DOI:
10.1039/D2RA07356K
(Paper)
RSC Adv., 2023,
13, 8779-8793
Dimerized fusion inhibitor peptides targeting the HR1–HR2 interaction of SARS-CoV-2†
Received
19th November 2022
, Accepted 27th February 2023
First published on 20th March 2023
Abstract
Membrane fusion is a critical and indispensable step in the replication cycles of viruses such as SARS-CoV-2 and human immunodeficiency virus type-1 (HIV-1). In this step, a trimer of the heptad repeat 1 (HR1) region interacts with the three HR2 regions and forms a 6-helix bundle (6-HB) structure to proceed with membrane fusion of the virus envelope and host cells. Recently, several researchers have developed potent peptidic SARS-CoV-2 fusion inhibitors based on the HR2 sequence and including some modifications. We have developed highly potent HIV-1 fusion inhibitors by dimerization of its HR2 peptides. Here, we report the development of dimerized HR2 peptides of SARS-CoV-2, which showed significantly higher antiviral activity than the corresponding monomers, suggesting that the dimerization strategy can facilitate the design of potent inhibitors of SARS-CoV-2.
Introduction
The pandemic of the novel COVID-19, which is caused by a positive-strand RNA virus SARS-CoV-2, continued in 2022. By July 2022, more than 560 million people had been infected with SARS-CoV-2 and more than 6 million people had died of the virus-induced disease. To date, more than 30 vaccines have been approved and clinically used worldwide to prevent SARS-CoV-2 infection and COVID-19 aggravation.1 Some drugs have been developed and authorized in many countries. These include Remdesivir, a repositioning inhibitor of RNA-dependent RNA polymerase (RdRp) from Ebola hemorrhagic fever, Molnupiravir, a novel SARS-CoV-2 RdRp targeting inhibitor, and Nirmatrelvir, aka Paxlovid, an inhibitor of the main protease (Mpro) of SARS-CoV-2.2–7 In order to develop drugs with different mechanisms of action and increase the repertory of drug choice, we have focused on fusion inhibitors which inhibit the interaction between heptad repeat 1 (HR1) and HR2, which plays an important role in a membrane fusion step.8,9 In the entry of SARS-CoV-2 into host cells, a spike protein, S1, on the viral surface binds to its receptor, angiotensin-converting enzyme 2 (ACE2), on the host cell surface (Fig. 1a). The S1 units of the host cell-attached viral particles are cleaved by exogeneous and membrane bound proteases such as trypsin and transmembrane protease, serine 2 (TMPRSS2), or cleaved by lysosomal protease such as cathepsin L after endocytosis. The resulting fusion peptides at N-termini of the S2 units are inserted into the host cell membrane (Fig. 1b). Then three HR2 regions interact with trimer of HR1 regions, and this complex forms a 6-helix bundle (6HB). This facilitates proceeding the membrane fusion of the viruses and the host cells.
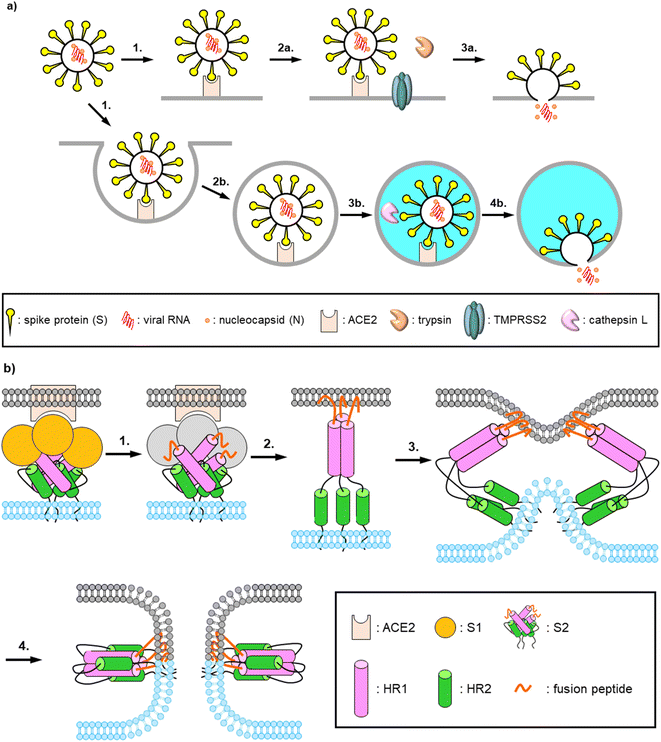 |
| | Fig. 1 Schematic representations of entry and fusion mechanisms of SARS-CoV-2. (a) Dual entry pathway of SARS-CoV-2 infection: (1) binding of spike proteins on the virus surface to the receptor, ACE2, on the host cell surface; (2a) processing of the S1 units by exogeneous and membrane bound proteases; (3a) membrane fusion and releasing viral RNA; (2b) internalization via endocytosis; (3b) endosomal acidification and processing of the S1 units by lysosomal proteases; (4b) membrane fusion and releasing virus RNA. (b) Membrane fusion mechanism of SARS-CoV-2: (1) receptor binding and processing of the S1 units by host proteases; (2) insertion of the exposed fusion peptides into the host cell membrane; (3) comformational changes of the S2 units with viral and host cell membrane approach; (4) formig 6-helix bundles and membrane fusion. | |
Recently, there have been many reports of development of fusion inhibitors based on the SARS-CoV-2 HR2 region, including the HR2 region peptide itself and its stapled peptides,10–13 and EK1, which was derived from the HR2 region of an α-coronavirus strain HCoV-OC43 and its C-terminally lipidated peptides.14,15 It is also reported that the HR2 region in the S2 unit is highly conserved among the existing mutants of SARS-CoV-2 from the alpha strain to the omicron strain (B.1.1.529).16 There is only one mutation (K1191N) of the HR2 region in the alpha strain. On the other hand, mutations of the S1 unit especially for the receptor binding domain (RBD) are frequently observed. Therefore, it seems to be difficult to develop universal anti-SARS-CoV-2 agents targeting the S1 unit. In fact, some of the developed neutralizing monoclonal antibodies dramatically decrease their potencies against the recent variants compared to that against the ancestral Wuhan strain.17–19 Therefore, it would be worth developing fusion inhibitors as universal anti-SARS-CoV-2 agents.
According to the virus classifications, human immunodeficiency virus type-1 (HIV-1) and SARS-CoV-2 both are positive-sense, single-stranded RNA viruses. These viruses have a similar fusion mechanism which includes the formation of 6-HB by interaction of a trimer of HR1 regions with three corresponding HR2 regions (Fig. 2a).8,9,15,20–23 In the past two decades, we have been developing anti-HIV-1 agents including fusion inhibitors,24–28 and have found that the C-terminal dimer of HIV-1 HR2 region (C34) based peptides demonstrated two orders of magnitude higher potency than the parent monomeric peptides (Fig. 2b).26,27 Consequently, we envisioned that this dimerization strategy can be applicable to the development of novel SARS-CoV-2 fusion inhibitors.
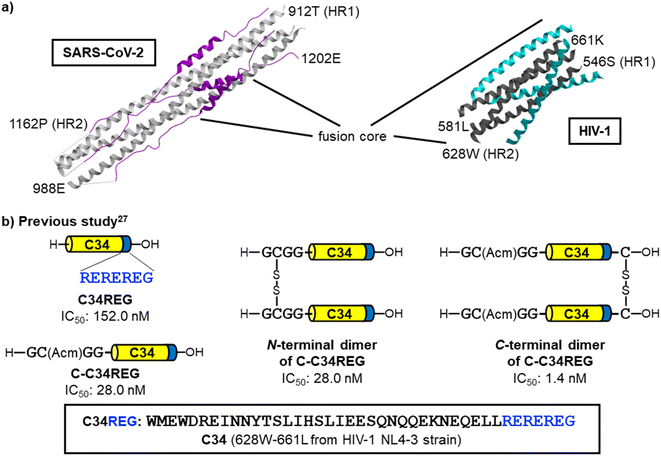 |
| | Fig. 2 Representation of the 6-helix bundle (6-HB) structures of SARS-CoV-2 and HIV-1 and the dimerized fusion inhibitors of HIV-1. (a) The SARS-CoV-2 HR1–HR2 crystal structure forming 6-HB (left, PDB code: 6LXT, white: HR1, purple: HR2) and the crystal structure of HIV-1 gp41 HR1 fragment N36 and HR2-derived fusion inhibitor SC34EK24 forming 6-HB (right, PDB code: 2Z2T, dark gray: HR1, cyan: HR2). (b) Our previous findings in the HIV-1 research;27 dimerization of HR2-derived peptides resulted highly potent fusion inhibitors. The dimerization at the C-terminus is preferable to that at the N-terminus. | |
Results and discussion
Design of fusion inhibitors
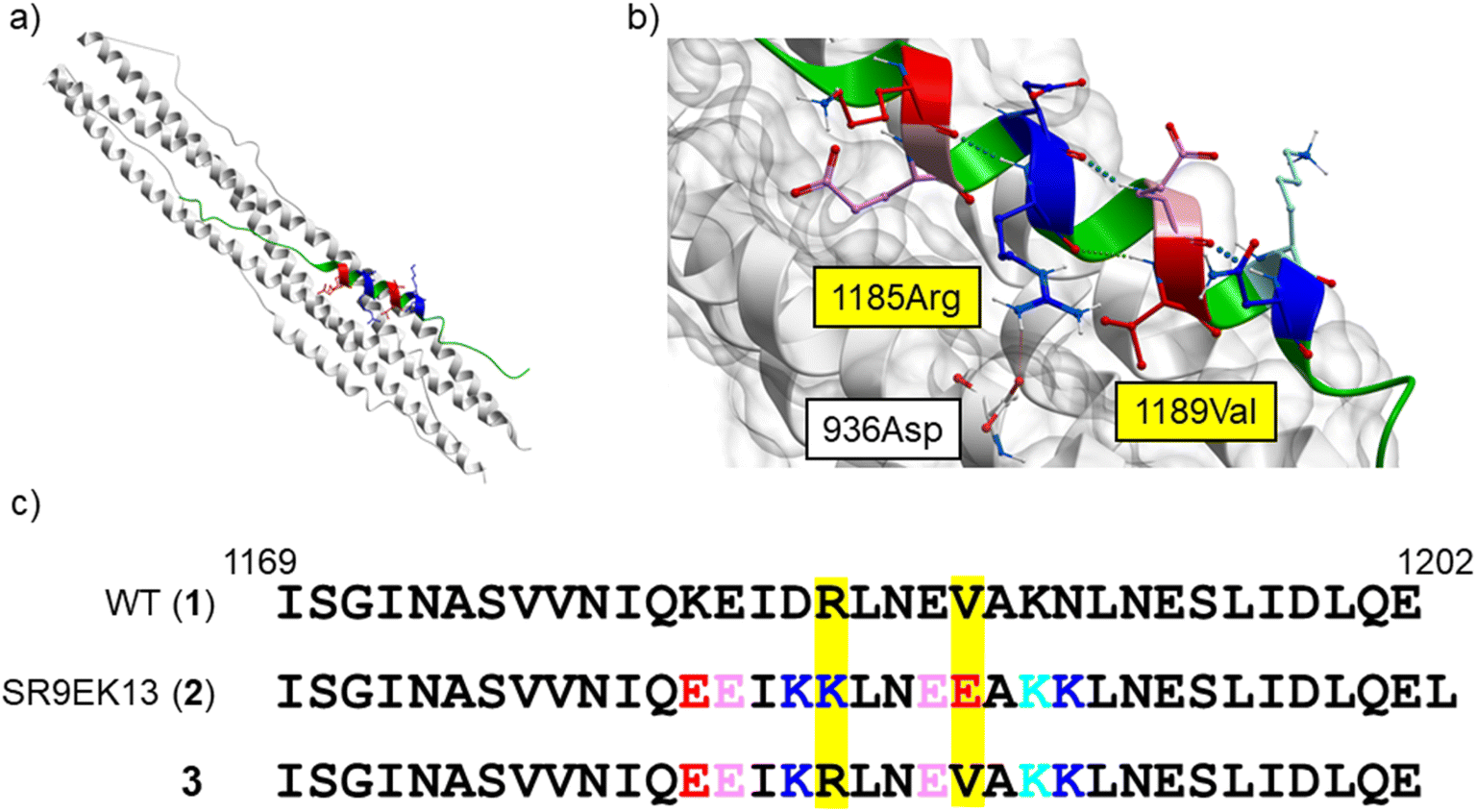
From a reported crystal structure of SARS-CoV-2 HR1–HR2, which leads to 6-HB (PDB code: 6LXT, Fig. 3),15 we extracted a 34-mer HR2 fragment (1169I–1202E) (1) as an initial sequence for the design of an inhibitor. A peptide correlated with 1, (1168D–1203L, a 36-mer), showed significant anti-SARS-CoV-2 activity in plaque reduction assays using the SARS-CoV-2 ancestral Wuhan strain/VeroE6 cells.12 Since SARS-CoV, which was reported in 2002 to cause severe acute respiratory syndrome (SARS),29,30 has an HR2 amino acid sequence identical with that of SARS-CoV-2,14,15 we also focused on a 35-mer peptide, SR9EK13 (2), which has been reported as a fusion inhibitor of SARS-CoV (Fig. 3).31 SR9EK13 (2) has 4 pairs of Glu (position i)-Lys (i + 3 position). Placement of Glu at the i position and Lys at the i + 3 or i + 4 position causes formation of a salt bridge between their sidechains, leading to a significant increase of α-helicity and aqueous solubility.24 According to the crystal structure of SARS-CoV-2 HR1–HR2, which forms 6-HB (PDB code: 6LXT, Fig. 3), 1185Arg and 1189Val of the HR2 region interact with the HR1 region. Therefore, an SR9EK13-modified 34-mer derivative, in which Lys17 and Glu21 were converted to wild type amino acids Arg and Val, respectively, with deletion of the C-terminal residue Leu35 (3), was considered. Based on these sequences, several monomeric peptides were designed.
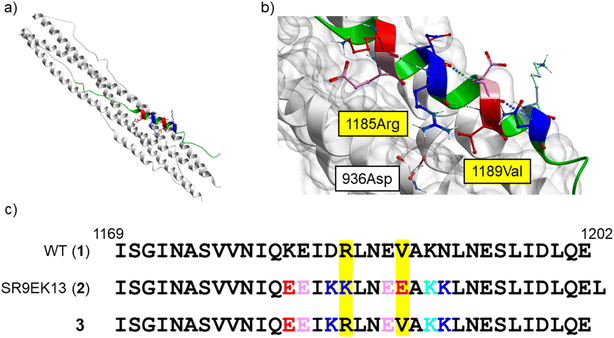 |
| | Fig. 3 (a) The SARS-CoV-2 HR1–HR2 crystal structure forming 6-HB (PDB code: 6LXT).15 (b) A focused structure around 1185Arg and 1189Val of the HR2 region. A hydrogen bond between 1185Arg (in HR2) and 936Asp (in HR1) is observed. HR1 region: white ribbon with grey surface; HR2 region: green, pink (Glu), red (mutated to Glu in SR9EK13 (2)), light blue (Lys), and blue (mutated to Lys in SR9EK13 (2)). (c) The sequences of SARS-CoV-2 HR2 (1169I–1202E) (1), SR9EK13, described with the corresponding position alignment (2) and its modified sequence (3). | |
Design of monomeric peptides
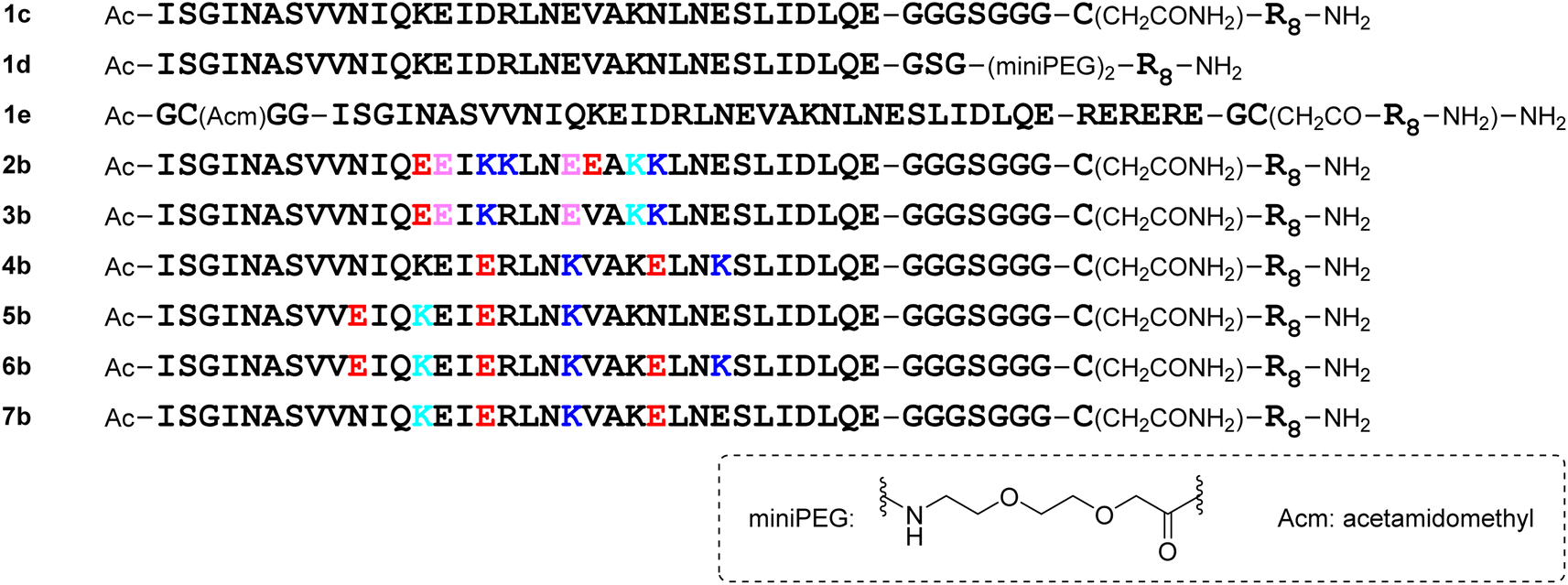
For the molecular design, all of the monomeric peptides were modified by N-terminal acetylation and C-terminal carboxyamidation. First, a peptide with the above 34-mer HR2 fragment (1169I–1202E) sequence (1a) was designed (Fig. 4). The HR2 region might have low aqueous solubility because of its hydrophobic region,11 and a hydrophilic tag sequence32 (RERERE)-attached HR2 peptide derivative (1b) was designed. As in the 34-mer HR2 fragment (1), the C-terminal residue Leu35 of a 35-mer peptide SR9EK13 (2) was deleted and its 34-mer derivative (2a) was designed. Derivative 3a, which corresponds to sequence 3, was also developed. Peptide 2a has 4 Glu–Lys pairs, and 3a has 2 such pairs. In addition, introduction of Glu–Lys pairs into peptide 1a was examined. Peptide 4a has 2 Glu–Lys pairs in positions 16–20 and 24–27 and 5a has these in positions 10–13 and 16–20. Peptide 6a possesses 3 Glu–Lys pairs in positions 10–13, 16–20 and 24–27, which includes the above Glu–Lys pairs of peptides 4a and 5a. Peptide 7a has the 2 reverse pairs: Lys–Glu in positions 13–16 and 20–24. Peptide 8a is an HCoV-OC43 HR2-derived peptide EK1, which has been reported as a pan-coronavirus fusion inhibitor peptide.14,15
 |
| | Fig. 4 Structures of designed monomeric peptides. E (red), K (blue): mutation introduction; E (pink), K (light blue): natural sequence; placement of E (i position) and K (i + 3 or i + 4 position) or placement of K (i position) and E (i + 3 or i + 4 position) leads to a salt bridge formation between their sidechains. | |
Design of octa-arginine-conjugated monomers
The addition of a cell-penetrating octa-arginyl moiety33 was attempted. Because our antiviral assay system uses TMPRSS2-expressing VeroE6 cells which allow viruses to enter via a cell surface route,34,35 cell membrane permeability might not be necessary, but the following peptides were designed to confirm their activity and increase aqueous solubility. Peptide 1c has an octa-arginyl group on the sidechain thiol group of the C-terminal Cys residue through a G3SG3-C linker attached to the C-terminus of peptide 1a (Fig. 5). Peptide 1d has an octa-arginyl group at the C-terminus through a GSG-(miniPEG)2 linker attached to peptide 1a. Peptide 1e has an octa-arginyl group on the sidechain thiol group of the C-terminal Cys residue through an RERERE-GC linker and a GC(Acm)GG sequence attached to peptide 1a. Peptides 2b, 3b, 4b, 5b, 6b and 7b have an octa-arginyl group on the sidechain thiol group of the C-terminal Cys residue through a G3SG3-C linker attached to the corresponding peptides 2a, 3a, 4a, 5a, 6a and 7a, respectively, are similar to peptide 1c.
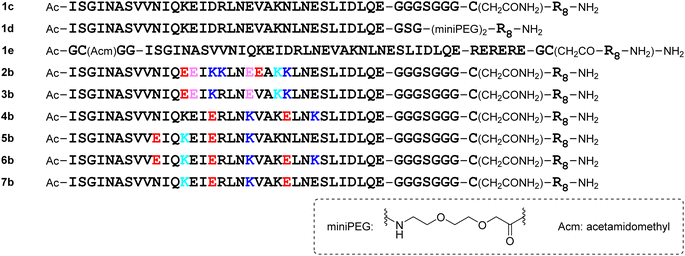 |
| | Fig. 5 Structures of octa-arginine (R8)-conjugated peptides. | |
Design of C-terminal dimers
Several dimeric derivatives of these monomeric peptides linked through a disulfide bridge at the C-terminus were constructed (Fig. 6). Dimers 1Ca and 1Cb are C-terminal disulfide dimers of peptide 1a with G3SG3-C and RERERE-GC linkers at the C-terminus, respectively. Dimer 1Cc is a C-terminal disulfide dimer of peptide 1a with an additional GC(Acm)GG sequence at the N-terminus and an RERERE-GC linker at the C-terminus. Dimers 2Ca, 2Cb and 2Cc are C-terminal disulfide dimers of peptide 2a with G3SG3-C, G3SG3-C-R4 and G3SG3-C-R8 linkers at the C-terminus, respectively. Dimers 3C, 4C, 5C, 6C and 7C are C-terminal disulfide dimers of peptides 3a, 4a, 5a, 6a and 7a, respectively with a G3SG3-C linker at the C-terminus. Dimer 8C is a C-terminal disulfide dimer of peptide 8a built through two Cys residues on the sidechain of the C-terminal Lys residue.
 |
| | Fig. 6 Structures of C-terminal dimers. | |
Design of N-terminal dimers
Several dimeric derivatives of the monomeric peptides linked through a disulfide bridge at the N-terminus were designed (Fig. 7). Dimers 1N, 2N, 3N, 6N and 7N are N-terminal disulfide dimers of peptides 1a, 2a, 3a, 6a and 7a, respectively with a C-aminohexanoic acid (Ahx) linker at the N-terminus. Dimers 4N, 5N and 8N are N-terminal disulfide dimers of peptides 4a, 5a and 8a, respectively with a CGSG linker at the N-terminus.
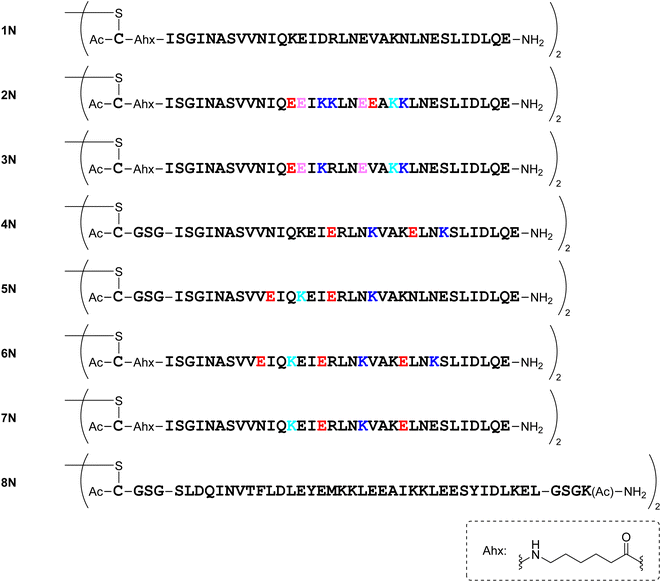 |
| | Fig. 7 Structures of N-terminal dimers. | |
Synthesis of compounds
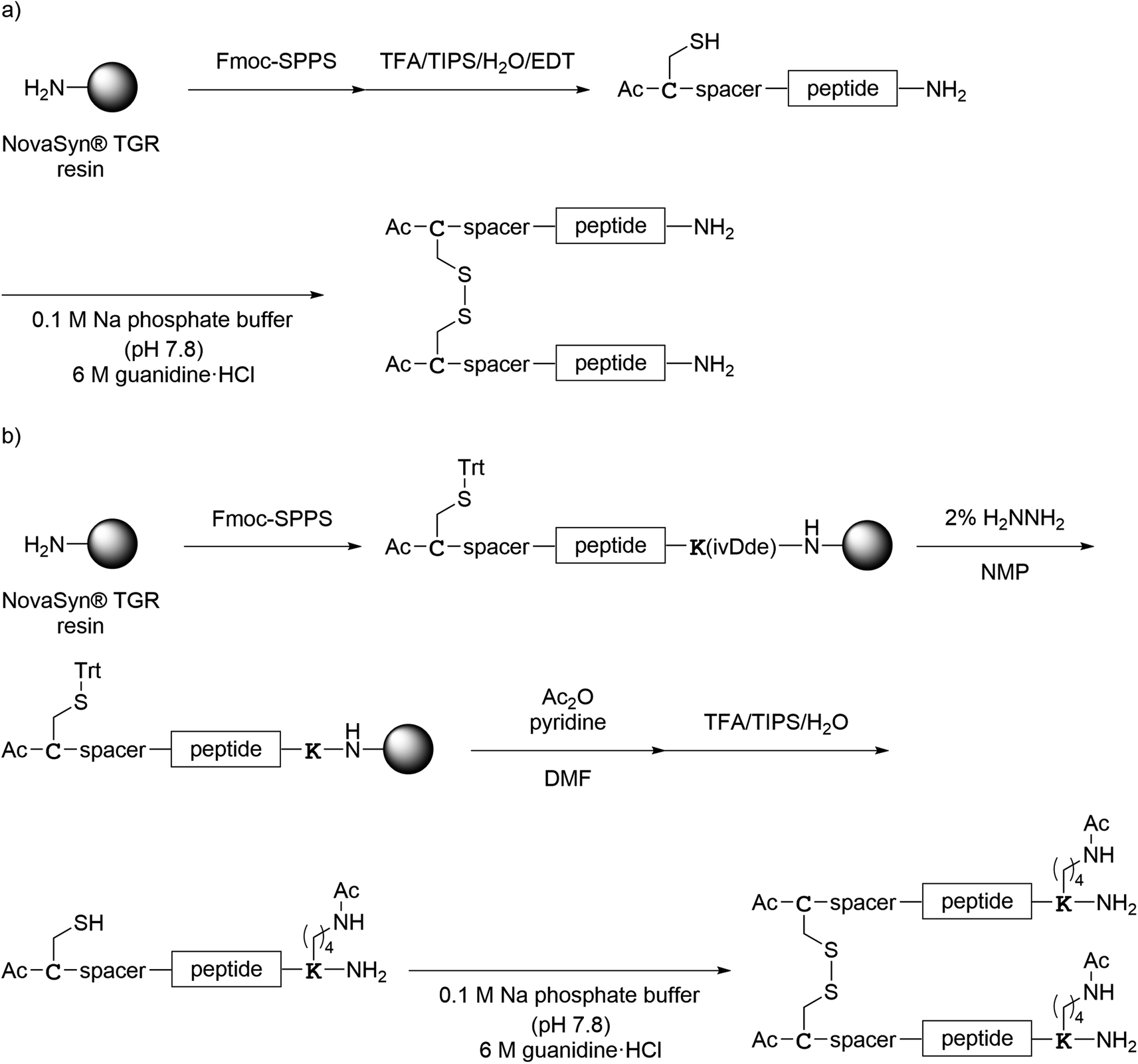
All monomer peptides and intermediate peptides for the dimer peptides were synthesized using 9-fluorenylmethyloxycarbonyl-based solid phase peptide synthesis (Fmoc-SPPS) methods. The protected peptide resins were constructed manually or with an automatic peptide synthesizer followed by acetylation of the N-terminal amino group. Treatment of the peptide resins with TFA cocktails (trifluoroacetic acid (TFA)/triisopropylsilane-(TIPS)/H2O or TFA/TIPS/H2O/ethanedithiol (EDT)) followed by purification using reverse-phase (RP)-high performance liquid chromatography (HPLC), afforded the corresponding monomers and intermediate peptides (Scheme 1). Several octa-arginine-conjugated peptides were synthesized using native chemical ligation (Scheme 2).36 All C- and N-terminal dimers were linked via disulfide bonds between two Cys sidechain thiols of the corresponding monomer peptides by air oxidation (Schemes 3 and 4).
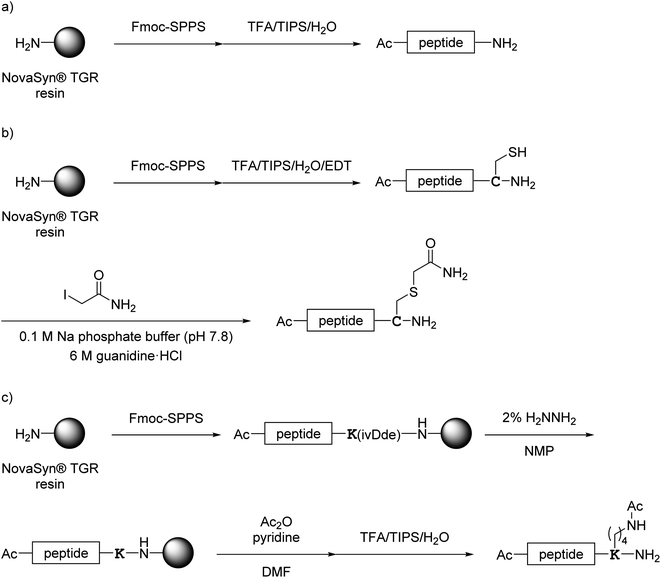 |
| | Scheme 1 Synthesis of monomer peptides shown in Fig. 4. (a) Preparation of 1a and 2a–7a; (b) preparation of 1b; (c) preparation of 8a. | |
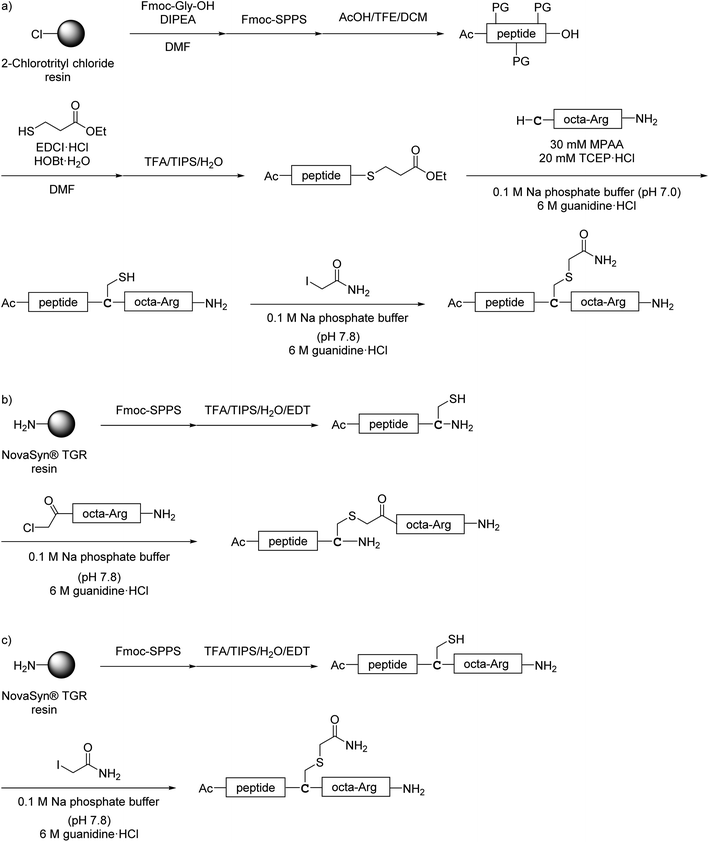 |
| | Scheme 2 Synthesis of octa-arginine-conjugated peptides shown in Fig. 5. (a) Preparation of 1c, 2b, 3b and 6b; (b) preparation of 1e; (c) preparation of 4b, 5b and 7b. Peptide 1d was synthesized as shown in Scheme 1a. | |
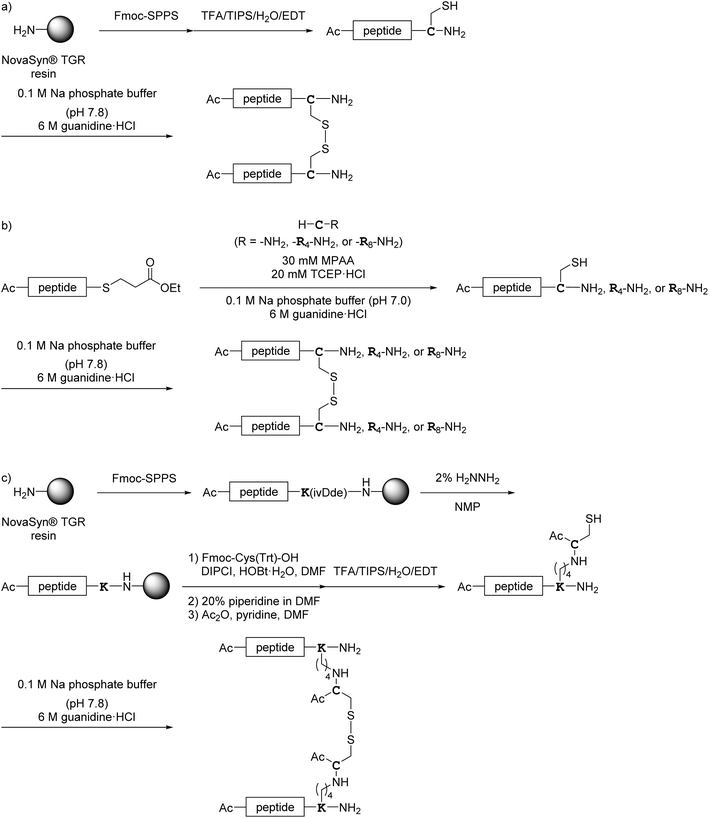 |
| | Scheme 3 Synthesis of the C-terminal dimers shown in Fig. 6. (a) Preparation of 1Ca, 1Cb, 1Cc, 4C, 5C and 7C; (b) preparation of 2Ca, 2Cb, 2Cc, 3C and 6C; (c) preparation of 8C. | |
 |
| | Scheme 4 Synthesis of the N-terminal dimers shown in Fig. 7. (a) Preparation of 1N–7N; (b) preparation of 8N. | |
Evaluation of anti-SARS-CoV-2 activity
In order to evaluate the anti-SARS-CoV-2 activity of the peptides, a plaque reduction assay37 using the SARS-CoV-2 ancestral Wuhan strain/VeroE6 cells was performed. The results of the assays are shown in Tables 1–4. Peptide 1a showed significant antiviral activity (Table 1). Peptide 1b showed slightly higher antiviral activity than peptide 1a, indicating the addition of a hydrophilic tag sequence (RERERE) has a positive effect on the increase of aqueous solubility and enhances antiviral activity. Peptide 2a showed less antiviral activity than peptide 1a. This suggests that Glu substitution for 1189Val or Lys substitution for 1185Arg is not suitable because 1185Arg and 1189Val of the HR2 region interact with the HR1 region (Fig. 3). Peptide 3a showed approximately 10 and 4 times higher antiviral activity than peptides 2a and 1a, respectively, suggesting that the return changing of Lys17 and Glu21 to wild type amino acids Arg and Val, respectively, is compatible with interaction with the HR1 region. Peptides 4a and 5a, which have 2 Glu–Lys pairs, exhibited half of the antiviral activity of peptide 3a, but both are more active than peptide 1a. Peptide 6a, which has 3 Glu–Lys pairs, showed remarkably less antiviral activity than peptides 1a, 4a and 5a. This indicates that substitution of 3 Glu–Lys pairs in a wide area at positions 10–13, 16–20 and 24–27 in peptide 6a is not appropriate because substitution of 2 Glu–Lys pairs at the same positions caused no problems in peptides 4a and 5a. Peptide 7a, which has the 2 reverse pairs: Lys–Glu in positions 13–16 and 20–24, also exhibited remarkably less antiviral activity than peptides 1a, 4a and 5a, suggesting that the introduction of reverse pairs, Lys–Glu, might be unsuitable. Peptide 8a, which is the HCoV-OC43 HR2-derived peptide, EK1 with significant broad antiviral activity against various corona viruses, exhibited significant but slightly diminished antiviral activity compared to peptide 1a. This result is consistent with previous reports from the other research groups.14
Table 1 Anti-SARS-CoV-2 activity of normal monomeric peptides (1a, 1b, 2a, 3a, 4a, 5a, 6a, 7a and 8a)
| Peptide |
IC50 (nM) |
Fold change to peptide 1ah |
| Experiment 1. Experiment 2. Experiment 3. Experiment 4. Experiment 5. Experiment 6. Experiment 7. The values of fold change to peptide 1a are represented as IC50test compound/IC501a from the same data set. |
| 1a |
206 ± 24.5a, 214b, 164c, 155d, 667e, 105f, 2530g |
1.00 |
| 1b |
68.3f |
0.648f |
| 2a |
629 ± 59.6a |
3.06a |
| 3a |
51.2b |
0.240b |
| 4a |
106d |
0.683d |
| 5a |
123d |
0.793d |
| 6a |
438c |
2.68c |
| 7a |
2280g |
0.901g |
| 8a |
152f |
1.44f |
Peptide 1c, which has an octa-arginyl group on the sidechain thiol group of the C-terminal Cys residue through a G3SG3-C linker attached to peptide 1a, showed less antiviral activity than peptide 1a, indicating that the addition of an octa-arginyl group at the C-terminus through a G3SG3-C linker has no positive effects on antiviral activity, and that nonspecific interaction of its octa-arginyl group might reduce affinity for the HR1 region (Table 2). Peptide 1d, which has an octa-arginyl group at the C-terminus through a GSG-(miniPEG)2 linker attached to peptide 1a, showed slightly higher antiviral activity than peptide 1a. Peptide 1e, which has an octa-arginyl group on the sidechain thiol group of the C-terminal Cys residue through an RERERE-GC linker attached to peptide 1a, exhibited twice the antiviral activity of peptide 1a, suggesting that the addition of an octa-arginyl group at the C-terminus through a GSG-(miniPEG)2 or RERERE-GC linker might not be responsible for its nonspecific interaction. Peptides 2b and 6b, which have an octa-arginyl group at the C-terminus through a G3SG3-C linker attached to peptides 2a and 6a, showed remarkably increased potency compared to peptides 2a and 6a, respectively, whereas peptides 3b, 4b, 5b and 7b, which have an octa-arginyl group at the C-terminus through a G3SG3-C linker attached to peptides 3a, 4a, 5a and 7a, showed decreased or retained the potency compared to peptides 3a, 4a, 5a and 7a, respectively. As a result, the addition of an octa-arginyl group at the C-terminus through a G3SG3-C linker might be effective for enhancement of the potency of only low-active parent peptides, but might cause a decrease in the potency of high-activity parent peptides. Enhancement of antiviral activity based on an increase of aqueous solubility due to the addition of an octa-arginyl group was not observed. This suggests that the positive effects of the addition of an octa-arginyl group are limited. Taken together, these data confirm that the addition of an octa-arginyl group for the introduction of the peptides into cells is not required because in this assay system, the virus enters cells via the TMPRSS2-processing cell surface pathway instead of by an endocytosis pathway.
Table 2 Anti-SARS-CoV-2 activity of octa-arginine-conjugated monomers (1c, 1d, 1e, 2b, 3b, 4b, 5b, 6b and 7b)
| Peptide |
IC50 (nM) |
Fold change to peptide 1ad |
| Experiment 4. Experiment 6. Experiment 7. The values of fold change to peptide 1a are represented as IC50test compound/IC501a from the same data set. |
| 1c |
357a |
2.30a |
| 1d |
1150c |
0.455c |
| 1e |
50.0b |
0.474b |
| 2b |
75.9b |
0.720b |
| 3b |
226a |
1.46a |
| 4b |
7480c |
2.96c |
| 5b |
25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 900c 900c |
10.2c |
| 6b |
135b |
0.868b |
| 7b |
2220c |
0.877c |
Dimer 1Ca, which is a C-terminal disulfide dimer of peptide 1a with a G3SG3-C linker at the C-terminus, exhibited slightly higher antiviral activity than the corresponding monomer (1a) although dimer 1Ca has twice the monomer peptide content (Table 3). Dimer 1Cb, which is a C-terminal disulfide dimer of peptide 1b at the C-terminus, exhibited slightly less antiviral activity than the corresponding monomer (1b) although dimer 1Cb has twice the monomer peptide content. Dimer 1Cc, which has an additional GC(Acm)GG sequence at the N-terminus of dimer 1Cb, showed less antiviral activity than peptide 1Cb. In terms of peptide contents, there was no advantage for dimerization even when using a linker of G3SG3-C (1Ca) or RERERE-GC (1Cb/1Cc) at the C-terminus. The additional GC(Acm)GG sequence at the N-terminus in dimer 1Cc was not appropriate.
Table 3 Anti-SARS-CoV-2 activity of C-terminal dimers (1Ca, 1Cb, 1Cc, 2Ca, 2Cb, 2Cc, 3C, 4C, 5C, 6C, 7C and 8C)
| Peptide |
IC50 (nM) |
Fold change to peptide 1ah |
Corresponding monomer (IC50 (nM)i) |
Fold change to the corresponding monomerj,k |
| Experiment 1. Experiment 2. Experiment 3. Experiment 4. Experiment 5. Experiment 6. Experiment 7. The values of fold change to peptide 1a are represented as IC50test compound/IC501a from the same data set. IC50 values from Tables 1 and 2. The values of fold change to the corresponding monomer are represented as IC50test compound/IC50corresponding monomer from the same data set. The values of fold change to the corresponding monomer are represented as (IC50test compound/IC501a)/(IC50corresponding monomer/IC501a) from two independent experiments. |
| 1Ca |
1080g |
0.427g |
— |
— |
| 1Cb |
93.3f |
0.885f |
1b (68.34f) |
1.37f,j |
| 1Cc |
231f |
2.19f |
— |
— |
| 2Ca |
112 ± 9.26a |
0.583a |
2a (629 ± 59.6a) |
0.191a,j |
| 2Cb |
270 ± 26.2a |
1.31a |
— |
— |
| 2Cc |
286 ± 23.6a |
1.39a |
2b (75.9f) |
1.93a,f,k |
| 3C |
753g |
0.298g |
3a (51.2b) |
1.24b,g,k |
| 4C |
155e |
0.233e |
4a (106d) |
0.341d,e,k |
| 5C |
259e |
0.389e |
5a (123d) |
0.491d,e,k |
| 6C |
135d |
0.868d |
6a (438c) |
0.324c,d,k |
| 7C |
213e |
0.320e |
7a (2280g) |
0.365e,g,k |
| 8C |
252c |
1.54c |
8a (152f) |
1.07c,f,k |
Dimers 2Ca, 2Cb and 2Cc, which are C-terminal disulfide dimers of peptide 2a with G3SG3-C, G3SG3-C-R4 and G3SG3-C-R8 linkers at the C-terminus, respectively, showed remarkably higher antiviral activity compared to the corresponding monomer (2a), but almost the same level of antiviral activity as peptide 1a. There is an evident advantage gained by dimerization, but no positive effects were observed for the addition of tetra-arginine/octa-arginine. Dimer 3C, a C-terminal disulfide dimer of peptide 3a with a G3SG3-C linker at the C-terminus, showed almost the same antiviral activity as the corresponding monomer (3a), in which the fold change to the corresponding monomer was calculated from two independent experiments in Table 3. However, 3C also clearly showed improved antiviral activity from that of the monomer 3a in the detailed evaluation (Table 5).
Dimers 4C, 5C, 6C and 7C, which are C-terminal disulfide dimers of peptides 4a, 5a, 6a and 7a, respectively with a G3SG3-C linker at the C-terminus, exhibited moderately to remarkably higher antiviral activity when compared to the corresponding monomers (4a, 5a, 6a and 7a), respectively, and peptide 1a. There is also an obvious advantage for dimerization. Dimer 8C, which is a C-terminal disulfide dimer of peptide 8a through two Cys residues on the sidechain of the C-terminal Lys residue, exhibited almost the same antiviral activity as the corresponding monomer (8a), but less antiviral activity than peptide 1a.
All of the dimers (2Ca, 3C, 4C, 5C, 6C and 7C) have significantly higher antiviral activity than the corresponding monomers (2a, 3a, 4a, 5a, 6a and 7a), respectively. Dimerized HR2 peptides at the C-termini with introduction of Glu–Lys pairs into peptide 1a clearly have effectiveness of dimerization leading to an increase in antiviral activity, although dimers of a wild type including a different corona virus HCoV-OC43 have no significant effectiveness following dimerization.
Dimers 1N, 2N and 7N, which are N-terminal disulfide dimers of peptides 1a, 2a and 7a, respectively with a C-aminohexanoic acid linker at the N-terminus, showed moderately higher antiviral activity than the corresponding monomers (1a, 2a and 7a), respectively (Table 4). Although slightly positive effects of the dimerization were observed, especially for dimers 2N and 7N, these dimers are unsuitable as lead compounds because the antiviral activity of the monomer peptides are relatively low. In addition, peptide contents of the dimers are twice that of the corresponding monomers. Dimers 3N, 4N, 5N and 6N, which are N-terminal disulfide dimers of peptides 3a, 4a, 5a and 6a, respectively with a C-aminohexanoic acid linker or a CGSG linker at the N-terminus, showed less or almost the same levels of antiviral activity compared to the corresponding monomers (3a, 4a, 5a and 6a), respectively. Considering the peptide contents of the dimers, the antiviral activities of these dimers are much lower, suggesting that dimerization offers no advantage. Dimer 8N, which is an N-terminal disulfide dimer of peptide 8a with a CGSG linker at the N-terminus, exhibited remarkably higher antiviral activity than the corresponding monomer (8a).
Table 4 Anti-SARS-CoV-2 activity of N-terminal dimers (1N, 2N, 3N, 4N, 5N, 6N, 7N and 8N)
| Peptide |
IC50 (nM) |
Fold change to peptide 1ag |
Corresponding monomer (IC50 (nM)h) |
Fold change to the corresponding monomeri,j |
| Experiment 1. Experiment 2. Experiment 3. Experiment 4. Experiment 6. Experiment 7. The values of fold change to peptide 1a are represented as IC50test compound/IC501a from the same data set. IC50 values from Tables 1 and 2. The values of fold change to the corresponding monomer are represented as IC50test compound/IC50corresponding monomer from the same data set. The values of fold change to the corresponding monomer are represented as (IC50test compound/IC501a)/(IC50corresponding monomer/IC501a) from two independent experiments. |
| 1N |
35.00e |
0.332e |
1a (105e) |
0.332e,i |
| 2N |
125 ± 9.78a |
0.608a |
2a (629 ± 59.6a) |
0.199a,i |
| 3N |
528b |
2.47b |
3a (51.2b) |
10.3b,i |
| 4N |
267d |
1.72d |
4a (106d) |
2.52d,i |
| 5N |
155d |
0.998d |
5a (123d) |
1.26d,i |
| 6N |
459d |
2.96d |
6a (438c) |
1.10c,d,j |
| 7N |
800f |
0.316f |
7a (2280f) |
0.351f,i |
| 8N |
41.3b |
0.193b |
8a (152e) |
0.134b,e,j |
HR2 peptides dimerized at the N-termini might be unsuitable as lead compounds with the exception of dimer 8N, which was derived from HCoV-OC43, a different corona virus.
CD analysis
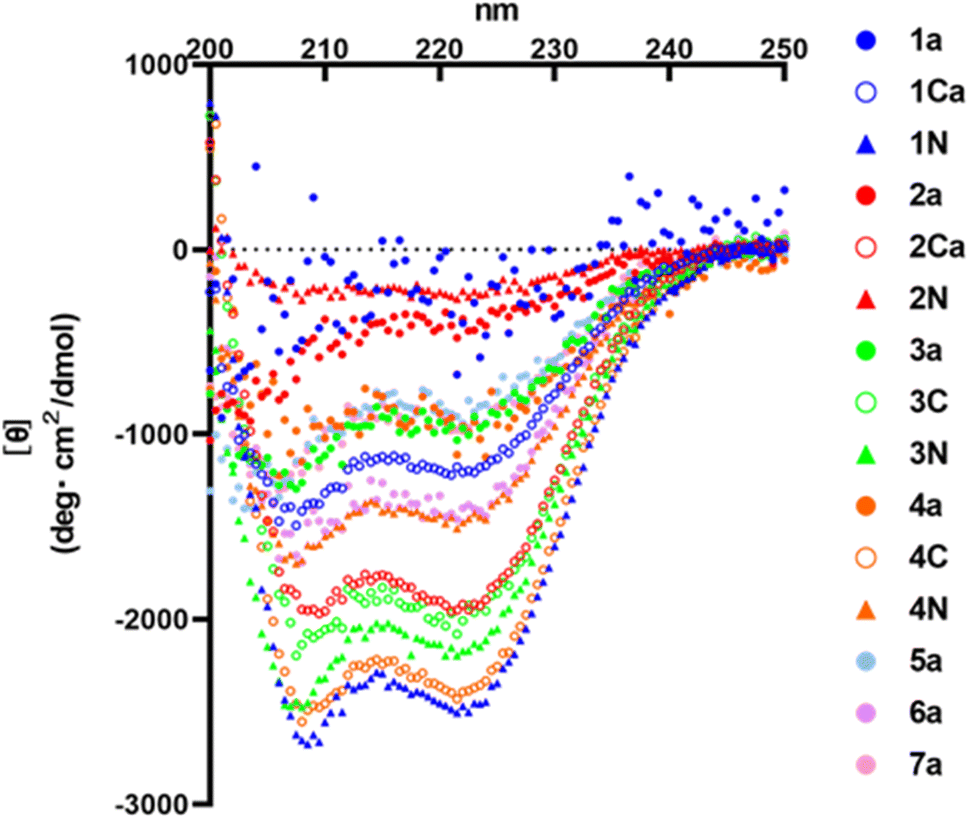
HR2 is an α-helical region. Therefore, α-helicity of HR2 fragment derivatives might have some effects on inhibitory activity. In this study, circular dichroism (CD) spectra of representative monomer peptides (1a, 2a, 3a, 4a, 5a, 6a and 7a), C-terminal dimers (1Ca, 2Ca, 3C and 4C) and N-terminal dimers (1N, 2N, 3N and 4N) were measured (Fig. 8). It was found that an initial lead monomer peptide (1a) or a peptide (2a) containing 4 Glu–Lys pairs did not show an α-helical structure. Introduction of Glu–Lys pairs or introduction of 4 pairs did not in any case induce helical formation, suggesting 4 pairs might be excessive. The other monomer peptides (3a, 4a, 5a and 7a) showed α-helical structures although their α-helicities were not very high. A 3 Glu–Lys pairs-containing monomer (6a) showed an α-helical structure of moderate strength, suggesting that the introduction of 3 Glu–Lys pairs into broad regions might be effective for enhancement of α-helicity. The C-terminal dimer (1Ca) exhibited an α-helical structure of moderate strength, and an N-terminal dimer (1N) exhibited a remarkably more pronounced α-helical structure than 1Ca, although the corresponding monomer (1a) did not show any α-helical structure. On the contrary, the C-terminal dimer (4C) showed a remarkably higher α-helical structure than 4N, which exhibited a moderately strong α-helical structure, although the corresponding monomer (4a) showed a weak α-helical structure. The C-terminal dimer (2Ca) showed a relatively high α-helical structure, although the corresponding monomer (2a) or its N-terminal dimer (2N) failed to show an α-helical structure. The C- and N-terminal dimers (3C and 3N) showed conspicuously high α-helical structures, although the corresponding monomer (3a) showed a weak α-helical structure. In general, the α-helicities of the dimers were significantly higher than those of the corresponding monomers. Taken together, introduction of 2 or 3 Glu–Lys pairs and dimerization at the C- or N-terminus generally caused an efficient increase in the α-helicity of the HR2 peptides.
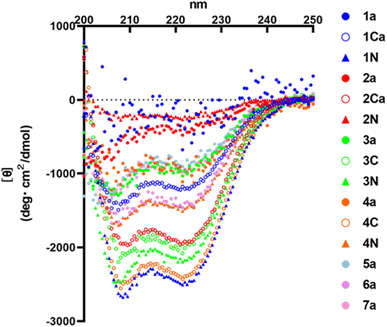 |
| | Fig. 8 CD spectra of the HR2 peptides. CD spectra were measured at 25 °C in PBS (50 mM Na phosphate buffer, 150 mM NaCl, pH 7.2). | |
In terms of the antiviral activity, peptide 3a is the most suitable monomer because it has potent antiviral activity and a weak but clear α-helical structure as a monomer, and amongst the synthesized peptides the C-terminal dimers (3C and 4C) are the most suitable derivatives. According to the previous report, HR2-derived peptides alone did not show α-helical structures, while these peptides showed α-helical structures in the presence of HR1-derived peptide.11 Therefore, it was considered that the HR2-derived peptides reported here would also form α-helical structures when these peptides bind to the HR1 region of SARS-CoV-2 to show their inhibitory activity.
Detailed evaluation of anti-SARS-CoV-2 activity of the selected peptides
Peptide 3a, the most suitable monomer, and the C-terminal dimers (3C and 4C), the most suitable derivatives, were statistically analyzed (Table 5). These activities were verified.
Table 5 Anti-SARS-CoV-2 activity of the hit peptides (1a, 3a, 3C and 4C)a
| Peptide |
IC50 (nM) |
Fold change to peptide 1a |
| The IC50 values represent the average IC50 value ± SEM (nM) from three independent experiments. |
| 1a |
1700 ± 433 |
1.00 |
| 3a |
955 ± 194 |
0.563 |
| 3C |
472 ± 189 |
0.278 |
| 4C |
575 ± 212 |
0.339 |
Conclusions
Development of new drugs with different mechanisms of action starting from existing drugs is necessary for increasing the repertory of therapeutic choice. In this study, we focused on and developed fusion inhibitors, which function at the steps of viral entry and fusion into cells. To date, we have developed HIV-1 fusion inhibitors based on dimerization of HR2 region peptides accompanied by introduction of Glu–Lys pairs.24 Since HIV-1 and SARS-CoV-2 have similar fusion mechanisms involving HR1-HR2 interactions,8,9,15,20–23 this strategy was applied to the development of inhibitors of SARS-CoV-2. Initially, an HR2 peptide (1a) was characterized as an inhibitor with significant antiviral activity against SARS-CoV-2 in spite the absence of significant α-helicity. Introduction of Glu–Lys pairs into derivatives of HR2 peptides significantly increased the antiviral activity and α-helicity in peptides 3a, 4a, 5a and 7a. Peptide 3a was determined to be the most suitable monomer. Effects of the addition of an octa-arginyl group on enhancement of antiviral activity might be limited for original peptides with low potency. There were no positive effects on original peptides with relatively high potency. Since an ancestral Wuhan strain of SARS-CoV-2 enters VeroE6 cells via the TMPRSS2-processing cell surface pathway instead of the endocytosis pathway in this assay system, the addition of an octa-arginyl group for the penetration of the peptides into cells is not required. However, it was recently reported that the SARS-CoV-2 omicron (B.1.529) strain prefers infection via an endosomal route rather than a cell surface route.38 Therefore, peptides modified with an octa-arginyl group might be useful for inhibition against universal strains of SARS-CoV-2.
HR2 peptides dimerized at the C-termini with introduction of Glu–Lys pairs into peptide 1a show the clear effectiveness of dimerization with an increase in antiviral activity. This phenomenon was observed in all of the dimers (2Ca, 3C, 4C, 5C, 6C and 7C), although dimers of a wild type including a different corona virus HCoV-OC43 showed no significant effectiveness of dimerization. Dimerized HR2 peptides at the N-termini might be unsuitable as lead compounds with the exception of dimer 8N derived from a different corona virus HCoV-OC43. In CD analysis, dimerization at the C- or N-terminus of monomer peptides with introduction of 2 or 3 Glu–Lys pairs caused an efficient increase in the α-helicity of HR2 peptides. Taken together, our data show that dimerization at the C-terminus is superior to dimerization at the N-terminus in case of HR2 peptide derivatives endowed with relatively high potency. Dimer 3C and 4C were the most suitable derivatives. In the present study, VeroE6 cells were used for plaque reduction assays, and living cells can be clearly observed 2 days after treatment with the synthesized HR2 peptides at concentrations below 100 μM. This suggests that these peptides might not have significant cytotoxicity, although precise investigation on cytotoxicity is required.
In our previous study on HIV-1 fusion inhibitors, the C-terminal dimer of the HIV-1 HR2 region peptide has significantly more potent antiviral activity than the N-terminal dimer and the corresponding monomer peptide.27 This phenomenon was also observed with SARS-CoV-2. It is confirmed that the C-terminal dimerization strategy might be useful for the design of inhibitors against viruses with fusion mechanism involving HR1-HR2 interactions.8,9,15,20–23 The obtained dimers (3C and 4C) are useful leads for antiviral agents for therapy of COVID-19. To date, new corona virus-caused infectious diseases, including SARS29,30 and middle east respiratory syndrome (MERS)39 have been repeated. Although COVID-19 may be stamped out, expansion of new infectious diseases caused by different corona viruses might be possibly of concern. Therefore, it is possible to apply the present strategy to the design of inhibitors against these diseases.
Experimental section
Compound synthesis
All materials and methods including general information, details of synthesis and characterization of the compounds are available in the ESI.† The synthetic methods for compounds are described in Schemes 1–4. The purity of the final compounds as measured by analytical HPLC was >95%.
Antiviral assay (plaque reduction assay)
One hundred plaque-forming units of SARS-CoV-2 (OMC-510)37 were mixed with peptides (or DMSO) in 200 μL of Dulbecco's Modified Eagle's Medium and the mixture was subjected to plaque-forming assays. VeroE6/TMPRSS2 cells35 were seeded in 24-well plates (1.5 × 105 cells per well) at 1 day before the assay and incubated with the virus/peptide mixture at 37 °C for 2 h. After removal of the mixture, cells were cultured with Eagle's minimum essential medium containing 1% methylcellulose 4000 cp (Fujifilm, Wako Pure Chemical Corporation, Japan) and 2% fetal bovine serum. Two days after infection, the cells were fixed with formaldehyde. Then, the plaques were visualized by staining with 1% crystal violet and counted.
CD spectroscopy
CD measurements were performed with a J-720 circular dichroism spectropolarimeter equipped with a thermo-regulator (JASCO, Japan). The wavelength dependence of molar ellipticity [θ] at 25 °C was monitored from 200 to 250 nm. The peptides were dissolved in PBS (50 mM sodium phosphate, 150 mM NaCl, pH 7.2) and measured at a concentration of 100 μM with the exception of peptides 1N (25 μM), 4a (50 μM) and 4C (50 μM). The spectral data were corrected by Spectra Manager v1.54.03 [Build 1] (JASCO) and analyzed by Microsoft Excel, and the Figure was prepared by GraphPad Prism 9.
Author contributions
Conceptualization, K. T. and H. T.; methodology, K. T. and Y. S.; validation, K. T. and Y. S.; formal analysis, K. T. and K. O.; investigation, K. T., K. O., Y. M, T. I., K. S., T. K. and A. E.; writing—original draft preparation, K. T.; writing—review and editing, H. T.; supervision, Y. S., T. N. and H. T. All authors have read and agreed to the published version of the manuscript.
Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
The authors deeply thank Dr Ryo Masuda and Prof. Takaki Koide (Waseda Univ.) for measuring CD spectra. This work was supported in part by JSPS KAKENHI Grant Numbers 20H03362 (H. T.) and 22K15243 (K. T.); Research Program on HIV/AIDS, Japan Agency for Medical Research and Development (AMED) JP21am0101098 and JP22ama121043 (Platform Project for Supporting Drug Discovery and Life Science Research, BINDS) (H. T.). This study was also supported financially by the research grant of Astellas Foundation for Research on Metabolic Disorders and Chugai Foundation for Innovative Drug Discovery Science: C-FINDs (K. T.). This research is based on the Cooperative Research Project of Research Center for Biomedical Engineering.
Notes and references
- World Health Organization, COVID-19 vaccine tracker and landscape, https://www.who.int/publications/m/item/draft-landscape-of-covid-19-candidate-vaccines, accessed on 2022-7-6 Search PubMed.
- J. Pardo, A. M. Shukla, G. Chamarthi and A. Gupte, Drugs Context, 2020, 9 DOI:10.7573/dic.2020-4-14.
- U.S. Food & Drug Administration, FDA approves first treatment for COVID-19, https://www.fda.gov/news-events/pressannouncements/fda-approves-first-treatment-covid-19, accessed on 2021-1-21 Search PubMed.
- Merck and Ridgeback's Investigational Oral Antiviral Molnupiravir Reduced the Risk of Hospitalization or Death by Approximately 50% Compared to Placebo for Patients with Mild or Moderate COVID-19 in Positive Interim Analysis of Phase 3 Study, Press Release from Merck & Co., Inc., 2021, accessed on 2022-5-12 Search PubMed.
- World Health Organization, WHO updates its treatment guidelines to include molnupiravir, March 3, 2022, https://www.who.int/news/item/03-03-2022-molnupiravir, accessed on 2022-5-12.
- J. A. McIntosh, T. Benkovics, S. M. Silverman, M. A. Huffman, J. Kong, P. E. Maligres, T. Itoh, H. Yang, D. Verma, W. Pan, H.-I. Ho, J. Vroom, A. M. Knight, J. A. Hurtak, A. Klapars, A. Fryszkowska, W. J. Morris, N. A. Strotman, G. S. Murphy, K. M. Maloney and P. S. Fier, ACS Cent. Sci., 2021, 7, 1980–1985 CrossRef CAS PubMed.
- D. R. Owen, C. M. N. Allerton, A. S. Anderson, L. Aschenbrenner, M. Avery, S. Berritt, B. Boras, R. D. Cardin, A. Carlo, K. J. Coffman, A. Dantonio, L. Di, H. Eng, R. Ferre, K. S. Gajiwala, S. A. Gibson, S. E. Greasley, B. L. Hurst, E. P. Kadar, A. S. Kalgutkar, J. C. Lee, J. Lee, W. Liu, S. W. Mason, S. Noell, J. J. Novak, R. S. Obach, K. Ogilvie, N. C. Patel, M. Pettersson, D. K. Rai, M. R. Reese, M. F. Sammons, J. G. Sathish, R. S. P. Singh, C. M. Steppan, A. E. Stewart, J. B. Tuttle, L. Updyke, P. R. Verhoest, L. Wei, Q. Yang and Y. Zhu, Science, 2021, 374, 1586–1593 CrossRef CAS PubMed.
- T. Tang, M. Bidon, J. A. Jaimes, G. R. Whittaker and S. Daniela, Antivir. Res., 2020, 178, 104792 CrossRef CAS PubMed.
- C. B. Jackson, M. Farzan, B. Chen and H. Choe, Nat. Rev. Mol. Cell Biol., 2022, 23, 3–20 CrossRef CAS PubMed.
- Y. Zhu, D. Yu, H. Yan, H. Chong and Y. He, J. Virol., 2020, 94, e00635-20 CrossRef PubMed.
- D. Yu, Y. Zhu, T. Jiao, T. Wu, X. Xiao, B. Qin, H. Chong, X. Lei, L. Ren, S. Cui, J. Wang and Y. He, Emerg. Microbes Infect., 2021, 10, 1227–1240 CrossRef CAS PubMed.
- M. Zheng, W. Cong, H. Peng, J. Qing, H. Shen, Y. Tang, C. Geng, S. Chen, Y. Zou, W.-D. Zhang, H.-G. Hu and X. Li, J. Med. Chem., 2021, 64, 17486–17495 CrossRef CAS PubMed.
- A. Dahal, J. J. Sonju, K. G. Kousoulas and S. D. Jois, J. Pept. Sci., 2022, 114, e24245 CAS.
- S. Xia, Y. Zhu, M. Liu, Q. Lan, W. Xu, Y. Wu, T. Ying, S. Liu, Z. Shi, S. Jiang and L. Lu, Cell. Mol. Immunol., 2020, 17, 765–767 CrossRef CAS PubMed.
- S. Xia, M. Liu, C. Wang, W. Xu, Q. Lan, S. Feng, F. Qi, L. Bao, L. Du, S. Liu, C. Qin, F. Sun, Z. Shi, Y. Zhu, S. Jiang and L. Lu, Cell Res., 2020, 30, 343–355 CrossRef CAS PubMed.
- V. Papanikolaou, A. Chrysovergis, V. Ragos, E. Tsiambas, S. Katsinis, A. Manoli, S. Papouliakos, D. Roukas, S. Mastronikolis, D. Peschos, A. Batistatou, E. Kyrodimos and N. Mastronikolis, Gene, 2022, 814, 146134 CrossRef CAS PubMed.
- E. Takashita, N. Kinoshita, S. Yamayoshi, Y. Sakai-Tagawa, S. Fujisaki, M. Ito, K. Iwatsuki-Horimoto, P. Halfmann, S. Watanabe, K. Maeda, M. Imai, H. Mitsuya, N. Ohmagari, M. Takeda, H. Hasegawa and Y. Kawaoka, N. Engl. J. Med., 2022, 386, 1475–1477 CrossRef PubMed.
- Y. Cao, A. Yisimayi, F. Jian, W. Song, T. Xiao, L. Wang, S. Du, J. Wang, Q. Li, X. Chen, Y. Yu, P. Wang, Z. Zhang, P. Liu, R. An, X. Hao, Y. Wang, J. Wang, R. Feng, H. Sun, L. Zhao, W. Zhang, D. Zhao, J. Zheng, L. Yu, C. Li, N. Zhang, R. Wang, X. Niu, S. Yang, X. Song, Y. Chai, Y. Hu, Y. Shi, L. Zheng, Z. Li, Q. Gu, F. Shao, W. Huang, R. Jin, Z. Shen, Y. Wang, X. Wang, J. Xiao and X. S. Xie, Nature, 2022, 608, 593–602 CrossRef CAS PubMed.
- Q. Wang, Y. Guo, S. Iketani, M. S. Nair, Z. Li, H. Mohri, M. Wang, J. Yu, A. D. Bowen, J. Y. Chang, J. G. Shah, N. Nguyen, Z. Chen, K. Meyers, M. T. Yin, M. E. Sobieszczyk, Z. Sheng, Y. Huang, L. Liu and D. D. Ho, Nature, 2022, 608, 603–608 CrossRef CAS PubMed.
- H. Tamamura, T. Kobayakawa and N. Ohashi, Springer Briefs in Pharmaceutical Science & Drug Development, Springer, Singapore, 2018, pp. 1−100 Search PubMed.
- E. O. Freed and M. A. Martin, J. Biol. Chem., 1995, 270, 23883–23886 CrossRef CAS PubMed.
- R. Wyatt and J. Sodroski, Science, 1998, 280, 1884–1888 CrossRef CAS PubMed.
- D. M. Eckert and P. S. Kim, Annu. Rev. Biochem., 2001, 70, 777–810 CrossRef CAS PubMed.
- A. Otaka, M. Nakamura, D. Nameki, E. Kodama, S. Uchiyama, S. Nakamura, H. Nakano, H. Tamamura, Y. Kobayashi, M. Matsuoka and N. Fujii, Angew. Chem., Int. Ed., 2002, 41, 2937–2940 CrossRef PubMed.
- W. Nomura, C. Hashimoto, A. Ohya, K. Miyauchi, E. Urano, T. Tanaka, T. Narumi, T. Nakahara, J. A. Komano, N. Yamamoto and H. Tamamura, ChemMedChem, 2012, 7, 205–208 CrossRef CAS PubMed.
- W. Nomura, C. Hashimoto, T. Suzuki, N. Ohashi, M. Fujino, T. Murakami, N. Yamamoto and H. Tamamura, Bioorg. Med. Chem., 2013, 21, 4452–4458 CrossRef CAS PubMed.
- T. Kobayakawa, K. Ebihara, Y. Honda, M. Fujino, W. Nomura, N. Yamamoto, T. Murakami and H. Tamamura, ChemBioChem, 2019, 20, 2101–2108 CrossRef CAS PubMed.
- T. Kobayakawa, K. Ebihara, K. Tsuji, T. Kawada, M. Fujino, Y. Honda, N. Ohashi, T. Murakami and H. Tamamura, Bioorg. Med. Chem., 2020, 28, 115812 CrossRef CAS PubMed.
- World Health Organization, Severe Acute Respiratory Syndrome (SARS), https://www.who.int/health-topics/severe-acute-respiratory-syndrome#tab=tab_1, accessed on 2022-7-6 Search PubMed.
- U. D. Parashar and L. J. Anderson, Int. J. Epidemiol., 2004, 33, 628–634 CrossRef PubMed.
- M. Ujike, H. Nishikawa, A. Otaka, N. Yamamoto, N. Yamamoto, M. Matsuoka, E. Kodama, N. Fujii and F. Taguchi, J. Virol., 2008, 82, 588–592 CrossRef CAS PubMed.
- W. Nomura, C. Hashimoto, A. Ohya, K. Miyauchi, E. Urano, T. Tanaka, T. Narumi, T. Nakahara, J. A. Komano, N. Yamamoto and H. Tamamura, ChemMedChem, 2012, 7, 205–208 CrossRef CAS PubMed.
- S. Futaki, T. Suzuki, W. Ohashi, T. Yagami, S. Tanaka, K. Ueda and Y. Sugiura, J. Biol. Chem., 2001, 276, 5836–5840 CrossRef CAS PubMed.
- M. Hoffmann, H. Kleine-Weber, S. Schroeder, N. Krüger, T. Herrler, S. Erichsen, T. S. Schiergens, G. Herrler, N.-H. Wu, A. Nitsche, M. A. Müller, C. Drosten and S. Pöhlmann, Cell, 2020, 181, 271–280 CrossRef CAS PubMed.
- S. Matsuyama, N. Nao, K. Shirato, M. Kawase, S. Saito, I. Takayama, N. Nagata, T. Sekizuka, H. Katoh, F. Kato, M. Sakata, M. Tahara, S. Kutsuna, N. Ohmagari, M. Kuroda, T. Suzuki, T. Kageyama and M. Takeda, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 7001–7003 CrossRef CAS PubMed.
- P. E. Dawson, T. W. Muir, I. Clark-Lewis and S. B. Kent, Science, 1994, 266, 776–779 CrossRef CAS PubMed.
- Y. Suzuki, T. Hishiki, A. Emi, S. Sakaguchi, R. Itamura, R. Yamamoto, T. Matsuzawa, K. Shimotohno, M. Mizokami, T. Nakano and N. Yamamoto, Biochem. Biophys. Res. Commun., 2021, 575, 36–41 CrossRef CAS PubMed.
- B. Meng, A. Abdullahi, I. A. T. M. Ferreira, N. Goonawardane, A. Saito, I. Kimura, D. Yamasoba, P. P. Gerber, S. Fatihi, S. Rathore, S. K. Zepeda, G. Papa, S. A. Kemp, T. Ikeda, M. Toyoda, T. S. Tan, J. Kuramochi, S. Mitsunaga, T. Ueno, K. Shirakawa, A. Takaori-Kondo, T. Brevini, D. L. Mallery, O. J. Charles, J. E. Bowen, A. Joshi, A. C. Walls, L. Jackson, D. Martin, K. G. C. Smith, J. Bradley, J. A. G. Briggs, J. Choi, E. Madissoon, K. B. Meyer, P. Mlcochova, L. Ceron-Gutierrez, R. Doffinger, S. A. Teichmann, A. J. Fisher, M. S. Pizzuto, A. de Marco, D. Corti, M. Hosmillo, J. H. Lee, L. C. James, L. Thukral, D. Veesler, A. Sigal, F. Sampaziotis, I. G. Goodfellow, N. J. Matheson, K. Sato and R. K. Gupta, CITIID-NIHR BioResource COVID-19 Collaboration; Genotype to Phenotype Japan (G2P-Japan) Consortium; Ecuador-COVID19 Consortium, Nature, 2022, 603, 706–714 CrossRef CAS PubMed.
- World Health Organization, Middle East respiratory syndrome coronavirus (MERS-CoV) – update, May 31, 2013, https://www.who.int/emergencies/disease-outbreak-news/item/2013_05_31_ncov-en, accessed on 2022-7-20.
Footnotes |
| † Electronic supplementary information (ESI) available: General information, details of synthesis of the compounds, characterization data of the compounds including MS and analytical HPLC charts to check the purity of the compounds, and data of their biological evaluation. See DOI: https://doi.org/10.1039/d2ra07356k |
| ‡ These authors contributed equally to this study. |
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Kofi Baffour-Awuah Owusu‡a,
Yutaro Miuraa,
Takahiro Ishiia,
Kouki Shinoharaa,
Takuya Kobayakawa
*a,
Kofi Baffour-Awuah Owusu‡a,
Yutaro Miuraa,
Takahiro Ishiia,
Kouki Shinoharaa,
Takuya Kobayakawa