
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePromoted glucose electrooxidation at Ni(OH)2/graphene layers exfoliated facilely from carbon waste material
Ahmed M. Abdelrahim ,
Muhammad G. Abd El-Moghny*,
Mohamed E. El-Shakre and
Mohamed S. El-Deab*
,
Muhammad G. Abd El-Moghny*,
Mohamed E. El-Shakre and
Mohamed S. El-Deab*
Department of Chemistry, Faculty of Science, Cairo University, Cairo, Egypt. E-mail: mugamal@cu.edu.eg; gmohamd@sci.cu.edu.eg; msaada@cu.edu.eg; msaada68@yahoo.com
First published on 10th January 2023
Abstract
Nowadays, the glucose electro-oxidation reaction (GOR) is considered one of the most important solutions for environmental pollution. The GOR is the anodic reaction in direct glucose fuel cells and hybrid water electrolysis. In this study, the GOR is boosted using a carbon support modified with Ni(OH)2 as a non-precious catalyst. The carbon support, with in situ generated graphene nanosheets having a large surface area, grooves, and surface functional groups, is prepared via a simple electrochemical treatment of the carbon rods of an exhausted zinc-carbon battery. Ni(OH)2 is electrodeposited on the surface of the functionalized exfoliated graphite rod (FEGR) via the dynamic hydrogen bubbling technique (DHBT) and tested for GOR. The thus-prepared Ni(OH)2/FEGR electrode is characterized by SEM, mapping EDX, HR-TEM, XRD, and XPS characterization tools. Ni(OH)2/FEGR displays an onset potential of 1.23 V vs. the reversible hydrogen electrode (RHE) and attains high current densities at lower potentials. Additionally, Ni(OH)2/FEGR showed prolonged stability toward GOR by supporting a constant current over a long electrolysis time. The enhanced catalytic performance is attributed to the superb ionic and electronic conductivity of the catalyst. Importantly, ionic conductivity increased, due to (i) a large surface area of in situ generated graphene layers, (ii) enhanced distribution of active material during deposition using DHBT, and (iii) increased hydrophilicity of the underlying substrate. Therefore, the Ni(OH)2/FEGR electrode can be used efficiently for GOR as a low-cost catalyst, achieving low onset potential and high current densities at low potentials.
1. Introduction
Recently, renewable energy technologies have attracted great attention due to the rapid depletion of fossil fuels and their limited resources, together with the related environmental drawbacks. Thus, an urgent need has emerged to increase the contribution of renewable energy sources (e.g., wind, solar and geothermal energy, fuel cells, etc.) to replace traditional fossil fuels.1–5 Nowadays, glucose solves many environmental problems via its use in fuel cells and hydrogen production. In this regard, direct glucose fuel cells (DGFCs) stand among the most efficient electrical energy conversion systems due to their unique advantages, i.e. glucose (as a fuel) is naturally abundant, cheap, non-toxic, easy to store and transport, eco-friendly, and high energy (glucose release 2.87 MJ mol−1 upon complete oxidation).6–9 Also, glucose electro–oxidation reaction (GOR) is used in hybrid water electrolysis instead of the oxygen evolution reaction (OER) to produce hydrogen at low cell voltage.10–13Recently, the usage of non-precious transition metal-based catalysts such as Cu, Ni, and Co toward GOR is a goal of research to replace the high-cost-based metals such as Pt and Au.14–16 Among the various catalysts based on transition metals, Ni(OH)2 materials had been proven to be promising catalyst for GOR due to their unique layered structure with large interlayer spacing that allows the electrolyte diffusion within the catalyst layers resulting in a boosted catalytic activity.17–20 Glucose can be oxidized into gluconolactone in the presence of Ni(OH)2 according to the following eqn (1) and (2):14
| Ni(OH)2 + OH− → NiOOH + H2O + e− | (1) |
| 2NiOOH + Glucose → gluconolactone + 2Ni(OH)2 | (2) |
Interestingly, Ni(OH)2 has two polymorphic crystal structures, α-Ni(OH)2 phase with hydrotalcite-like structure and β-Ni(OH)2 with brucite-like structure (see Scheme 1).21–23 α-Ni(OH)2 has a higher d-spacing value with the turbostratically crystallized structure due to the presence of the water molecules and/or anions (e.g. SO42−, Cl−, NO3−, and CH3COO−) between the hydroxide layers.21–23 β-Ni(OH)2 has a lower theoretical oxidation potential than α-Ni(OH)2 and thus has higher catalytic activity toward GOR into gluconolactone, as demonstrated by El-Nagar.14 However, it is sometimes found that the α-Ni(OH)2 showed higher catalytic activity than β-NiOOH because of its morphology and the large d-spacing that facilitates the access of electrolyte ions to its active sites thus being converted to the γ-NiOOH which oxidizes the glucose. Guo-Xiu Tong24 prepared α-Ni(OH)2 with flower-like morphology with high surface area and higher catalytic activity toward GOR than β-Ni(OH)2 microsphere. Also, Pan lu25 proved that rose-like nanostructured α-Ni(OH)2 prepared by a hydrothermal method has enhanced the catalytic activity toward GOR.
 | ||
| Scheme 1 Schematic illustration of Ni(OH)2/NiOOH phase transformations (Bode diagram). Note that the A represents the possible anions (e.g., SO42−, Cl−, NO3−, and CH3COO−) that can be incorporated between the layers of hydroxide during synthesis.21,42 | ||
Although Ni(OH)2 has a higher catalytic behavior toward GOR, it has some drawbacks such as low electrical conductivity (∼10−17 S cm−1), low ionic conductivity, and low durability. Therefore, research efforts are directed to investigate substrate materials, which act as catalyst support, characterized by a high electrical conductivity, and high surface area with a porous structure to overcome these problems.15,26
Carbon-based materials are promising catalyst support due to their unique characteristics such as (i) high surface area and porous structure, (ii) high electronic conductivity, and (iii) wide operating potential window which is a very important parameter for the electrode performance, and (iv) excellent physical, mechanical and electrochemical properties.27–31 Graphene is one of the most important used carbon-based materials in energy devices due to its unrivaled advantages, unique 2D structure with a huge gravimetric surface area (∼2600 m2 g−1), ultra-high electrical conductivity, very high charge carrier mobility (∼200![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cm2 V−1 s−1), excellent mechanical and physical properties, outstanding thermal conductivity, and being non-toxic material.29,30,32–36
000 cm2 V−1 s−1), excellent mechanical and physical properties, outstanding thermal conductivity, and being non-toxic material.29,30,32–36
Therefore, the researchers were motivated to prepare high-quality graphene using different methods; micromechanical exfoliation, thermal decomposition of SiC, chemical vapor deposition (CVD), and chemical and electrochemical exfoliation. However, micromechanical exfoliation displays high-quality graphene production unless this method is not suitable for large-scale productions. CVD is a well-known method to produce graphene on a large scale, but this method requires a highly expensive substrate during the preparation process which limits its use in commercial applications.30,32,37,38 On the other hand, Hummer developed a cost-effective method based on chemical exfoliation replacing the high-cost CVD and thermal decomposition methods. Hummers' method is based on two sequential steps. Firstly, the production of graphene oxide (GO) via exfoliation of graphite in the strong oxidants and acids followed by the reduction of the produced GO using chemical or thermal way.30,38 Interestingly, the complete exfoliation Hummers' method has many disadvantages such as being sophisticated and time-consuming, using harsh materials, and requiring a binder which hinders the catalyst performance by increasing resistance rather than the binder's high cost.27,33,34,37
To solve this issue, many researchers have proposed the partial exfoliation of graphite using an electrochemical oxidation setup. Advantageously, electrochemical partial exfoliation has many advantages such as being a simple, safe, controllable, binder-free, low-cost, and eco-friendly method. This method produces functionalized graphene sheets as top layers electrically connected to the graphite (bottom layer). The resulting highly conductive three-dimensional functionalized exfoliated graphene can be used as excellent support to accommodate the redox-active materials to enhance their electrical conductivity.28,29,32
Over and above, preparation techniques play an essential role in determining the catalytic activity of the electrode material. Several techniques have been reported to synthesize metal-based materials. The electrochemical deposition technique has attracted much attention due to being simple, controllable, cost-effective, and free binder method. Specifically, the dynamic hydrogen bubbling technique (DHBT) is considered a promising method for electrodeposition due to being simple and fast, producing high surface area by developing a porous structure, and being controllable.39–41
Therefore, the current research focuses on the preparation of non-precious catalysts on the surface of supporting materials with characteristic properties. Interestingly, seeking supporting materials with characteristic properties from waste materials is still challenging.
Herein, graphene layers with a large surface area are prepared from waste material via a facile method. Electrochemical partial exfoliation of the recycled graphite rod (RGR) is done to produce the functionalized exfoliated graphite rod (FEGR) to act as excellent support for Ni(OH)2. The FEGR boosted the catalytic activity of Ni(OH)2 towards GOR. The preparation of catalyst, Ni(OH)2/FEGR, has many advantages such as being a simple, safe, controllable, low-cost, binder-free, and eco-friendly method. Additionally, the FEGR support enhances the hydrogen evolution reaction during electrodeposition by DHBT providing homogeneous distribution of active sites. As a result, Ni(OH)2/FEGR boosted the GOR by displaying a lower onset potential (1.23 V vs. RHE) and by attaining high current densities at a lower potentials. Also, Ni(OH)2/FEGR electrode displays excellent stability toward GOR. Additionally, at any current density, the potential of the GOR is much lower than that of OER. Therefore, the boosted GOR on the surface of the Ni(OH)2/FEGR can be used in many applications such as DGFCs, and hybrid water electrolysis. The superior performance of Ni(OH)2/FEGR catalyst is attributed to (a) the increased exposed active sites, (b) the enhanced diffusion of the electrolytes by increasing the surface hydrophilicity, (c) the enhanced electronic conductivity of the catalyst, and (d) the superb stability.
2. Experimental
2.1. Chemicals and electrodes preparation
(1) Electrodeposition of Ni metal using DHBT. During the deposition of Ni, the RGR and FEGR are used as working electrodes, and the graphite rod and SCE are serving as the counter and reference electrodes, respectively. The deposition was done by allowing the passage of 20 Coulombs (C) at −2 V in the aqueous solution containing 0.1 M NiCl2 and 1 M NH4Cl at room temperature (25 °C ± 1).
(2) Electrochemical passivation of the obtained Ni metal is carried out in an aqueous solution of 0.5 M NaOH potentiodynamically by cycling the potential from −0.1 to 0.7 V vs. SCE for 30 cycles using a potential scan rate of 50 mV s−1.
2.2. Materials characterization
Surface morphology, chemical composition, and structure of the various modified electrodes were examined by field emission scanning electron microscope (FE-SEM, QUANTA FEG 250) coupled with an energy dispersive X-ray spectrometer (EDX) unit and high-resolution transmission electron microscopy (HR-TEM) (JEOL, JEM-2100, Tokyo, Japan). X-ray photoelectron spectroscopy (XPS), using monochromatic X-ray Al Kα radiation, Thermo Fisher Scientific. Additionally, the crystalline structures were characterized using X-ray diffraction (XRD) with Cu Kα radiation, STOE STADI. Inductively coupled plasma-optical emission spectrometer (ICP-OES) measurements were carried out (ICP-OES Optima 2000 DV PerkinElmer Microwave CRM Mars 5) to obtain the amount of active material on each electrode.2.3. Electrochemical measurements
The electrochemical measurements were investigated using a three-electrode cell configuration at room temperature (25 °C ± 1) and a Biologic potentiostat (model VSP-300). Electrocatalytic performance was measured via, cyclic voltammetry (CV), linear sweep voltammetry (LSV), chronoamperometry (CA), and electrochemical impedance spectroscopy (EIS) (from 100 kHz to 20 mHz) in a 0.5 M NaOH aqueous solution containing a known amount of glucose.3. Results and discussion
The surface morphology was inspected using SEM analysis. From Fig. 1, while the surface of RGR displays smooth surface with small percentage of defects (composed mainly of carbon and small percentage of oxygen) (see Fig. 1A), the FEGR is characterized by the presence of exfoliated graphene layers and uniformly distributed grooves atop the graphite rod's surface (see Fig. 1B). Also, as a result of the oxidation, the FEGR displays highly distributed oxygenated functional groups (see Fig. 1B, elemental mapping EDX analysis). Because of the characteristic morphology of FEGR, it can be used as promising support to accommodate a large number of active materials that can be used efficiently toward GOR. | ||
| Fig. 1 SEM images with different magnifications and elemental mapping EDX analysis of (A) RGR support, and (B) FEGR support. | ||
Firstly, the enhancement of the FEGR support can be obvious by providing a large surface area that can be available to ingest the large quantity of catalyst layer. As shown in Fig. 2A, the calculated specific capacitance (Cs) of FEGR using eqn (3) is 5.5 mF cm−2 which is greater than the Cs of RGR (0.21 mF cm−2) by 26 times.
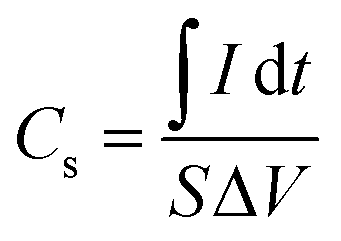 | (3) |
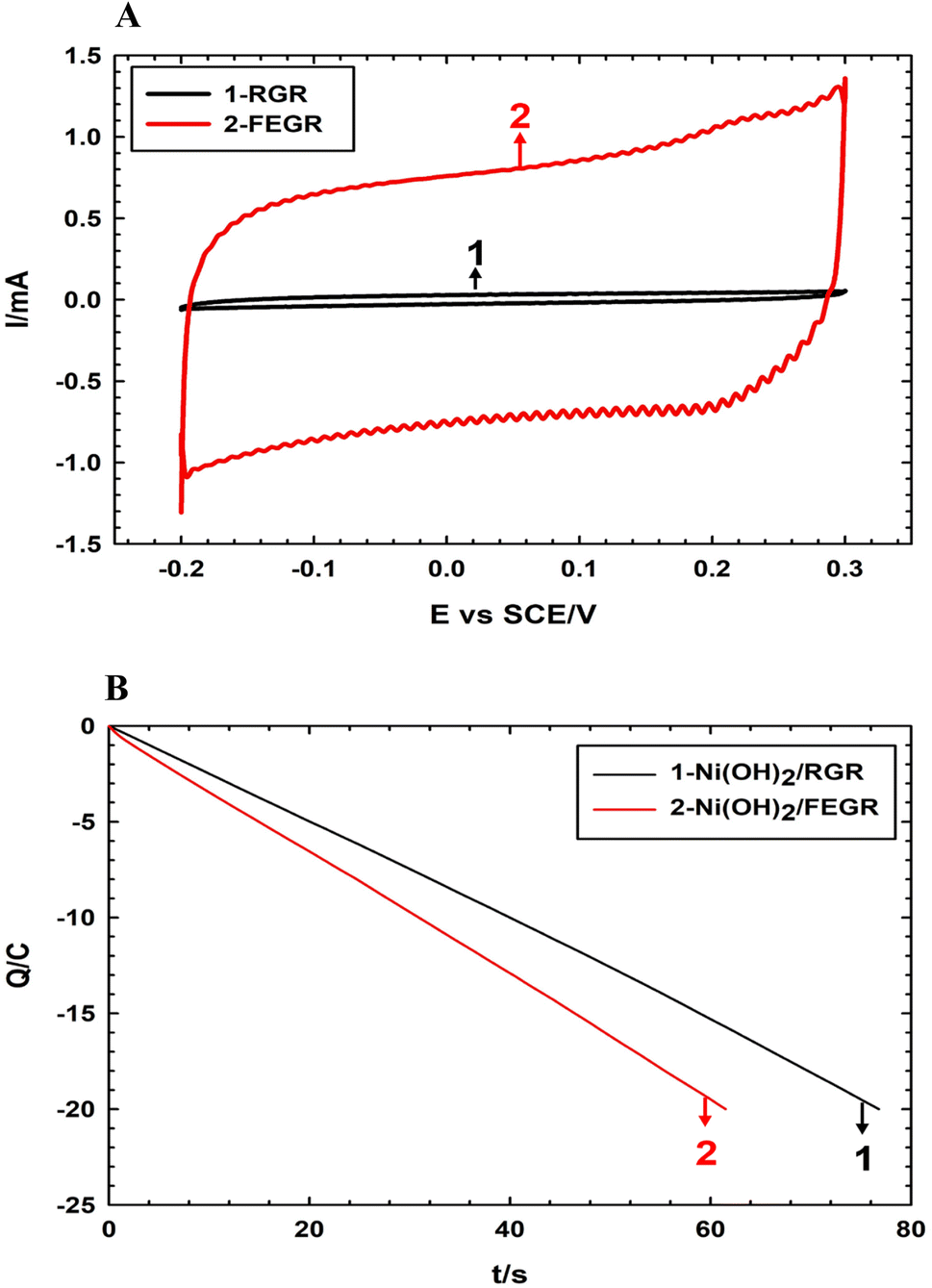 | ||
| Fig. 2 (A) CVs obtained at RGR and FEGR supports measured in 0.5 M NaOH solution at a potential scan rate of 100 mV s−1. (B) Controlled potential coulometry recorded at −2 V vs. SCE during electrodeposition of Ni(OH)2 films on the surface of FEGR (red line) and RGR (black line) supports. | ||
The high specific capacitance of the FEGR support is reflected in, the large adsorption capacity of this large activated electrode's surface to ingest a large number of electrolyte species by supplying a rough surface, a large number of the homogeneous-distributed grooves on the surface of the electrode, in addition to the large produced number of expanded graphene sheets.
Secondly, the FEGR electrode minimizes the coagulation of the catalyst species during its electrodeposition using the DHBT. During metal electrodeposition (see eqn (4)) using DHBT, H2 bubbles are formed from the electrolysis of water and ammonium chloride due to higher overpotential applied according to eqn (5) and (6), respectively, which bubbles off the growing deposited metal (eqn (4)), disrupting the growth mechanism.44,45
| Mn+(aq) + ne− → M(s) | (4) |
| 2H+(aq) + 2e− → H2 | (5) |
| 2NH4+ + 2e− → H2 + 2NH3 | (6) |
So, the coagulation nature of the prepared film of the catalyst layer can be minimized by increasing the rate of hydrogen evolution at the expense of the metal deposition. During the deposition of Ni(OH)2/RGR and Ni(OH)2/FEGR, a controlled amount of charge (20 C) is allowed to pass. The presence of more functional groups over graphene layers, and grooves enhance the rate of electrolysis, and this is obvious from Fig. 2B. The number of Columbus passed during electrolysis with the time is displayed in Fig. 2B. As shown, the time needed to prepare the Ni(OH)2 film on the surface of FEGR is shorter than that needed for deposition on the surface of RGR. This confirms the enhancement of the electrolysis rate. That is, the number of Columbus consumed for the evolution of hydrogen on the FEGR support increased more than that on the RGR support which led to the better distribution of deposited Ni(OH)2 on the surface of FEGR support (c.f. Fig. 3).
 | ||
| Fig. 3 SEM images with different magnifications elemental mapping EDX analysis of (A) Ni(OH)2/RGR electrode, and (B) Ni(OH)2/FEGR electrode. | ||
Firstly, in this regard, ICP measurements are carried out to confirm that the consumed amount of Columbus for hydrogen evolution increased on FEGR. The amounts of electrodeposited Ni(OH)2 on the FEGR and RGR are 0.7 and 1.3 mg, respectively. Secondly, from mapping EDX (Fig. 3), the mass percentage of Ni obtained on the surface of FEGR support (12.64%) is less than that on the RGR support (31.59%) which is consistent with ICP analysis. Additionally, the mass percentage of elemental oxygen of FEGR, Ni(OH)2/RGR, and Ni(OH)2/FEGR is 26.92%, 6.04%, and 12.64%, respectively. The high O% on the surface of the FEGR is a result of the exfoliation step during its preparation.21 On the other hand, a noticeable decrease of O% on Ni(OH)2/RGR, and Ni(OH)2/FEGR electrodes is due to the coverage of Ni(OH)2 on the surface of the support.
Furthermore, from Fig. 3A and B, Ni(OH)2 on both RGR and FEGR electrodes, respectively, displays spherical nanoparticle morphology. But the distribution of these spherical nanoparticles on the FEGR is in a homogeneous uniform matter which is better than that on the RGR as depicted from SEM and mapping EDX analysis (see Fig. 3A and B). Moreover, the HR-TEM analysis of the Ni(OH)2/FEGR electrode as the best one towards GOR (c.f. Fig. 8 and 9) is displayed in Fig. 4. HR-TEM is performed to highlight the morphology and investigate the crystalline structure of the proposed catalyst. As shown in Fig. 4A, the TEM image of Ni(OH)2/FEGR agrees with the morphology obtained from the SEM analysis. Also, from images with high magnifications (Fig. 4B and C), we can safely conclude that the spherical nanoparticles of Ni(OH)2 are deposited on the exfoliated graphene layers. Importantly, Fig. 4D displays the lattice fringes that confirms the crystalline nature of the Ni(OH)2 active particles. Additionally, Fig. 4E displays the selected area electron diffraction (SAED) of Ni(OH)2 active particles. The bright spots around the ring indicate the polycrystalline nature of the Ni(OH)2. The measured d-spacing values from the obtained rings in Fig. 4E are 7.6, 4.6, 3.9, and 2.3 Å. While the d-spacing values of 7.6 and 3.9 Å are nearly equal to the values corresponding to α-Ni(OH)2 (JCPDS No. 41–1424),46,47 the values of 4.6 and 2.3 Å indicate the presence of some facets of β-Ni(OH)2 (JCPDS No. 14–0117).46,48 So, one can conclude that Ni(OH)2 is electrodeposited with mixed α-, and β-phases that is further confirmed by the XRD analysis (c.f. Fig. 6). Moreover, Fig. 4F displays the SAED pattern of selected regions of metal-free graphene layers. From Fig. 4F, the obtained ring that contains bright spots displays a d-spacing value of 2.5 Å confirming the presence of graphene layers.49,50
 | ||
| Fig. 4 HR-TEM images of Ni(OH)2/FEGR electrode with different magnifications (A–D). The corresponding SAED pattern of: (E) Ni(OH)2, and (F) graphene layers. | ||
XPS analyses are carried out to investigate the oxidation states of Ni in RGR and FEGR electrodes. From the enlarged view of Ni 2p of both electrodes, as shown in Fig. 5A and D, the observed peaks at binding energies around 856 and 873 eV confirm that Ni is present as Ni(OH)2. Also, the presence of the two satellite peaks at binding energies of 861 and 880 eV confirms the formation of Ni(OH)2.51–55 Additionally, from the deconvoluted O 1s spectra of both Ni(OH)2/RGR and Ni(OH)2/FEGR electrodes, Fig. 5B and E, the main peak around 531 eV confirms the formation of nickel hydroxide.51 By comparing the other peaks of the O 1s of both electrodes, Ni(OH)2/RGR displays two other peaks for C–OH and adsorbed H2O. The Ni(OH)2/FEGR electrode displays four other peaks for C–OH, C(O), C(O)O, and adsorbed H2O. The presence of the C(O), C(O)O peaks in the Ni(OH)2/FEGR electrodes that do not exist in the Ni(OH)2/RGR electrode is due to the oxidation which occurred during the preparation of the FEGR support, the electro exfoliation step.21 Moreover, the oxygenated functional groups that exist in the C 1s spectra of both electrodes, as shown in Fig. 5C and F, which is consistent with the O 1s spectra (Fig. 5B and E).21,54 The occurrence of more oxygenated functional groups on the surface of the Ni(OH)2/FEGR consistency with the elemental mapping EDX analysis of Ni(OH)2/FEGR displays higher oxygen percentage compared to the Ni(OH)2/RGR.
 | ||
| Fig. 5 XPS spectra of (A) Ni 2p, (B) O 1s, and (C) C 1s for Ni(OH)2/RGR electrode. XPS spectra of (D) Ni 2p, (E) O 1s, and (F) C 1s for Ni(OH)2/FEGR electrode. | ||
Also, the XRD analyses are carried out to investigate the phase of Ni(OH)2. XRD patterns of the as-prepared electrodes, i.e., Ni(OH)2/RGR and Ni(OH)2/FEGR are depicted in Fig. 6. Both electrodes are composed of mixed phases (alpha and beta). While the corresponding peaks of β-Ni(OH)2 were observed at 36°, 37°, 52°, and 63° (JCPDS No. 14–0117),46,48 the corresponding peaks of α-Ni(OH)2 appeared at 12°, 21°, 23°, and 40° (JCPDS No. 41–1424).46,47 Additionally, peaks due to the hexagonal graphitic structure of RGR and FEGR support are detected around 2θ of 25°, 42°, 43°, 77°, and 83°.56,57
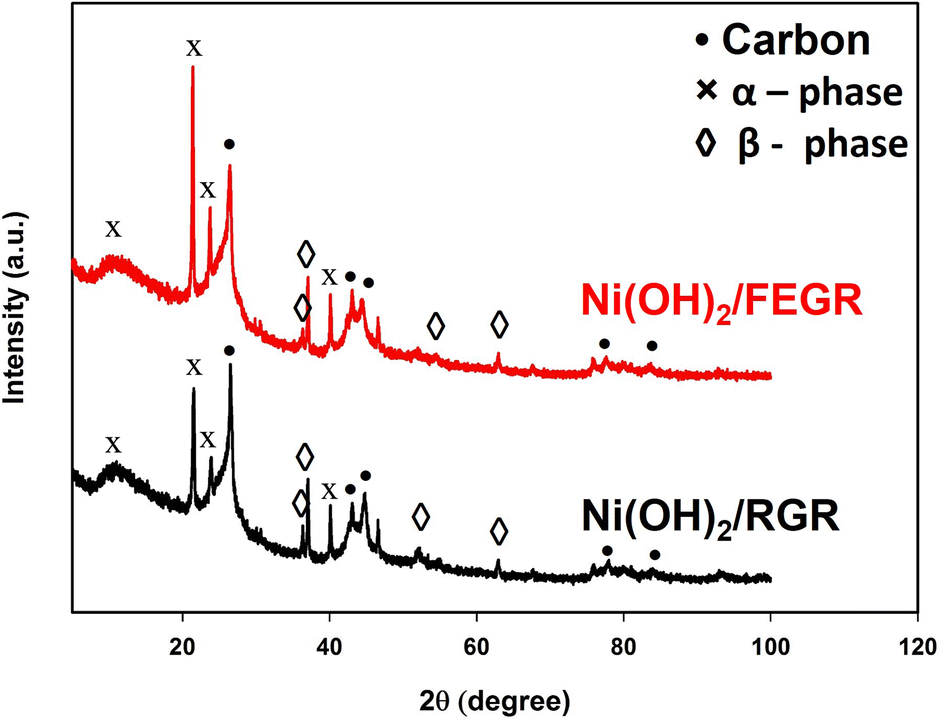 | ||
| Fig. 6 XRD patterns of Ni(OH)2/RGR (black line), and Ni(OH)2/FEGR (red line) electrodes. | ||
As predicted from the physical analysis of both Ni(OH)2/RGR and Ni(OH)2/FEGR electrodes, Ni(OH)2/FEGR has unique properties that promote the catalytic activity toward GOR. Fig. 7A displays the CVs of the prepared electrodes that show characteristic redox peaks within the selected potential window resulting from the transformation of the α- and β-Ni–OH to β- and γ-Ni–OOH, respectively. The position of the peaks on the CV indicates that the transformation of Ni(OH)2 to NiOOH is a diffusion-controlled process.2,21 To confirm that this transformation is a diffusion-controlled process, the study of the effect of potential scan rate (v) on the current peak (Ip) was done. By plotting Ip against the v0.5 (see Fig. 7B), a linear relation is obtained with R2 equals 0.9999 which confirms that the transformation of the Ni(OH)2 to NiOOH is a diffusion-controlled process.58,59 The large difference in CVs currents between the Ni(OH)2/RGR, and Ni(OH)2/FEGR indicates that the Ni(OH)2/FEGR possesses a higher electrochemical active site on its surface than Ni(OH)2/RGR electrode that in turn contributes larger in the electrocatalysis of glucose oxidation with greater extent. As mentioned above the higher capacity of the Ni(OH)2/FEGR is due to (i) the FEGR support providing a large surface area to accommodate a large number of active sites on its surface, (ii) the presence of the grooves and oxygenated functional groups enhances the rate and the consumed amount of the hydrogen evolution that prevent the coagulation of catalyst’ active sites on the surface of the substrate, and (iii) the presence of exfoliated graphene layers that enhance the electronic conductivity. To confirm that the active sites are well distributed on the surface of the FEGR support electrochemically, the following relation (eqn (7)) was used:
| q = q∞ + av−0.5 | (7) |
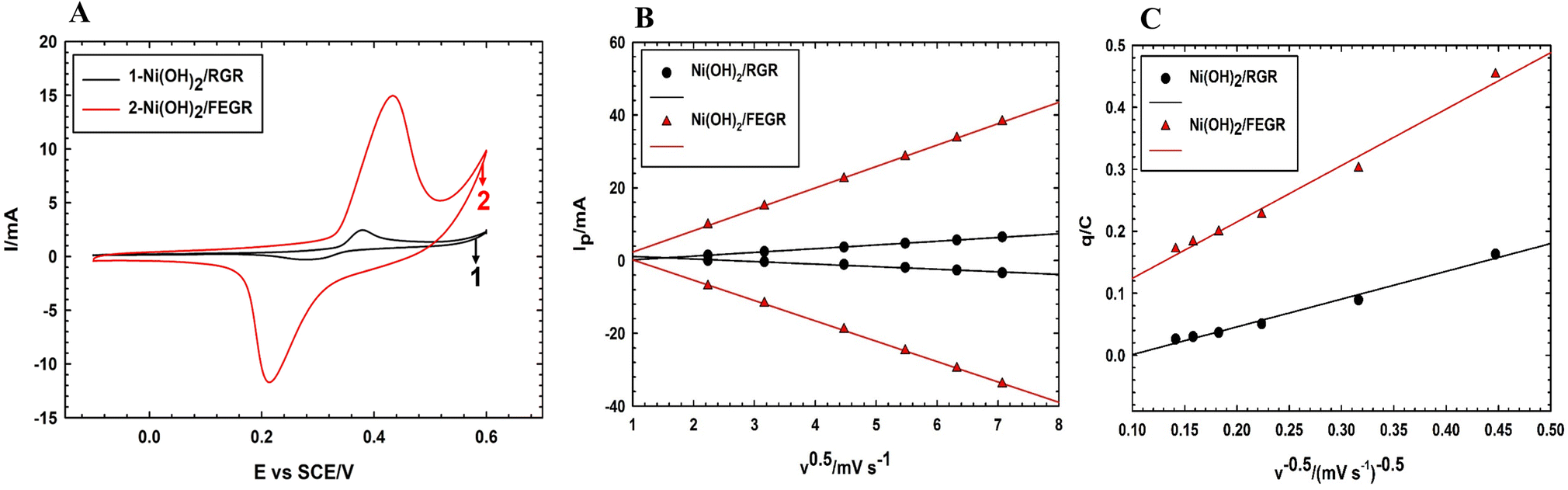 | ||
| Fig. 7 (A) CVs of Ni(OH)2/RGR, and (B) Ni(OH)2/FEGR electrodes measured in 0.5 M NaOH solution at a potential scan rate of 10 mV s−1. (B) The variation of anodic and cathodic peaks current with the square root of potential scan rate (ν0.5) for Ni(OH)2/RGR, and Ni(OH)2/FEGR electrodes measured in 0.5 M NaOH solution. (C) The variation of q with the reciprocal of the square root of the potential scan rate (ν−0.5) measured in 0.5 M NaOH solution. | ||
As indicated in Fig. 8A and B, the GOR is markedly enhanced on the surface of the Ni(OH)2/FEGR electrode compared with Ni(OH)2/RGR electrode. Ni(OH)2/FEGR has a larger current than Ni(OH)2/RGR and this is attributed to the accessibility of the large active sites to oxidize a large number of glucose molecules. Although the Ni(OH)2/FEGR electrode has a lower loading amount of Ni than that of the Ni(OH)2/RGR electrode as indicated above from ICP measurements and mapping EDX analysis, the Ni(OH)2/FEGR has a higher degree of efficiency toward the GOR due to possessing a high number of available active sites that distributed homogeneously with a low degree of coagulation compared to Ni distribution in case of Ni(OH)2/RGR electrode. Importantly from the reduction peaks of both electrodes after the oxidation of glucose, the reduction peak of Ni(OH)2/RGR disappears in comparison with its blank due to all active sites (small active sites due to the coagulation nature of the prepared catalyst) are consumed during the GOR. On the other hand, the reduction peak of Ni(OH)2/FEGR is minimized at a small concentration of glucose and disappears at high glucose concentration in comparison with its blank which confirms that there is an excess of active sites that contribute to the glucose oxidation more at high glucose concentration. Fig. 8C and D displayed the resulting current due to glucose oxidation only by subtracting the total current (current due to the transformation of the α- and β-Ni–OH to β- and γ-Ni–OOH and glucose oxidation) from the blank current (current due to the transformation of α- and β-Ni–OH to β- and γ-Ni–OOH only). Table 1 summarizes the results of Fig. 8C and D. Peak current due to glucose oxidation at the surface of Ni(OH)2/FEGR is higher than Ni(OH)2/RGR and the difference between current peaks increased by increasing glucose oxidation which confirms there is a more oxidation degree for glucose at higher concentration due to the more contribution of active sites of Ni(OH)2/FEGR catalyst (see Fig. 8E). From Fig. 8F, by comparing the degree of oxidation of glucose over the selected potential window, the amount of Columbus relating due to the oxidation of glucose at the surface of the Ni(OH)2/FEGR is higher than that of Ni(OH)2/RGR and displays the same trend of the Fig. 8E confirms more oxidation of glucose molecules at the surface of the Ni(OH)2/FEGR. The number of glucose molecules oxidized per catalytic active sites is defined as the turnover number (TON). TON is directly proportional to the current (at specific potential) produced due to fuel oxidation per number of active sites of the prepared catalyst.61,62 It is found that the TON of Ni(OH)2/FEGR at 0.55 V is two times the TON of Ni(OH)2/RGR which is consistent with the above results.
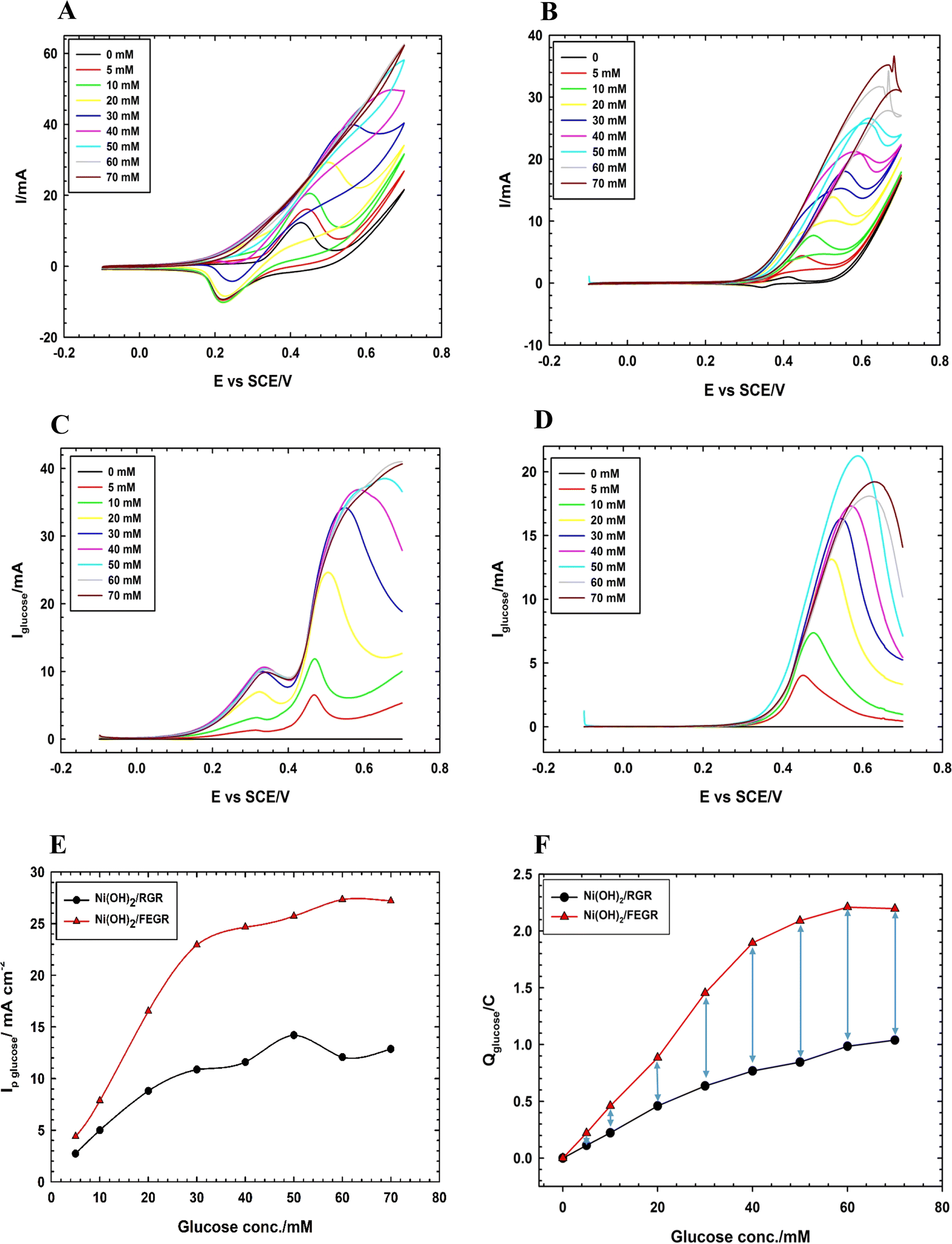 | ||
| Fig. 8 CVs of (A) Ni(OH)2/FEGR, and (B) Ni(OH)2/RGR electrodes. LSVs of (C) Ni(OH)2/FEGR and (D) Ni(OH)2/RGR electrodes. (E) Variation of peak current due to glucose oxidation with the used glucose concentration at Ni(OH)2/FEGR (red line) and Ni(OH)2/RGR (black line) electrodes. (F) Variation of the amount of Columbus due to glucose oxidation with the used glucose concentration at Ni(OH)2/FEGR (red line) and Ni(OH)2/RGR (black line) electrodes. Note that: all measurements were operated in 0.5 M NaOH solution containing various concentrations of glucose (0, 5, 10, 20, 30, 40, 50, 60, and 70 mM) at a potential scan rate of 10 mV s−1. | ||
| Glucose conc. | Ni(OH)2/RGR | Ni(OH)2/FEGR | ||||
|---|---|---|---|---|---|---|
| Ip/mA cm−2 | Ip/A g−1 | Q/mC | Ip/mA cm−2 | Ip/A g−1 | Q/mC | |
| 5 | 2.73 | 3 | 112.5 | 4.4 | 9.3 | 220.6 |
| 10 | 5 | 5.6 | 223 | 7.9 | 16.7 | 459.9 |
| 20 | 8.8 | 9.9 | 459.5 | 16.5 | 35.1 | 884.7 |
| 30 | 10.9 | 12.2 | 636.3 | 22.9 | 48.6 | 1453.9 |
| 40 | 11.6 | 13 | 767.8 | 24.7 | 52.3 | 1894.6 |
| 50 | 14.2 | 15.9 | 845.3 | 25.7 | 54.6 | 2089.7 |
| 60 | 12.1 | 13.6 | 985 | 27.3 | 58 | 2209.9 |
| 70 | 12.9 | 14.4 | 103.9 | 27.2 | 57.7 | 2196.1 |
Moreover, Ni(OH)2/FEGR has a lower onset potential (0.18 V vs. SCE) than that of Ni(OH)2/RGR (0.3 V) and this is mainly due to an increase in the electronic conductivity of the prepared electrode. The electronic conductivity of the Ni(OH)2/FEGR is higher than that of the Ni(OH)2/RGR due to the presence of exfoliated graphene layers. Exfoliation delivers the electron very fast to the surface of the active material and facilitates the transformation of Ni(OH)2 to NiOOH that in turn oxidizes glucose to gluconolactone according to eqn (1) and (2). To confirm that the Ni(OH)2/FEGR has a higher electronic conductivity, the EIS measurements firstly at open circuit potential were performed. The Nyquist plots are displayed in Fig. 9A and it is clear that the electrochemical equivalent series resistance (ESR) for the Ni(OH)2/FEGR is smaller than that of Ni(OH)2/RGR indicating that the Ni(OH)2/FEGR has a higher electronic conductivity.21,63,64 Also, Ni(OH)2/FEGR has a more vertical line compared to Ni(OH)2/RGR which confirms that the Ni(OH)2/FEGR has a higher capacity value than Ni(OH)2/RGR due to the uniform distribution of a large number of active sites on the surface of the FEGR support as mentioned above and concluded from CVs of both electrodes.65 Additionally, the admittance plot, see inset of Fig. 9A, displayed an increment consistent with the ESR because the admittance, as known, is the inverse of the impedance. So, the GOR is expected to have a lower charge transfer resistance on the surface of Ni(OH)2/FEGR than Ni(OH)2/RGR. Clearly, from the Nyquist plots operated in the presence of 30 mM glucose for both electrodes at 0.3 V vs. SCE (Fig. 9B) and the data resulting from the fitted equivalent circuit (see inset of Fig. 9B), the charge transfer resistance on the surface of Ni(OH)2/FEGR (15 Ohm) is much lower than that of the Ni(OH)2/RGR (157 Ohm) which reflected the lower onset potential of the Ni(OH)2/FEGR compared to Ni(OH)2/RGR. Not only the onset potential and Ip are factors that are enhanced on the surface of the Ni(OH)2/FEGR but also the durability of the prepared electrode is outstanding compared to that of the Ni(OH)2/RGR. From Fig. 9C, both electrodes start displaying a large decaying in the response to the gain current and this is due to the oxidation of a large number of glucose molecules near the surface of both electrodes. But Ni(OH)2/RGR continues to decay compared with the Ni(OH)2/FEGR that displays superb durability by showing a constant current response with time for about 3 h. The excellent stability of the Ni(OH)2/FEGR may be attributed to the strong interaction between the Ni(OH)2 and FEGR support throughout oxygen functional groups. Additionally, the persistence of morphology of the Ni(OH)2 active particles after continuous operation for 3 h towards GOR reveals the robustness and insignificant distortion of the catalyst reflecting the remarkable stability of the catalyst (see Fig. 10A and B). From Fig. 10A and B, the Ni(OH)2 active particles have the same morphology (nanosphere) as before the long-term stability operation. Moreover, the active particles are still uniformly distributed over the exfoliated graphene layers (see Fig. 10C). As a general conclusion, the Ni(OH)2/FEGR has a premium catalytic performance than the Ni(OH)2/RGR due to: (i) the ionic conductivity is increased by making the most active sites available for ease of interaction with hydroxide ions and hence oxidation of glucose, and (ii) the electronic conductivity is also increased that in turn delivers the electron very fast to active sites and hence glucose molecules are oxidized efficiently. Additionally, the prepared catalyst is a promising catalyst due to its facile preparation via partial electrochemical exfoliation that led to the formation of graphene layers easily without any binder or using any harsh chemicals used in the preparation of catalyst-based graphene support applied toward glucose oxidation. Moreover, the prepared catalyst is an excellent catalyst for GOR compared to other reported catalysts (see Table 2).
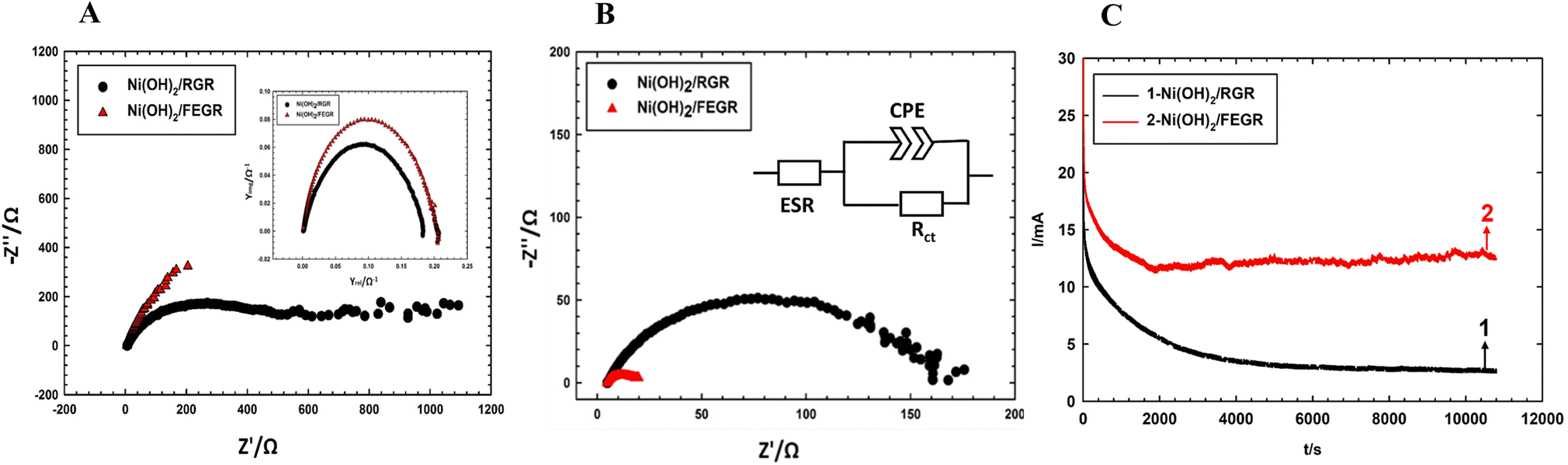 | ||
| Fig. 9 (A) Nyquist plots of Ni(OH)2/RGR (black line) and Ni(OH)2/FEGR (red line) electrodes at OCP and their insets admittance plots of Ni(OH)2/RGR (black line) and Ni(OH)2/FEGR (red line) electrodes measured in 0.5 M NaOH solution. (B) Nyquist plots of Ni(OH)2/RGR (black line) and Ni(OH)2/FEGR (red line) electrodes measured in 0.5 M NaOH solution containing 30 mM glucose at 0.3 V vs. SCE. (C) Chronoamperograms for GOR were obtained at Ni(OH)2/RGR (black line) and Ni(OH)2/FEGR (red line) electrodes measured in 0.5 M NaOH solution containing 30 mM glucose at 0.55 V for 3 h. | ||
 | ||
| Fig. 10 SEM images of Ni(OH)2/FEGR after GOR long-term operation for 3 h of continuous operation at 0.55 V measured in 0.5 M NaOH solution containing 30 mM glucose at different magnifications (A and B). (C) The corresponding elemental mapping-EDX analysis of Ni(OH)2/FEGR electrode. | ||
| Material | Supporting electrolyte | Glucose | Current density (mA cm−2) | Scan rate (mV s−1) | Onset potential (mV) vs. SCE | Ref. |
|---|---|---|---|---|---|---|
| Ni4CO2/AC | 3 M KOH | 1 M | 77 | 10 | 229 | 17 |
| Ni–Co-rGO | 3 M KOH | 1 M | 13.5 | 50 | 211 | 66 |
| NiOx/MnOx/GNs/GC | 0.5 M NaOH | 20 mM | 170 | 100 | 320 | 67 |
| Nano Ni/Ti | 0.5 M NaOH | 10 mM | 29 | 10 | 410 | 68 |
| Nano NiOx/GC | 0.3 M KOH | 10 mM | 12 | 100 | 330 | 69 |
| NiCuO/ITO | 1 M KOH | 30 mM | 0.425 | 100 | 105 | 70 |
| NiOx/MnOx/GC | 0.5 M NaOH | 20 mM | 14 | 100 | 266 | 71 |
| Ni6/AB | 0.1 M KOH | 5 mM | 2.5 | 10 | 241 | 72 |
| NiFe2O4/GC | 0.5 M NaOH | 60 mM | 0.29 | 200 | 292 | 73 |
| Ni(OH)2/FEGR | 0.5 M NaOH | 30 mM | 22.9 | 10 | 180 | This work |
4. Conclusion
Ni(OH)2 active material is deposited on in situ generated FEGR. FEGR support enhanced GOR by increasing the ionic conductivity of Ni(OH)2 via providing a large activated surface area (26 times than RGR) that is characterized by defective surface, high oxygenated surface functional groups, and exfoliated graphene layers. In addition, FEGR increases the hydrogen evolution reaction during the cathodic electrodeposition of Ni resulting in a better distribution of active material. Ionic conductivity is reflected in the high peak current due to the oxidation of a large number of glucose molecules even if Ni(OH)2/FEGR has a low mass loading compared to Ni(OH)2/RGR. Also, the electronic conductivity contributes to the increment of peak current by delivering the electrons very fast to the active material which transfers a large number of Ni(OH)2 to NiOOH in a short time and hence oxidizes a large number of glucose molecules. Additionally, the electronic conductivity plays an important role in the oxidation of glucose at a lower onset potential (120 mV lower than Ni(OH)2/RGR). The exfoliated graphene layers increase the electronic conductivity of the prepared Ni(OH)2/FEGR electrode (as revealed by EIS measurements) which in turn assists the oxidation at lower overpotential. Moreover, the FEGR support boosted the stability of Ni(OH)2 toward the GOR reaction by supplying a constant current for a long time of electrolysis.Conflicts of interest
There are no conflicts to declare.References
- M. Bahari, M. A. Malmberg, D. M. Brown, S. H. Nazari, R. S. Lewis, G. D. Watt and J. N. Harb, Appl. Energy, 2020, 261, 114382 CrossRef CAS.
- A. M. Abdelrahim, M. G. Abd El-Moghny, M. E. El-Shakre and M. S. El-Deab, Electrochim. Acta, 2021, 378, 137991 CrossRef CAS.
- A. H. Abu-Ghazala, H. H. Abdelhady, A. A. Mazhar and M. S. El-Deab, Renewable Energy, 2022, 200, 1120–1133 CrossRef CAS.
- Y. Liu, C. Liu, X. Yu, H. Osgood and G. Wu, Phys. Chem. Chem. Phys., 2017, 19, 330–339 RSC.
- O. A. Mawlid, H. H. Abdelhady and M. S. El-Deab, Energy Convers. Manage., 2022, 273, 116435 CrossRef CAS.
- A. Khoobi and M. Salavati-Niasari, Energy, 2019, 178, 50–56 CrossRef CAS.
- H. Bensalah, S. A. Younssi, M. Ouammou, A. Gurlo and M. F. Bekheet, J. Environ. Chem. Eng., 2020, 8, 103807 CrossRef CAS.
- Y. Chen, K. P. Prasad, X. Wang, H. Pang, R. Yan, A. Than, M. B. Chan-Park and P. Chen, Phys. Chem. Chem. Phys., 2013, 15, 9170–9176 RSC.
- M. Irfan, I. U. Khan, J. Wang, Y. Li and X. Liu, RSC Adv., 2020, 10, 6444–6451 RSC.
- Y. Xin, F. Wang, L. Chen, Y. Li and K. Shen, Green Chem., 2022, 24, 6544–6555 RSC.
- W.-J. Liu, Z. Xu, D. Zhao, X.-Q. Pan, H.-C. Li, X. Hu, Z.-Y. Fan, W.-K. Wang, G.-H. Zhao and S. Jin, Nat. Commun., 2020, 11, 1–11 CrossRef.
- M. R. Rizk, M. G. Abd El-Moghny, A. Mazhar, M. S. El-Deab and B. E. El-Anadouli, Sustainable Energy Fuels, 2021, 5, 986–994 RSC.
- A. M. Abdelrahim, M. G. Abd El-Moghny, M. E. El-Shakre and M. S. El-Deab, Electrochim. Acta, 2023, 440, 141726 CrossRef.
- G. A. El-Nagar, I. Derr, A. Fetyan and C. Roth, Appl. Catal., B, 2017, 204, 185–199 CrossRef CAS.
- R. Fu, Y. Lu, Y. Ding, L. Li, Z. Ren, X. Si and Q. Wu, Microchem. J., 2019, 150, 104106 CrossRef CAS.
- S. Kukunuri, M. R. Krishnan and S. Sampath, Phys. Chem. Chem. Phys., 2015, 17, 23448–23459 RSC.
- M. Gao, X. Liu, M. Irfan, J. Shi, X. Wang and P. Zhang, Int. J. Hydrogen Energy, 2018, 43, 1805–1815 CrossRef CAS.
- A. Eshghi, Int. J. Hydrogen Energy, 2017, 42, 15064–15072 CrossRef CAS.
- M. A. Goda, M. G. Abd El-Moghny and M. S. El-Deab, J. Electrochem. Soc., 2020, 167, 064522 CrossRef CAS.
- Y. Wu, X. Yang, S. Liu, Y. Xing, J. Peng, Y. Peng, G. Ni and X. Jin, RSC Adv., 2020, 10, 39447–39454 RSC.
- A. M. Abdelrahim, M. G. Abd El-Moghny and M. S. El-Deab, RSC Adv., 2021, 11, 26258–26272 RSC.
- T. Nguyen, M. Boudard, M. J. Carmezim and M. F. Montemor, Sci. Rep., 2017, 7, 1–10 CrossRef PubMed.
- L. Liu, Y. Hou, Y. Gao, N. Yang, J. Liu and X. Wang, Electrochim. Acta, 2019, 295, 340–346 CrossRef CAS.
- G.-X. X. Tong, F.-T. T. Liu, W.-H. H. Wu, J.-P. P. Shen, X. Hu and Y. Liang, CrystEngComm, 2012, 14, 5963–5973 RSC.
- P. Lu, Y. Lei, S. Lu, Q. Wang and Q. Liu, Anal. Chim. Acta, 2015, 880, 42–51 CrossRef CAS PubMed.
- J. Xing, S. Wu and K. Y. S. Ng, RSC Adv., 2015, 5, 88780–88786 RSC.
- X. L. Shilei Xie, S. I. Liu and F. Cheng, ChemElectroChem, 2108, 5, 571–582 Search PubMed.
- Y. Song, T. Liu, F. Qian, C. Zhu, B. Yao, E. Duoss, C. Spadaccini, M. Worsley and Y. Li, J. Colloid Interface Sci., 2018, 509, 529–545 CrossRef.
- C. J. Raj, R. Manikandan, W. G. Lee, W. J. Cho, K. H. Yu and B. C. Kim, Electrochim. Acta, 2018, 283, 1543–1550 CrossRef.
- D. Wei, L. Grande, V. Chundi, R. White, C. Bower, P. Andrew and T. Ryhänen, Chem. Commun., 2012, 48, 1239–1241 RSC.
- M. E. Ghaith, M. G. Abd El-Moghny, G. A. El-Nagar, H. H. Alalawy, M. E. El-Shakre and M. S. El-Deab, RSC Adv., 2023, 13, 895–905 RSC.
- S. Yang, M. R. Lohe, K. Müllen and X. Feng, Adv. Funct. Mater., 2016, 28, 6213–6221 CAS.
- Y. Song, D. Y. Feng, T. Y. Liu, Y. Li and X. X. Liu, Nanoscale, 2015, 7, 3581–3587 RSC.
- H. Y. Li, Y. Yu, L. L. Liu, L. L. Liu and Y. Wu, Electrochim. Acta, 2017, 228, 553–561 CrossRef CAS.
- Y. Song, J. L. Xu and X. X. Liu, J. Power Sources, 2014, 249, 48–58 CrossRef CAS.
- J. Hou, Y. Shao, M. W. Ellis, R. B. Moore and B. Yi, Phys. Chem. Chem. Phys., 2011, 13, 15384–15402 RSC.
- J. I. Paredes and J. M. Munuera, J. Mater. Chem. A, 2017, 5, 7228–7242 RSC.
- J. Liu, C. K. Poh, D. Zhan, L. Lai, S. H. Lim, L. Wang, X. Liu, N. Gopal Sahoo, C. Li, Z. Shen and J. Lin, Nano Energy, 2013, 2, 377–386 CrossRef CAS.
- I. O. Baibars, M. G. Abd El-Moghny and M. S. El-Deab, Electrochim. Acta, 2022, 410, 139992 CrossRef CAS.
- M. R. Rizk, M. G. Abd El-Moghny, H. H. Abdelhady, W. M. Ragheb, A. H. Mohamed, H. F. Fouad, M. Mohsen, A. S. Kamel and M. S. El-Deab, Int. J. Hydrogen Energy, 2022, 47, 32145–32157 CrossRef CAS.
- I. O. Baibars, M. G. Abd El-Moghny and M. S. El-Deab, J Environ Chem Eng, 2022, 10, 108736 CrossRef CAS.
- D. S. Hall, D. J. Lockwood, C. Bock and B. R. MacDougall, Proc. R. Soc. A, 2015, 471, 20140792 CrossRef PubMed.
- A. M. Abdelrahim, M. G. Abd El-Moghny, M. E. El-Shakre and M. S. El-Deab, J Energy Storage, 2023, 57, 106218 CrossRef.
- B. J. Plowman, L. A. Jones and S. K. Bhargava, Chem. Commun., 2015, 51, 4331–4346 RSC.
- H. Ashassi-Sorkhabi and P. La’le Badakhshan, Appl. Surf. Sci., 2017, 419, 165–176 CrossRef CAS.
- W. He, X. Li, S. An, T. Li, Y. Zhang and J. Cui, Sci. Rep., 2019, 9, 1–8 CrossRef.
- D. Yang, P. Liu, Y. Gao, H. Wu, Y. Cao, Q. Xiao and H. Li, J. Mater. Chem., 2012, 22, 7224–7231 RSC.
- L.-J. Zhou, X. Huang, H. Chen, P. Jin, G.-D. Li and X. Zou, Dalton Trans., 2015, 44, 11592–11600 RSC.
- P. C. Ooi, M. A. S. Mohammad Haniff, M. F. Mohd Razip Wee, B. T. Goh, C. F. Dee, M. A. Mohamed and B. Y. Majlis, Sci. Rep., 2019, 9, 1–8 CrossRef.
- T. Fan, W. Zeng, W. Tang, C. Yuan, S. Tong, K. Cai, Y. Liu, W. Huang, Y. Min and A. J. Epstein, Nanoscale Res. Lett., 2015, 10, 1–8 CrossRef.
- M. R. Rizk and M. G. Abd El-Moghny, Int. J. Hydrogen Energy, 2021, 46, 645–655 CrossRef.
- L. Hou, Y. Shi, C. Wu, Y. Zhang, Y. Ma, X. Sun, J. Sun, X. Zhang and C. Yuan, Adv. Funct. Mater., 2018, 28, 1705921 CrossRef.
- H. Chen, L. Hu, M. Chen, Y. Yan and L. Wu, Adv. Funct. Mater., 2014, 24, 934–942 CrossRef.
- Y. Chen, W. K. Pang, H. Bai, T. Zhou, Y. Liu, S. Li and Z. Guo, Nano Lett., 2017, 17, 429–436 CrossRef CAS.
- T.-H. Wu and B.-W. Hou, Catal. Sci. Technol., 2021, 11, 4294–4300 RSC.
- J. Zhang, J. Chen, H. Yang, J. Fan, F. Zhou, Y. Wang, G. Wang and R. Wang, J. Solid State Electrochem., 2017, 21, 2975–2984 CrossRef CAS.
- M. Kazazi, A. R. Sedighi and M. A. Mokhtari, Appl. Surf. Sci., 2018, 441, 251–257 CrossRef CAS.
- Z. Sun, X. Cai, D. Y. Feng, Z. H. Huang, Y. Song and X. X. Liu, ChemElectroChem, 2018, 5, 1501–1508 CrossRef CAS.
- Z. Wei, J. Yuan, S. Tang, D. Wu and L. Wu, J. Colloid Interface Sci., 2019, 542, 15–22 CrossRef CAS.
- Y. Wang, Z. Yin, G. Yan, Z. Wang, X. Li, H. Guo and J. Wang, Electrochim. Acta, 2020, 336, 135734 CrossRef CAS.
- Y. Zhang, Y. Gu, S. Lin, J. Wei, Z. Wang, C. Wang, Y. Du and W. Ye, Electrochim. Acta, 2011, 56, 8746–8751 CrossRef CAS.
- G. A. El-Nagar and C. Roth, Phys. Chem. Chem. Phys., 2017, 19, 2537–2548 RSC.
- Y. Song, D. Y. Feng, T. Y. Liu, Y. Li and X. X. Liu, Nanoscale, 2015, 7, 3581–3587 RSC.
- Y. Xie and Y. Zhou, J. Mater. Res., 2019, 34, 2472–2481 CrossRef CAS.
- B. E. Conway, Electrochemical supercapacitors scientific fundamentals and technological applications, springer US, 1999 Search PubMed.
- M. Irfan, X. Liu, S. Li, I. U. Khan, Y. Li, J. Wang, X. Wang, X. Du, G. Wang and P. Zhang, Renewable Energy, 2020, 155, 1118–1126 CrossRef CAS.
- A. M. Ahmed, S. Y. Sayed, G. A. El-Nagar, W. M. Morsi, M. S. El-Deab and B. E. El-Anadouli, J. Electroanal. Chem., 2019, 835, 313–323 CrossRef CAS.
- Q. Yi, W. Huang, W. Yu, L. Li and X. Liu, Electroanalysis, 2008, 20, 2016–2022 CrossRef.
- A. S. Danial, M. M. Saleh, S. A. Salih and M. I. Awad, J. Power Sources, 2015, 293, 101–108 CrossRef.
- M. Cao, H. Cao, W. Meng, Q. Wang, Y. Bi, X. Liang, H. Yang, L. Zhang, M.-F. Lang and J. Sun, Int. J. Hydrogen Energy, 2021, 46, 28527–28536 CrossRef.
- S. M. El-Refaei, M. I. Awad, B. E. El-Anadouli and M. M. Saleh, Electrochim. Acta, 2013, 92, 460–467 CrossRef.
- X. Gao, W. Feng, Y. Xu, Y. Jiang, C. Huang, Y. Yi, A. Guo, X. Qiu and W. Chen, Nanoscale Res. Lett., 2020, 15, 1–9 CrossRef PubMed.
- F. A. Mohammed, M. M. Khalaf, I. M. A. Mohamed, M. M. Saleh and H. M. Abd El-Lateef, Microchem. J., 2020, 153, 104507 CrossRef.
| This journal is © The Royal Society of Chemistry 2023 |