DOI:
10.1039/D2RA07612H
(Paper)
RSC Adv., 2023,
13, 2024-2035
Sequential hydrogenation of nitroaromatics to alicyclic amines via highly-dispersed Ru–Pd nanoparticles anchored on air-exfoliated C3N4 nanosheets†
Received
30th November 2022
, Accepted 22nd December 2022
First published on 11th January 2023
Abstract
Developing efficient and green catalytic systems is highly desired in the syntheses of alicyclic amines via hydrogenation of nitroaromatics. Herein, we developed Ru–Pd dual active site catalysts in which Ru and Pd species were anchored and highly dispersed on air-exfoliated carbon nitride (Ru–Pd/C3N4-air). As-prepared catalysts were employed in the hydrogenation of nitrobenzene (NB) to cyclohexylamine (CHA). Compared with single Ru or Pd based catalysts, Ru–Pd dual active site catalysts obtained a higher CHA production rate of 26.7 mol CHA mol−1 Ru·Pd h−1 at 80 °C and 3 MPa H2. The activation energy for the hydrogenation of the nitro group and benzene ring was calculated as 26.26 kJ mol−1 and 66.30 kJ mol−1, respectively. Intrinsic kinetic studies demonstrated that Pd was the dominant metal for hydrogenation of nitro group, while Ru was dominant for benzene ring. Thereinto, the corresponding non-dominant metals enhanced activation and dissociation of H2, thereby improving catalytic activity significantly. This excellent performance of Ru–Pd catalysts could be attributed to highly dispersed Ru–Nx and Pd–Nx at a nanoscale distance, which was conducive to metal-assisted hydrogenation. Stability investigation showed that the performance of Ru–Pd catalysts could be essentially maintained at a high level. Additionally, the substrate scope could be successfully extended to hydrogenation of other nitroaromatics with different substituents.
Introduction
Alicyclic amines are critical foundational materials for the synthesis of curing agents, coatings, dyestuff, high-end isocyanates, etc., generally produced by the hydrogenation of nitroaromatics.1–3 Due to different adsorption behaviors of nitro group and benzene ring, the production of alicyclic amine is often divided into two hydrogenation processes and conducted step by step in industries.4–7 However, multiple steps mean different catalysts, fickle conditions, and massive equipment, directly resulting in high cost, complex operation, and low security. Therefore, coupling multiple successive hydrogenation processes in a single vessel or catalyst under similar and even identical conditions is critical to efficient synthesis of alicyclic amines, which has become a great research interest in the past decades. Basically, Ru based catalysts have demonstrated huge potential in one-step hydrogenation of nitroaromatics to alicyclic amines as well as its derivates.4,8–10 However, competitive adsorption between the nitro group and benzene ring on single Ru active site limits efficiency to a great extent and also requires severe reaction conditions. A convincing explanation for this could be scaling relationship limit (SRL).11 Namely, a solo active site determines a fixed adsorption mode, and it is almost impossible to optimally adjust the adsorption behavior of every intermediate on the solo site simultaneously. Therefore, introducing dual active sites is a promising solution to break the SRL and aid in the efficient synthesis of alicyclic amines from nitroaromatics.
For selective hydrogenation of the nitro group, some noble metal catalytic systems with high activity of >99.9% conversion and selectivity have been developed, including Ru, Pt, and Pd.12–15 Among them, Pd based catalysts have been demonstrated to have prominent catalytic activity and product selectivity for hydrogenation of nitro group under mild conditions even at room temperature.16–19 These mild catalytic systems lead to a rather green process, but more efficient systems are still highly desired to be developed. By contrast, the key to the hydrogenation of benzene ring is to regulate electronic properties of active centre and adsorption behavior of substrate. Generally, the electronic structure and properties of benzene ring could be influenced by substituent groups. Particularly, for aromatic amines, the formed p–π conjugation via nitrogen in amino and benzene ring has a relatively high energy barrier, which makes activation of benzene ring more difficult.20 Dual-active site synergistic catalysis is proposed to decrease the energy barrier in the hydrogenation of benzene ring, where Ru enhances dissociation of H2 while support as an active centre to regulate the adsorption behavior of benzene ring.21 Additionally, the excessive adsorption of amino groups on metal active sites usually triggers side reactions like deamination and condensation and even leads to the deactivation of catalyst.8 In order to inhibit these undesired side reactions and regulate the adsorption behavior of amino groups, alkaline liquors, such as NaOH and NaNO2, are always introduced to the catalytic system to regulate adsorption behavior of amino groups. However, the alkaline additives also bring potential environmental risks.22,23
In recent years, heteroatom-doped carbon materials and a series of derivatives have attracted much attention in heterogeneous catalysis.24 C3N4, as a special type of N-doped carbon material, exhibited excellent performance in aromatic ring hydrogenation because its N species provide not only environment for metal coordination but also abundant alkaline sites for side reaction inhibition. By virtue of the above advantages of C3N4, Ru nanoparticles were anchored on C3N4 to catalyse hydrogenation of 4,4′-diaminodiphenylmethane (MDA) in an alkali-free system.25 The alkali species on this solid alkali support exhibited equivalent effects as hydroxyl, which is regarded as an environmentally friendly substitute for traditional alkali liquors.
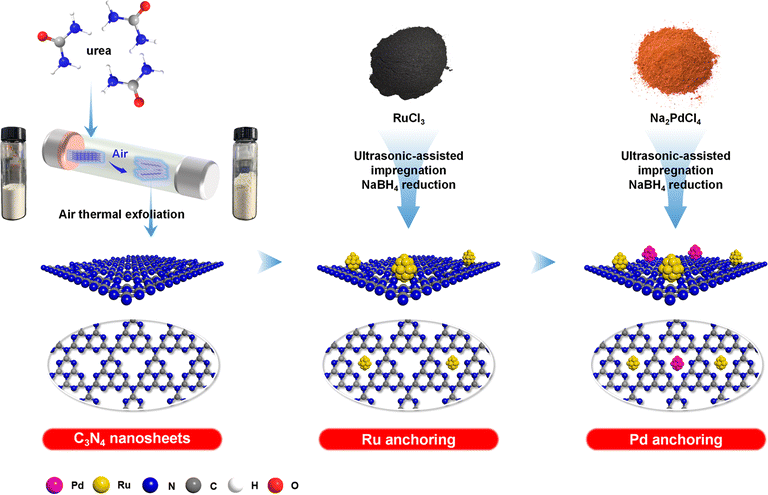
Herein, a highly dispersed Ru–Pd dual active site catalyst was prepared by facile ultrasonic-assisted impregnation, where air-exfoliated C3N4 was used as support with Pd and Ru as active sites for the hydrogenation of nitro group and benzene ring, respectively (Scheme 1). Catalyst textural properties and morphological features were characterized by BET and SEM to show the superiority of air thermal exfoliation. TEM and EDS were employed to elucidate the particle distribution of Ru and Pd on the catalyst surface. The interactions between metals and support were investigated by XPS. Additionally, the ability of dissociation and activation of H2 under mild conditions was characterized by H2-TPD. Based on the above analysis, the structure–performance relationship was analysed. By operando IR, the hydrogenation pathway of NB to CHA was investigated. Kinetic experiments systematically calculated the activation energy of tandem hydrogenation and revealed the phenomenon of metal-assisted hydrogenation. Eventually, the stability and versatility of the Ru–Pd catalyst were studied.
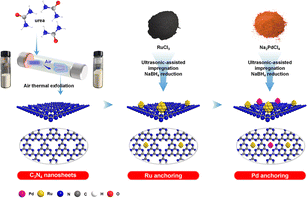 |
| | Scheme 1 Schematic illustration of catalyst preparation via sequential anchoring of Ru–Pd dual metal sites on C3N4 nanosheets. | |
Experimental section
Materials
Ruthenium(III) chloride anhydrous (RuCl3), sodium tetrachloropalladate (Na2PdCl4), nitrobenzene (NB, 99%), tetrahydrofuran (THF, 99%), N,N-dimethylformamide (DMF, 99%), cyclohexane (99%), ethanol (99%), methanol (99%), aniline (AN, 99.5%), cyclohexylamine (CHA, 99.5%), o-nitrotoluene (99%), m-nitrotoluene (99%), p-nitrotoluene (99%), 4-ethylnitrobenzene (99%), 2,6-dimethylnitrobenzene (99%), o-nitrophenol (99%), p-nitrophenol (99%), 3-nitroaniline (99%), and 4-nitroaniline (99%) were purchased from Shanghai Aladdin Biochemical Technology Co., Ltd. Sodium borohydride (99%) and urea (99%) were supplied by Sinopharm Chemical Reagents Co., Ltd., China. All chemicals were directly used as received unless otherwise specified.
Catalyst preparation
Preparation of bulk C3N4. 200 g urea was placed in crucible with cover, which was placed in muffle furnace and calcined under air atmosphere at 600 °C for 4 h. The obtained yellow solid was washed with deionized water and ethanol three times to remove all residual species like ammonia absorbed on the surface. After filtering and drying at 110 °C for 12 h, bulk C3N4 was obtained.
Exfoliation of C3N4. 4 g of prepared bulk C3N4 was placed in a crucible without any covers and then was calcined in tube furnace at 550 °C for 2 h. The obtained solid was denoted as C3N4-air.
Preparation of Ru–Pd/C3N4-air. All catalysts were prepared by ultrasonic-assisted impregnation method. Ru and Pd species were anchored on C3N4-air successively. Typically, a dosage of RuCl3 was dissolved in deionized water to obtain 10 mL Ru3+-contained precursor, while 10 mL Pd2+-contained precursor was obtained after dissolving a dosage of Na2PdCl4. A certain amount of C3N4-air was dispersed in deionized water by ultrasonic treatment for 0.5 h to form a suspension, where the obtained Ru3+-contained precursor was added and proceeded with ultrasonic treatment for 1 h to obtain Ru3+-contained suspension. Subsequently, excessive NaBH4 was dissolved in deionized water and added to the above Ru3+-contained suspension dropwise and proceeded with ultrasonic treatment for 2 h to reduce Ru3+. In the same way, the previously obtained Pd2+-contained precursor was added into the above suspension and reduced Pd2+ by an excessive dosage of NaBH4 solution after the same ultrasonic treatment. After filtration and drying at 100 °C for 12 h, Ru–Pd/C3N4-air catalyst was obtained. According to different mass fractions of supported Ru and Pd, the catalysts were denoted as “x%Ru–y%Pd/C3N4-air” (where x and y stand for the mass fraction of Ru and Pd, respectively). The Ru/C3N4-air and Pd/C3N4-air were prepared following the same procedures, but only a single metal precursor was added. In addition, Ru–Pd/C3N4 was prepared also using the same procedures, except C3N4-air was replaced by bulk C3N4.
Catalyst characterization
N2 physical adsorption–desorption was carried out on Quantachrome Autosorb Adsorption Instrument to measure the specific surface area, pore volume, and pore diameter of catalysts. Before measurement, the sample was subjected to a degassing procedure at 200 °C in vacuum for 4 h to remove possible physically adsorbed impurities. The composition and crystal structure of the catalyst was investigated by X-ray diffraction (XRD) conducted on the Panalytical Empyrean diffractometer. The morphology of catalysts and supports were investigated by scanning electron microscope TESCAN MIRA4 (SEM) and transmission electron microscope (TEM), as well as TEM-HAADF, EDX element mapping, and line-scanning carried out on field-emission transmission electron microscopy (FEI Talos F200×) operating at 200 kV. X-ray photoelectron spectroscope (XPS) was used to analyze the chemical state of elements on the sample surface, which was conducted on an ultrahigh vacuum using an ESCALAB 250Xi spectrometer with Al Kα radiation (hν = 1486.6 eV) and a multichannel detector. The ability of the catalyst for H2 activation and dissociation was studied by H2-TPD carried out on a temperature-programmed chemisorption instrument equipped with low temperature attachment (AutoChem II 2920). The operando IR was carried out in a tailor-made autoclave, and all FTIR spectra were collected on a Mettler Toledo ReactIR 15 equipped with a liquid N2 detector and a 9.5 mm AgX fiber probe. The detected IR spectra were recorded every 5 s and analyzed by ICIR 7.1 software.
Catalyst performance
The catalyst performance was evaluated in a 50 mL autoclave. Typically, nitrobenzene (1 mmol), THF (10 mL), and catalyst (25 wt% based on the substrate) were added to an autoclave and sealed. After that, the autoclave was flushed with N2 three times to remove air, then charged with 3 MPa H2 at room temperature. The hydrogenation reaction was carried out at 80 °C for 4 h under magnetic stirring (550 rpm). In particular, the kinetic experiments of hydrogenation of nitro group and benzene ring were carried out in a 250 mL autoclave, which can ensure continuous charge with H2 in the case of pressure loss caused by batch production of the reaction liquid. It was worth noting that once the pressure was lower than that desired during the experiment, H2 was supplemented as soon as possible. To guarantee the experiments were carried out in intrinsic kinetic control, for hydrogenation of the nitro group, typically, nitrobenzene (70 mmol), THF (105 mL), and 1.5%Ru–1.5%Pd/C3N4-air (1 wt% based on the substrate) were added into an autoclave and sealed. After that, the autoclave was flushed with N2 three times to remove the air and then heated to the desired temperature under stirring conditions (550 rpm). When the temperature was stabilized, 1 MPa H2 was charged into the autoclave. The reaction liquid was collected at appropriate intervals, and the composition was detected by GC-MS. The kinetic experiments of benzene ring hydrogenation were almost the same as that of the nitro group. Hence, only considering the hydrogenation of benzene ring, aniline was used as substrate. Typically, aniline (10 mmol), THF (100 mL), and 1.5%Ru–1.5%Pd/C3N4-air (25 wt% based on the substrate) were added into the autoclave and then charged with 3 MPa H2.
Results and discussion
Catalyst characterization
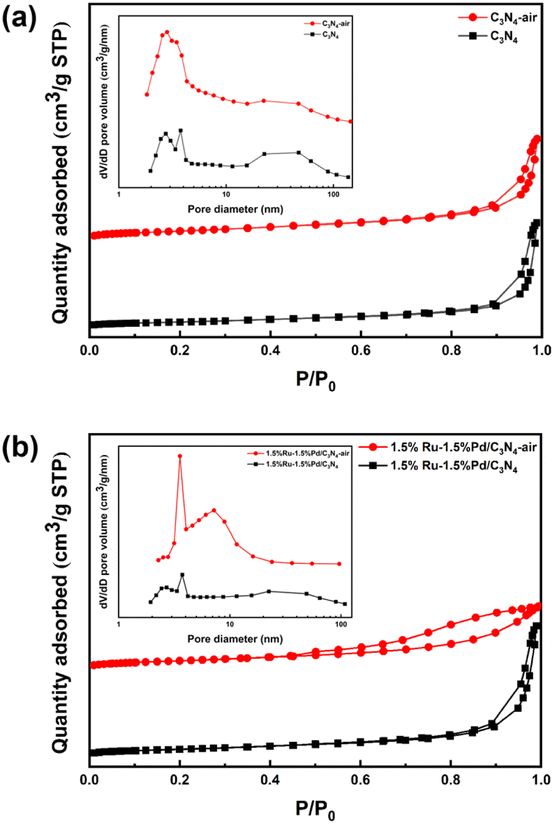
The surface properties were studied here by N2 physisorption. Fig. 1 shows the N2 adsorption–desorption isotherms and pore size distribution. The sorption isotherm of C3N4 and C3N4-air both exhibited typical IV curves with H3 hysteresis loop in the P/P0 range of 0.8–1.0 and no obvious saturated adsorption platform was observed, which means C3N4 was a kind of mesoporous material with lamellar structure (Fig. 1a). For pore diameter, the mesoporous proportion of C3N4-air in the range of 2–4 nm was significantly larger than that of C3N4, which could be attributed to the lamella separation caused by air thermal exfoliation, resulting in formation of many slit pores. After Ru and Pd species were loaded, 1.5%Ru–1.5%Pd/C3N4 and 1.5%Ru–1.5%Pd/C3N4-air still maintained IV curves, which indicated that introducing metals did not destroy the mesoporous structure of C3N4. However, 1.5%Ru–1.5%Pd/C3N4-air showed an H4 hysteresis loop indicating the existence of narrow slit pores26 (Fig. 1b). From the pictures of catalysts, it was visible that C3N4-air was fluffier and showed an increase in volume after air thermal exfoliation (Fig. S1†). Table S1† summarized the structural properties of supports and Ru–Pd catalysts. Clearly, the air thermal exfoliation led to a noteworthy increase in specific surface area from 85.48 m2 g−1 of C3N4 to 137.22 m2 g−1 of C3N4-air. With the emergence of abundant slit pores, the average pore diameter decreased from 22.24 nm of C3N4 to 13.65 nm of C3N4-air. Moreover, due to the occupation of Ru and Pd particles, the pore volume decreased from 0.47 cm3 g−1 of C3N4-air to 0.23 cm3 g−1 of 1.5%Ru–1.5%Pd/C3N4-air. Thus, air thermal exfoliation could increase the specific surface area leading to better accessibility of the substrate to active sites.
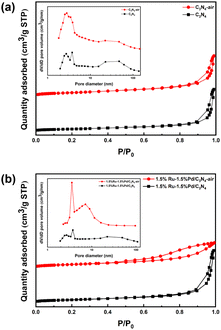 |
| | Fig. 1 N2 adsorption–desorption isotherms and pore size distribution. (a) C3N4 and C3N4-air, (b) 1.5%Ru–1.5%Pd/C3N4 and 1.5%Ru–1.5%Pd/C3N4-air. | |
The crystal structure of catalysts was studied by X-ray diffraction (XRD), as shown in Fig. 2. The 1.5%Ru–1.5%Pd/C3N4-air and 1.5%Ru–1.5%Pd/C3N4 both presented two typical C3N4 characteristic peaks centered at 13.1° and 27.7°. The peak of 13.1° could be ascribed to the repeated tri-s-triazine unit in the C3N4 plane, while 27.7° is the inter-planar stacking peak of conjugated heptazine units of C3N4 material.27,28 The results indicated that the loading of Ru and Pd as well as air thermal exfoliation did not destroy the inherent graphite-like lamellar structure of C3N4. Additionally, no other distinguishable characteristic peaks of Ru or Pd were observed, which implied high dispersion of Ru and Pd species. A similar phenomenon was also observed and reported by Liu et al.29
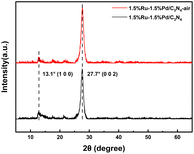 |
| | Fig. 2 XRD patterns of 1.5%Ru–1.5%Pd/C3N4 and 1.5%Ru–1.5%Pd/C3N4-air. | |
Scanning electron microscope (SEM) was employed to characterize morphological features of supports and catalysts. As shown in Fig. 3, C3N4 appeared as a large dense particle with lots of sheets stacked and squeezed together, resulting in a few exposed pores (Fig. 3a and b). Nevertheless, after air thermal exfoliation, C3N4-air exhibited a structure of nanosheets separated from each other (Fig. 3c and d). Due to the bending and stacking of nanosheets, plenty of disordered connected pores with different sizes were formed, leading to the increase in the specific surface area of C3N4-air, which was consistent with the results of N2 physisorption. Because of ultrasonic-assisted impregnation, the dense particle structure of C3N4 was partially destroyed to form many pores, but some massive structures still remained (Fig. S2†).
 |
| | Fig. 3 SEM images with different resolutions. (a and b) C3N4, (c and d) C3N4-air. | |
Transmission electron microscope (TEM) further supplemented the morphology features of catalysts, as shown in Fig. 4, 1.5%Ru–1.5%Pd/C3N4-air emerged as nanosheet-shaped arrays with cataphracted stack and crimp, which was beneficial for increasing the specific surface area (Fig. 4a). The particle size was calculated as 2.8 ± 0.6 without any agglomerations observed (Fig. 4b). The HR-TEM image of 1.5%Ru–1.5%Pd/C3N4-air clearly showed the lattice fringe of 0.206 nm and 0.224 nm, which were attributed to the (1 0 1) facet of Ru and (1 1 1) facet of Pd, respectively (Fig. 4c). EDS mapping of 1.5%Ru–1.5%Pd/C3N4-air demonstrated that C, N, Ru, and Pd were the main elements and highly dispersed (Fig. S3†). Under ultrasonic-assisted impregnation, Pd species and Ru species were uniformly distributed on the C3N4-air surface (Fig. 4d–g). By comparing the HAADF-STEM image with the results of element mapping, the Ru NPs and Pd NPs could be identified and marked as a yellow circle and blue circle, respectively, where a distance of 5 nm between Pd NPs and Ru NPs was measured (Fig. 4h). It was reported that the phenomenon of H2 spillover between two different species at a nano-scale distance of less than 15 nm could occur.30,31 Hydrogen spillover is well-known in heterogeneous catalytic hydrogenation, wherein the migration of hydrogen atoms between multiple components is supposed to enhance the mass transfer of H2, thereby facilitating the whole catalysis process.32
 |
| | Fig. 4 Morphology characterization of 1.5%Ru–1.5%Pd/C3N4-air. (a) TEM image, (b) particle size distribution, (c) HR-TEM image, (d–g) EDS element mapping, (h) HAADF-STEM. | |
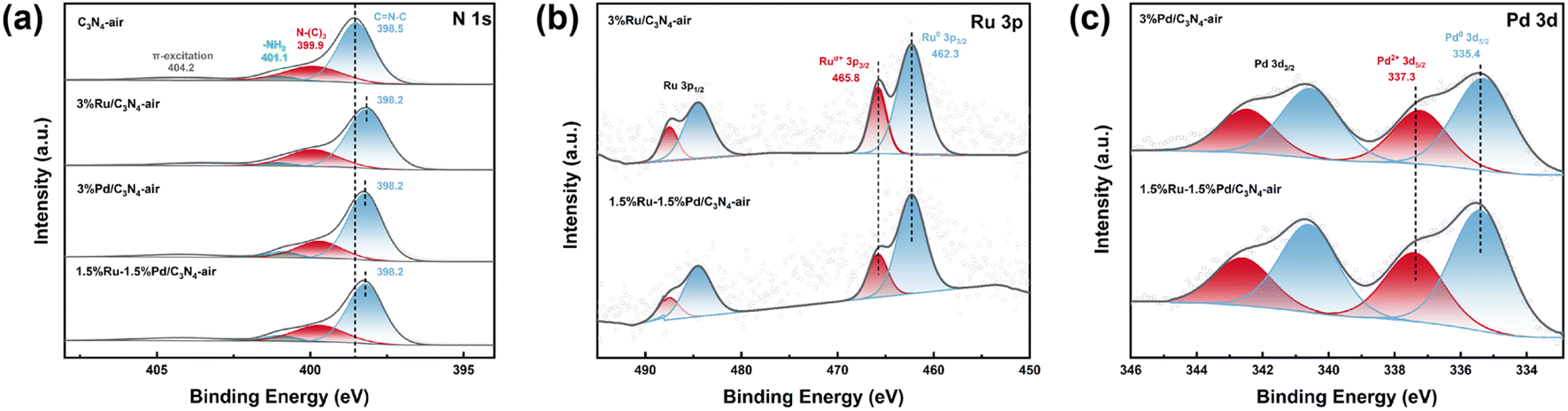
X-ray photoelectron spectroscope (XPS) was employed to elucidate the surface composition of the catalyst and valence states of elements, and the results are shown in Fig. 5. The N 1s high-resolution spectra of C3N4-air was deconvoluted into four peaks at around ∼398.5 eV, ∼399.9 eV, ∼401.1 eV, and ∼404.2 eV, which could be assigned to the heterocyclic N in triazine also named as pyridine N (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–C), tertiary nitrogen group or bridged N (N–(C)3), amino functional group (–NH2), and charge effect (π-excitation), respectively26–28 (Fig. 5a). Thereinto, the emergence of –NH2 was from the incomplete polymerization of urea. Notably, compared with ∼398.5 eV of C3N4-air, the pyridine N characteristic peaks of 3%Ru/C3N4-air, 3%Pd/C3N4-air, and 1.5%Ru–1.5%Pd/C3N4-air are all centred at ∼398.2 eV. This 0.3 eV binding energy (BE) decrease after metal anchoring suggested strong interactions between metals and C3N4-air due to electron shift from the metal species to pyridine N. Therefore, the Ru–Nx and Pd–Nx active sites were likely to be formed.27,28,33 The metal–N bonding is reported as a kind of Strong Metal–Support Interaction (SMSI), which could confine metal species on the support, thereby immobilizing metals in their original positions and restraining their agglomeration and mobility.34,35 Additionally, the peak of ∼398.2 eV might include the contribution from nitrogen in metal–N bonding, which was assigned to Ru–Nx and Pd–Nx. Because the binding energy of nitrogen in metal–N bond is similar to pyridine N, it is difficult to differentiate them quantitatively. Moreover, Fig. 5b shows the high-resolution spectrum of Ru 3p. The peaks of ∼462.3 eV and ∼465.8 eV were attributed to Ru0 3p3/2 and Ruσ+ 3p3/2, while those of ∼484.5 eV and ∼487.5 eV were corresponding to Ru0 3p1/2 and Ruσ+ 3p1/2, respectively.25,36,37
N–C), tertiary nitrogen group or bridged N (N–(C)3), amino functional group (–NH2), and charge effect (π-excitation), respectively26–28 (Fig. 5a). Thereinto, the emergence of –NH2 was from the incomplete polymerization of urea. Notably, compared with ∼398.5 eV of C3N4-air, the pyridine N characteristic peaks of 3%Ru/C3N4-air, 3%Pd/C3N4-air, and 1.5%Ru–1.5%Pd/C3N4-air are all centred at ∼398.2 eV. This 0.3 eV binding energy (BE) decrease after metal anchoring suggested strong interactions between metals and C3N4-air due to electron shift from the metal species to pyridine N. Therefore, the Ru–Nx and Pd–Nx active sites were likely to be formed.27,28,33 The metal–N bonding is reported as a kind of Strong Metal–Support Interaction (SMSI), which could confine metal species on the support, thereby immobilizing metals in their original positions and restraining their agglomeration and mobility.34,35 Additionally, the peak of ∼398.2 eV might include the contribution from nitrogen in metal–N bonding, which was assigned to Ru–Nx and Pd–Nx. Because the binding energy of nitrogen in metal–N bond is similar to pyridine N, it is difficult to differentiate them quantitatively. Moreover, Fig. 5b shows the high-resolution spectrum of Ru 3p. The peaks of ∼462.3 eV and ∼465.8 eV were attributed to Ru0 3p3/2 and Ruσ+ 3p3/2, while those of ∼484.5 eV and ∼487.5 eV were corresponding to Ru0 3p1/2 and Ruσ+ 3p1/2, respectively.25,36,37
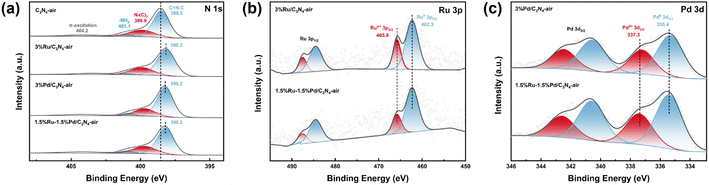 |
| | Fig. 5 XPS spectrum of catalysts. | |
The XPS fitting data are summarized in Tables S2–S4.† The ratio of Ru0/Ruσ+ was 1.96 in both 3%Ru/C3N4-air and 1.5%Ru–1.5%Pd/C3N4-air, which indicated that the introduction of Pd species hardly influenced Ru species. Ru was first anchored on C3N4-air to form Ru–Nx. Subsequently, the Pd was introduced and coordinated with pyridine N to form Pd–Nx. Fig. 5c shows the high-resolution spectra of Pd 3d. The peaks of ∼335.4 eV, ∼340.6 eV, ∼337.3 eV, and ∼342.5 eV correspond to Pd0 3d5/2, Pd2+ 3d5/2, Pd0 3d3/2, and Pd2+ 3d3/2, respectively.16,17,28 There was no credible electron shift observed in 3%Pd/C3N4-air and 1.5%Ru–1.5%Pd/C3N4-air. Besides, the ratio of Pd0/Pd2+ in 3%Pd/C3N4-air and 1.5%Ru–1.5%Pd/C3N4-air were rather close, 1.32 and 1.37, respectively. Therefore, Ru–Nx and Pd–Nx are relatively independent dual metal sites, which were conducive to high dispersion of Ru and Pd species at a nanoscale distance consistent with the results of TEM.
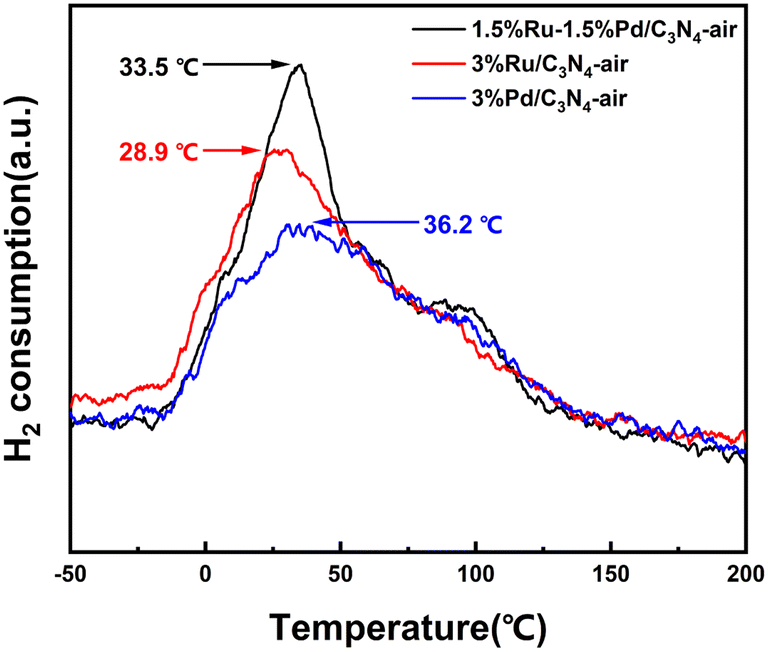
It was universally reported that H2-TPD is convinced in evaluating the ability of the catalyst to dissociate and activate H2.30 As displayed in Fig. 6, H2 was desorbed from the catalyst surface at a mild temperature, which indicated H2 could be activated and dissociated easily by the catalysts. 3%Ru/C3N4-air and 3%Pd/C3N4-air exhibited desorption peaks centred at 28.9 °C and 36.2 °C, respectively. By contrast, the desorption peak of 1.5%Ru–1.5%Pd/C3N4-air was centred at 33.5 °C, which implied the interaction between H2 and catalyst in 1.5%Ru–1.5%Pd/C3N4-air was stronger than 3%Ru/C3N4-air but weaker than 3%Pd/C3N4-air. In addition, the integration of the desorption peak area describes the amount of chemisorbed hydrogen quantitatively. Ru–Pd catalyst exhibited a relatively higher peak area, which suggested that Ru–Pd catalyst enabled activation and dissociation of more H2 than single Ru and Pd based catalysts under the same conditions.
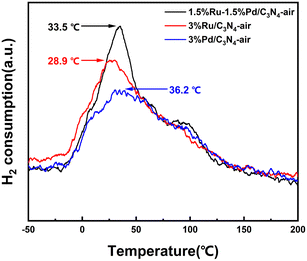 |
| | Fig. 6 H2-TPD profiles of 1.5%Ru–1.5%Pd/C3N4-air, 3%Ru/C3N4-air, and 3%Pd/C3N4-air. | |
Catalyst evaluation
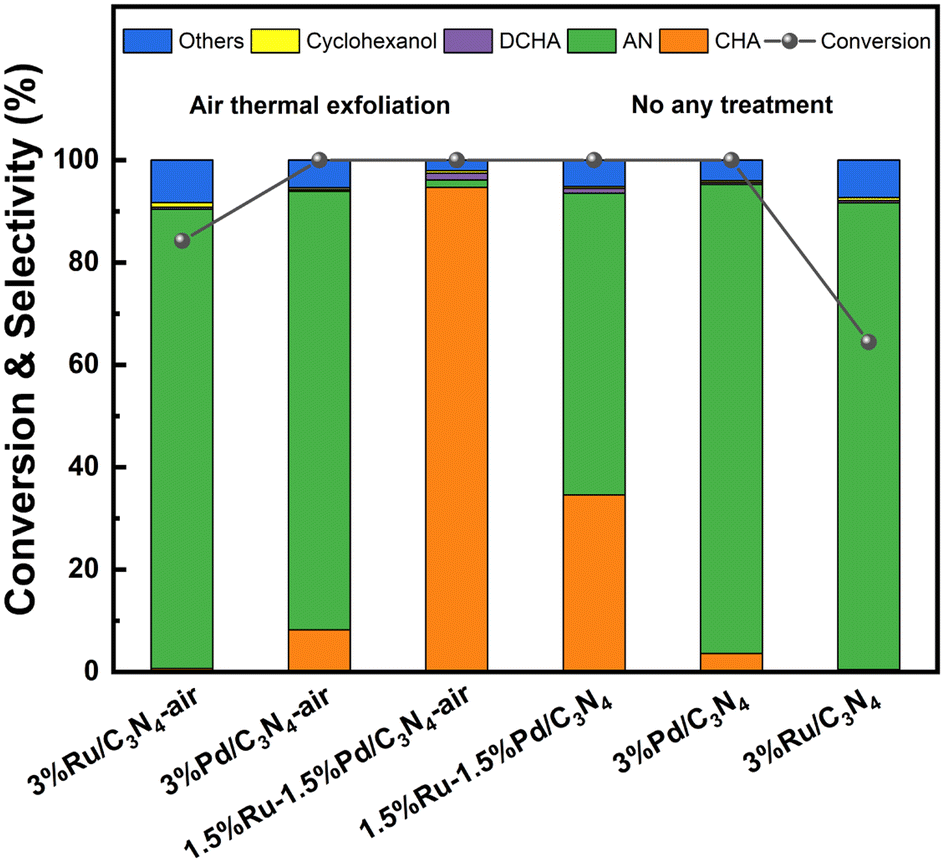
The catalyst performance for hydrogenation of nitroaromatics to alicyclic amines was evaluated by NB to CHA as the probe reaction. As reported, the side reactions in the hydrogenation of nitroaromatics included dehalogenation, deamination, condensation, etc. Based on GC-MS analysis, the main by-products in this work were detected as dicyclohexylamine (DCHA) and cyclohexanol (Fig. S4†). Some intermediates and a few products were collected as others. Systematically, the effects of Ru and Pd loading, solvent, temperature, pressure, and time were investigated. Firstly, the loading of Ru and Pd was studied. In most cases, 3 wt% active component loading was appropriate for noble metal supported catalysts; therefore, the total loading of Ru and Pd for all prepared catalysts was 3 wt%, as shown in Fig. S5.† Notably, compared with single 3 wt% Ru and Pd catalysts, as well as physically mixed 1.5%Ru/C3N4-air and 1.5%Pd/C3N4-air, 1.5%Ru–1.5%Pd/C3N4-air showed superiority in tandem hydrogenation from NB to CHA with 100% NB conversion, 96.8% CHA selectivity, and CHA production rate (defined as mole number of generated CHA per mole active component per h, i.e., mol CHA/mol Ru·Pd/h) of 26.70 h−1 (Fig. 7), which is consistent with the literature reported that the synergistic effect was optimum when the ratio of Ru to Pd was 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.38 Therefore, 1.5%Ru–1.5%Pd/C3N4-air was used in the following investigation. It was worth noting that the physically mixed Ru–Pd catalyst showed a CHA production rate of 16.90 h−1, which was higher than both Ru and Pd based single metal catalysts but lower than the Ru–Pd catalyst. This elucidated the proposed dual active sites were practical for the sequential hydrogenation of NB to CHA, and there was an interaction between Ru and Pd that enhances the hydrogenation process. Considering the possible effect of protons, several commonly used polar aprotic solvents (DMF, cyclohexane, THF) and polar protic solvents (ethanol, methanol, water) were selected to investigate the solvent effect for hydrogenation of NB to CHA (Fig. S6a†). CHA was the major product except for DMF as the solvent, which is consistent with previously reported.12 The formylation of DMF might be the reason why CHA is not produced. As for cyclohexane, the selectivity of generated by-product DCHA reached as high as 7.6% with that of desired product CHA reduced to 79.5%.9 In terms of alcohol solvents, ethanol showed a higher CHA selectivity of 90.1% than 68.8% of methanol, which was attributed to higher H2 solubility in ethanol. The CHA selectivity in water was 80.7% because of deamination between water and CHA. By contrast, the highest CHA selectivity of 96.8% could be obtained in THF because of good solubility of H2 in it. Then, the effect of reaction parameters, e.g., reaction temperature, pressure, and time, on tandem hydrogenation of NB was further investigated, and the results in Fig. S6b–d† showed that under the optimum conditions of 80 °C and 3 MPa H2 for 3 h, 1.5%Ru–1.5%Pd/C3N4-air exhibited excellent performance with 100.0% NB conversion and 96.8% CHA selectivity.
:1.38 Therefore, 1.5%Ru–1.5%Pd/C3N4-air was used in the following investigation. It was worth noting that the physically mixed Ru–Pd catalyst showed a CHA production rate of 16.90 h−1, which was higher than both Ru and Pd based single metal catalysts but lower than the Ru–Pd catalyst. This elucidated the proposed dual active sites were practical for the sequential hydrogenation of NB to CHA, and there was an interaction between Ru and Pd that enhances the hydrogenation process. Considering the possible effect of protons, several commonly used polar aprotic solvents (DMF, cyclohexane, THF) and polar protic solvents (ethanol, methanol, water) were selected to investigate the solvent effect for hydrogenation of NB to CHA (Fig. S6a†). CHA was the major product except for DMF as the solvent, which is consistent with previously reported.12 The formylation of DMF might be the reason why CHA is not produced. As for cyclohexane, the selectivity of generated by-product DCHA reached as high as 7.6% with that of desired product CHA reduced to 79.5%.9 In terms of alcohol solvents, ethanol showed a higher CHA selectivity of 90.1% than 68.8% of methanol, which was attributed to higher H2 solubility in ethanol. The CHA selectivity in water was 80.7% because of deamination between water and CHA. By contrast, the highest CHA selectivity of 96.8% could be obtained in THF because of good solubility of H2 in it. Then, the effect of reaction parameters, e.g., reaction temperature, pressure, and time, on tandem hydrogenation of NB was further investigated, and the results in Fig. S6b–d† showed that under the optimum conditions of 80 °C and 3 MPa H2 for 3 h, 1.5%Ru–1.5%Pd/C3N4-air exhibited excellent performance with 100.0% NB conversion and 96.8% CHA selectivity.
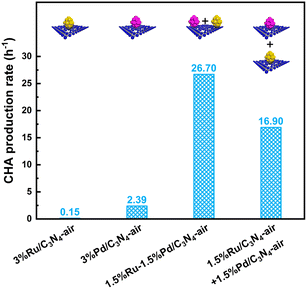 |
| | Fig. 7 CHA production rate of Ru–Pd catalyst, single Ru/Pd based catalyst, and physically mixed Ru and Pd based catalyst. | |
Structure–performance relationship
Clearly, 1.5%Ru–1.5%Pd/C3N4-air performed best in the hydrogenation of nitrobenzene. So, aiming at 1.5%Ru–1.5%Pd/C3N4-air, the structure–performance was studied here. Support modification is a promising way to enhance catalyst performance. As shown in Table S1,† a series of Ru–Pd catalysts with different supports were evaluated. Compared with 100.0% NB conversion and 34.6% CHA selectivity of 1.5%Ru–1.5%Pd/C3N4, 100.0% NB conversion and 96.8% CHA selectivity was obtained over 1.5%Ru–1.5%Pd/C3N4-air catalyst. For all catalysts, the NB conversion and CHA selectivity increased after air thermal exfoliation, as shown in Fig. 8, which confirmed the universality of air thermal exfoliation in promoting activity. The structural properties of C3N4-air could be responsible for this. Similar to previous results of N2 physisorption, SEM, and TEM characterization, the bulk C3N4 appeared as a large dense particle with stacked and squeezed nanosheets, while C3N4-air exhibited a lamella-separated nanosheet structure with higher specific surface area and numerous connected slit pores with different sizes, resulting in high dispersion of active sites and further facilitating contact between substrates and active sites. By contrast, 1.5%Ru–1.5%Pd/C showed 100.0% NB conversion but only 7.5% CHA selectivity, which could be attributed to side reactions caused by excessive adsorption of amino groups. So, alkaline liquors were always added to regulate the adsorption behavior of amino groups in industry. It has been reported that C3N4 is a kind of solid alkali rich in alkaline N sites, which could provide an alkaline environment to inhibit side reactions like deamination, condensation, etc. Moreover, XPS suggested the existence of Pd–Nx and Ru–Nx, which were crucial for successive hydrogenation of nitro group and benzene ring. The dispersion of Ru and Pd species should also be taken into consideration. Anchoring multiple metals in nano-scale is expected to facilitate the activation and dissociation process of H2 via H2 spillover taking place on the catalyst surface.32 Thus, highly dispersed Ru and Pd species at a nanoscale distance made H2 spillover possible on the C3N4 surface. To be precise, the Ru–Pd catalyst had ternary active sites rather than merely dual active sites. Pd–Nx and Ru–Nx active sites catalyze hydrogenation of nitro group and benzene ring effectively. Meanwhile, the alkaline N site played a synchronous role in side reaction inhibition. Because our previous studies have given an insight into the superiority of C3N4 as support, we focused more on the effects of Ru–Pd dual active sites herein.
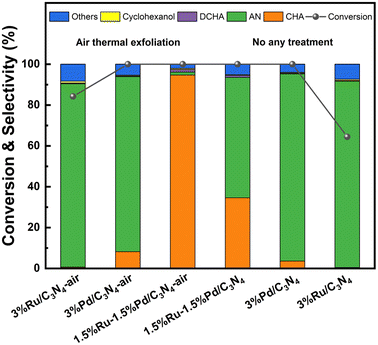 |
| | Fig. 8 Effects of air thermal exfoliation. Reaction conditions: 1 mmol NB, 25 wt% catalyst (based on the substrate), 10 mL THF, 80 °C, 3 MPa H2, 4 h. | |
Hydrogenation mechanism
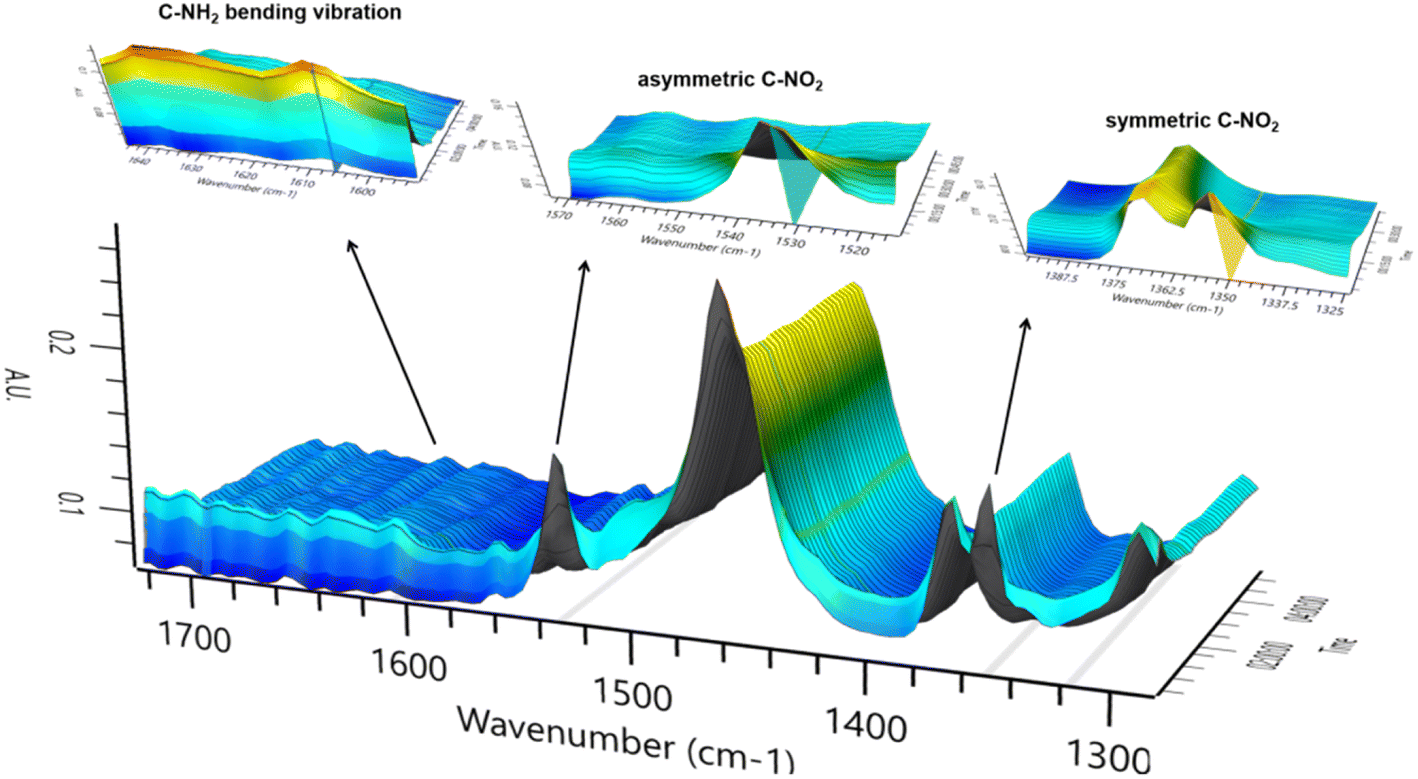
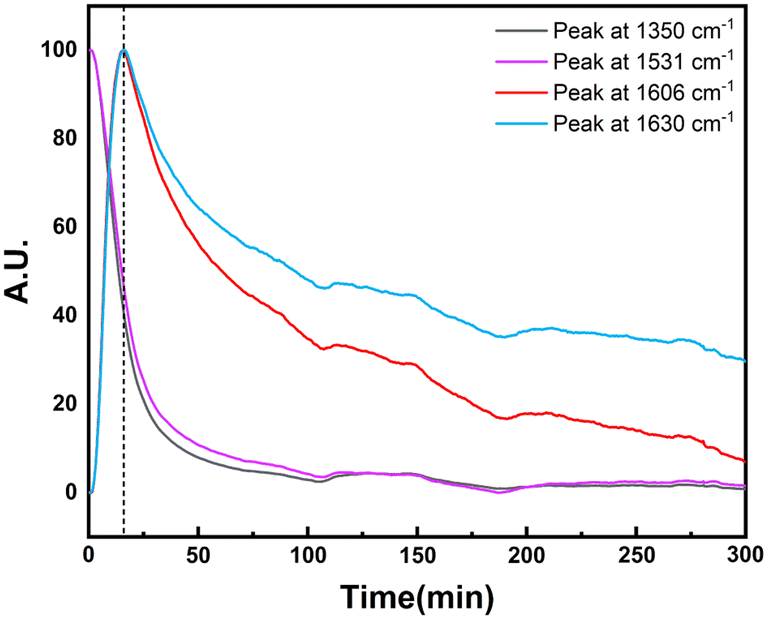
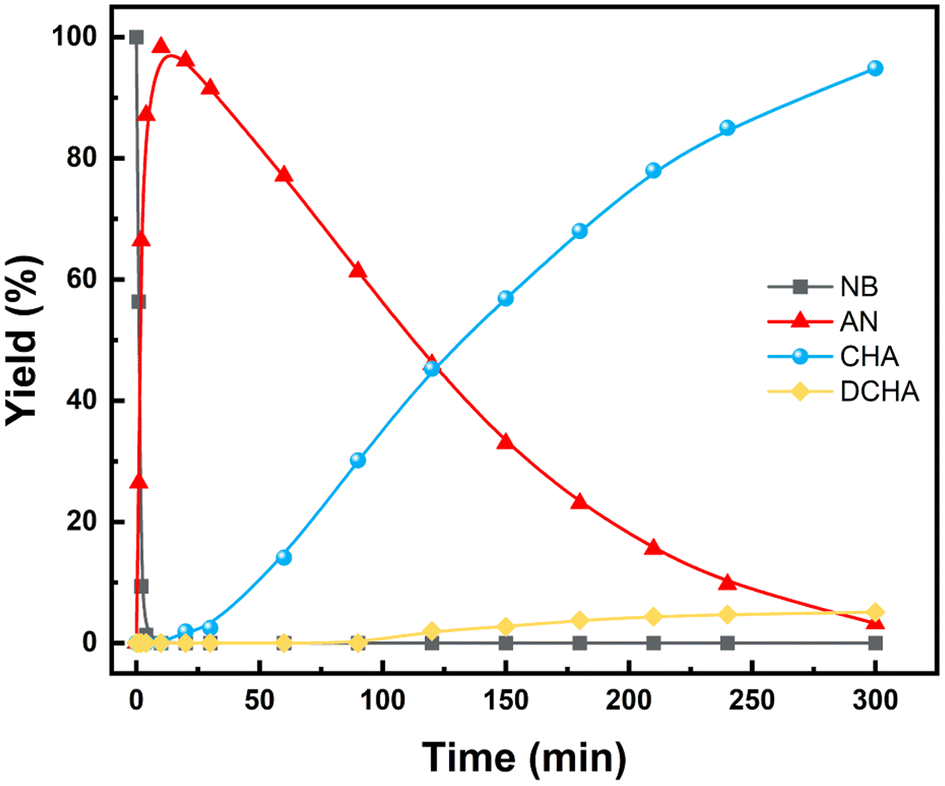
In order to explore the catalytic hydrogenation mechanism, operando infrared (IR) was employed to understand the hydrogenation process of NB to CHA. As displayed in Fig. 9, distinctly, two strong peaks at 1350 cm−1 and 1531 cm−1 were assigned to the symmetric and asymmetric C–NO2 stretch in NB,39 whose intensity decreased dramatically in 15 min, indicating NB was fully converted in such a relatively short period of time. With characteristic peaks of nitro group weakening, the double peaks of primary amine at 1606 cm−1 and 1630 cm−1 emerged and increased significantly, which demonstrated the conversion of –NO2 to –NH2.39 Therefore, AN was the direct hydrogenation product of NB at this moment. Besides, the conversion of nitro group was too rapid to capture any characteristics of intermediates like nitroso compounds, which indicated considerably high activity of Ru–Pd catalyst in the hydrogenation of nitro group. However, with further hydrogenation after 15 min, the characteristic peaks of –NH2 at 1606 cm−1 and 1630 cm−1 decreased gradually, which implied the hydrogenation from benzene ring to naphthene.40 The double peaks at 1606 cm−1 and 1630 cm−1 also contained the contribution of benzene ring skeleton vibration.41,42 Therefore, the skeleton vibration became weaker with the hydrogenation of benzene ring, which reflected as characteristic peaks weakening at peaks of 1606 cm−1 and 1630 cm−1, i.e., hydrogenation of AN to CHA. The intensity of the above characteristic peaks varying with time is shown in Fig. 10. Clearly, the hydrogenation of NB to CHA was proved as a tandem process. Besides, the hydrogenation of nitro group was prior to the hydrogenation of benzene ring, which was the rate-determining step in this tandem process. Fig. 11 showed the yields varied with time. The conversion of NB was also completed in approximately 15 min consistent with the results of operando IR.
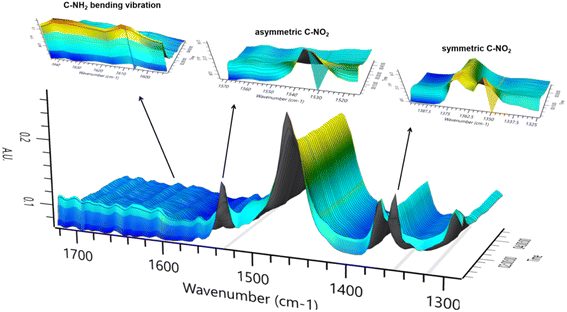 |
| | Fig. 9 Characteristic peaks of operando IR spectrum. | |
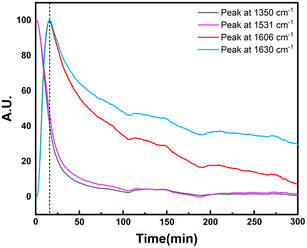 |
| | Fig. 10 Effect of time on operando IR characteristic peak strength. | |
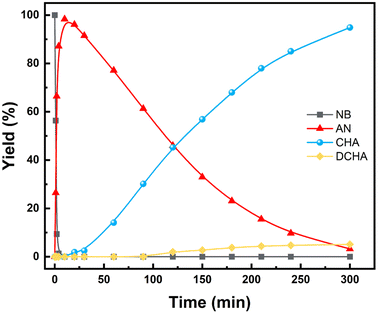 |
| | Fig. 11 Effect of time on product yields (reaction condition: 10 mmol NB, 25 wt% catalyst (based on substrate), 100 mL THF, 80 °C, 3 MPa H2. Reaction was carried out in a 250 mL autoclave.). | |
Based on the results of operando IR and the GC-MS results of an incomplete hydrogenation (Fig. S7†), a reaction pathway was proposed as in Fig. S8.† Through the breaking of N–O bond and formation of N–H bond, nitro group was converted into amino, and AN was generated first. With further hydrogenation of benzene ring, AN was converted into CHA. The whole hydrogenation process was accompanied by the deamination of AN and condensation of CHA. Accordingly, cyclohexanol and DCHA were the main side products that could be detected in the catalytic system.
Hydrogenation kinetics
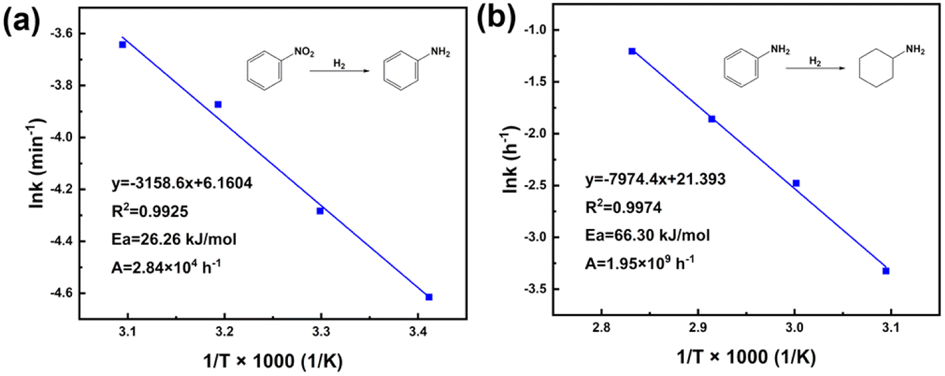
First and foremost, the effects of diffusion should be eliminated before starting the experiments of intrinsic kinetics. Basically, catalyst particle size is regarded as the main factor of internal diffusion, while agitation speed determines external diffusion. Therefore, the prepared catalysts were sieved into different particle sizes to investigate the size effect on nitrobenzene conversion, as well as the effect of agitation speed, shown in Fig. S9.† Clearly, the internal diffusion was obviously the dominant factor in mass transfer. Hence, when particle size was less than 150 μm and agitation speed was 550 rpm, the effects of internal and external diffusion were deemed to be negligible. As for the kinetics model, Pandit et al.,43 Milian et al.,44 and Yu et al.45 have reported that the hydrogenation of nitrobenzene is a first-order reaction, which could be expressed in the form of −ln(1 − X) = kt that fitted our system. In order to obtain intrinsic kinetic data, the hydrogenation of nitro group was carried out under 20, 30, 40, and 50 °C, respectively. Fig. S10a† shows the plots of −ln(1 − X) vs. time of NB hydrogenation, presenting a linear relationship. The results of the linear fit of −ln(1−X) vs. time are summarized in Table S5.† All the obtained linearly dependent coefficients R2 were larger than 0.99, which meant the proposed kinetic model was valid. The slope of the regression equation obtained by linear fitting stood for rate constant k at different temperatures. With increasing temperature, the value of k increased, which meant a faster hydrogenation process consistent with the optimization of temperature. Fig. 12a shows the plot of lnk vs. T−1; according to the Arrhenius equation, the slope of the line was the value of −Ea/R, and the intercept was lnA. Therefore, the activation energy Ea could be calculated as 26.26 kJ mol−1 and pre-exponential factor A was 2.84 × 104 h−1. Furthermore, the relationship between rate constant k and temperature T (20–50 °C) could be expressed in the form of the Arrhenius equation: k = 2.84 × 104 exp(−3158.6 T−1). Compared with other reported hydrogenation of nitrobenzene to aniline, 1.5%Ru–1.5%Pd/C3N4-air significantly decreased the activation energy. The relevant results are listed in Table S6.† Accordingly, for hydrogenation of benzene ring, the experiment was carried out at 50, 60, 70, and 80 °C, respectively. Fig. S10b† shows the plots of −ln(1−X) vs. time of aniline hydrogenation, and Table S7† lists the results of the linear fit, where all the values of R2 were larger than 0.99. Briefly, as shown in Fig. 12b, the value of −Ea/R equals to −7974.8, and lnA equals to 21.393. Therefore, the activation energy can be calculated as 66.30 kJ mol−1, and the pre-exponential factor is 1.95 × 109 h−1. The Arrhenius equation of rate constant k and temperature T (50–80 °C) could be expressed as: k = 1.95 × 109 exp(−7974.4 T−1). The relevant kinetic data of hydrogenation of aniline suggested the activation energy of aniline was generally 50–80 kJ mol−1, as collected in Table S8.† So, the above calculated result of 66.30 kJ mol−1 is reasonable.
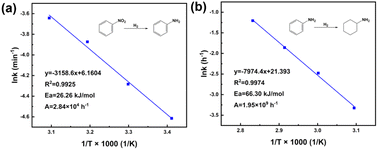 |
| | Fig. 12 Plot of lnk vs. T−1 over 1.5%Ru–1.5%Pd/C3N4-air catalyst. (a) Nitrobenzene hydrogenation, (b) aniline hydrogenation. | |
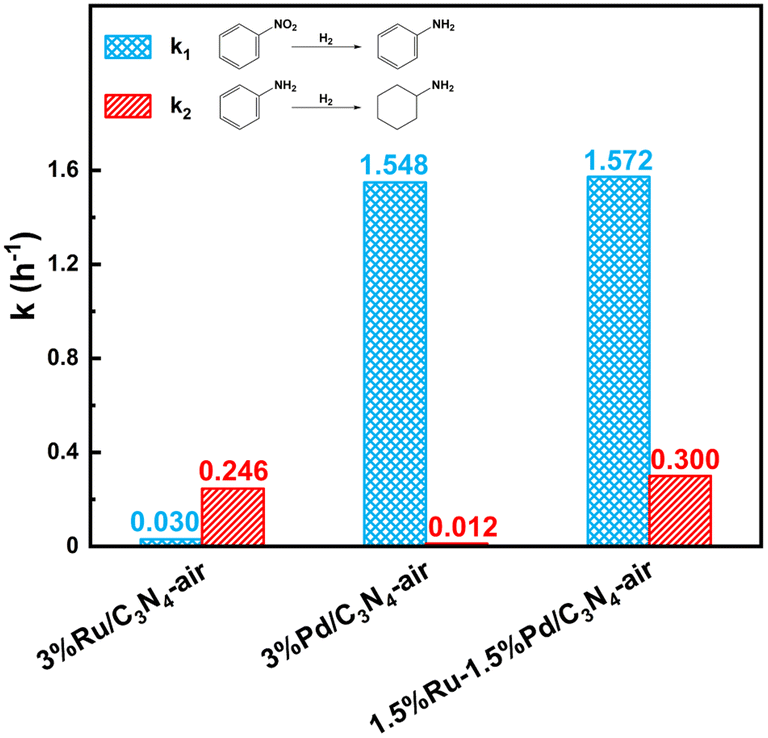
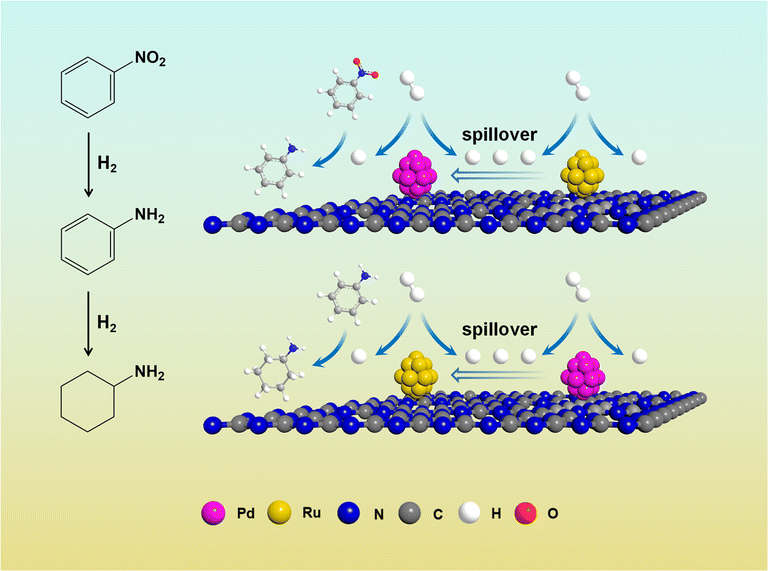
Here, metal-assisted hydrogenation was testified by the kinetics of single metal-based catalysts and Ru–Pd catalyst. Based on the kinetics of hydrogenation of NB (Fig. S11†), Fig. S12a† showed the plots of −ln(1−X) versus time, and linear fit results of rate constants are listed in Table S9.† Fig. 13 shows the comparison of the hydrogenation rate constant over different catalysts. Clearly, 3%Pd/C3N4-air manifested a higher rate constant of 1.548 h−1, while 3%Ru/C3N4-air showed poor activity in the hydrogenation of nitro group with a rate constant of 0.030 h−1. Therefore, Pd species were deemed to be conducive to the hydrogenation of nitro groups. Conventionally, higher metal loading means more active sites exposed; thus, the rate constant will be relatively higher. Interestingly, the rate constant of 1.5%Ru–1.5%Pd/C3N4-air was as high as 1.572 h−1 and even slightly higher than that of 3%Pd/C3N4-air. The results of H2-TPD have confirmed that Ru species could activate and dissociate H2 under mild temperatures in both single Ru based catalyst and bimetallic catalysts. These results illuminated the role of 1.5%Ru in 1.5%Ru–1.5%Pd/C3N4-air assisted in activating and dissociating H2 instead of any chemisorption effects on the substrate, while Pd as the dominant metal played a role in chemical adsorption and desorption of substrate as well as activation and dissociation of H2. Therefore, H2 was dissociated via Ru, and the H2 spillover effect probably took place simultaneously from Ru to Pd, which facilitated the nitro hydrogenation mainly on Pd nanoparticles. This process was called metal-assisted hydrogenation, illustrated in Fig. 14. For the hydrogenation of aniline, the same phenomenon was also observed, shown as Fig. S11b and Fig. 12b, Table S10† lists the results of linear fitting of rate constant. On the contrary, 3%Ru/C3N4-air catalyst showed a significantly higher rate constant of 0.246 h−1 than 0.012 h−1 of 3%Pd/C3N4-air catalyst, which elucidated Ru based catalyst was a better choice for hydrogenation of benzene ring. Equally, the rate constant of 1.5%Ru–1.5%Pd/C3N4-air reached 0.300 h−1, almost the same, even slightly higher than that of 3%Ru/C3N4-air. Thus, the 1.5%Pd in 1.5%Ru–1.5%Pd/C3N4-air assisted the metal and Ru was the dominant one. This metal-assisted hydrogenation was regarded as the critical factor in enhancing catalytic activity in tandem hydrogenation.
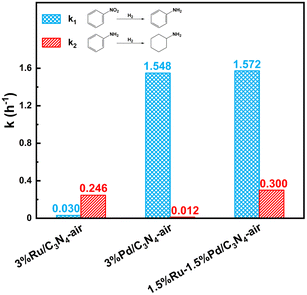 |
| | Fig. 13 Hydrogenation rate constant of nitro group (k1) and benzene ring (k2) over different catalysts. | |
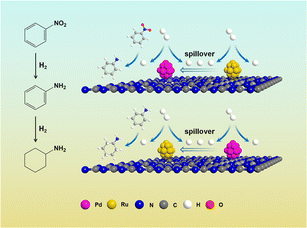 |
| | Fig. 14 Illustration of metal-assisted hydrogenation in one-pot synthesis of NB to CHA. | |
Catalyst recyclability and versatility
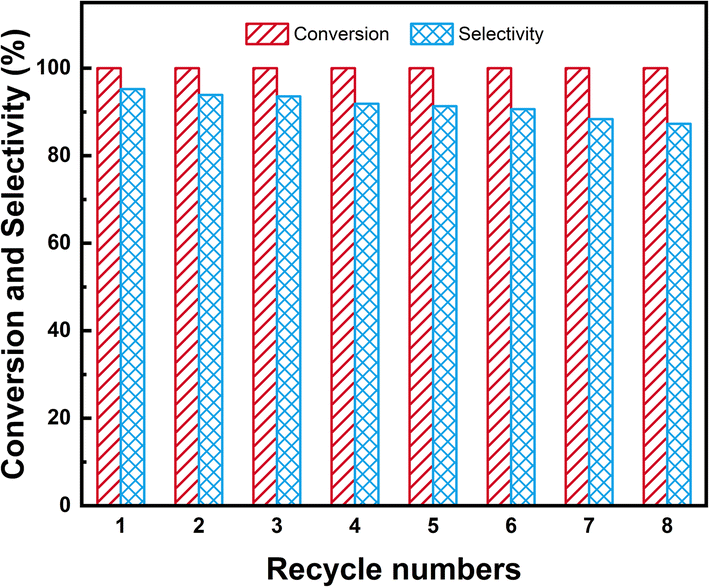
Under optimal reaction conditions of 80 °C, 3 MPa H2, the recyclability of 1.5%Ru–1.5%Pd/C3N4-air was investigated in batch-wise recycling. The results are shown in Fig. 15. It was worth noting that 1.5%Ru–1.5%Pd/C3N4-air virtually remained unchanged at a high level of 100.0% NB conversion in 8-time recycles, which demonstrated Ru–Pd catalyst had superior activity in hydrogenation of nitro group. By contrast, the selectivity of CHA gently decreased from an initial 95.2% to a final 87.3%, but it was still at a relatively high level. According to the TEM image of the used catalyst (Fig. S13†), this slight decrease could be ascribed to a small part of agglomeration at the edge of the catalyst. Overall, the Ru–Pd catalyst exhibited excellent activity and good selectivity in the sequential hydrogenation of NB to CHA, which could be attributed to the strong metal–support interaction between Ru–Pd and C3N4-air that could reduce the possibility of huge agglomeration of active metal and maintain the catalytic activity.34
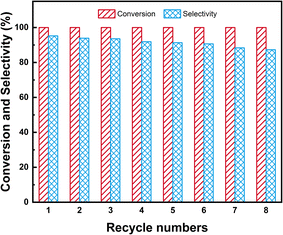 |
| | Fig. 15 Recyclability of 1.5%Ru–1.5%Pd/C3N4-air. Reaction conditions: 1 mmol substrate, 25 wt% catalyst based on substrate, 10 mL THF, 80 °C, 3 MPa H2, 3 h. | |
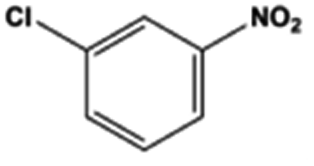
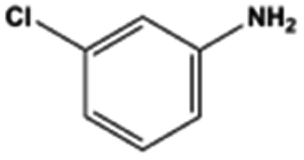
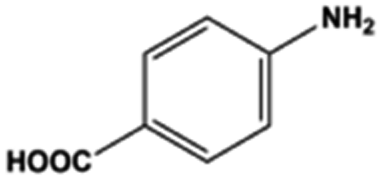
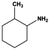
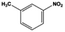
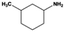
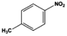
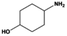
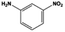
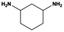
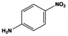
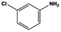
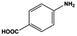
The general substrate scope of 1.5%Ru–1.5%Pd/C3N4-air catalyst was investigated for sequential hydrogenation of nitroaromatics with electron-donating and electron-withdrawing substituents. The results are listed in Table 1 and Fig. S14.† Definitely, the efficiency of tandem hydrogenation varied with different substituent groups. For extended nitroaromatics with alkyl-substituted including methyl, ethyl, and even dimethyl, >97.0% yield was obtained over 1.5%Ru–1.5%Pd/C3N4-air catalyst (Entries 2–6, Table 1). Therefore, compared with only nitro-substituted nitrobenzene, alkyl nitrobenzene could also achieve tandem hydrogenation of nitro group and benzene ring but took more time. For other nitroaromatics with strong electron-donating groups like nitrophenol, the dehydroxylation reaction occurred easily and the main by-product CHA could be detected. However, 1.5%Ru–1.5%Pd/C3N4-air still performed well with >90.0% product yield (Entries 7 and 8, Table 1). Another typical nitroaromatic with a strong electron-donating group was nitroaniline. The hydrogenation of nitroaniline was relatively more difficult. However, >90.0% yield was still obtained for 3-nitroaniline and 4-nitroaniline over Ru–Pd catalyst (Entries 9 and 10, Table 1). This was probably because of the strong interaction between the amino group and catalyst, which resulted in some of the active sites were occupied by the amino group reducing the hydrogenation rate of benzene ring. As for nitroaromatics with electron-withdrawing groups, typically like chlorine and carboxyl, 1.5%Ru–1.5%Pd/C3N4-air showed excellent performance only in the hydrogenation of nitro group. For the hydrogenation of 3-nitrochlorobenzene, the main product was 3-chloroaniline obtained by hydrogenation of nitro group and by-products were aniline and cyclohexylamine (Entry 11, Table 1). Therefore, it could be considered that the hydrogenation of benzene ring would occur after the dehalogenation side reaction. The reason might be that the activation energy of dehalogenation is lower than that of benzene ring hydrogenation. Moreover, for the hydrogenation of 4-nitrobenzoic acid, the detected main product was 4-aminobenzoic acid, with a selectivity of 96.2% (Entry 12, Table 1). The only by-product was from the condensation reaction, and no hydrogenation product of hydrogenation was detected. The nitro and carboxyl, as electron withdrawing groups, might make it more difficult to activate the benzene ring. All the above results indicated good versatility of as-prepared Ru–Pd catalyst in tandem hydrogenation of nitroaromatics to alicyclic amines.
Table 1 Versatility of 1.5%Ru–1.5%Pd/C3N4-air catalyst on tandem hydrogenation of nitroaromaticsa
| Entry |
Substrate |
Product |
T(h) |
Conv.b (%) |
Sel.b (%) |
| Reaction conditions: 1 mmol substrate, 25 wt% catalyst (based on the substrate), 10 mL THF, 80 °C, 3 MPa H2. Conversion of substrate and selectivity of the product were detected by GC-MS. Only the results of nitrobenzene were calculated by the internal standard method, the others by the area normalization method. |
| 1 |
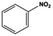 |
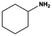 |
3 |
100.0c |
96.8c |
| 2 |
 |
 |
8 |
100.0 |
99.0 |
| 3 |
 |
 |
8 |
100.0 |
97.5 |
| 4 |
 |
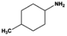 |
8 |
100.0 |
98.7 |
| 5 |
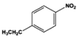 |
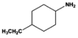 |
10 |
100.0 |
97.3 |
| 6 |
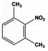 |
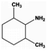 |
14 |
100.0 |
98.0 |
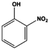
| 7 |
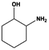
 |
 |
12 |
100.0 |
94.6 |
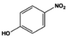
| 8 |
 |
 |
12 |
100.0 |
90.1 |
| 9 |
 |
 |
18 |
100.0 |
93.9 |
| 10 |
 |
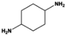 |
24 |
100.0 |
91.1 |
| 11 |
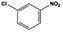 |
 |
2 |
100.0 |
72.1 |
| 12 |
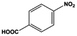 |
 |
8 |
100.0 |
96.2 |
Conclusion
In summary, a Ru–Pd/C3N4-air with Ru and Pd dual active sites as well as alkaline N sites was prepared by facile ultrasonic-impregnation, which enabled sequential hydrogenation of nitroaromatics to alicyclic amines under mild conditions. Air-exfoliated C3N4-air nanosheets showed higher specific area than bulk C3N4, which increased the catalyst activity. The particle size of the Ru–Pd catalyst was 2.8 ± 0.6 nm, where Pd NPs and Ru NPs were highly dispersed at a nano-scale distance. The formed Ru–Nx and Pd–Nx species were conducive to high dispersion of Ru and Pd. Ru–Pd catalyst enabled the activation and dissociation of H2 at 33.5 °C, which was beneficial to metal-assisted hydrogenation. Under optimum reaction conditions, prepared 1.5%Ru–1.5%Pd/C3N4-air exhibited excellent performance with the almost completed conversion of NB and 96.8% selectivity of CHA, which could be attributed to highly dispersed Ru–Nx and Pd–Nx species. C3N4 as support was beneficial to reduce by-products, mainly including cyclohexanol and DCHA, which could be ascribed to the existence of alkali N species. Moreover, the hydrogenation of NB to CHA was demonstrated as a tandem process with aniline as an intermediate by operando IR, and hydrogenation of the nitro group was prior to the benzene ring. Intrinsic kinetic studies demonstrated that the activation energies of nitro group and benzene ring were 26.26 kJ mol−1 and 66.30 kJ mol−1, respectively, which directly gave evidence that hydrogenation of the benzene ring was the rate-determining step. Importantly, kinetics also revealed the phenomenon of metal-assisted hydrogenation. Thereinto, Pd and Ru were the dominant metals for hydrogenation of the nitro group and benzene ring, respectively; meanwhile, the corresponding non-dominant metal could enhance activation and dissociation of H2, thereby improving catalytic activity. Additionally, the stability of Ru–Pd catalyst essentially remained at a high level after recycling for 8 times, and its substrate scope could be extended to other nitroaromatics with different substituent groups. This work is expected to provide an environmentally friendly catalytic system for the selective hydrogenation of nitroaromatics to alicyclic amines.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was financially supported by the Strategic Priority Research Program of the Chinese Academy of Sciences (Grant No. XDA 29030201).
Notes and references
- T. Zhang, Z. Xie, L. Jiang, W. Zhao, S. Cao, B. Wang, R. Si, R. Zhang, Y. Liu and Z. Zhao, Chem. Eng. J., 2022, 443, 136416 CrossRef CAS.
- E. H. Boymans, P. T. Witte and D. Vogt, Catal. Sci. Technol., 2015, 5, 176–183 RSC.
- W. Li, J. Artz, C. Broicher, K. Junge, H. Hartmann, A. Besmehn, R. Palkovits and M. Beller, Catal. Sci. Technol., 2019, 9, 157–162 RSC.
- S. G. Oh, V. Mishra, J. K. Cho, B.-J. Kim, H. S. Kim, Y.-W. Suh, H. Lee, H. S. Park and Y. J. Kim, Catal. Commun., 2014, 43, 79–83 CrossRef CAS.
- T. Solos, N. Methiritthikul, C. Homla-or, P. Duangchan, K. Choojun and T. Sooknoi, Catal. Sci. Technol., 2022, 12, 5053–5066 RSC.
- N. Abuhafez, H. Ruffin, R. Kamaraj, C. Bruneau and R. Gramage-Doria, Catal. Sci. Technol., 2021, 11, 5772–5776 RSC.
- X. Zhu, S. Liang, S. Chen, X. Liu and R. Li, Catal. Sci. Technol., 2020, 10, 8332–8338 RSC.
- M. R. Axet, S. Conejero and I. C. Gerber, ACS Appl. Nano Mater., 2018, 1, 5885–5894 CrossRef CAS.
- P. Tomkins, E. Gebauer-Henke, W. Leitner and T. E. Mueller, ACS Catal., 2015, 5, 203–209 CrossRef CAS.
- F. Leng, I. C. Gerber, P. Lecante, S. Moldovan, M. Girleanu, M. R. Axet and P. Serp, ACS Catal., 2016, 6, 6018–6024 CrossRef CAS.
- L. Li, K. Yuan and Y. Chen, Acc. Mater. Res., 2022, 3, 584–596 CrossRef CAS.
- H. Yang, L. Wang, S. Xu, X. Hui, Y. Cao, P. He, Y. Li and H. Li, Chem. Eng. J., 2022, 431, 133863 CrossRef CAS.
- Q. Guan, C. Zhu, Y. Lin, E. I. Vovk, X. Zhou, Y. Yang, H. Yu, L. Cao, H. Wang, X. Zhang, X. Liu, M. Zhang, S. Wei, W.-X. Li and J. Lu, Nat. Catal., 2021, 4, 840–849 CrossRef CAS.
- K. Shen, L. Chen, J. Long, W. Zhong and Y. Li, ACS Catal., 2015, 5, 5264–5271 CrossRef CAS.
- Z. Li, M. Zhang, X. Dong, S. Ji, L. Zhang, L. Leng, H. Li, J. H. Horton, Q. Xu and J. Zhu, Appl. Catal., B, 2022, 313, 121462 CrossRef CAS.
- S. Zhang, C.-R. Chang, Z.-Q. Huang, J. Li, Z. Wu, Y. Ma, Z. Zhang, Y. Wang and Y. Qu, J. Am. Chem. Soc., 2016, 138, 2629–2637 CrossRef CAS PubMed.
- X. Xu, J. Luo, L. Li, D. Zhang, Y. Wang and G. Li, Green Chem., 2018, 20, 2038–2046 RSC.
- Z. Li, W. Wei, H. Li, S. Li, L. Leng, M. Zhang, J. H. Horton, D. Wang, W. Sun, C. Guo, W. Wu and J. Wang, ACS Nano, 2021, 15, 10175–10184 CrossRef PubMed.
- Z. Li, L. Leng, X. Lu, M. Zhang, Q. Xu, J. H. Horton and J. Zhu, Nano Res., 2022, 15, 3114–3121 CrossRef CAS.
- S. Yang, G. Xu, S. Shi, H. Xin, J. Gao and Z. An, Catal. Commun., 2019, 123, 105–108 CrossRef CAS.
- X. Xue, J. Liu, D. Rao, S. Xu, W. Bing, B. Wang, S. He and M. Wei, Catal. Sci. Technol., 2017, 7, 650–657 RSC.
- G. Mink and L. Horvath, React. Kinet. Catal. Lett., 1998, 65, 59–65 CrossRef CAS.
- X. Li, Z. Wang, S. Mao, Y. Chen, M. Tang, H. Li and Y. Wang, Chin. J. Chem., 2018, 36, 1191–1196 CrossRef CAS.
- Y. Song, S. Wang, W. Chen, S. Li, G. Feng, W. Wei and Y. Sun, ChemSusChem, 2020, 13, 293–297 CrossRef CAS PubMed.
- H. Yang, L. Wang, S. Xu, Y. Cao, P. He, J. Chen, Z. Zheng and H. Li, Green Energy Environ., 2022, 7, 1361–1376 CrossRef.
- Y. Huang, S. Yang, M. Jiang, J. Li, L. Peng, C. Cao and W. Song, Cryst. Growth Des., 2020, 20, 7526–7532 CrossRef CAS.
- G. N. Li, L. Li, H. Y. Yuan, H. F. Wang, H. R. Zeng and J. L. Shi, J. Colloid Interface Sci., 2017, 495, 19–26 CrossRef CAS PubMed.
- C. Chang, Y. Fu, M. Hu, C. Wang, G. Shan and L. Zhu, Appl. Catal., B, 2013, 142–143, 553–560 CrossRef CAS.
- H. Liu, P. Wu, H. Li, Z. Chen, L. Wang, X. Zeng, Y. Zhu, Y. Jiang, X. Liao, B. S. Haynes, J. Ye, C. Stampfl and J. Huang, Appl. Catal., B, 2019, 259, 118026 CrossRef CAS.
- H. Zou, J. Dai, J. Suo, R. Ettelaie, Y. Li, N. Xue, R. Wang and H. Yang, Nat. Commun., 2021, 12, 4968 CrossRef CAS PubMed.
- W. Karim, C. Spreafico, A. Kleibert, J. Gobrecht, J. VandeVondele, Y. Ekinci and J. A. van Bokhoven, Nature, 2017, 541, 68–71 CrossRef CAS PubMed.
- M. Xiong, Z. Gao and Y. Qin, ACS Catal., 2021, 11, 3159–3172 CrossRef CAS.
- B. Wang, Y. Yue, C. Jin, J. Lu, S. Wang, L. Yu, L. Guo, R. Li, Z.-T. Hu, Z. Pan, J. Zhao and X. Li, Appl. Catal., B, 2020, 272, 118944 CrossRef CAS.
- J. H. Dong, Q. Fu, H. B. Li, J. P. Xiao, B. Yang, B. S. Zhang, Y. X. Bai, T. Y. Song, R. K. Zhang, L. J. Gao, J. Cai, H. Zhang, Z. Liu and X. H. Bao, J. Am. Chem. Soc., 2020, 142, 17167–17174 CrossRef CAS PubMed.
- X. Lu, C. Guo, M. Zhang, L. Leng, J. Hugh Horton, W. Wu and Z. Li, Nano Res., 2021, 14, 4347–4355 CrossRef CAS.
- J. Y. Cao, F. G. Han, L. G. Wang, X. Y. Huang, Y. Cao, P. He, H. H. Yang, J. Q. Chen and H. Q. Li, RSC Adv., 2020, 10, 16515–16525 RSC.
- W. Jiang, J.-P. Cao, C. Zhu, M. Zhao, Z.-H. Ni, X.-Y. Zhao, J.-X. Xie, L. Zhao, Y.-P. Zhao and H.-C. Bai, Appl. Catal., B, 2022, 307, 121137 CrossRef CAS.
- C. Chaudhari, K. Sato, Y. Ikeda, K. Terada, N. Abe and K. Nagaoka, New J. Chem., 2021, 45, 9743–9746 RSC.
- U. Hartfelder, C. Kartusch, J. Sa and J. A. van Bokhoven, Catal. Commun., 2012, 27, 83–87 CrossRef CAS.
- C. Boulho, E. K. Gibson, M. I. McAllister, R. Moss and D. Lennon, Top. Catal., 2020, 63, 386–393 CrossRef CAS.
- L. J. Shorthouse, Y. Jugnet and J. C. Bertolini, Catal. Today, 2001, 70, 33–42 CrossRef CAS.
- Q. Y. Cheng, W. Li, M. H. Zhang, N. J. Guan and K. Y. Tao, Chin. J. Catal., 2001, 22, 326–330 CAS.
- B. R. Patil, A. H. Bari, D. V. Pinjari and A. B. Pandit, Ind. Eng. Chem. Res., 2017, 56, 11034–11041 CrossRef CAS.
- R. Milian, L. Liu, M. Boronat and A. Corma, J. Catal., 2018, 364, 19–30 CrossRef.
- C. Huang, X. Wang, F. Yu, B. Yuan, C. Xie and S. Yu, Res. Chem. Intermed., 2018, 44, 13–26 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *abce,
Shuang Xua,
Yan Caoa,
Ziqiang Hana and
Huiquan Li
*abce,
Shuang Xua,
Yan Caoa,
Ziqiang Hana and
Huiquan Li