overpotential of 54 mV at −10 mA cm−2 with excellent durability of 120 h.231 The most recent work involves doping of Ru onto NiCoP inducing numerous Ni and Co vacancies leading to abundant number of active sites and increased conductivity after electrochemical activation. Dr Jun Kim and group232 proved Ru doped NiCoP showed excellent OER catalytic performance with Tafel slope value of 42.7 mVdec−1. Multicomponent heterostructures Co–Mo–P/Zn–Co–S in alkaline medium act as bifunctional catalyst for HER and OER displaying outstanding activity that outperforms the state of the art catalyst because of their large surface area, synergistic effect of the hybrid material and augmented electron and mass transfer.
6.3.3. Transition metal sulphides and selenides. The intrinsic electro activity, excellent performance due their metallic/semiconducting nature, variety of crystal phases, flexibility for incoporperation of dopants make transition metal disulphides (TMDs) excellent HER and OER electrocatalysts. The efficient HER electrocatalyst among MoS2, WS2, NbS2 and Nb1.35S2 was studied by Jieun et al. and the disulphide with excess niobium showcase excellent activity and lowest onset potential.233 MoS2 & WS2 deposition on sulphur doped graphene had good HER activity and Antonia et al.234 confirmed that presence of the doped graphene was responsible for the efficiency. Among the many single atom dopants of MoS2, ruthenium doped MoS2 exhibit the highest HER activity. HER performance was also studied for gold encapsulated MoS2 shells and it was concluded the core size of Au matters. Low Tafel slope value and overpotential was exhibited by the largest Au core.235 Whereas Co doped WS2 (ref. 236) also showed HER activity and the presence of surface macropores due to the use of hydrogen peroxide, helped facilitate the proton and electrolyte diffusion. By supporting CoS2 nanowires on graphdiyne, the unsaturated sulphurs in (211) plane of CoS2 becomes activated, diminishing the energy barriers of electron transfer caused by the synergistic effect between the planar unsaturated sulphur and the sp1 hybridized carbons of graphdiyne.237 Graphdiyne-CoS2 heterojunction nanocomposite with low overpotential of 97 mV at 10 mA cm−2 exhibit superior HER catalytic performance. Nickel based sulphide being easy to prepare, cheap and having electroactive sites are considered promising alternatives for noble metal catalysts. Its conductivity and metallicity is due to continuous Ni–Ni bonds. Recently, by rational cationic-doped strategy, Zn was doped on Ni3S2 nanosheet arrays supported on Ni foam (Zn–Ni3S2/NF)238 and it was revealed that the electronic structure is modulated on doping with Zn altering the ΔGH* thus exhibiting HER catalytic performance the interaction involved contributing to this electro activity is further confirmed XPS and Raman spectrum. XiaoqingMao measured the electrochemical tests for oxygen evolution and concluded that Co and Fe doped Ni3S4 shows better performance that than single cation doped and pure Ni3S4 with the overall water splitting of 1.47 V @ 10 mA cm−2.239 Among four different morphologies, Gong et al. concluded that 3D NiCo2S4 nanostructures had the excellent OER catalytic activity.240 Mn-doped NiCo2S4,241 sea urchin-shaped nanospheres catalyst grown on nickel foam also exhibit OER activity and have overpotential 40 mV less than NiCo2S4/NF and Tafel slope lower than pure NiCo2S4. Mingjin Cui synthesised and studied the electrocatalytic OER properties of quinary high entropy metal sulfide (CrMnFeCoNi)Sx in 1 M KOH.242 The catalyst was observed to outperform most MxSy materials because of the synergistic effect of the metals caused by the transfer of charge state between the different metallic sites, established from computational results. The quinary HEMS nanoparticles had low overpotential of 295 mV at a current density of 100 mA cm−2 and excellent stability. Yu Ma designed a 2D bimetallic sulfoselenide nanosheets (NiSe0.1MoS6.4/NF-P123) via one step hydrothermal method. Inorder to tailor the nanosheet, morphology-controlling reagents such as Mo7O246− and Pluronic P123 are used.242,243 The synergistic effect of Ni and Mo accompanied by presence of S and Se contribute to electronic properties and elevate HER kinetics and behave as outstanding non-metal based HER electrocatalysts with the excellent stability up to 18 h.Selenium centres have localized polarization induced partial negative charges enabling them to capture protons and improve HER. Moreover the coordination step with hydroxides compared to P and S site; Se site is less affected, allowing the dioxygen molecule delivery to be quicker, making them more efficient than sulphides and phosphides. The primary preparation method to synthesis Se based electrocatalyst is to use hydrothermal or solvothermal method for precursor followed by selenization by selenium powder or its composite and the different mono/bi and multimetallic transition metal selenides, their HER and OER aspects was reported by Xia & group. Increasing the Se content on NiSe2 reduced the overpotential and lower the Tafel slope value in case of NiSe2 nanosheet for HER.244 The morphology optimization also counts as NiSe2 nanowire had higher HER activity over NiSe2 nanoparticle. Hui Lui doped P anion onto NiSe2 nanosheet in (Fig. 21), studied their HER catalytic kinetics in alkaline medium and noticed improved activity with Tafel slope of 61.3 mV dec−1 and a low overpotential of 86 mV at 10 mA cm−2 with overall water splitting at 1.62 V @ 10 mA cm−2 and durability of 4 h.245 Bimetallic NiFe selenide based metal–organic framework is also studied for their efficiency in oxygen evolution reaction. The synergistic effects of Ni and Fe increased the OER electrocatalytic activity Fe2NiSe4 phase.246 NiFe–Se/CFP compared with monometallic FeSe2/CFP and NiSe2/CFP have lower overpotential of 281 mV at 10 mA cm−2 and shows a small Tafel slope of 40.93 mV dec−1. Heterostructural NiSe2/FeSe2 nanoparticles surpass the OER performance of the state of the art catalyst RuO2 with overpotential of 274 mV at 40 mA cm−2 and Tafel slope value of 57.07 mV dec−1, owing to the heterointerface interaction between NiSe2 and FeSe2 which reduces the Gibbs free energy of intermediates having oxygen from 3.15 eV for NiSe2 to 2.14 eV for NiSe2/FeSe2 heterostructures which is confirmed from the DFT calculations. Yuan Huang et al. reported phosphorus doping of the fe electronic structure in iron–nickel selenide nanosheets as OER electrocatalysts.247
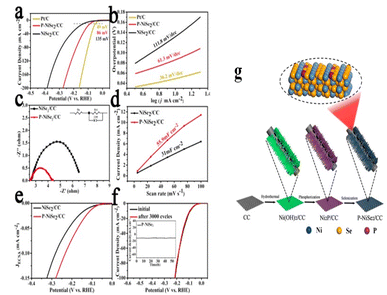 |
| | Fig. 21 (a) Polarization curves of P-NiSe2/CC, NiSe2/CC, Pt/C, and CC electrodes in 1.0 M KOH, (b) corresponding Tafel plots of P-NiSe2/CC, NiSe2/CC, and Pt/C electrodes, (c) Nyquist plots of P-NiSe2/CC and NiSe2/CC electrodes and inset has equivalent circuit, (d) the Cdl values of P-NiSe2/CC and NiSe2/CC electrodes at scanning rate of 5, 25, 50, 75 and 100 mV s. (e) Based on the electrochemical active surface area, the specific activity of P-NiSe2/CC and NiSe2/CC electrodes are depicted (f) durability testing showing the P-NiSe2/CC electrode's first and 3000th polarization curves; inset shows a time-dependent current density curve recorded on the P-NiSe2/CC electrode for 55 hours at a constant overpotential of 10 mV (vs. RHE). (g) Fabrication process schematic illustration. Reproduced with permission.245 Copyright 2021, Elsevier. | |
The number of active sites increased on doping with P as P-Ni0.75Fe0.25Se2 had higher electrochemical double layer capacitance (Cdl) value of 6.25 mF cm−2 when compared to Ni0.75Fe0.25Se2 with 4.13 mF cm−2 and the RuO2 with 5.92 mF cm−2. The OER activity also follow the similar trend as that of the capacitances making them ideal electrocatalyst. Shuyuan Pan et al. synthesised heterostructured electrocatalyst having boosted HER activity comprising of metallic Rh and Rh3Se4 prepared by partial selenization of Rh/C at 300C with overpotential 32 mV and 29 mV at 10 mA cm−2 in both acidic and alkaline environments respectively.248 Rhodium increased the electric density near Fermi level thereby improving the electronic conductivity.
6.3.4. Noble metals and their composites. Electrocatalyst based on platinum, palladium, ruthenium, rhodium and iridium noble metals are commonly acknowledged as outstanding electrocatalyst for electrochemical water splitting. While Ru/Ir oxides are excellent OER catalyst and HER activity is showed by Pt and Pt based nanomaterials. However because to their expensive amount and lack of abundance, cost effective approaches such as substitution of these noble metals, doping of low amount of noble metals which act as active centres into non-noble electrocatalyst are being taken into consideration. During proton exchange membrane fuel cell operation, the catalyst deteriorates gradually, preventing sudden proton exchange membrane fuel cell failure. since the anode is exposed to high potentials (>1.5 V), the stability of the electrocatalyst at the anode is crucial. In a few studies, the state-of-the-art iridium dissolution during anodic treatment has been documented. The mechanism of iridium dissolution has been proposed by Cherevko et al.249 Because soluble iridium (III) complexes, such as Ir(OH)3 or Ir2O3, are well known for their instability and have a relatively high solubility at low pH, were accounted for the dissolution of iridium catalyst. The authors claim that the formation pathway of Ir(III) complexes i.e. the dissolution of hydrous iridium oxide in the region of OER is explained below. | | |
2IrO2(OH) + 2H2O → 2Ir(OH)3 + O2
| (87) |
| | |
2IrO2(OH) → 2HIrO2 + O2
| (88) |
The entire oxygen evolution reaction pathway, forming a closed cycle, can be rewritten as follows for the second reaction:
| | |
HIrO2 → IrO2 + H+ + e−
| (89) |
| | |
IrO2 + H2O → IrO2(OH) + H+ + e−
| (90) |
| | |
2IrO2(OH) → 2HIrO2 + O2
| (91) |
Additionally, the oxidizing ability of iridium is correlated with the rate at which anode catalyst dissolves. The catalyst dissolution process may be slowed down by a lower oxidation state caused by interactions between the metal and the metal oxide support. Under high current densities (4 A cm2), it has been noted that the dissolution and migration of iridium particles worsen significantly and significant portion of the iridium catalyst particles was seen to diffuse into the membrane after a lengthy test.250
Doping of ultralow Ru into Co MoF followed by annealing in air led to conversion of hollow Ru–Co MOF derivative into (Ru–Co)Ox nanosheets which consist of Co3O4 and RuO2.251 The nanosheets provide active sites and Ru induces modulation in electronic structures that helps strengthen the adsorption of Oads and weaken the Hads adsorption resulting in outstanding OER and HER activity. Li et al. introduced a single atom Ru into MOF and observed that the electronic structure was tailored upon the doping. H* and H2O, binding strength are altered owing to the excellent HER activity. Ir4+ doped NiFe layered double hydroxide (LDH) as efficient HER electrocatalyst was reported by Chen and coworkers, and the improved dissolution kinetics of water molecules is was found to contributed by doped Ir.252 Fan et al. fabricated atomic Ir doped NiCo LDH and the EPR (electron paramagnetic resonance) spectra confirmed the elevation in production of oxygen vacancies after incorporation of Ir.253 There was also increase in active sites, promoting both HER and OER performances. Bifunctional catalyst Ir doped NiV(OH)2 developed by Du et al., carried out both theoretical and experimental calculations and concluded that Ir, V and O modified the electronic structure as well as adsorption energies of intermediated and water molecules acting as HER and OER electrocatalysts.254 Zhu et al. observed that rhodium atom improves electrical conductivity of CoFe LDH by reducing energy barriers and induce Fe vacancies which favor both HER and OER activity, as they have overpotential of 28 mV for the hydrogen evolution reaction and 245 mV for the oxygen evolution reaction at 10 mA cm−2.255
2 Dimensional Ru nanosheets were developed by Kong et al.256 via simple solvothermal method that exhibited overall water splitting. 2D nanostructures are highly appreciated as catalysts as there is exposure of nearly all atoms which act as active sites and favor interfacial charge transfer. Compared to rhodium nanocrystal with tetrahedra and concave tetrahedra, rhodium nanosheets delivered better electrocatalytic activity towards both HER and OER. This is because the electrochemical surface area (ECSA) of Rh tetrahedra is 32.5 m2 g−1, Rh concave tetrahedra is 18.9 m2 g−1, while Rh nanosheets have the highest at 65.3 m2 g−1. 2D PdCu nanosheets facilitate electron transfer due to 3D their architecture and synergistic effect between Pd & Cu, thus exhibiting HER performance.257 Multi metallic ultrathin nanorings have seen to have improved electron kinetics and efficiency and thus Guo and coworkers demonstrated that PtPd based multi metallic nanorings have excellent OER activity. Pt deposition on 2D Pd3Pb enhanced the electrocatalytic HER performance in (Fig. 22) due to the improved H* binding on Pt attributed charge transfer between Pd3Pb.258,259 2D Ir nanosheets developed by Yamauchi et al. demonstrated better oxygen evolution activity having an overpotential of 240 mV at 10 mA cm−2.260 Table S1† shows the comparison of bi-functional OER and HER activities of various s, p & d block metal catalyst & noble metals based on performance and lifetime.
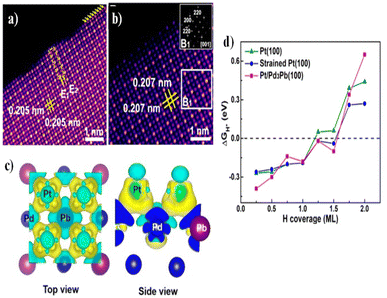 |
| | Fig. 22 Atomic-resolution HAADF-STEM images of the (A) Pd3Pb and (B) AL-Pt/Pd3Pb. (C) Pt and Pd3Pb charge density differences in AL-Pt/Pd3Pb. (D) The calculated average binding energy of H* at various hydrogen coverages on Pt(100), strained Pt(100), and Pt/Pd3Pb(100) Reproduced with permission258 Copyright 2019, American Chemical Society. | |
7. Conclusion
Fuel cell technology is a more environmentally friendly method of generating electrical energy from various fuels, including hydrogen, oxygen, methanol, formic acid, borohydride, etc. The greenest approach to creating electrical power is via a simple process employing H2 and O2 as fuel. The electrochemical redox process generates water as a byproduct. If the H2 provided is as pure as feasible, the fuel cell's efficiency can be as high as attainable. The current technique for H2 production of coal reforming technology isn't the best option for getting the cleanest H2. Electrochemical water splitting is a better method of generating vast amounts of hydrogen in a short period of time with the most outstanding possible purity, thereby minimizing the impact on the environment. The key to improving water electrolysis efficiency and lowering costs is the creation of high-efficiency, low-cost electrocatalysts with low overpotential and minimum value of Tafel slope for HER & OER. The state-of-the-art electrocatalyst Pt is for HER, and in the case of OER, the state-of-the-art catalysts are iridium or ruthenium. Their compounds were considered and continue to be ideal catalysts. PGM catalysts have demonstrated good electrocatalytic activity in both HER and OER to date. Their morphology also matters as noble metals with nanostructure and controlled crystal form has been widely studied to get higher performance to optimize the usage of these rare resources. Modifying, composing, or alloying PGMs with specific materials on purpose has led to enhanced electrochemical activity. Recently there has been a greater emphasis on using inexpensive non-noble metals to replace PGM catalysts. Fabrication of non-noble metal electrocatalysts with nanostructures with different morphologies to increase their surface area and conductivity also accompanied by support and different precursor templates are extensively being studied. In this review, other mechanisms of HER and OER, parameters involved in electrochemical studies, and many of the metal and metalloids based electrocatalysts that have been recently reported are detailed. Their electrochemical activities in different electrolytes in accordance with their physiochemical characterizations are briefly explained. There are several challenges while switching from conventional combustion engines to fuel cells where hydrogen is considered a fuel. Most HER/OER electrocatalysts have been investigated in current density ranging from 10 to 100 mA cm−2. However, their operation in high current densities like 2 A cm−2 in water electrolyzer is still challenging, and these conditions require much more advanced electrochemical techniques besides CV and Tafel slopes. Another major problem is that O2 may diffuse into the cathode chamber, combine with the generated hydrogen and be converted back to the water. This worsens if operated at high current densities. Another reason for opting for low current density is to decrease the ohmic resistance of the diaphragm and liquid electrolyte. Experimental studies convey that HER activity is excellent in acidic medium whereas OER activity in alkaline medium. Still, combining their activity is drastically lower with weak stability and insufficient corrosion resistance. Many groups are working to rectify this issue and develop a versatile material in all pH.
Thus, many efforts are put across the globe to develop new materials to improve the performance of the already developed catalysts for HER and OER processes. The investigations are directed to the synthesis of inexpensive, sustainable and environmental friendly bifunctional catalysts that could carry out both HER and OER activities in acidic, alkaline, and near-neutral mediums and their structural changes, chemical adsorption processes, and coordination environment can be studied by in situ techniques such as XPS, in situ operando Raman, XAS which help in the upcoming studies.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
The author wishes to acknowledge the basic support from the National Institute of Technology Puducherry, Karaikal, India.
Notes and references
- J. L. Liu and S. Bashir, Advanced Nanomaterials and Their Applications in Renewable Energy, Elsevier Inc., Amsterdam, 2015, pp. 1–438 Search PubMed.
- T. R. Cook, D. K. Dogutan, S. Y. Reece, Y. Surendranath, T. S. Teets and D. G. Nocera, Chem. Rev., 2010, 110, 6474–6502 CrossRef CAS PubMed.
- S. Chu, Y. Cui and N. Liu, Nat. Mater., 2017, 16, 16–22 CrossRef PubMed.
- S. Chu and A. Majumdar, Nature, 2012, 488, 294–303 CrossRef CAS PubMed.
- P. P. Edwards, V. L. Kuznetsov, W. I. David and N. P. Brandon, Energy Policy, 2008, 36, 4356–4362 CrossRef.
- I. Roger, M. A. Shipman and M. D. Symes, Nat. Rev. Chem., 2017, 1, 1–13 CrossRef.
- J. M. Ogden, Phys. Today, 2002, 55, 69 CrossRef CAS.
- Y. Gong, Y. Zhi, Y. Lin, T. Zhou, J. Li, F. Jiao and W. Wang, Dalton Trans., 2019, 48, 6718–6729 RSC.
- W. Wang, M. Xu, X. Xu, W. Zhou and Z. Shao, Angew. Chem., Int. Ed., 2020, 59, 136–152 CrossRef CAS PubMed.
- Y. Yan, T. He, B. Zhao, K. Qi, H. Liu and B. Y. Xia, J. Mater. Chem. A, 2018, 6, 15905–15926 RSC.
- Q. Hu, G. Li, G. Li, X. Liu, B. Zhu, X. Chai, Q. Zhang, J. Liu and C. He, Adv. Energy Mater., 2019, 9, 1803867 CrossRef.
- N. Armaroli and V. Balzani, ChemSusChem, 2011, 4, 21–36 CrossRef CAS PubMed.
- Y.-J. Tang, H.-J. Zhu, L.-Z. Dong, A.-M. Zhang, S.-L. Li, J. Liu and Y.-Q. Lan, Appl. Catal., B, 2019, 245, 528–535 CrossRef CAS.
- H. Cao, X. Gong, T. Liu, F. Xiao, X. Lv, J. Zhou and L. Gai, J. Alloys Compd., 2019, 797, 341–347 CrossRef CAS.
- A. P. Simpson and A. E. Lutz, Int. J. Hydrogen Energy, 2007, 32, 4811–4820 CrossRef CAS.
- D. K. Liguras, D. I. Kondarides and X. E. Verykios, Appl. Catal., B, 2003, 43, 345–354 CrossRef CAS.
- L. Barelli, G. Bidini, F. Gallorini and S. Servili, Energy, 2008, 33, 554–570 CrossRef CAS.
- J. F. Callejas, J. M. McEnaney, C. G. Read, J. C. Crompton, A. J. Biacchi, E. J. Popczun, T. R. Gordon, N. S. Lewis and R. E. Schaak, ACS Nano, 2014, 8, 11101–11107 CrossRef CAS PubMed.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC.
- R. Shwetharani, M. Sakar, C. Fernando, V. Binas and R. G. Balakrishna, Catal. Sci. Technol., 2019, 9, 12–46 RSC.
- Z. Chen, T. F. Jaramillo, T. G. Deutsch, A. Kleiman-Shwarsctein, A. J. Forman, N. Gaillard, R. Garland, K. Takanabe, C. Heske and M. Sunkara, J. Mater. Res., 2010, 25, 3–16 CrossRef CAS.
- K. Tedsree, T. Li, S. Jones, C. W. A. Chan, K. M. K. Yu, P. A. Bagot, E. A. Marquis, G. D. Smith and S. C. E. Tsang, Nat. Nanotechnol., 2011, 6, 302–307 CrossRef CAS PubMed.
- B. H. Liu, Z. P. Li and S. Suda, J. Alloys Compd., 2006, 415, 288–293 CrossRef CAS.
- Y. Kojima, K. Suzuki, K. Fukumoto and M. Sasaki, Int. J. Hydrogen Energy, 2002, 27, 1029–1034 CrossRef CAS.
- J.-H. Kim, H. Lee, S.-C. Han, H.-S. Kim, M.-S. Song and J.-Y. Lee, Int. J. Hydrogen Energy, 2004, 29, 263–267 CrossRef.
- U. Demirci, O. Akdim and P. Miele, Int. J. Hydrogen Energy, 2009, 34, 2638–2645 CrossRef CAS.
- D. Xu, H. Zhang and W. Ye, Catal. Commun., 2007, 8, 1767–1771 CrossRef CAS.
- Y. Jiao, Y. Zheng, M. Jaroniec and S. Z. Qiao, Chem. Soc. Rev., 2015, 44, 2060–2086 RSC.
- B. You and Y. Sun, ChemPlusChem, 2016, 81, 1045–1055 CrossRef CAS PubMed.
- Y. P. Zhu, C. Guo, Y. Zheng and S.-Z. Qiao, Acc. Chem. Res., 2017, 50, 915–923 CrossRef CAS PubMed.
- Y. Yan, B. Y. Xia, B. Zhao and X. Wang, J. Mater. Chem. A, 2016, 4, 17587–17603 RSC.
- D. V. Schroeder, An Introduction to Thermal Physics, Addison-Wesley Publishing Company, 1999, pp. 1–214 Search PubMed.
- C. G. Morales-Guio, L.-A. Stern and X. Hu, Chem. Soc. Rev., 2014, 43, 6555–6569 RSC.
- X. Zou and Y. Zhang, Chem. Soc. Rev., 2015, 44, 5148–5180 RSC.
- Y. Shi and B. Zhang, Chem. Soc. Rev., 2016, 45, 1529–1541 RSC.
- S. Anantharaj, P. E. Karthik, B. Subramanian and S. Kundu, ACS Catal., 2016, 6, 4660–4672 CrossRef CAS.
- C. Wang, K. E. DeKrafft and W. Lin, J. Am. Chem. Soc., 2012, 134, 7211–7214 CrossRef CAS PubMed.
- A. R. Zeradjanin, J. Masa, I. Spanos and R. Schlögl, Front. Energy Res., 2021, 8, 613092 CrossRef.
- J. Rossmeisl, Z.-W. Qu, H. Zhu, G.-J. Kroes and J. K. Nørskov, J. Electroanal. Chem., 2007, 607, 83–89 CrossRef CAS.
- V. Alves, L. Da Silva, J. Boodts and S. Trasatti, Electrochim. Acta, 1994, 39, 1585–1589 CrossRef CAS.
- C. C. McCrory, S. Jung, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2013, 135, 16977–16987 CrossRef CAS PubMed.
- K. Zeng and D. Zhang, Prog. Energy Combust. Sci., 2010, 36, 307–326 CrossRef CAS.
- X. Xia, N. Deng, G. Cui, J. Xie, X. Shi, Y. Zhao, Q. Wang, W. Wang and B. Tang, Chem. Commun., 2015, 51, 10899–10902 RSC.
- J. Peng, W. Dong, Z. Wang, Y. Meng, W. Liu, P. Song and Z. Liu, Mater. Today Adv., 2020, 8, 100081 CrossRef.
- M. G. de Chialvo and A. Chialvo, J. Braz. Chem. Soc., 1994, 5, 137–143 CrossRef.
- M. Zeng and Y. Li, J. Mater. Chem. A, 2015, 3, 14942–14962 RSC.
- M. G. de Chialvo and A. Chialvo, J. Electroanal. Chem., 1994, 372, 209–223 CrossRef.
- S. Gupta, M. K. Patel, A. Miotello and N. Patel, Adv. Funct. Mater., 2020, 30, 1906481 CrossRef CAS.
- A. J. Bard and L. R. Faulkner, Electrochem., 2001, 2, 580–632 Search PubMed.
- L. Peter, Platinum Met. Rev., 2002, 46, 15–17 Search PubMed.
- Y.-P. Zhu, X. Xu, H. Su, Y.-P. Liu, T. Chen and Z.-Y. Yuan, ACS Appl. Mater. Interfaces, 2015, 7, 28369–28376 CrossRef CAS PubMed.
- C. Zhang, H. Nan, H. Tian and W. Zheng, J. Alloys Compd., 2020, 838, 155685 CrossRef CAS.
- S. Anantharaj, S. R. Ede, K. Sakthikumar, K. Karthick, S. Mishra and S. Kundu, ACS Catal., 2016, 6, 8069–8097 CrossRef CAS.
- Y. Zhan, M. Lu, S. Yang, C. Xu, Z. Liu and J. Y. Lee, ChemCatChem, 2016, 8, 372–379 CrossRef CAS.
- H. Osgood, S. V. Devaguptapu, H. Xu, J. Cho and G. Wu, Nano Today, 2016, 11, 601–625 CrossRef CAS.
- Y. Matsumoto and E. Sato, Mater. Chem. Phys., 1986, 14, 397–426 CrossRef CAS.
- J. O. M. Bockris and T. Otagawa, J. Electrochem. Soc., 1984, 131, 290 CrossRef CAS.
- E. Fabbri, A. Habereder, K. Waltar, R. Kötz and T. J. Schmidt, Catal. Sci. Technol., 2014, 4, 3800–3821 RSC.
- I. C. Man, H. Y. Su, F. Calle-Vallejo, H. A. Hansen, J. I. Martínez, N. G. Inoglu, J. Kitchin, T. F. Jaramillo, J. K. Nørskov and J. Rossmeisl, ChemCatChem, 2011, 3, 1159–1165 CrossRef CAS.
- J. S. Yoo, X. Rong, Y. Liu and A. M. Kolpak, ACS Catal., 2018, 8, 4628–4636 CrossRef CAS.
- J. T. Mefford, X. Rong, A. M. Abakumov, W. G. Hardin, S. Dai, A. M. Kolpak, K. P. Johnston and K. J. Stevenson, Nat. Commun., 2016, 7, 1–11 Search PubMed.
- A. Grimaud, O. Diaz-Morales, B. Han, W. T. Hong, Y.-L. Lee, L. Giordano, K. A. Stoerzinger, M. Koper and Y. Shao-Horn, Nat. Chem., 2017, 9, 457–465 CrossRef CAS PubMed.
- R. L. Doyle and M. E. Lyons, in Photoelectrochemical solar fuel production, Springer, 2016, pp. 41–104 Search PubMed.
- J. Wang, X. Yue, Y. Yang, S. Sirisomboonchai, P. Wang, X. Ma, A. Abudula and G. Guan, J. Alloys Compd., 2020, 819, 153346 CrossRef CAS.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef PubMed.
- J. O. M. Bockris, J. Chem. Phys., 1956, 24, 817–827 CrossRef CAS.
- P. E. Karthik, H. Rajan, V. R. Jothi, B.-I. Sang and S. C. Yi, J. Hazard. Mater., 2022, 421, 126687 CrossRef CAS PubMed.
- S. Anantharaj, S. Ede, K. Karthick, S. S. Sankar, K. Sangeetha, P. Karthik and S. Kundu, Energy Environ. Sci., 2018, 11, 744–771 RSC.
- J. Kibsgaard and I. Chorkendorff, Nat. Energy, 2019, 4, 430–433 CrossRef.
- S. Anantharaj, S. Ede, K. Sakthikumar, K. Karthick, S. Mishra, S. Kundu and A. Catal, Adv. Mater., 2016, 28, 9266–9291 CrossRef PubMed.
- S. Anantharaj, K. Karthick and S. Kundu, Mater. Today Energy, 2017, 6, 1–26 CrossRef.
- L. D. S. Munoz, A. Bergel, D. Féron and R. Basseguy, Int. J. Hydrogen Energy, 2010, 35, 8561–8568 CrossRef.
- R. H. Coridan, A. C. Nielander, S. A. Francis, M. T. McDowell, V. Dix, S. M. Chatman and N. S. Lewis, Energy Environ. Sci., 2015, 8, 2886–2901 RSC.
- C. C. McCrory, S. Jung, I. M. Ferrer, S. M. Chatman, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2015, 137, 4347–4357 CrossRef CAS PubMed.
- M. Hussain, J. Baschuk, X. Li and I. Dincer, Int. J. Therm. Sci., 2005, 44, 903–911 CrossRef CAS.
- R. Alam and J. Q. Shang, J. Water Process. Eng., 2016, 12, 78–88 CrossRef.
- P. C. Vesborg, B. Seger and I. Chorkendorff, J. Phys. Chem. Lett., 2015, 6, 951–957 CrossRef CAS PubMed.
- M. Xiao, Y. Miao, Y. Tian and Y. Yan, Electrochim. Acta, 2015, 165, 206–210 CrossRef CAS.
- C. Xiang, K. M. Papadantonakis and N. S. Lewis, Mater. Horiz., 2016, 3, 169–173 RSC.
- B. Han, S. M. Steen III, J. Mo and F.-Y. Zhang, Int. J. Hydrogen Energy, 2015, 40, 7006–7016 CrossRef CAS.
- M. Ni, M. K. Leung and D. Y. Leung, Proceeding of the WHEC, 2006, vol. 16 Search PubMed.
- M. Wang, Z. Wang, X. Gong and Z. Guo, Renewable Sustainable Energy Rev., 2014, 29, 573–588 CrossRef CAS.
- A. L. Santos, M.-J. Cebola and D. M. Santos, Energies, 2021, 14, 3193 CrossRef CAS.
- E. Fabbri and T. J. Schmidt, ACS catal., 2018, 8, 9765–9774 CrossRef CAS.
- S. Cherevko, A. R. Zeradjanin, A. A. Topalov, N. Kulyk, I. Katsounaros and K. J. Mayrhofer, ChemCatChem, 2014, 6, 2219–2223 CrossRef CAS.
- S. Anantharaj, S. Kundu and S. Noda, J. Mater. Chem. A, 2020, 8, 4174–4192 RSC.
- Y. Hou, M. R. Lohe, J. Zhang, S. Liu, X. Zhuang and X. Feng, Energy Environ. Sci., 2016, 9, 478–483 RSC.
- Z. Lu, W. Xu, W. Zhu, Q. Yang, X. Lei, J. Liu, Y. Li, X. Sun and X. Duan, Chem. Commun., 2014, 50, 6479–6482 RSC.
- H. Liang, F. Meng, M. Cabán-Acevedo, L. Li, A. Forticaux, L. Xiu, Z. Wang and S. Jin, Nano Lett., 2015, 15, 1421–1427 CrossRef CAS PubMed.
- J. M. V. Nsanzimana, V. Reddu, Y. Peng, Z. Huang, C. Wang and X. Wang, Chem.–Eur. J., 2018, 24, 18502–18511 CrossRef CAS PubMed.
- Y. Liang, X. Sun, A. M. Asiri and Y. He, Nanotechnology, 2016, 27, 12LT01 CrossRef PubMed.
- K. A. Lewinski, D. van der Vliet and S. M. Luopa, ECS Trans., 2015, 69, 893 CrossRef CAS.
- T. Shinagawa, A. T. Garcia-Esparza and K. Takanabe, Sci. Rep., 2015, 5, 1–21 Search PubMed.
- D. Koley and A. J. Bard, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 11522–11527 CrossRef CAS PubMed.
- J. M. Bockris and E. Potter, J. Electrochem. Soc., 1952, 99, 169 CrossRef CAS.
- S. Anantharaj and S. Kundu, ACS Energy Lett., 2019, 4, 1260–1264 CrossRef CAS.
- Y. Guo, T. Park, J. W. Yi, J. Henzie, J. Kim, Z. Wang, B. Jiang, Y. Bando, Y. Sugahara and J. Tang, Adv. Mater., 2019, 31, 1807134 CrossRef PubMed.
- K. Liu, H. Zhong, F. Meng, X. Zhang, J. Yan and Q. Jiang, Mater. Chem. Front., 2017, 1, 2155–2173 RSC.
- H. Vrubel, T. Moehl, M. Grätzel and X. Hu, Chem. Commun., 2013, 49, 8985–8987 RSC.
- S. Anantharaj, P. Karthik and S. Kundu, J. Mater. Chem. A, 2015, 3, 24463–24478 RSC.
- S. Anantharaj, P. Karthik and S. Kundu, Catal. Sci. Technol., 2017, 7, 882–893 RSC.
- T. N. Kumar, S. Sivabalan, N. Chandrasekaran and K. L. Phani, Chem. Commun., 2015, 51, 1922–1925 RSC.
- S.-X. Guo, Y. Liu, A. M. Bond, J. Zhang, P. E. Karthik, I. Maheshwaran, S. S. Kumar and K. L. N. Phani, Phys. Chem. Chem. Phys., 2014, 16, 19035–19045 RSC.
- S. Mulkapuri, A. Ravi, R. Nasani, S. K. Kurapati and S. K. Das, Inorg. Chem., 2022, 61, 13868–13882 CrossRef CAS PubMed.
- B. Xiong, L. Chen and J. Shi, ACS Catal., 2018, 8, 3688–3707 CrossRef CAS.
- S. S. Kumar and V. Himabindu, Mater. Sci. Energy Technol., 2019, 2, 442–454 Search PubMed.
- J. Chi and H. Yu, Chin. J. Catal., 2018, 39, 390–394 CrossRef CAS.
- M. Gao, W. Sheng, Z. Zhuang, Q. Fang, S. Gu, J. Jiang and Y. Yan, J. Am. Chem. Soc., 2014, 136, 7077–7084 CrossRef CAS PubMed.
- J. Xie and Y.-C. Lu, Nat. Commun., 2020, 11, 1–4 CrossRef PubMed.
- R. Subbaraman, D. Tripkovic, D. Strmcnik, K.-C. Chang, M. Uchimura, A. P. Paulikas, V. Stamenkovic and N. M. Markovic, Science, 2011, 334, 1256–1260 CrossRef CAS PubMed.
- Z. Jiang, W. Zhou, A. Hong, M. Guo, X. Luo and C. Yuan, ACS Energy Lett., 2019, 4, 2830–2835 CrossRef CAS.
- X. Ren, C. Wei, Y. Sun, X. Liu, F. Meng, X. Meng, S. Sun, S. Xi, Y. Du and Z. Bi, Adv. Mater., 2020, 32, 2001292 CrossRef CAS PubMed.
- S. W. Lee, C. Carlton, M. Risch, Y. Surendranath, S. Chen, S. Furutsuki, A. Yamada, D. G. Nocera and Y. Shao-Horn, J. Am. Chem. Soc., 2012, 134, 16959–16962 CrossRef CAS PubMed.
- T. Maiyalagan, K. A. Jarvis, S. Therese, P. J. Ferreira and A. Manthiram, Nat. Commun., 2014, 5, 1–8 Search PubMed.
- Z. Lu, H. Wang, D. Kong, K. Yan, P.-C. Hsu, G. Zheng, H. Yao, Z. Liang, X. Sun and Y. Cui, Nat. Commun., 2014, 5, 1–7 CAS.
- M. Gong, Y. Li, H. Wang, Y. Liang, J. Z. Wu, J. Zhou, J. Wang, T. Regier, F. Wei and H. Dai, J. Am. Chem. Soc., 2013, 135, 8452–8455 CrossRef CAS PubMed.
- J. Wang, L. Li, H. Tian, Y. Zhang, X. Che and G. Li, ACS Appl. Mater. Interfaces, 2017, 9, 7100–7107 CrossRef CAS PubMed.
- Z. Li, S. Wang, Y. Tian, B. Li, H. jun Yan, S. Zhang, Z. Liu, Q. Zhang, Y. Lin and L. Chen, Chem. Commun., 2020, 56, 1749–1752 RSC.
- J. Xu, S. Wang, C. Yang, T. Li, Q. Liu and X. Kong, Chem. Eng. J., 2021, 421, 129741 CrossRef CAS.
- M. Sakthivel, S. Bhandari and J.-F. Drillet, ECS Electrochem. Lett., 2015, 4, A56 CrossRef CAS.
- D. Chen, C. Chen, Z. M. Baiyee, Z. Shao and F. Ciucci, Chem. Rev., 2015, 115, 9869–9921 CrossRef CAS PubMed.
- X. Cheng, E. Fabbri, M. Nachtegaal, I. E. Castelli, M. El Kazzi, R. Haumont, N. Marzari and T. J. Schmidt, Chem. Mater., 2015, 27, 7662–7672 CrossRef CAS.
- Y. Shi, R. Xie, X. Liu, N. Zhang, C. Aruta and N. Yang, Phys. Chem. Chem. Phys., 2019, 21, 16230–16239 RSC.
- W. Zhou, M. Zhao, F. Liang, S. C. Smith and Z. Zhu, Mater. Horiz., 2015, 2, 495–501 RSC.
- A. A. Eskandrani, S. M. Ali and H. M. Al-Otaibi, Int. J. Mol. Sci., 2020, 21, 3785 CrossRef CAS PubMed.
- M. Li, L. Zhuang, Z. Wang, A. ur Rehman, L. X. Liu and Z. Zhu, J. Colloid Interface Sci., 2019, 553, 813–819 CrossRef CAS PubMed.
- D. Lee, Y.-L. Lee, A. Grimaud, W. T. Hong, M. D. Biegalski, D. Morgan and Y. Shao-Horn, J. Mater. Chem. A, 2014, 2, 6480–6487 RSC.
- X. Xu, Y. Pan, Y. Zhong, L. Ge and Z. Shao, Composites, Part B, 2020, 198, 108214 CrossRef CAS.
- S. Gholamrezaei, M. Ghanbari, O. Amiri, M. Salavati-Niasari and L. K. Foong, Ultrason. Sonochem., 2020, 61, 104829 CrossRef CAS PubMed.
- K. J. May, Surface structural changes of perovskite oxides during oxygen evolution in alkaline electrolyte, Massachusetts Institute of Technology, 2013, pp. 1–69 Search PubMed.
- C.-F. Chen, G. King, R. M. Dickerson, P. A. Papin, S. Gupta, W. R. Kellogg and G. Wu, Nano Energy, 2015, 13, 423–432 CrossRef CAS.
- R. Zhang, C. Tang, R. Kong, G. Du, A. M. Asiri, L. Chen and X. Sun, Nanoscale, 2017, 9, 4793–4800 RSC.
- C. Zhang, R. D. Fagan, R. D. Smith, S. A. Moore, C. P. Berlinguette and S. Trudel, J. Mater. Chem. A, 2015, 3, 756–761 RSC.
- Z. Li, W. Niu, L. Zhou and Y. Yang, ACS Energy Lett., 2018, 3, 892–898 CrossRef CAS.
- L. Ma, Z. Liu, T. Chen, Y. Liu and G. Fang, Electrochim. Acta, 2020, 355, 136777 CrossRef CAS.
- H. Zhang, Q. Liu, J. Xu, L. Wei, Q. Liu and X. Kong, Inorg. Chem., 2019, 58, 8267–8270 CrossRef CAS PubMed.
- J. Y. Chen, J. T. Miller, J. B. Gerken and S. S. Stahl, Energy Environ. Sci., 2014, 7, 1382–1386 RSC.
- X. Wang, P. Sun, H. Lu, K. Tang, Q. Li, C. Wang, Z. Mao, T. Ali and C. Yan, Small, 2019, 15, 1804886 CrossRef PubMed.
- A. Kakoria, B. Devi, A. Anand, A. Halder, R. R. Koner and S. Sinha-Ray, ACS Appl. Nano Mater., 2018, 2, 64–74 CrossRef.
- S.-C. Lim, C.-Y. Chan, K.-T. Chen and H.-Y. Tuan, Electrochim. Acta, 2019, 297, 288–296 CrossRef CAS.
- S.-C. Lim, C.-Y. Chan, K.-T. Chen and H.-Y. Tuan, Nanoscale, 2019, 11, 8518–8527 RSC.
- N. Wang, H. Lei, Z. Zhang, J. Li, W. Zhang and R. Cao, Chem. Sci., 2019, 10, 2308–2314 RSC.
- T. Jing, D. Liang, M. Deng, S. Cai and X. Qi, ACS Appl. Nano Mater., 2021, 4, 7125–7133 CrossRef CAS.
- Q. Zhang, Y. Fan, W. Wang, N. Liu and J. Guan, ChemElectroChem, 2020, 7, 118–123 CrossRef CAS.
- S. Zhang, J. Zhang, P. Liang, C. Zhang, T. Kou and Z. Zhang, J. Power Sources, 2021, 497, 229895 CrossRef CAS.
- J. Wang, X. Zheng, Y. Cao, L. Li, C. Zhong, Y. Deng, X. Han and W. Hu, ACS Appl. Mater. Interfaces, 2020, 12, 8115–8123 CrossRef CAS PubMed.
- P. Ilanchezhiyan, G. Mohan Kumar, S. Tamilselvan, T. W. Kang and D. Y. Kim, Int. J. Energy Res., 2020, 44, 6819–6827 CrossRef CAS.
- U. Shamraiz, A. Badshah, A. Alfantazi, H. Hussain, B. Raza, S. Ullah and I. R. Green, J. Electrochem.
Soc., 2021, 168, 066510 CrossRef CAS.
- J. Qian, T.-T. Li, Y. Hu and S. Huang, Chem. Commun., 2017, 53, 13027–13030 RSC.
- P. Ciocci, J.-F. Lemineur, J.-M. Noël, C. Combellas and F. Kanoufi, Electrochim. Acta, 2021, 386, 138498 CrossRef CAS.
- J. Jian, X. Kou, H. Wang, L. Chang, L. Zhang, S. Gao, Y. Xu and H. Yuan, ACS Appl. Mater. Interfaces, 2021, 13, 42861–42869 CrossRef CAS PubMed.
- J. Yu, F. X. Ma, Y. Du, P. P. Wang, C. Y. Xu and L. Zhen, ChemElectroChem, 2017, 4, 594–600 CrossRef CAS.
- L. Lang, Y. Shi, J. Wang, F.-B. Wang and X.-H. Xia, ACS Appl. Mater. Interfaces, 2015, 7, 9098–9102 CrossRef CAS PubMed.
- Y. Liu, H. Lu and X. Kou, Int. J. Hydrogen Energy, 2019, 44, 8099–8108 CrossRef CAS.
- F. Liu, W. He, Y. Li, F. Wang, J. Zhang, X. Xu, Y. Xue, C. Tang, H. Liu and J. Zhang, Appl. Surf. Sci., 2021, 546, 149101 CrossRef CAS.
- S. Ravula, C. Zhang, J. B. Essner, J. D. Robertson, J. Lin and G. A. Baker, ACS Appl. Mater. Interfaces, 2017, 9, 8065–8074 CrossRef CAS PubMed.
- Y. Akinay, H. C. Kazici, I. N. Akkuş and F. Salman, Ceram. Int., 2021, 47, 29515–29524 CrossRef CAS.
- R. Syah, A. Ahmad, A. Davarpanah, M. Elveny, D. Ramdan, M. D. Albaqami and M. Ouladsmane, Catalysts, 2021, 11, 1099 CrossRef CAS.
- S. Wang, W. Xue, Y. Fang, Y. Li, L. Yan, W. Wang and R. Zhao, J. Colloid Interface Sci., 2020, 573, 150–157 CrossRef CAS PubMed.
- T.-W. Chen, P. Kalimuthu, G. Anushya, S.-M. Chen, R. Ramachandran, V. Mariyappan and D. C. Muthumala, Materials, 2021, 14, 2976 CrossRef CAS PubMed.
- S. Khatun and P. Roy, Chem. Commun., 2020, 56, 7293–7296 RSC.
- C. Duan, H. Liu, Z. Huang, H. Qiao, Y. Zhou, G. Liao, Y. Liu and X. Qi, J. Sol-Gel Sci. Technol., 2021, 99, 132–139 CrossRef CAS.
- Y. Surendranath, M. Dinca and D. G. Nocera, J. Am. Chem. Soc., 2009, 131, 2615–2620 CrossRef CAS PubMed.
- M. Dincă, Y. Surendranath and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 10337–10341 CrossRef PubMed.
- M. W. Kanan and D. G. Nocera, Science, 2008, 321, 1072–1075 CrossRef CAS PubMed.
- S. Gupta, N. Patel, A. Miotello and D. Kothari, J. Power Sources, 2015, 279, 620–625 CrossRef CAS.
- H. Vrubel and X. Hu, Angew. Chem., Int. Ed., 2012, 51, 12703–12706 CrossRef CAS PubMed.
- X. Liu, G. He, H. Liu, Y. Zhu, J. Xiao and L. Han, J. Alloys Compd., 2022, 893, 162208 CrossRef CAS.
- Z. Chen, R. Zheng, M. Graś, W. Wei, G. Lota, H. Chen and B.-J. Ni, Appl. Catal., B, 2021, 288, 120037 CrossRef CAS.
- M. Bat-Erdene, M. Batmunkh, B. Sainbileg, M. Hayashi, A. S. Bati, J. Qin, H. Zhao, Y. L. Zhong and J. G. Shapter, Small, 2021, 17, 2102218 CrossRef CAS PubMed.
- D. Shi, B. Chang, Z. Ai, H. Jiang, F. Chen, Y. Shao, J. Shen, Y. Wu and X. Hao, Nanoscale, 2021, 13, 2849–2854 RSC.
- T. Zhang, F. Song, Y. Qian, H. Gao, J. Shaw and Y. Rao, ACS Appl. Energy Mater., 2021, 4, 5434–5442 CrossRef CAS.
- I.-K. Ahn, S.-Y. Lee, H. G. Kim, G.-B. Lee, J.-H. Lee, M. Kim and Y.-C. Joo, RSC Adv., 2021, 11, 8198–8206 RSC.
- W. Hao, D. Yao, Q. Xu, R. Wang, C. Zhang, Y. Guo, R. Sun, M. Huang and Z. Chen, Appl. Catal., B, 2021, 292, 120188 CrossRef CAS.
- H. Li, B. Ren, W. Liu, L. Jing, R. Y. Tay, S. H. Tsang, L. Ricardez-Sandoval, A. Yu and E. H. T. Teo, Nano Energy, 2021, 88, 106246 CrossRef CAS.
- Y. He, T. L. Wang, M. Zhang, T. W. Wang, L. F. Wu, L. Zeng, X. Wang, M. Boubeche, S. Wang and K. Yan, Small, 2021, 17, 2006153 CrossRef CAS PubMed.
- B. Shen, Y. He, Z. He, Z. Wang, Y. Jiang and H. Gao, J. Colloid Interface Sci., 2022, 605, 637–647 CrossRef CAS PubMed.
- E. Chiani, S. N. Azizi and S. Ghasemi, Int. J. Hydrogen Energy, 2021, 46, 25468–25485 CrossRef CAS.
- X. Chen, H. Zhang and Y. Zhang, Colloids Surf., A, 2021, 630, 127628 CrossRef CAS.
- M. Fan, B. Zhang, L. Wang, Z. Li, X. Liang, X. Ai and X. Zou, Chem. Commun., 2021, 57, 3889–3892 RSC.
- J. N. Hausmann, R. A. Khalaniya, C. Das, I. Remy-Speckmann, S. Berendts, A. V. Shevelkov, M. Driess and P. W. Menezes, Chem. Commun., 2021, 57, 2184–2187 RSC.
- S. Shen, Y. Gan, X.-x. Xue, J. Wei, L.-m. Tang, K. Chen and Y. Feng, Appl. Phys. Express, 2019, 12, 075502 CrossRef.
- J. A. Gauthier, L. A. King, F. T. Stults, R. A. Flores, J. Kibsgaard, Y. N. Regmi, K. Chan and T. F. Jaramillo, J. Phys. Chem. C, 2019, 123, 24007–24012 CrossRef CAS.
- S. Santisouk, P. Sengdala, X. Jiang, X.-X. Xue, K.-Q. Chen and Y. Feng, ACS Omega, 2021, 6, 13124–13133 CrossRef CAS PubMed.
- C.-Y. Chan, C.-H. Chang and H.-Y. Tuan, J. Colloid Interface Sci., 2021, 584, 729–737 CrossRef CAS PubMed.
- S. Lu, H. L. Huynh, F. Lou, K. Guo and Z. Yu, Nanoscale, 2021, 13, 12885–12895 RSC.
- L. Yang, H. Xu, H. Liu, D. Cheng and D. Cao, Small Methods, 2019, 3, 1900113 CrossRef.
- Q. U. Khan, M. W. Ishaq, N. Begum, K. Khan, U. Khan, A. T. Khan, M. A. U. Din, M. Idrees and L. Zhu, Int. J. Hydrogen Energy, 2021, 46, 18729–18739 CrossRef CAS.
- H. Kwon, D. Bae, D. Won, H. Kim, G. Kim, J. Cho, H. J. Park, H. Baik, A. R. Jeong and C.-H. Lin, ACS Nano, 2021, 15, 6540–6550 CrossRef CAS PubMed.
- R. Riedel, Handbook of Ceramic Hard Materials, WILEY-VCH Verlag GmbH, 2000, pp. 1–1020 Search PubMed.
- J. Zhu, L. Hu, P. Zhao, L. Y. S. Lee and K.-Y. Wong, Chem. Rev., 2019, 120, 851–918 CrossRef PubMed.
- F. Yu, Y. Gao, Z. Lang, Y. Ma, L. Yin, J. Du, H. Tan, Y. Wang and Y. Li, Nanoscale, 2018, 10, 6080–6087 RSC.
- L. Zhang, X. Ding, M. Cong, Y. Wang and X. Zhang, Int. J. Hydrogen Energy, 2019, 44, 9203–9209 CrossRef CAS.
- J. Hou, B. Zhang, Z. Li, S. Cao, Y. Sun, Y. Wu, Z. Gao and L. Sun, ACS Catal., 2018, 8, 4612–4621 CrossRef CAS.
- D. Jana, H. K. Kolli, S. Sabnam and S. K. Das, Chem. Commun., 2021, 57, 9910–9913 RSC.
- T. Ahmed, M. A. Asghar, A. Ali, Z. Akhter, S. Ali, I. Ullah, T. Nisar, V. Wagner, S. Touseef and A. Hussain, J. Alloys Compd., 2022, 909, 164709 CrossRef CAS.
- S. Mulkapuri, A. Ravi and S. K. Das, Chem. Mater., 2022, 34, 3624–3636 CrossRef CAS.
- S. Mulkapuri, A. Ravi, S. Mukhopadhyay, S. K. Kurapati, V. Siby and S. K. Das, Inorganic Chemistry Frontiers, 2022 Search PubMed.
- H. Liu, C. Liu, X. Zong, K. Chen, W. Yao, Y. Su, H. Qian, Y. Wang, S. Chou and Z. Hu, ChemistrySelect, 2022, 7, e202202382 CAS.
- U. Khan, A. Nairan, J. Gao and Q. Zhang, Small Struct., 2022, 2200109 CrossRef.
- N. Zaman, T. Noor and N. Iqbal, RSC Adv., 2021, 11, 21904–21925 RSC.
- B. Zhang, Y. Zheng, T. Ma, C. Yang, Y. Peng, Z. Zhou, M. Zhou, S. Li, Y. Wang and C. Cheng, Adv. Mater., 2021, 33, 2006042 CrossRef CAS PubMed.
- M. Nemiwal, V. Gosu, T. C. Zhang and D. Kumar, Int. J. Hydrogen Energy, 2021, 46, 10216–10238 CrossRef CAS.
- H. B. Wu, B. Y. Xia, L. Yu, X.-Y. Yu and X. W. D. Lou, Nat. Commun., 2015, 6, 1–8 Search PubMed.
- Z. Kou, W. Zang, W. Pei, L. Zheng, S. Zhou, S. Zhang, L. Zhang and J. Wang, J. Mater. Chem. A, 2020, 8, 3071–3082 RSC.
- L. Ma, L. R. L. Ting, V. Molinari, C. Giordano and B. S. Yeo, J. Mater. Chem. A, 2015, 3, 8361–8368 RSC.
- Z. Kou, L. Zhang, Y. Ma, X. Liu, W. Zang, J. Zhang, S. Huang, Y. Du, A. K. Cheetham and J. Wang, Appl. Catal., B, 2019, 243, 678–685 CrossRef CAS.
- H. Wang, S. Zhu, J. Deng, W. Zhang, Y. Feng and J. Ma, Chin. Chem. Lett., 2021, 32, 291–298 CrossRef CAS.
- X. Xing, R. Liu, K. Cao, U. Kaiser and C. Streb, Chem.–Eur. J., 2019, 25, 11098–11104 CrossRef CAS PubMed.
- M. Y. Zu, C. Wang, L. Zhang, L. R. Zheng and H. G. Yang, Mater. Horiz., 2019, 6, 115–121 RSC.
- H. Yu, X. Yang, X. Xiao, M. Chen, Q. Zhang, L. Huang, J. Wu, T. Li, S. Chen and L. Song, Adv. Mater., 2018, 30, 1805655 CrossRef PubMed.
- P. Chen, J. Ye, H. Wang, L. Ouyang and M. Zhu, J. Alloys Compd., 2021, 883, 160833 CrossRef CAS.
- B. Chang, J. Yang, Y. Shao, L. Zhang, W. Fan, B. Huang, Y. Wu and X. Hao, ChemSusChem, 2018, 11, 3198–3207 CrossRef CAS PubMed.
- B. Cao, G. M. Veith, J. C. Neuefeind, R. R. Adzic and P. G. Khalifah, J. Am. Chem. Soc., 2013, 135, 19186–19192 CrossRef CAS PubMed.
- Z. Cui, G. Fu, Y. Li and J. B. Goodenough, Angew. Chem., Int. Ed., 2017, 56, 9901–9905 CrossRef CAS PubMed.
- Y. Liu, F. Li, H. Yang, J. Li, P. Ma, Y. Zhu and J. Ma, ChemSusChem, 2018, 11, 2358–2366 CrossRef CAS PubMed.
- Z. Zheng, X. Du, Y. Wang, C. M. Li and T. Qi, ACS Sustainable Chem. Eng., 2018, 6, 11473–11479 CrossRef CAS.
- K. Meng, S. Wen, L. Liu, Z. Jia, Y. Wang, Z. Shao and T. Qi, ACS Appl. Energy Mater., 2019, 2, 2854–2861 CrossRef CAS.
- J. Chang, K. Li, Z. Wu, J. Ge, C. Liu and W. Xing, ACS Appl. Mater. Interfaces, 2018, 10, 26303–26311 CrossRef CAS PubMed.
- L. Yan, B. Zhang, J. Zhu, Z. Liu, H. Zhang and Y. Li, J. Mater. Chem. A, 2019, 7, 22453–22462 RSC.
- E. Cao, Z. Chen, H. Wu, P. Yu, Y. Wang, F. Xiao, S. Chen, S. Du, Y. Xie and Y. Wu, Angew. Chem., Int. Ed., 2020, 59, 4154–4160 CrossRef CAS PubMed.
- L. Zhang, X. Wang, X. Zheng, L. Peng, J. Shen, R. Xiang, Z. Deng, L. Li, H. Chen and Z. Wei, ACS Appl. Energy Mater., 2018, 1, 5482–5489 CAS.
- H. Sun, Y. Min, W. Yang, Y. Lian, L. Lin, K. Feng, Z. Deng, M. Chen, J. Zhong and L. Xu, ACS Catal., 2019, 9, 8882–8892 CrossRef CAS.
- B. You, Y. Zhang, Y. Jiao, K. Davey and S. Z. Qiao, Angew. Chem., 2019, 131, 11922–11926 CrossRef.
- S. Xu, Y. Qi, Y. Lu, S. Sun, Y. Liu and D. Jiang, Int. J. Hydrogen Energy, 2021, 46, 26391–26401 CrossRef CAS.
- D. Huo, F. Song, J. Hu, J. Yuan, L. Niu and A.-j. Wang, Int. J. Hydrogen Energy, 2021, 46, 8519–8530 CrossRef CAS.
- X. Zhong, K. Huang, Y. Zhang, Y. Wang and S. Feng, ChemSusChem, 2021, 14, 2188–2197 CrossRef CAS PubMed.
- W. Xiao, L. Zhang, D. Bukhvalov, Z. Chen, Z. Zou, L. Shang, X. Yang, D. Yan, F. Han and T. Zhang, Nano Energy, 2020, 70, 104445 CrossRef CAS.
- Y. Li, B. Jia, B. Chen, Q. Liu, M. Cai, Z. Xue, Y. Fan, H.-P. Wang, C.-Y. Su and G. Li, Dalton Trans., 2018, 47, 14679–14685 RSC.
- H. Feng, X. Sun, X. Guan, D. Zheng, W. Tian, C. Li, C. Li, M. Yan and Y. Yao, FlatChem, 2021, 26, 100225 CrossRef CAS.
- Q. He, D. Tian, H. Jiang, D. Cao, S. Wei, D. Liu, P. Song, Y. Lin and L. Song, Adv. Mater., 2020, 32, 1906972 CrossRef CAS PubMed.
- M. Shamloofard and S. Shahrokhian, Nanoscale, 2021, 13, 17576–17591 RSC.
- J. Yang, A. R. Mohmad, Y. Wang, R. Fullon, X. Song, F. Zhao, I. Bozkurt, M. Augustin, E. J. Santos and H. S. Shin, Nat. Mater., 2019, 18, 1309–1314 CrossRef CAS PubMed.
- A. Kagkoura, M. Pelaez-Fernandez, R. Arenal and N. Tagmatarchis, Nanoscale Adv., 2019, 1, 1489–1496 RSC.
- R. Bar-Ziv, P. Ranjan, A. Lavie, A. Jain, S. Garai, A. Bar Hen, R. Popovitz-Biro, R. Tenne, R. Arenal and A. Ramasubramaniam, ACS Appl. Energy Mater., 2019, 2, 6043–6050 CrossRef CAS.
- A. Maiti and S. K. Srivastava, J. Mater. Chem. A, 2018, 6, 19712–19726 RSC.
- W. Xie, K. Liu, G. Shi, X. Fu, X. Chen, Z. Fan, M. Liu, M. Yuan and M. Wang, J. Energy Chem., 2021, 60, 272–278 CrossRef CAS.
- W. He, H. Liu, J. Cheng, J. Mao, C. Chen, Q. Hao, J. Zhao, C. Liu, Y. Li and L. Liang, Nanoscale, 2021, 13, 10127–10132 RSC.
- X. Mao, Y. Liu, Z. Chen, Y. Fan and P. Shen, Chem. Eng. J., 2022, 427, 130742 CrossRef CAS.
- Y. Gong, J. Wang, Y. Lin, Z. Yang, H. Pan and Z. Xu, Appl. Surf. Sci., 2019, 476, 600–607 CrossRef CAS.
- X. Yu, S. Xu, X. Liu, X. Cheng, Y. Du and Q. Wu, J. Alloys Compd., 2021, 878, 160388 CrossRef CAS.
- M. Cui, C. Yang, B. Li, Q. Dong, M. Wu, S. Hwang, H. Xie, X. Wang, G. Wang and L. Hu, Adv. Energy Mater., 2021, 11, 2002887 CrossRef CAS.
- Y. Ma, W.-L. Yu, R.-Y. Fan, J.-Y. Fu, M.-X. Li, H.-J. Liu, L. Wang, J.-F. Yu, Y.-M. Chai and B. Dong, Colloids Surf., A, 2021, 628, 127228 CrossRef CAS.
- X. Xia, L. Wang, N. Sui, V. L. Colvin and W. Y. William, Nanoscale, 2020, 12, 12249–12262 RSC.
- H. Liu, K. Wang, W. He, X. Zheng, T. Gong, Y. Li, J. Zhao, J. Zhang and L. Liang, Int. J. Hydrogen Energy, 2021, 46, 1967–1975 CrossRef CAS.
- Y. Guo, C. Zhang, J. Zhang, K. Dastafkan, K. Wang, C. Zhao and Z. Shi, ACS Sustainable Chem. Eng., 2021, 9, 2047–2056 CrossRef CAS.
- Y. Huang, L. W. Jiang, B. Y. Shi, K. M. Ryan and J. J. Wang, Adv. Sci., 2021, 8, 2101775 CrossRef CAS PubMed.
- S. Pan, S. Ma, C. Chang, X. Long, K. Qu and Z. Yang, Mater. Today Phys., 2021, 18, 100401 CrossRef CAS.
- S. Cherevko, S. Geiger, O. Kasian, A. Mingers and K. J. Mayrhofer, J. Electroanal. Chem., 2016, 774, 102–110 CrossRef CAS.
- Q. Feng, G. Liu, B. Wei, Z. Zhang, H. Li and H. Wang, J. Power Sources, 2017, 366, 33–55 CrossRef CAS.
- G. Li, K. Zheng, W. Li, Y. He and C. Xu, ACS Appl. Mater. Interfaces, 2020, 12, 51437–51447 CrossRef CAS PubMed.
- Q.-Q. Chen, C.-C. Hou, C.-J. Wang, X. Yang, R. Shi and Y. Chen, Chem. Commun., 2018, 54, 6400–6403 RSC.
- R. Fan, Q. Mu, Z. Wei, Y. Peng and M. Shen, J. Mater. Chem. A, 2020, 8, 9871–9881 RSC.
- S. Li, C. Xi, Y.-Z. Jin, D. Wu, J.-Q. Wang, T. Liu, H.-B. Wang, C.-K. Dong, H. Liu and S. A. Kulinich, ACS Energy Lett., 2019, 4, 1823–1829 CrossRef CAS.
- K. Zhu, J. Chen, W. Wang, J. Liao, J. Dong, M. O. L. Chee, N. Wang, P. Dong, P. M. Ajayan and S. Gao, Adv. Funct. Mater., 2020, 30, 2003556 CrossRef CAS.
- X. Kong, K. Xu, C. Zhang, J. Dai, S. Norooz Oliaee, L. Li, X. Zeng, C. Wu and Z. Peng, ACS Catal., 2016, 6, 1487–1492 CrossRef CAS.
- L. Tian, Z. Li, M. Song and J. Li, Nanoscale, 2021, 13, 12088–12101 RSC.
- Y. Yao, in Controllable Synthesis and Atomic Scale Regulation of Noble Metal Catalysts, Springer, 2022, pp. 93–116 Search PubMed.
- H. Xu, H. Shang, C. Wang and Y. Du, Small, 2021, 17, 2005092 CrossRef CAS PubMed.
- B. Jiang, Y. Guo, J. Kim, A. E. Whitten, K. Wood, K. Kani, A. E. Rowan, J. Henzie and Y. Yamauchi, J. Am. Chem. Soc., 2018, 140, 12434–12441 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a
*a



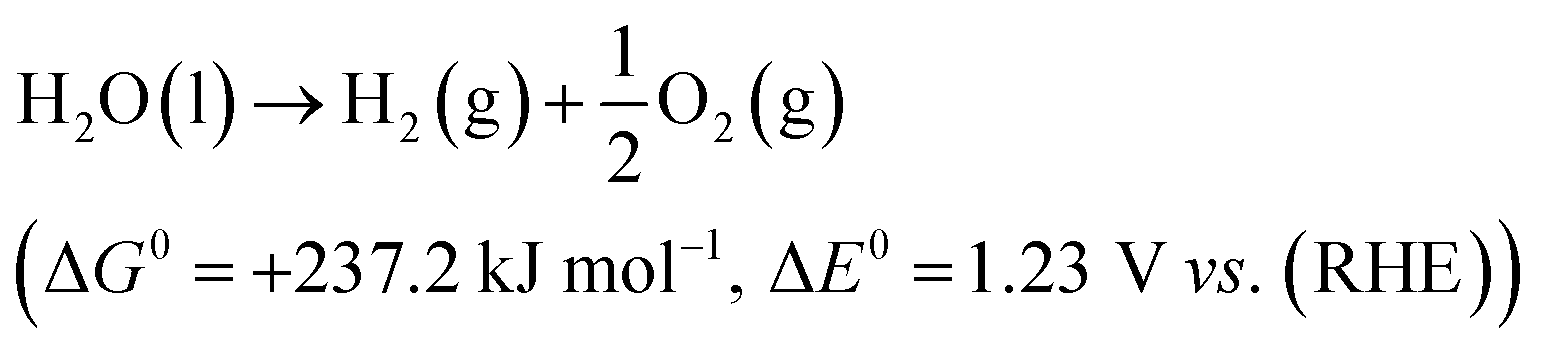
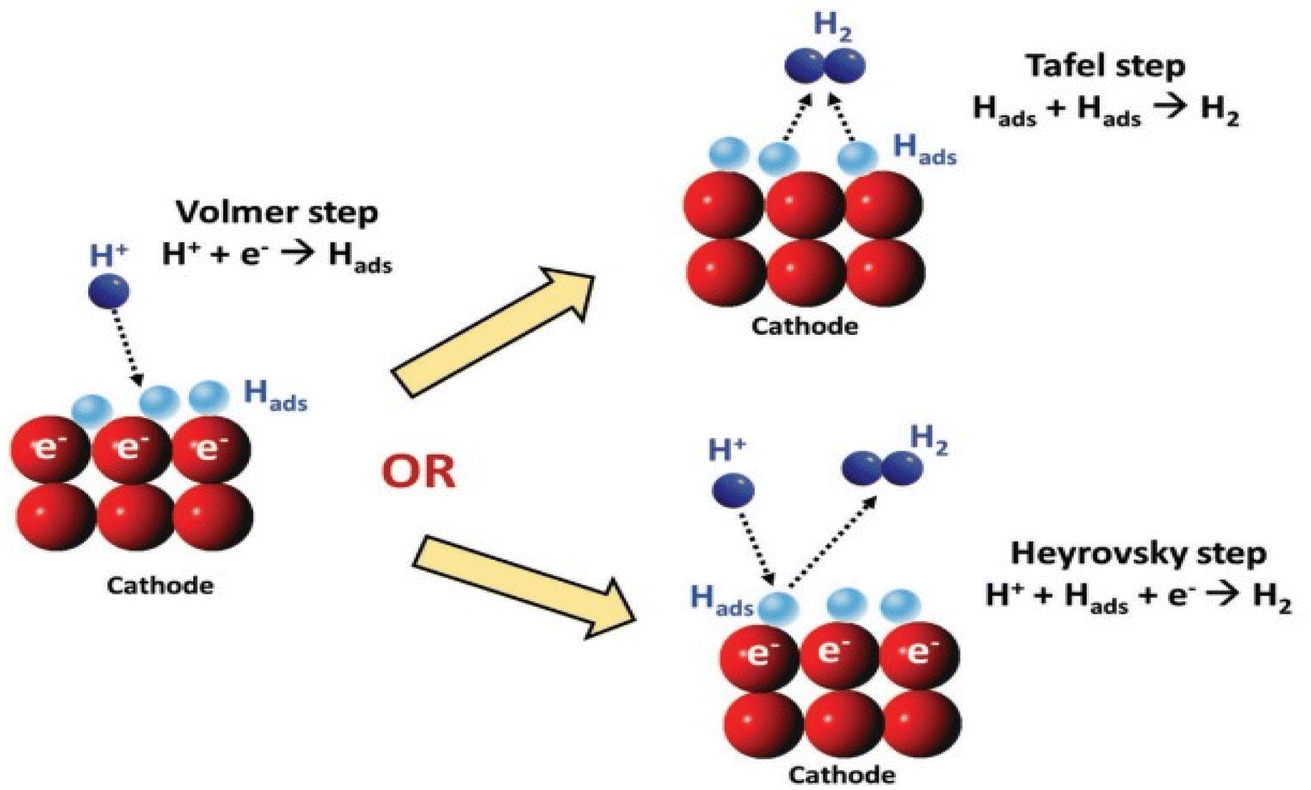
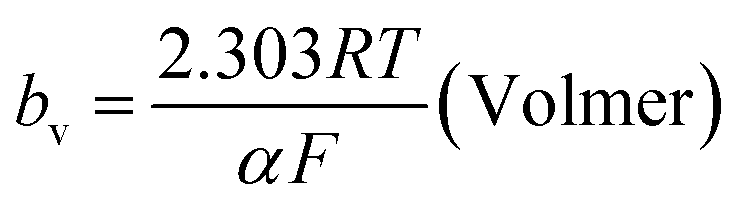
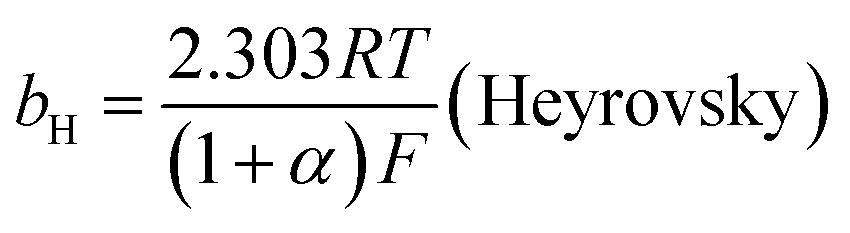
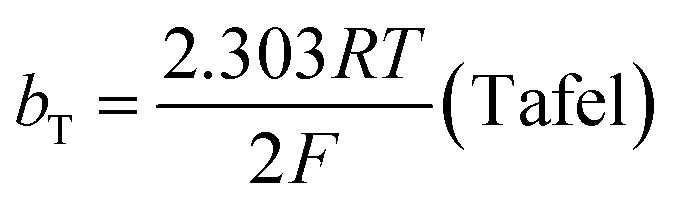
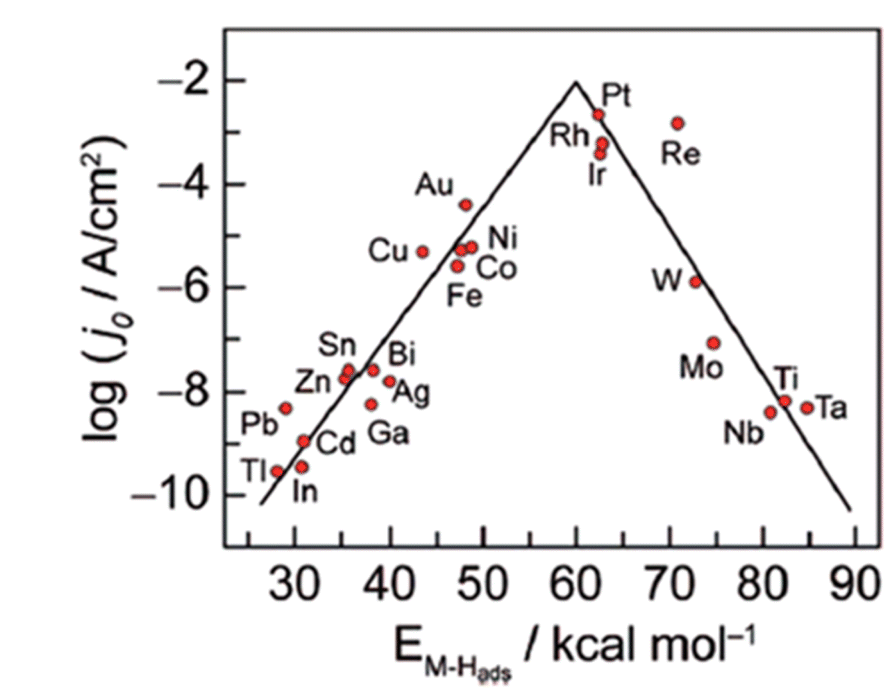
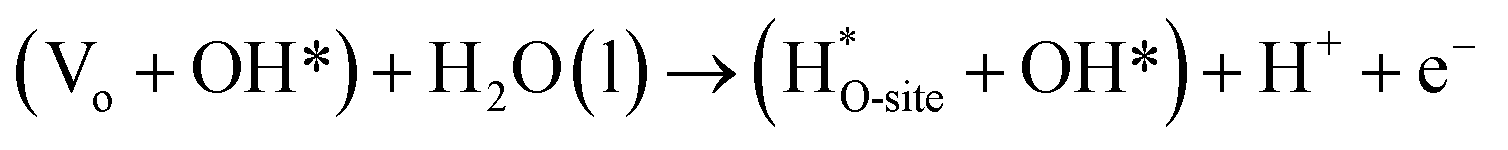
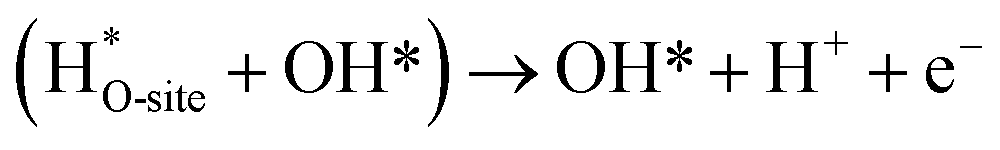
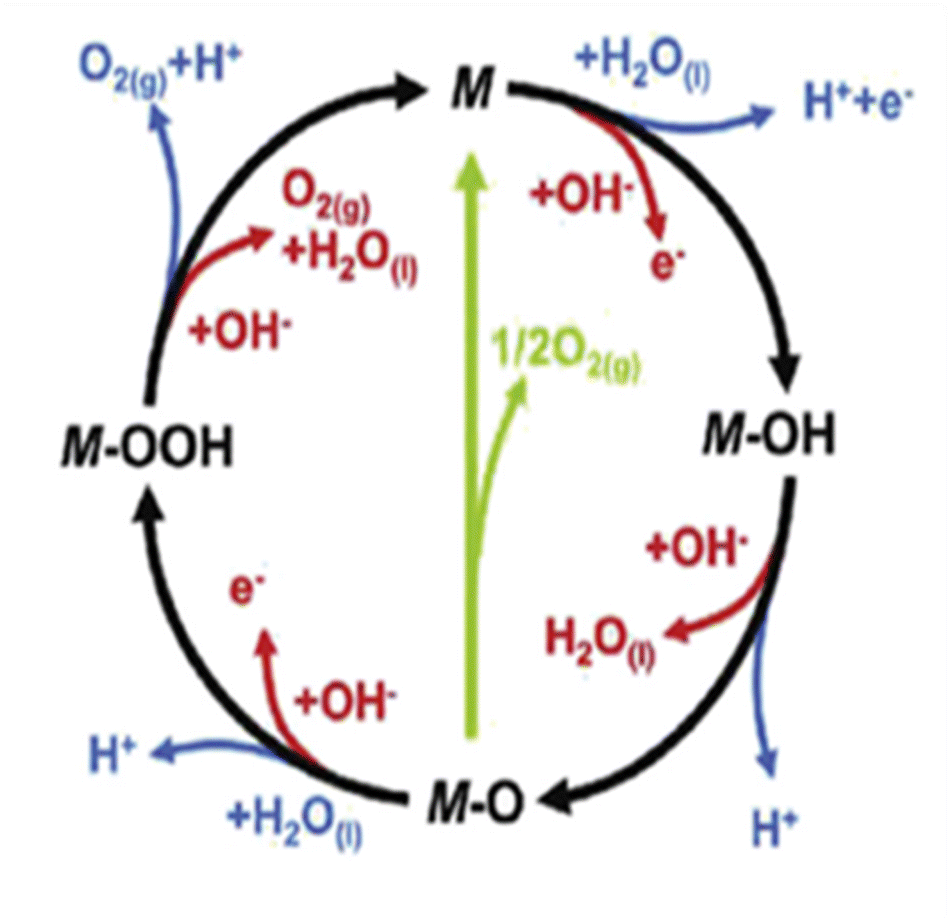
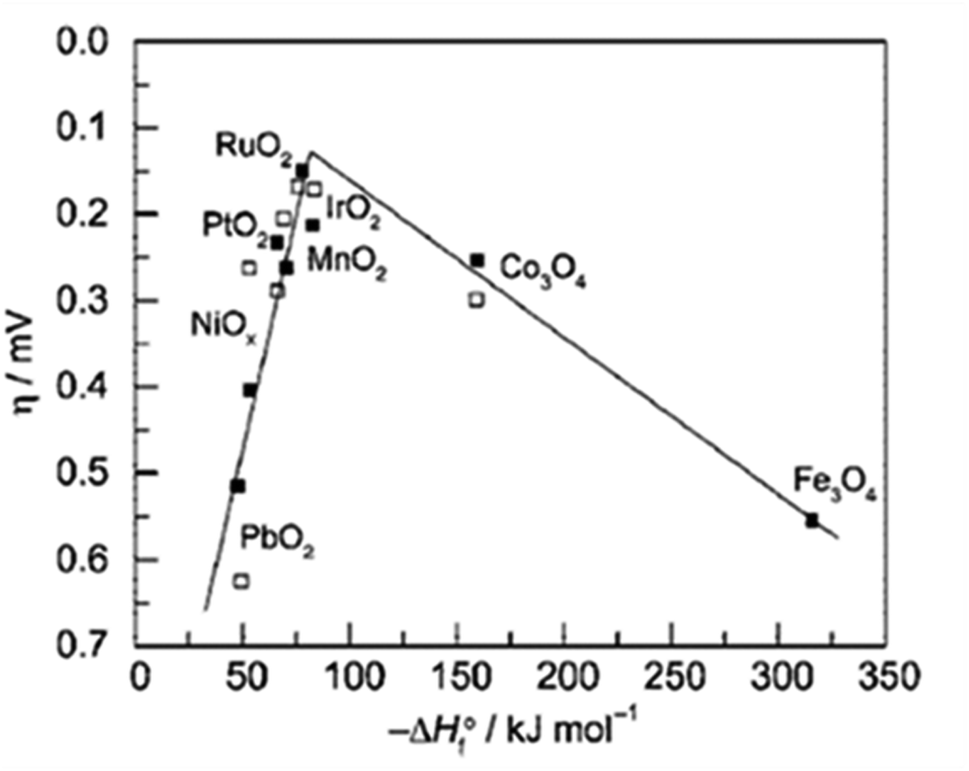
 ) and basic (
) and basic ( ) solutions. Measurement of overpotential in relation to 0.1 mA cm2 current density. Reproduced with permission2 Copyright 2010, American Chemical Society.
) solutions. Measurement of overpotential in relation to 0.1 mA cm2 current density. Reproduced with permission2 Copyright 2010, American Chemical Society.