DOI:
10.1039/D2RA07554G
(Paper)
RSC Adv., 2023,
13, 2213-2219
Continuous CO2 capture and methanation over Ni–Ca/Al2O3 dual functional materials†
Received
28th November 2022
, Accepted 23rd December 2022
First published on 12th January 2023
Abstract
Although Ni–Ca-based dual functional materials (DFMs) have been examined for CO2 capture and reduction with H2 (CCR) for the synthesis of CH4, their performance has generally been investigated using single reactors in an oxygen-free environment. In addition, continuous CCR operations have scarcely been investigated. In this study, continuous CCR for the production of CH4 was investigated using a double reactor system over Al2O3-supported Ni–Ca DFMs in the presence of O2. We found that a high Ca loading (Ni(10)–Ca(30)/Al2O3, 10 wt% Ni, and 30 wt% CaO) was necessary for reaction efficiency under isothermal conditions at 450 °C. The optimized DFM exhibited an excellent performance (46% CO2 conversion, 45% CH4 yield, and 97% CH4 selectivity, respectively) and good stability over 24 h. The structure and CCR activity of Ni(10)–Ca(30)/Al2O3 were studied using X-ray diffraction (XRD), scanning transmission electron microscopy (STEM), energy-dispersive X-ray spectrometry (EDS), temperature-programmed desorption (TPD), and temperature-programmed surface reaction (TPSR) techniques.
Introduction
As a major component of greenhouse gases, CO2 is a significant contributor to global warming. To reduce CO2 emissions and establish a low-carbon society, carbon capture and utilization (CCU) strategies, including CO2 capture and reduction (CCR) using H2 or hydrocarbons, provide valuable approaches.1 Among these, CO2 capture and methanation is an effective protocol for the synthesis of carbon-neutral CH4.2 Alkali and alkaline earth metals are widely used for CO2 capture, owing to their high capacity for CO2 uptake and the ability to form metal carbonates. To promote carbonate hydrogenation, transition metals, such as Ru, Pt, Fe, Ni, and Cu are utilized. Thus, dual functional materials (DFMs) consisting of alkali/earth alkaline metal salts and transition metal species have attracted considerable interest in recent years.3
CaO is generally used for CO2 capture, and Ni is particularly promising owing to its low price.4 Therefore, Ni–Ca DFMs hold promise for CO2 capture and methanation processes. For example, Yang et al.5 found that CO2 conversion and CH4 selectivity were higher with the use of Ni/CaO–Al2O3 than with that of Ni/Al2O3 as a result of surface coverage by CO2-derived species on the CaO–Al2O3 surface. Alipour et al.6 reported that the capacity for CO2 capture was improved noticeably when CaO, MgO, or BaO was loaded onto a Ni/Al2O3 catalyst. Bermejo-Lopez et al.4 investigated the effect of Ni content in DFMs on CCR using CaO as a sorbent. They established that the maximum CH4 formation (142 μmol g−1) at 520 °C was achieved using 15Ni15Ca (15 wt% Ni, and 15 wt% CaO). Sun et al.7 studied the effect of the interactions between the Ni active sites and CaO sorbents on the CCR process. They found that CO2 conversion and CH4 selectivity using 1% Ni/CeO2–CaO, obtained by physical mixing of 1% Ni/CeO2 and CaO in a mass ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, increased significantly to 62% and 84%, respectively, when the distance between the catalytic sites and sorbents was increased to a suitable scale. Although CCR over DFMs aims to utilize diluted CO2 in air or flue gas, most previous studies on Ni-based DFMs were conducted under O2-free conditions.4–7 Recently, Kuramoto and co-workers8 studied the CCR activity of Ni-based DFMs comprising Na, K, or Ca, in the presence of oxygen. They found that when the operational pressure was increased from 0.1 to 0.9 MPa using 400 ppm CO2, CH4 production over Ni–Na/Al2O3 increased from 111 to 160 μmol g−1. Although pressure elevation effectively enhances the CCR performance, mild reaction conditions are preferable from an economic viewpoint.
:1, increased significantly to 62% and 84%, respectively, when the distance between the catalytic sites and sorbents was increased to a suitable scale. Although CCR over DFMs aims to utilize diluted CO2 in air or flue gas, most previous studies on Ni-based DFMs were conducted under O2-free conditions.4–7 Recently, Kuramoto and co-workers8 studied the CCR activity of Ni-based DFMs comprising Na, K, or Ca, in the presence of oxygen. They found that when the operational pressure was increased from 0.1 to 0.9 MPa using 400 ppm CO2, CH4 production over Ni–Na/Al2O3 increased from 111 to 160 μmol g−1. Although pressure elevation effectively enhances the CCR performance, mild reaction conditions are preferable from an economic viewpoint.
Recently, we developed DFMs comprising Na-modified Pt NPs on Al2O3 as effective DFMs for CO2 capture in the presence of O2 and reduction with H2 to generate CO.9 A continuous CCR operation was achieved using a double reactor system, whereby the valves on the top and bottom were controlled to switch the gas supply. This protocol was proposed by Urakawa et al. in the first time.10 In this study, continuous CCR was investigated using Al2O3-supported Ni–Ca DFMs. We optimized the loading amounts of Ni and Ca and found that a high Ca loading (Ni = 10 wt% and CaO = 30 wt%, Ni(10)–Ca(30)/Al2O3) was optimal for CCR under isothermal conditions at 450 °C. Characterization of the high Ca-content Ni–Ca/Al2O3 DFM revealed the formation of a Ca–Al mixed oxide phase derived from the mayenite (Ca12Al14O33) structure.11 The impact of mixed oxide formation on CO2 adsorption and desorption, as well as on the hydrogenation of adsorbed CO2 was discussed.
Experimental
DFM preparation
Ni–Ca/Al2O3 DFMs were synthesized using the wetness impregnation method. The γ-Al2O3 support was obtained by calcination of boehmite (γ-AlOOH, Sasol Chemicals) at 900 °C for 3 h. An appropriate amount of aqueous Ca(NO3)2·H2O (AR 98.5%, FUJIFILM Wako Pure Chemical Corporation) was stirred for 3 h with calcined γ-Al2O3 for impregnation (Ca: 6, 20, 30, 40, and 50 wt%). The resultant suspension of Al2O3 and Ca(NO3)2 was then evaporated at 50 °C using a vacuum pump, followed by drying at 100 °C overnight. The Ca/Al2O3 support was obtained after calcination at 600 °C for 2 h. Next, Ca–Al2O3 was impregnated with Ni(NO3)2·6H2O (AR > 99%, FUJIFILM Wako Pure Chemical Corporation) (Ni: 1, 5, 10, and 15 wt%) as follows. A Ca/Al2O3 suspension in Ni precursor solution was stirred at room temperature for 30 min, and then the mixture was evaporated, dried, and calcined, as described for the Ca–Al2O3 support preparation procedure. Finally, Ni–Ca/Al2O3 DFMs with varying Ca loadings were obtained, denoted as Ni(x)–Ca(y)/Al2O3 (where x and y are the loadings of Ni and Ca, respectively).
For the synthesis of Ca12Al14O33, mixed aqueous solutions of Ca(NO3)2·H2O and γ-AlOOH in a molar ratio of 12:7 were continuously stirred at room temperature for 3 h and then evaporated at 50 °C using a vacuum pump, followed by drying at 100 °C overnight. The dried powder was calcined at 1050 °C for 2 h to obtain Ca12Al14O33. Finally, Ni/Ca12Al14O33 was obtained using the above-described procedure for the synthesis of Ni–Ca/Al2O3 DFMs.
Characterization
Powder X-ray diffraction (XRD) measurements were carried out on a Rigaku MiniFlex II/AP diffractometer with Cu Kα radiation. High-Angle Annular Dark Field Scanning Transmission Electron Microscopy (HAADF-STEM) images were recorded on a FEI Titan G2 microscope equipped with an energy dispersive X-ray (EDX) analyzer. The specific surface area was calculated using Brunauer–Emmet–Teller (BET) theory over the range P/P0 = 0.1–0.3. Temperature programmed desorption of carbon dioxide (CO2 TPD) was performed on a vertical quartz fixed-bed flow reactor connected with a mass spectrometer (Microtrac BEL Corp.). A 100 mg of sample was put on quartz wool in the middle of the reactor. The reactor set in an electric tube furnace and purged under N2 flow (95 mL min−1) for 30 min at 450 °C, and then cool down the sample to room temperature. Next, the 1% CO2/N2 (100 mL min−1) mixed gases fed into the reactor for 30 min and then flowed N2 for 15 min. After that, the TPD profile was obtained by heating the sample from 30 to 650 °C in N2 flow with elevating temperature by 10 °C min−1. Temperature program surface reaction (TPSR) was performed on the same equipment as described for TPD measurement. First, sample with 100 mg was put on quartz wool in the middle of the reactor. The reactor set in an electric tube furnace and pretreatment under 10% H2/N2 flow (100 mL min−1) for 30 min at 450 °C. The sample was cooled down to room temperature in subsequently. Next, 1% CO2/10% O2/N2 (100 mL min−1) flow was fed into the reactor for 30 min and then flowed N2 for 15 min. Finally, TPSR profile was obtained by heating the sample from 30 to 650 °C by 10 °C min−1 with 5% H2/N2 flow (100 mL min−1).
Continuous CCR operation
CCR was performed in continuous separated fixed-bed flow reactors (Fig. 1). The similar reaction system has been developed in our previous study.9 Two vertical quartz reactors were utilized (reactors A and B), and 100 mg of the sample was placed on quartz wool in the middle of the reactor. Two sides of the tube were filled with sea sand. Both reactors were equipped with the same DFM and placed in an electric tube furnace. Reactors were heated to 500 °C under N2 flow (90 mL min−1), followed by the introduction of 10% H2/N2 (100 mL min−1) for pretreatment over 20 min at 500 °C. The two timer-control 4-way valves were switched simultaneously to continuously collect the effluent gases containing uncaptured CO2 (effluent 1) and generated CO and CH4, and desorbed CO2 (effluent 2) from each outlet line. The compositions of effluents 1 and 2 were monitored employing Fourier transform infrared (FTIR) spectroscopy (JASCO FT/IR-4600) using gas cells. The background spectrum was acquired once the temperature decreased to 450 °C after H2 pretreatment followed by N2 purging. Next, 100 mL min−1 of 1% CO2/10% O2/N2 was fed to reactor A for 30 s, and then the gas feed was switched to 100 mL min−1 of pure H2 for 30 s. Reactor B underwent reverse treatment; thus, 100 mL min−1 of pure H2 was fed into reactor B for 30 s, and then the gas was switched to 100 mL min−1 of 1% CO2/10% O2/N2 for 30 s. CCR was also conducted using a single reactor for comparison. Thus, only one reactor was equipped with a DFM (100 mg) and the other reactor was filled with pure sea sand. The subsequent steps implemented for the single reactor were the same as those described for the continuous operation of the double reactor system. The CO2 capture amount (qCO2), amount of generated CO and CH4 (denoted as qCO, qCH4, respectively), selectivity for CO (SelCO), and conversion of captured CO2 (ConvcapCO2) were calculated as follows:| |
 | (1) |
| |
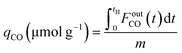 | (2) |
| |
 | (3) |
| |
 | (4) |
| |
 | (5) |
where  and
and 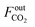 are the CO2 molar flow rates at the column inlet and outlet, respectively; FoutCO and
are the CO2 molar flow rates at the column inlet and outlet, respectively; FoutCO and  are the CO and CH4 molar flow rates at the column outlet, respectively; m is the DFM mass; ta is the duration of the adsorption stage; tH denotes the duration of the H2 reduction process.
are the CO and CH4 molar flow rates at the column outlet, respectively; m is the DFM mass; ta is the duration of the adsorption stage; tH denotes the duration of the H2 reduction process.
 |
| | Fig. 1 Diagram of the continuous-operation CCR double reactor system. | |
Results and discussion
DFMs comprising different loading amounts of Ni and Ca on Al2O3 were screened to determine the optimal composition for the continuous CCR system. First, the effect of Ca content was studied, while the Ni loading was kept constant at 10 wt% (CaO: 0, 6, 20, 30, and 40 wt%). As shown in Fig. 2, Ni(10)/Al2O3 without Ca adsorbed a moderate amount of CO2 (132 μmol g−1), but CH4 production was low (2 μmol g−1). The CO2 capture amount gradually increased from 228 to 390 μmol g−1 as the Ca content increased from 6 to 40 wt% whereas CH4 formation was the highest (153 μmol g−1) for Ni(10)–Ca(30)/Al2O3. This result illustrates the importance of optimizing the Ca loading on Ni–Ca/Al2O3 DFMs. The CO2 conversion, CH4 yield, and CH4 selectivity in CCR using Ni(10)–Ca(30)/Al2O3 were 46%, 45%, and 97%, respectively. These values were comparable with those for reported Ni–Ca DFMs (Table S1†). Next, Ni(x)–Ca(30)/Al2O3 containing varying Ni loadings (Ni: 5, 10, and 20 wt%) were also prepared and tested for continuous CCR (Fig. 2). A Ni loading of 10 wt% was found to be optimal for both CO2 adsorption and CH4 formation. Thus, the optimal loading amounts of Ca and Ni were 30 and 10 wt%, respectively, for continuous CCR operation under isothermal conditions at 450 °C. Furthermore, 30 wt% CaO was loaded onto Ni(10)/Al2O3 and then applied to continuous CCR to investigate the effect of the Ni and Ca loading sequence (Fig. 2). The amounts of CO2 captured and CH4 formed over Ca(30)–Ni(10)/Al2O3 were 217 and 64 μmol g−1, respectively. These values are lower than those for Ni(10)–Ca(30)/Al2O3 (340 and 153 μmol g−1), suggesting the importance of the loading sequence of Ni and Ca onto Al2O3. Thus, Ni(10)–Ca(30)/Al2O3 was applied in further investigations in this study.
 |
| | Fig. 2 Effluent gas compositions for continuous CCR operation over Ni(x)–Ca(y)/Al2O3 with varying Ca and Ni loadings. Conditions: 100 mg sample for each reactor, 450 °C, 100 mL min−1 of 1% CO2/10% O2/N2 for 30 s, then switched to 100 mL min−1 of H2 for 30 s. | |
Next, the comparison of continuous CCRs using double and single reactor systems using Ni(10)–Ca(30)/Al2O3 is shown in Fig. S1.† The effluent concentration profiles of uncaptured CO2, generated CH4 and CO, and desorbed CO2 during continuous CCR are shown in Fig. S1a.† The concentration of uncaptured CO2 ranged from 1100 to 2200 ppm. The highest concentration of formed CH4 was 4600 ppm, with a selectivity of 97%, and the amount of CO formed was <150 ppm. The concentration of desorbed CO2 ranged from 1300 to 2300 ppm. In the single reactor system containing Ni(10)–Ca(30)/Al2O3 (Fig. S1b†), the concentration of uncaptured CO2 (100–7500 ppm) was higher than that in the double reactors. The desorbed CO2 in the single reactor ranged from 200 to 2700 ppm, whereas the highest concentrations of CH4 (680 ppm) and CO (40 ppm) were considerably lower than those in the double reactors. These results indicate that CO2 capture and methanation efficiency were significantly improved using a double reactor system.
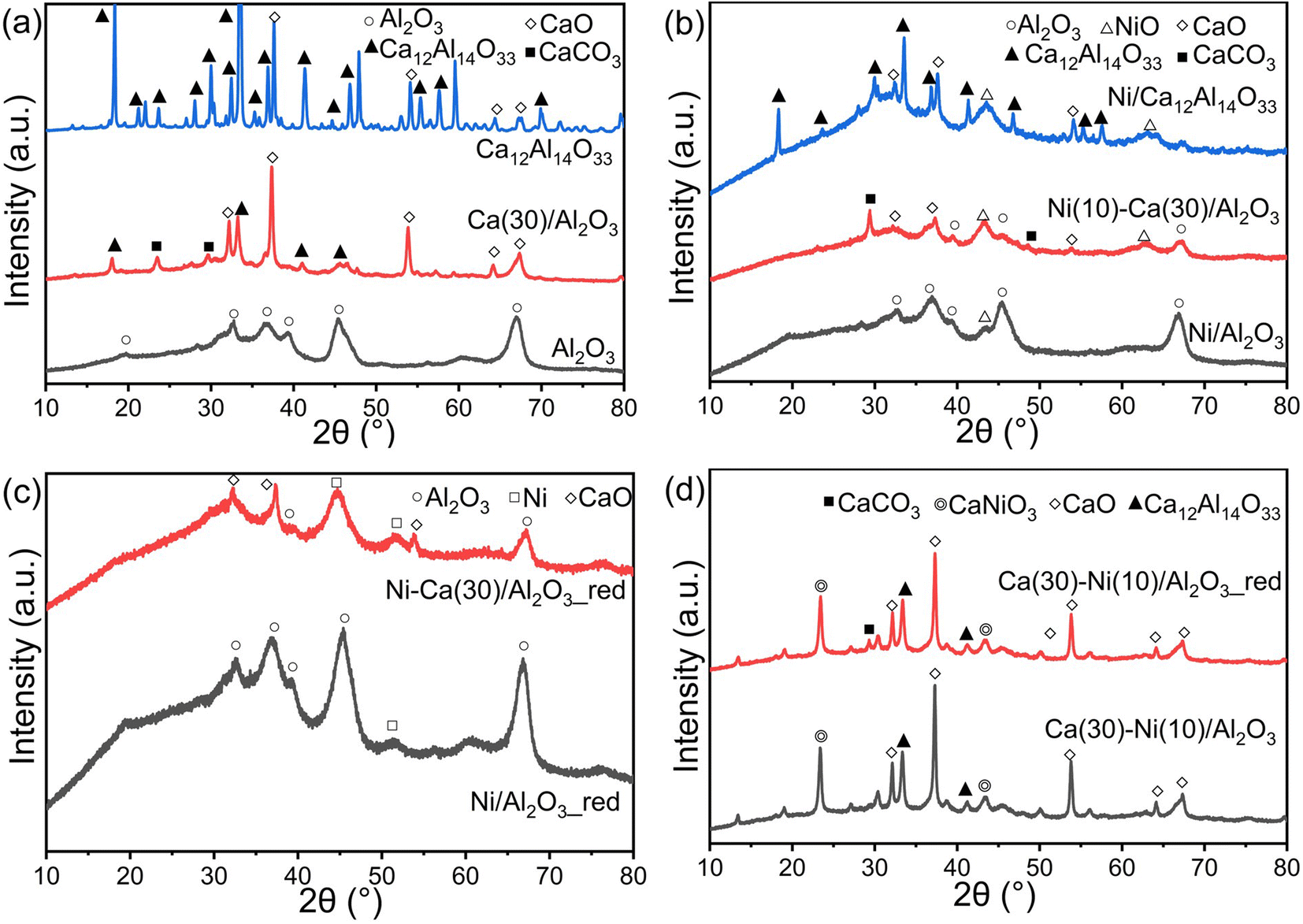
Fig. 3a shows XRD patterns of Al2O3 and Ca(30)/Al2O3 without Ni. Peaks at 19.6°, 31.9°, 37.6°, 39.5°, 45.8°, 60.5°, and 66.8° were observed for the Al2O3 support, assignable to the γ-Al2O3 phase (JCPDS No. 50-0741). For Ca(30)/Al2O3, peaks arising from the CaO (JCPDS No. 37-1497) and CaCO3 (JCPDS No. 17-0763) phases were observed. In addition, the diffraction pattern of Ca12Al14O33 (mayenite, JCPDS No. 48-1882) appeared, and the γ-Al2O3 phase almost disappeared, suggesting that γ-Al2O3 was possibly converted to Ca12Al14O33. A pure Ca12Al14O33 phase was also synthesized using calcium nitrate and boehmite as precursors in a suitable stoichiometric ratio and calcination at 1050 °C. The XRD patterns indicated that the prepared sample largely comprised Ca12Al14O33 (Fig. 3a) and a very small amount of CaO. This verified the formation of the Ca12Al14O33 structure over Ca(30)/Al2O3. Fig. 3b shows the XRD patterns of Ni-loaded Al2O3, Ca(30)/Al2O3, and Ca12Al14O33. For calcined Ni(10)/Al2O3, in addition to the Al2O3 peaks, a peak appeared at 43.3°, which was attributed to the (200) facet of NiO (JCPDS No. 47-1049). A comparison of the XRD patterns of Ni(10)/Al2O3 and Ni(10)–Ca(30)/Al2O3 revealed that the intensity of the NiO peak was higher for the latter, indicating that Ca-loaded Al2O3 promoted NiO crystal growth. Notably, the Ca12Al14O33 phase was not detected after Ni loading, whereas the CaO and CaCO3 phases were still present in Ni(10)–Ca(30)/Al2O3. For Ni(10)/Ca12Al14O33, the diffraction peaks assignable to the Ca12Al14O33 phase decreased dramatically, and a new phase did not appear. These results suggest that Ni loading likely induced the transformation of the Ca12Al14O33 phase to amorphous structures. After H2 reduction at 500 °C, the Ni metal phase (JCPDS No. 04-0850) appeared in both Ni(10)/Al2O3 and Ni(10)–Ca(30)/Al2O3 (Fig. 3c), indicating that NiO was converted to Ni metal under H2 flow. When 10 wt% of Ni was supported on Al2O3 first, followed by the introduction of 30 wt% of CaO onto Ni/Al2O3 Ca(30–Ni(10)/Al2O3), a diffraction pattern indicative of CaNiO3 (ID: mvc-3998) was observed with peaks assignable to CaO, CaCO3, and Ca12Al14O33 phases (Fig. 3d). The CaNiO3 phase was maintained after H2 treatment at 500 °C, and peaks attributable to Ni metal did not appear, suggesting that Ca–Ni composite oxides are difficult to reduce to Ni metal at CCR operation temperature (450 °C). The formation of CaNiO3 possibly led to a decrease in the amount of Ni active species, resulting in the inferior CCR performance of Ca(30)–Ni(10)/Al2O3 compared to that of Ni(10)–Ca(30)/Al2O3.
 |
| | Fig. 3 XRD patterns of (a) Al2O3, Ca(30)/Al2O3, and Ca12Al14O33 without Ni loading; (b) calcined Ni(10)/Al2O3, Ni(10)–Ca(30)/Al2O3 and Ni/Ca12Al14O33; (c) reduced Ni(10)/Al2O3 and Ni(10)–Ca(30)/Al2O3; (d) calcined and reduced Ca(30)–Ni(10)/Al2O3. | |
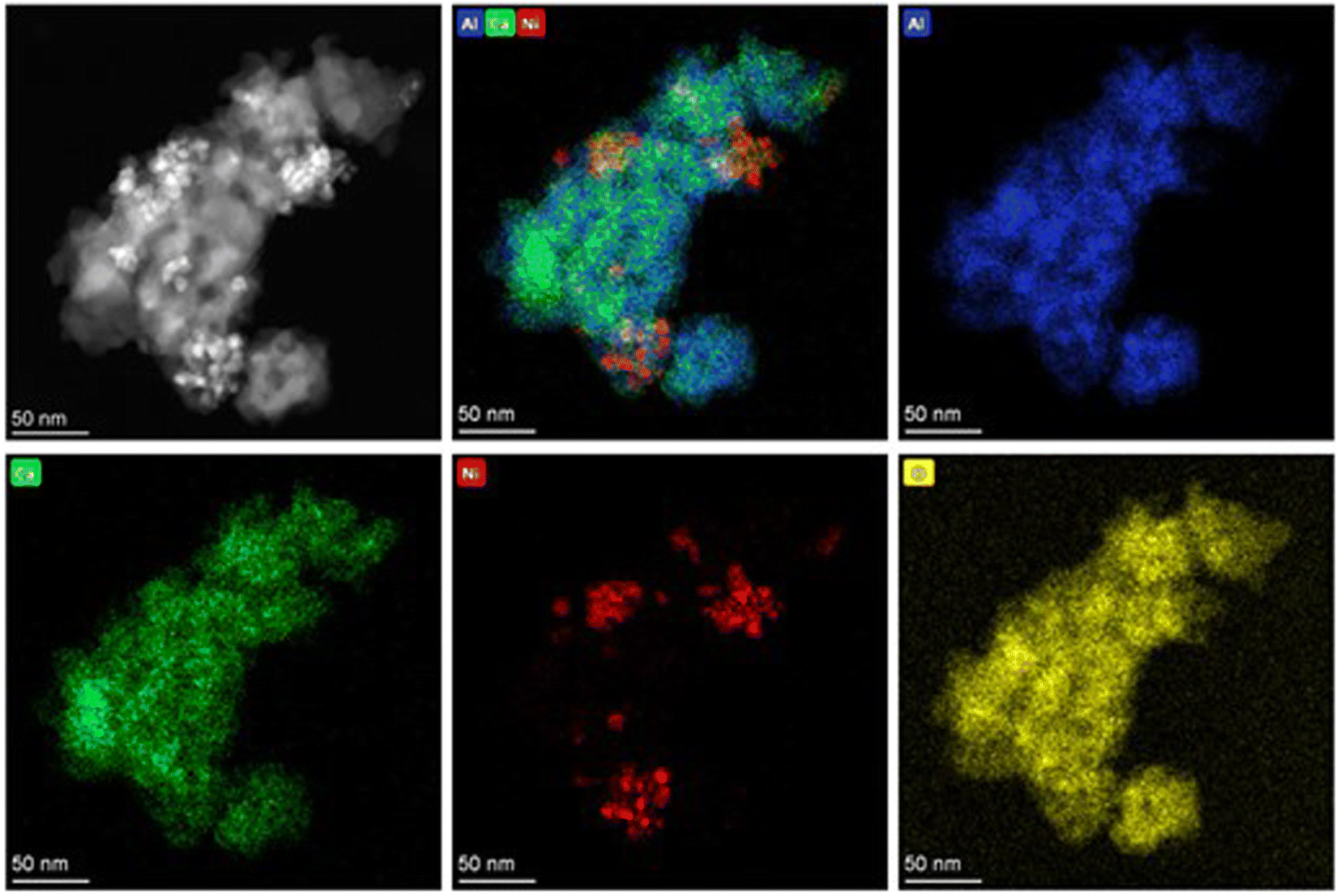
The specific surface areas of Ni(10)/Al2O3, Ni(10)–Ca(30)/Al2O3, and Ca(30)/Al2O3 were 154, 37, and 34 m2 g−1, respectively. Ni(10)–Ca(30)/Al2O3 and Ca(30)/Al2O3 have similar specific surface areas, which are appreciably lower than that of Ni(10)/Al2O3. This is possibly due to the structural transformation of γ-Al2O3 to Ca12Al14O33 phase. The similar surface area values were (20–40 m2 g−1) obtained in the solid-phase synthesis of Ca12Al14O33 in the previous literature.12 The morphology and distribution of the Ni nanoparticles on Ni(10)/Al2O3 and Ni(10)–Ca(30)/Al2O3 were characterized using STEM and EDS mapping (Fig. S2† and 4). In the case of Ni(10)/Al2O3, the Ni nanoparticles were dispersed on Al2O3 with an average size of 6.9 nm (Fig. S2†), whereas for Ni(10)–Ca(30)/Al2O3, the Ni nanoparticles were aggregated and the particle size increased (Fig. 4), which is consistent with the XRD results.
 |
| | Fig. 4 STEM images and EDS mapping of (a) Ni(10)/Al2O3 and (b) Ni(10)–Ca(30)/Al2O3. | |
CO2 TPD measurements were conducted to explore the CO2 desorption properties of Ni(10)/Al2O3, Ca(30)/Al2O3, Ni(10)–Ca(30)/Al2O3, Ca12Al14O33, and Ni(10)/Ca12Al14O33 (Fig. 5). Ni(10)/Al2O3 gave rise to two desorption peaks at 80 and 160 °C, possibly arising from the physical adsorption of CO2 onto the surface of Al2O3. In contrast, the TPD profiles for Ni(10)–Ca(30)/Al2O3 and Ca(30)/Al2O3 displayed a dominant CO2 desorption peak during the temperature increase from 400 to 600 °C. A similar desorption peak was observed for Ni(10)/Ca12Al14O33 and Ca12Al14O33, implying that this CO2 desorption was derived from adsorbed CO2 over Ca–Al mixed oxides.
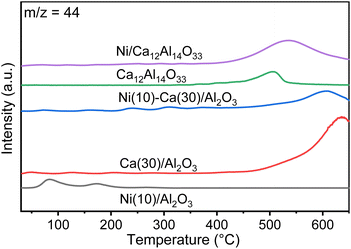 |
| | Fig. 5 CO2 TPD profiles for Ni(10)/Al2O3, Ca(30)/Al2O3, Ni(10)–Ca(30)/Al2O3, Ca12Al14O33, and Ni(10)/Ca12Al14O33. Conditions: 100 mg of DFM, N2 pretreatment at 450 °C for 30 min, followed by cooling down to room temperature and capture using 1% CO2/N2 (100 mL min−1), and then an increase in temperature to 650 °C under pure N2 (100 mL min−1). | |
TPSR measurements were conducted using Ni(10)/Al2O3, Ni(10)–Ca(30)/Al2O3, and Ni(10)/Ca12Al14O33 to obtain insights into the formation of CH4 through the reduction of adsorbed CO2 with H2 (Fig. 6). Fig. 6a shows the CO2 desorption peaks during reduction with H2. Compared to the CO2 TPD profiles (Fig. 5), the temperature for the CO2 desorption peaks over Ni(10)/Al2O3 was similar to that of the CO2 adsorption peaks, while for Ni(10)–Ca(30)/Al2O3 and Ni(10)/Ca12Al14O33, the CO2 desorption peaks were nearly absent at temperatures above 400 °C. Fig. 6b shows the CH4 (m/z = 16) formation profiles. For Ni(10)/Al2O3, the CH4 formation temperature was similar to that of CO2 desorption, implying that once the adsorbed CO2 species were desorbed, the desorbed CO2 was converted to CH4 through reduction with H2. For Ni(10)–Ca(30)/Al2O3 and Ni(10)/Ca12Al14O33, the CH4 formation peaks were clearly observed at 225, 290, and 345 °C, being lower than the temperature range for CO2 desorption in their CO2 TPD profiles. This strongly indicates that the adsorbed CO2 species were directly converted to CH4. Additional CH4 formation peaks were observed at 175 and 520 °C for Ni(10)–Ca(30)/Al2O3. The CH4 formation peak below 200 °C resulted from the activity of Ni supported on Al2O3, and the peak at 520 °C possibly appeared due to the Ni supported on calcium oxide or carbonate species. Considering the CCR operation temperature of 450 °C, the Ni species supported on amorphous Ca–Al mixed oxides most likely played the dominant role in the reaction. The performance of Ni/Ca12Al14O33 in continuous CCR verified this hypothesis, whereby high CO2 capture and CH4 formation concentrations were obtained (Fig. S3†).
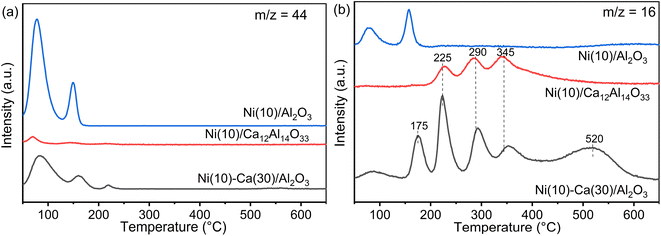 |
| | Fig. 6 TPSR profiles of (a) CO2 and (b) CH4 for Ni(10)/Al2O3, Ni(10)–Ca(30)/Al2O3, and Ni (10)/Ca12Al14O33. Conditions: 100 mg of DFM, H2 pretreatment at 450 °C for 30 min, followed by cooling to room temperature and capture under 1% CO2/10% O2/N2 (100 mL min−1), and then an increase in temperature to 650 °C under 5% H2/N2 (100 mL min−1). | |
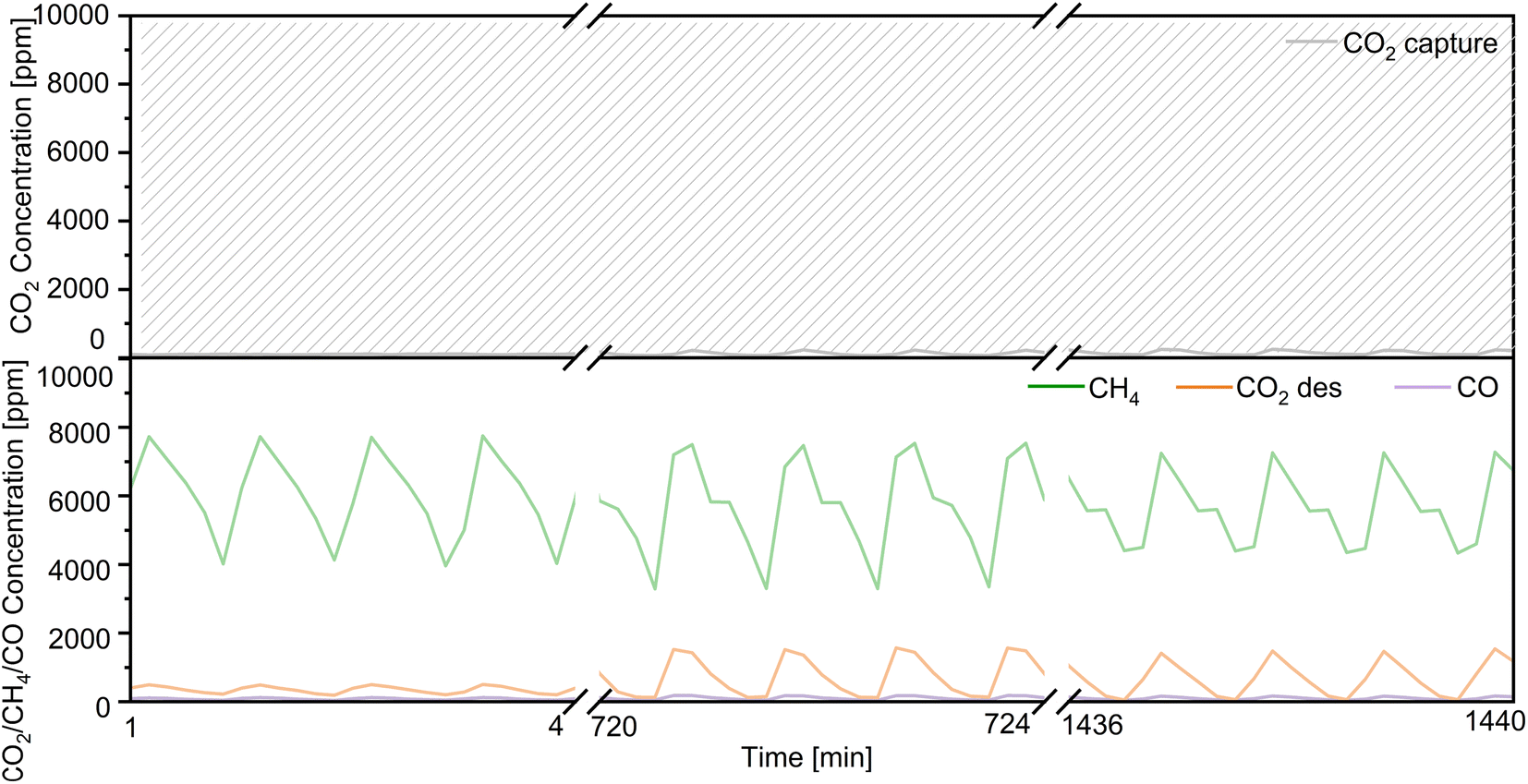
Regarding the utilization of low-concentration CO2, we decreased the flow rate of CO2 in the mixed gases from 1% CO2/10% O2/N2 to 0.1% CO2/10% O2/N2 (100 mL min−1) over Ni(10)–Ca(30)/Al2O3. As shown in Fig. 7, the concentration of uncaptured CO2 was approximately 350 ppm. The highest concentration of produced CH4 was 965 ppm; the concentrations of both the produced CO and desorbed CO2 were below 30 ppm. The conversion of captured CO2 and the selectivity for CH4 were 94% and 95%, respectively, indicating that Ni(10)–Ca(30)/Al2O3 favored the conversion of low concentrations of CO2. A long-term CCR operation experiment was also conducted using 300 mg of Ni(10)–Ca(30)/Al2O3 with flowing 1% CO2/10% O2/N2 for 24 h (Fig. 8). Initially, the produced CH4 concentration ranged from 3200 to 7800 ppm. After 24 h, the highest concentration of CH4 was 7400 ppm. The highest CO and uncaptured CO2 concentrations were below 150 and 250 ppm, respectively, for all the reaction times. The desorbed CO2 concentration ranged from 0 to 1500 ppm in the whole reaction process. Meanwhile, after reaction for 24 h, the CO2 conversion and CH4 yield were stable at 60% and 59%, respectively. These results indicate that the Ni(10)–Ca(30)/Al2O3 DFM not showed obvious deterioration in the CCR performance for at least 24 h, which exhibited a good durability. The effect of 20% water vapor on continuous CCR operation over Ni(10)–Ca(30)/Al2O3 was investigated to evaluate the potential for application to real natural gas power plant effluent (Fig. S4†). The uncaptured CO2 concentration was maintained between 1000 and 2000 ppm; however, the maximum concentration of formed CH4 decreased to 2700 ppm. Therefore, water removal from exhaust gas is necessary for real-world applications.
 |
| | Fig. 7 Effluent gas composition during continuous CCR operation over Ni(10)–Ca(30)/Al2O3 using a decreased CO2 concentration of 1000 ppm in the gas mixture. Conditions: 100 mg of DFM, 450 °C, 100 mL min−1 of 0.1% CO2/10% O2/N2 for 30 s, switched to 100 mL min−1 of H2 for 30 s. | |
 |
| | Fig. 8 Long-term continuous CCR operation using the double reactor system for 24 h. Conditions: 300 mg of Ni(10)–Ca(30)/Al2O3 for each reactor, 450 °C, 100 mL min−1 of 1% CO2/10% O2/N2 for 30 s, switched to 100 mL min−1 of H2 for 30 s. | |
Conclusion
Continuous CO2 capture and methanation reactions over Ni–Ca based DFMs were studied using double reactors in the presence of oxygen. The utilization of a double reactor system increased the amounts of CO2 captured and CH4 produced when compared to the performance of a single reactor system. A relatively high Ca loading (30 wt%) in the DFM was found to be the most effective for continuous CCR operation under isothermal conditions at 450 °C; thus, the Ni(10)–Ca(30)/Al2O3 sample displayed an excellent activity and good durability, maintaining its high CCR performance for at least 24 h. The XRD results revealed that a high Ca loading on Al2O3 induced the formation of Ca12Al14O33. Subsequent Ni loading resulted in the transformation of Ca12Al14O33 into amorphous structures, which was responsible for the favorable performance of Ni(10)–Ca(30)/Al2O3, as indicated by TPD and TPSR measurements. The order of Ca and Ni introduction also affected the structure of the Ni–Ca DFM and its CCR performance, and better results were obtained when Ni was introduced last.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This study was financially supported by KAKENHI (Grant No. JP20H02518, JP20H02775, and JP21H04626) from the Japan Society for the Promotion of Science (JSPS) and by the Japanese Ministry of Education, Culture, Sports, Science, and Technology (MEXT) within the projects “Integrated Research Consortium on Chemical Sciences (IRCCS)”. This study was also supported by the JST-CREST project JPMJCR17J3 and obtained from a project, “Moonshot Research and Development Program” (JPNP18016), commissioned by the New Energy and Industrial Technology Development Organization (NEDO). Z. M. thanks a JACI Prize for Encouraging Young Researcher. L. L. acknowledges a JSPS postdoctoral fellowship (No. P22049). S. M. acknowledges a JST SPRING fellowship (No. JPMJSP2119). The authors sincerely thank the technical division of the Institute for Catalysis (Hokkaido University) for manufacturing experimental equipment as well as the technical staff at the Open Facility of Hokkaido University for their assistance.
References
- S. Sun, H. Sun, P. T. Williams and C. Wu, Sustainable Energy Fuels, 2021, 5, 4546–4559 RSC.
- J. Lin, C. Ma, Q. Wang, Y. Xu, G. Ma, J. Wang, H. Wang, C. Dong, C. Zhang and M. Ding, Appl. Catal., B, 2019, 243, 262–272 CrossRef CAS.
- I. S. Omodolor, H. O. Otor, J. A. Andonegui, B. J. Allen and A. C. Alba-Rubio, Ind. Eng. Chem. Res., 2020, 59, 17612–17631 CrossRef CAS.
- A. Bermejo-Lopez, B. Pereda-Ayo, J. A. Gonzalez-Marcos and J. R. Gonzalez-Velasco, J. CO2 Util., 2019, 34, 576–587 CrossRef CAS.
- W. Yang, Y. Feng and W. Chu, Int. J. Chem. Eng., 2016, 2016, 1–8 CAS.
- Z. Alipour, M. Rezaei and F. Meshkani, J. Ind. Eng. Chem., 2014, 20, 2858–2863 CrossRef CAS.
- H. Sun, Y. Wang, S. Xu, A. I. Osman, G. Stenning, J. Han, S. Sun, D. Rooney, P. T. Williams, F. Wang and C. Wu, Fuel, 2021, 286, 119308 CrossRef CAS.
- F. Kosaka, Y. Liu, S. Y. Chen, T. Mochizuki, H. Takagi, A. Urakawa and K. Kuramoto, ACS Sustainable Chem. Eng., 2021, 9, 3452–3463 CrossRef CAS.
- L. Li, S. Miyazaki, S. Yasumura, K. W. Ting, T. Toyao, Z. Maeno and K. Shimizu, ACS Catal., 2022, 12, 2639–2650 CrossRef CAS.
- L. F. Bobadilla, J. M. Riesco-García, G. Penelás-Pérez and A. Urakawa, J. CO2 Util., 2016, 14, 106–111 CrossRef CAS.
- A. R. Keshavarz and M. Soleimani, Res. Chem. Intermed., 2018, 44, 1485–1503 CrossRef CAS.
- C. Sriwong, C. Phrompet, W. Tuichai, A. Karaphun, K. Kurosaki and C. Ruttanapun, Sci. Rep., 2022, 10, 11077 CrossRef PubMed.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  a,
Zen Maeno
a,
Zen Maeno




 and
and  are the CO2 molar flow rates at the column inlet and outlet, respectively; FoutCO and
are the CO2 molar flow rates at the column inlet and outlet, respectively; FoutCO and  are the CO and CH4 molar flow rates at the column outlet, respectively; m is the DFM mass; ta is the duration of the adsorption stage; tH denotes the duration of the H2 reduction process.
are the CO and CH4 molar flow rates at the column outlet, respectively; m is the DFM mass; ta is the duration of the adsorption stage; tH denotes the duration of the H2 reduction process.