
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePreferential formation of specific hexose and heptose in the formose reaction under microwave irradiation†
Akihito Hashidzume *a,
Toru Imaia,
Nanako Deguchia,
Takashi Tanibayashia,
Takumi Ikedaa,
Tomohiro Michitakaa,
Satoki Kuwaharaa,
Masaki Nakahataa,
Yuri Kamonb and
Yasuto Todokoroc
*a,
Toru Imaia,
Nanako Deguchia,
Takashi Tanibayashia,
Takumi Ikedaa,
Tomohiro Michitakaa,
Satoki Kuwaharaa,
Masaki Nakahataa,
Yuri Kamonb and
Yasuto Todokoroc
aDepartment of Macromolecular Science, Graduate School of Science, Osaka University, 1-1 Machikaneyama-cho, Toyonaka, Osaka 560-0043, Japan. E-mail: hashidzume@chem.sci.osaka-u.ac.jp
bAdministrative Department, Graduate School of Science, Osaka University, 1-1 Machikaneyama-cho, Toyonaka, Osaka 560-0043, Japan
cAnalytical Instrument Facility, Graduate School of Science, Osaka University, 1-1 Machikaneyama-cho, Toyonaka, Osaka 560-0043, Japan
First published on 30th January 2023
Abstract
To realize sustainable societies, the production of organic compounds not based on fossil resources should be developed. Thus, C1 chemistry, utilizing one-carbon compounds as starting materials, has been of increasing importance. In particular, the formose reaction is promising because the reaction produces sugars (monosaccharides) from formaldehyde under basic conditions. On the other hand, since microwave (MW) induces the rotational motion of molecules, MW irradiation often improves the selectivity and efficiency of reactions. In this study, the formose reaction under MW irradiation was thus investigated under various conditions. The formose reaction proceeded very fast using 1.0 mol per kg formaldehyde and 55 mmol per kg calcium hydroxide (Ca(OH)2) as a catalyst at a high set temperature (150 °C) for a short time (1 min) to form preferentially specific hexose and heptose. The major products were isolated by thin layer chromatography and characterized by mass spectroscopy and NMR. These characterization data elucidated that the hexose and heptose were 2-hydroxymethyl-1,2,4,5-tetrahydroxy-3-pentanone (C6*) and 2,4-bis(hydroxymethyl)-1,2,4,5-tetrahydroxy-3-pentanone (C7*), respectively. On the basis of these observations, as well as density functional theory calculations, a plausible reaction pathway was also discussed; once 1,3-dihydroxyacetone is formed, consecutive aldol reactions favorably occur to form C6* and C7*.
1 Introduction
Today, huge amounts of various organic compounds are produced by the petrochemical industry from fossil resources.1 The production of organic compounds not based on fossil resources should be developed to realize sustainable societies. In these decades, biomass has been used for the production of biofuel and bioplastics.2–9 On the other hand, C1 chemistry, utilizing one-carbon compounds (e.g., carbon dioxide, formic acid, formaldehyde and methanol) as starting materials, has been of increasing importance.10–17 Since formaldehyde has the molecular formula CH2O, it can be oligomerized to form sugars (monosaccharides), of which the general molecular formula is (CH2O)n. In 1861, Butlerow18 first reported the formation of sugar-like products called “formose” from an aqueous solution of formaldehyde at higher temperatures under basic conditions. This reaction is called the “formose reaction”. Previous studies have demonstrated that the formose reaction usually yields a complicated mixture of sugars and sugar alcohols containing nonnatural branched and L-isomers.19–26 Some research groups have worked on a controlled formose reaction, aiming at selective formation of specific sugars or sugar alcohols. Shigemasa et al.27 first isolated a sugar alcohol of seven carbons (2,4-bis(hydroxymethyl)pentane-1,2,3,4,5-pentaol) from a mixture of formose reaction carried out in water at 60 °C by switching the catalyst from calcium hydroxide (Ca(OH)2) to potassium hydroxide (KOH) at the end of induction period. Matsumoto et al.28 selectively obtained a triose (1,3-dihydoxyacetone) by formose reaction carried out at 100 °C in ethanol using 3-ethylbenzothiazoline bromide as a catalyst in the presence of triethylamine. More recently, some research groups have reported that formose reaction yields preferably pentoses in the presence of borate,29 silicate minerals30 and hydroxyapatite.31 We also reported a few examples of controlled formose reaction in reverse micelles and in the presence of boronic acid compounds.32–34 The formose reaction in reverse micelles was accelerated to yield ethylene glycol as the major product.32 The formose reaction in the presence of phenylboronic acid produced favorably triose and tetrose, whereas the reaction in the presence of a boronic acid polymer formed preferably sugar alcohols of carbon numbers from six to eight.33,34Since microwave (MW) induces the rotational motion of molecules, MW irradiation allows very fast heating to improve the selectivity and efficiency of reactions.35–41 To the best of our knowledge, although MW was utilized for formose reaction in an example, the authors did not describe experimental details or characterization of the products.42 Thus, this study deals with formose reaction carried out using MW irradiation under varying conditions, and indicates that formose reaction proceeds very fast using Ca(OH)2 as a catalyst at a high set temperature (150 °C) for a short time (1 min), resulting in the preferential formation of specific sugars. The major products, i.e., hexose and heptose, were isolated by thin layer chromatography (TLC) and characterized by electrospray ionization mass spectroscopy (ESI-MS) and NMR. On the basis of these observations, a plausible reaction pathway is also discussed.
2 Experimental section
2.1 Materials
An aqueous solution of formaldehyde (36 wt%), barium carbonate (BaCO3) and N,N,N′,N′-tetraethylethylenediamine were purchased from Sigma-Aldrich (St. Louis, MO). Calcium hydroxide (Ca(OH)2), silver(I) oxide (Ag2O), alumina (Al2O3), imidazole, pyridine and acetylacetone were purchased from FUJIFILM Wako Pure Chemical Corp. (Osaka, Japan). Barium hydroxide octahydrate (Ba(OH)2·8H2O), sodium hydroxide (NaOH), potassium hydroxide (KOH), triethylamine, 2-picoline, 3-picoline, acetic acid and ammonium acetate were purchased from Nacalai Tesque, Inc. (Kyoto, Japan). Lithium oxide monohydrate (LiOH·H2O), 2-(dimethylamino)ethanol and acetonitrile for HPLC were purchased from Kishida Chemical Co., Ltd. (Osaka, Japan). Magnesium hydroxide (Mg(OH)2), 1,4-diazabicyclo[2.2.2]octane (DABCO) and N-methylpiperidine were purchased from Kanto Chemical Co. Inc. (Tokyo, Japan). Amberlite® IR-120 H and IRA-910 Cl were purchased from Sigma-Aldrich (St. Louis, MO). Amberlite® IRA-910 Cl was used after exchanging Cl− ions to OH− ions (Amberlite® IRA-910 OH) by washing with 1.0 M NaOH. For thin layer chromatography (TLC), Merck precoated TLC plates (silica gel 60 F254) were used. Water was purified with a Millipore Milli-Q system. Other reagents were used without further purification.2.2 Measurements
The UV-visible absorption spectra were recorded on a HITACHI U-4100 spectrometer. 1H NMR spectra were measured on a JEOL JNM ECS400 or ECA500 spectrometer using D2O as a solvent at 30 °C. Chemical shifts were referenced to acetonitrile signal (δ = 2.08 ppm). 1H NMR, 13C NMR, heteronuclear single-quantum correlation (HSQC) and heteronuclear multiple bond correlation (HMBC) spectra were recorded on a Bruker AVANCE700 spectrometer using D2O as a solvent at 25 °C. Acetonitrile was used as an internal standard and chemical shifts were referenced to the signal due to the methyl carbon of acetonitrile (δ = 1.47 ppm). High performance liquid chromatography (HPLC) measurements for the formose samples were carried out on a Jasco LC-200 Plus system equipped with a Jasco PU-2080 pump and an RI-2031 detector. A Shodex Asahipak 5NH2P-50 4E column was used, and a mixed solvent of acetonitrile and water (3/1, v/v) was used as eluent with a flow rate of 0.6 mL min−1 and the preset temperature of column oven was 40 °C. Liquid chromatography-mass spectroscopy (LC-MS) measurements were performed on a Bruker Daltonics micrOTOF-QIII compact connected with a Shimadzu Prominence UFLC system. In the LC system, Shimadzu LC-20AD pumps and a Shodex 5NH2P-50 4E column were equipped, and a mixed solvent of acetonitrile and water (3/1, v/v) containing 0.1 vol% formic acid was used as an eluent at a flow rate of 0.6 mL min−1. Electrospray-ionization mass spectroscopy (ESI-MS) data were recorded in a positive mode on a Thermo Fisher Scientific LTQ-Orbitrap-XL, controlled by the XCARIBUR 2.1 software package. The condition of ionization was set to the following parameters; ion spray voltage at 3.5 kV, ion spray temperature at 100 °C and ion transfer tube temperature at 275 °C. Mixed solvent of methanol and water (1/1, v/v) was used as a solvent. Internal calibration of ESI-MS was carried out using the monoisotopic peaks of sodium adducted ion of diethyl phthalate (m/z 314.1410), protonated ion of di-2-ethylhexyl phthalate (m/z 391.2843) and sodium adducted ion of di-2-ethylhexyl-phthalate (m/z 413.2662). Scanning electron microscopy (SEM) images were obtained on a JEOL JSM-7600F scanning electron microscope. Ca(OH)2 crystallites were directly mounted on a conductive tape. The morphology of Ca(OH)2 crystallites was scanned at an accelerating voltage of 5.0 kV with the LEI mode.2.3 Formose reaction under microwave irradiation
A typical procedure is described below. An aqueous solution of formaldehyde (36 wt%) was diluted to 1.0 mol kg−1 with water. A predetermined amount of Ca(OH)2 was added to the diluted aqueous formaldehyde solution (5.0 mL) in a 2–5 mL vial. After the reaction mixture was shaken for 1 min and stirred for 5 min, the reaction was carried out under microwave (MW) irradiation at 2.45 GHz using a Biotage Initiator+ microwave synthesizer at a set temperature of 150 °C for 1 min. The very high absorption level was used. The other absorption levels, i.e., normal, high and low, were also tested, but the results were practically the same. After heating under MW irradiation, the reaction was quenched by cooling with a water bath for 1 min and an ice-water bath for 5 min. A portion (60 μL) of the reaction mixture was taken for the acetylacetone method, which is used to determine the concentration of formaldehyde based on the quantitative formation of 3,5-diacetyl-1,4-dihydrolutidine from formaldehyde and acetylacetone.43 The experiments were done at least three times, and the errors of conversion were smaller than ca. 10%. The products were purified by treatment with ion-exchange resins (Amberlite® IR-120 H and IRA-910 OH) for 30 min each. After filtration and freeze-drying of the solvent, the product was recovered. The products were also purified by TLC on a silica gel plate using a mixed solvent of 1-butanol, ethanol and water (9/3/1, v/v/v) as an eluent. The spots were detected by p-anisaldehyde staining.2.4 Density functional theory calculations
To evaluate the total energy for the sugars, density functional theory (DFT) calculations were carried out for the isomers of trioses, tetroses and pentoses using the Gaussian 09 program.44 In all the calculations, DFT with B3LYP functional was used and 6-311++G(2d,p) basis sets were applied for the hydrogen, carbon and oxygen atoms. All the geometries of the model systems were fully optimized, in which the requested convergence condition of difference on root mean square (RMS) density matrix, maximum density matrix and energy were 10−8 (in atomic unit), 10−6 (in atomic unit) and 10−6 Hartee, respectively.3 Results and discussion
Our preliminary results on formose reaction under MW irradiation using various catalysts elucidated that Ca(OH)2 is the most efficient catalyst (see ESI†). Formose reaction under MW irradiation using Ca(OH)2 as a catalyst was thus carried out to optimize the conditions. We conducted formose reaction by irradiating an aqueous solution of formaldehyde (1.0 mol kg−1, 5 mL) with MW at 2.45 GHz on a Biotage Initiator+ microwave synthesizer. Conversions were determined by the acetylacetone method.43 Fig. 1A compares the conversions at varying Ca(OH)2 concentrations for formose reaction under MW irradiation at a set temperature of 150 °C for 1, 3 and 5 min. It should be noted here that the conversion was practically independent of the reaction time under these conditions. As the Ca(OH)2 concentration ([Ca(OH)2]) was increased from 0 to 40 mmol kg−1, the conversion gradually increased from 0 to 20%. As [Ca(OH)2] was further increased, the conversion increased rapidly and reached a quantitative one at 55 mmol kg−1. Fig. 1B compares the time-conversion plots for formose reaction under MW irradiation at set temperatures of 150, 100 and 60 °C. At 150 °C, the conversion increased rapidly to a quantitative one within 1 min (see also Fig. S2 in ESI†). Using a 2–5 mL vial, the temperature of reaction mixture (5 mL) increased in a sigmoidal manner to 150 °C in ca. 1 min, as it can be seen in Fig. S3A in ESI.† When the temperature was set at 100 °C, the conversion increased more slowly and reached ca. 90% after 30 min. On the other hand, when the temperature was set at 60 °C, the conversion increased slowly to ca. 20% in the first 120 min and then leveled off.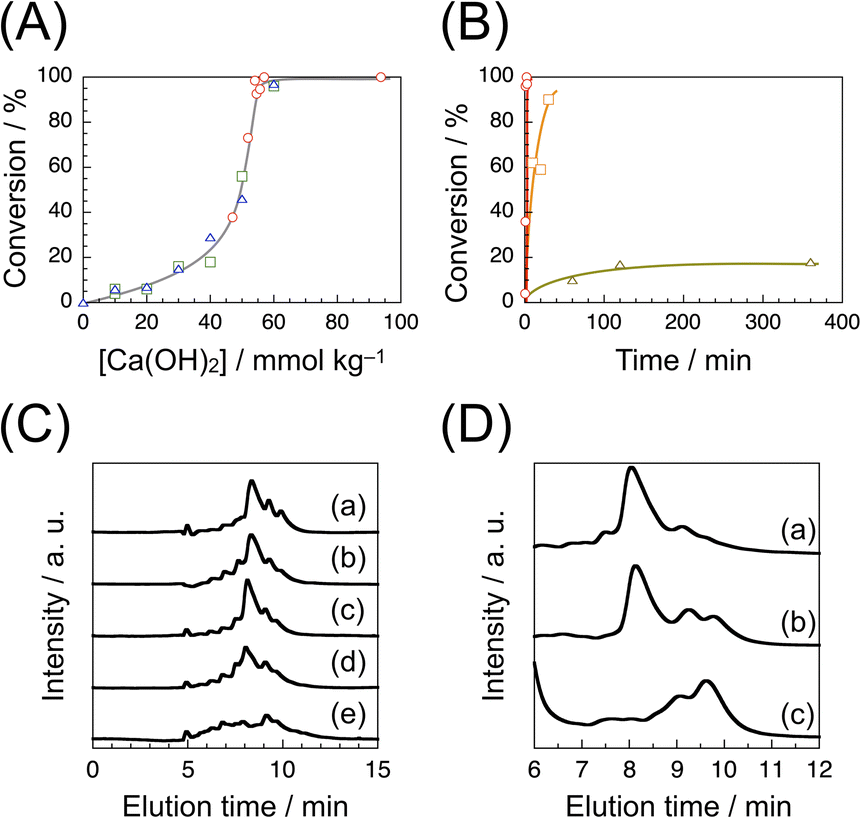 | ||
| Fig. 1 Formose reaction under MW irradiation using 1.0 mol per kg formaldehyde and Ca(OH)2 under various conditions. (A) The conversion of formose reaction carried out at a set temperature of 150 °C for 1 (red circle), 3 (green square), and 5 min (blue triangle) using varying concentrations of Ca(OH)2. (B) Time-conversion plots for formose reaction carried out using 55 mmol per kg Ca(OH)2 at set temperatures of 150 (red circle), 100 (orange square), and 60 °C (dark yellow triangle). (C) HPLC charts for the products of formose reaction carried out at a set temperature of 150 °C for 1 min using (a) 47, (b) 54, (c) 55, (d) 57, and (e) 94 mmol per kg Ca(OH)2. (D) HPLC charts for the products of formose reaction carried out using 55 mmol per kg Ca(OH)2 at set temperatures of (a) 150, (b) 100, and (c) 60 °C for 1, 20, and 360 min, respectively. | ||
The products of formose reaction under MW irradiation at a set temperature of 150 °C for 1 min at varying Ca(OH)2 concentrations were treated with ion exchange resins and characterized by HPLC (Fig. 1C). In the cases of formose reaction using 47, 54 and 55 mmol per kg Ca(OH)2, the HPLC charts are similar, which show a major signal at ca. 8.2 min. As it can be seen in Fig. 1A, the conversions were determined to be 37.8, 98.5 and 92.6% at [Ca(OH)2] = 47, 54 and 55 mmol kg−1, respectively. These observations indicate that the major product was formed even at lower conversions. At 57 mmol kg−1 (100% conversion), the HPLC chart also contains the major signal at ca. 8.2 min as well as rather significant minor signals, indicative of a lower selectivity. It should be noted here that the HPLC chart for [Ca(OH)2] = 94 mmol kg−1 (100% conversion) does not exhibit a significant signal at ca. 8.2 min, indicating that the major product was decomposed at a high Ca(OH)2 concentration in 1 min. The products obtained by formose reaction under MW irradiation at set temperatures of 150, 100 and 60 °C were also treated with ion exchange resins and characterized by HPLC (Fig. 1D). The HPLC chart for the product obtained at 150 °C after 1 min exhibits a large signal at ca. 8.2 min. The HPLC chart for the product obtained at 100 °C after 30 min also shows a large signal at ca. 8.2 min, but minor signals in the elution time region of 9–10 min are stronger, indicative of a lower selectivity than that at 150 °C. The HPLC chart for the product after 120 min at 60 °C contains a few larger signals in the elution time region of 9–10 min without any significant signals at ca. 8 min. The product of the formose reaction under MW irradiation at a set temperature of 150 °C for 45 s was characterized by LC-MS (Fig. S4 in ESI†). The LC-MS data indicate that the major signals at ca. 8.2 min exhibit MS signals at m/z = 203.07 and 233.08 and minor signals at ca. 9.8 and 10.5 min show MS signals at m/z = 205.08 and 235.09, respectively. Since these MS signals are due to sodium adducts, it can be concluded that the major signal is ascribable to hexose (C6H12O6) and heptose (C7H14O7) and the minor signals are assignable to sugar alcohols of six and seven carbons (C6H14O6 and C7H16O7), respectively (it is known that sugar alcohols are the major byproduct of formose reaction, which are formed through Cannizzaro reaction.19–23).
The hexose and heptose were purified by TLC on a silica gel plate using a mixed solvent of 1-butanol, ethanol and water (9/3/1, v/v/v) as an eluent (Fig. 2A and B). The yields of hexose and heptose were ca. 1.7 and 2.0 mg (ca. 0.37 and 0.44%), respectively, using three batches of formose reaction (3 × 150 mg of formaldehyde).45 To improve the yields of hexose and heptose, the reaction should be carried out on a larger scale, although it was not possible to scale up using a Biotage Initiator+ microwave synthesizer (the conversions were much lower on 10 and 20 mL scales using a 10–20 mL vial presumably because of the lack of power of the microwave synthesizer). The structures of hexose and heptose were characterized by 1H NMR, 13C NMR, HSQC and HMBC using D2O as solvent. As it can be seen in Fig. 2C, the 1H NMR spectrum for the heptose exhibits two doublets at ca. 4.0 and 3.7 ppm. The 13C NMR for the heptose indicates three signals at ca. 215.1, 86.1 and 64.6 ppm (Fig. 2C). The HSQC and HMBC spectra exhibit significant correlation signals (Figs. S5A and B in ESI†). These NMR data have confirmed that the heptose is 2,4-bis(hydroxymethyl)-1,2,4,5-tetrahydroxy-3-pentanone (C7*), as it can be seen in Fig. 2. On the other hand, the 1H NMR spectrum for the hexose shows signals at ca. 4.9, 4.0, 3.9–3.8 and 3.6 ppm with the ratio of area intensities of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:3:2 (Fig. 2C). The 1H NMR signal at ca. 4.9 ppm is ascribed to the methine proton, and the signals at ca. 4.0, 3.9–3.8 and 3.6 ppm are assignable to the methylene protons. In the 13C NMR spectrum for the hexose, there are six signals at ca. 216.3, 84.8, 76.5, 65.5, 64.4 and 62.8 ppm (Fig. 2D). These NMR data as well as the HSQC and HMBC spectra (Figs. S5C and D in ESI†) have confirmed that the hexose is 2-hydroxymethyl-1,2,4,5-tetrahydroxy-3-pentanone (C6*), as shown in Fig. 2. Previously, Shigemasa et al.46,47 obtained the same heptose C7* by formose reaction, in which, e.g., a solution of formaldehyde in methanol (2.5 M) was warmed in the presence of barium chloride at 60 °C for 20–25 min with adjusting the solution pH to 12.0 with KOH. It is thus likely that the heptose C7* with a branched and highly symmetric structure is preferentially formed in formose reaction. It is noteworthy that the present formose reaction under MW irradiation requires a much simpler procedure than those by Shigemasa et al.46,47
:1:3:2 (Fig. 2C). The 1H NMR signal at ca. 4.9 ppm is ascribed to the methine proton, and the signals at ca. 4.0, 3.9–3.8 and 3.6 ppm are assignable to the methylene protons. In the 13C NMR spectrum for the hexose, there are six signals at ca. 216.3, 84.8, 76.5, 65.5, 64.4 and 62.8 ppm (Fig. 2D). These NMR data as well as the HSQC and HMBC spectra (Figs. S5C and D in ESI†) have confirmed that the hexose is 2-hydroxymethyl-1,2,4,5-tetrahydroxy-3-pentanone (C6*), as shown in Fig. 2. Previously, Shigemasa et al.46,47 obtained the same heptose C7* by formose reaction, in which, e.g., a solution of formaldehyde in methanol (2.5 M) was warmed in the presence of barium chloride at 60 °C for 20–25 min with adjusting the solution pH to 12.0 with KOH. It is thus likely that the heptose C7* with a branched and highly symmetric structure is preferentially formed in formose reaction. It is noteworthy that the present formose reaction under MW irradiation requires a much simpler procedure than those by Shigemasa et al.46,47
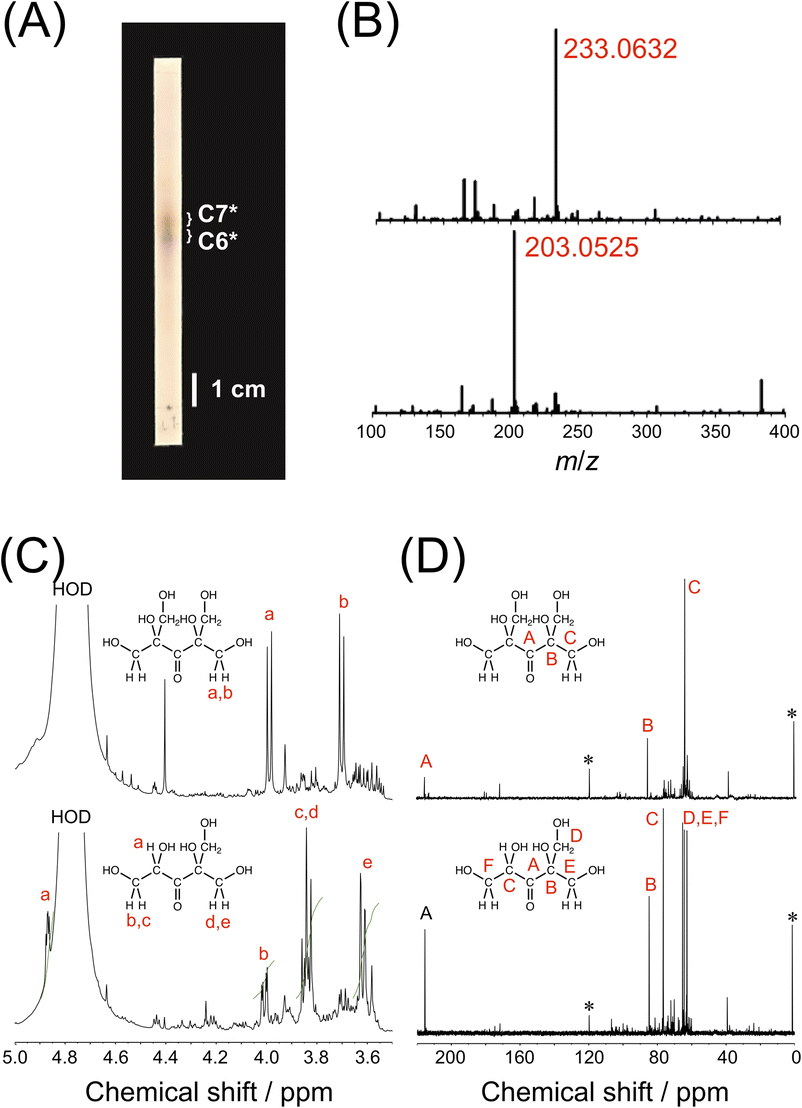 | ||
| Fig. 2 Purification and characterization of the major products (heptose C7* and hexose C6*) of formose reaction under MW irradiation using 1.0 mol per kg formaldehyde and 55 mmol per kg Ca(OH)2 at a set temperature of 150 °C for 1 min. (A) A photograph of a TLC plate for separation of the major products. (B) MS for C7* (upper) and C6* (lower). (C) 1H NMR spectra for C7* (upper) and C6* (lower) (D2O). (D) 13C NMR spectra for C7* (upper) and C6* (lower) (D2O). | ||
Here, we discuss how C6* and C7* are preferentially formed. In the present formose reaction, high temperature (150 °C) and short reaction time (1 min) are critical, in which decomposition reactions (e.g., retro-aldol reaction and Cannizzaro reaction) are significantly suppressed. C6* and C7* were also formed preferentially in formose reaction by heating with an oil bath thermostated at 150 °C for a short time (see ESI†), indicating that the type of heat source (MW or an oil bath) is not important. Since C7* obtained in this study is formed by aldol reaction of C6*, these products should be on the same reaction pathway. It is thus necessary to elucidate which pentose is the precursor of C6* and how the pentose is formed. Here we assume that the major reaction is aldol reaction of the sugars formed with formaldehyde (C1) because C6* and C7* were obtained preferentially even at lower conversions (Fig. 1C). Fig. 3 exhibits reaction pathways through acyloin condensation, aldol reaction with C1 and isomerization via enediol to produce all the possible pentoses, where retro-aldol reaction or Cannizzaro reaction are not included.48 Fig. 3 contains C1, glycolaldehyde (C2), two trioses (glyceraldehyde (C3-1) and 1,3-dihydroxyacetone (C3-2)), four tetroses ((2R,3R)- and (2R,3S)-2,3,4-trihydroxybutanal (C4-1RR and C4-1RS), 1,3,4-trihydroxy-2-butanone (C4-2) and 2-hydroxymethyl-2,3-dihydroxypropanal (C4-1b2)) and twelve pentoses ((2R,3R,4R)-, (2R,3R,4S)-, (2R,3S,4R)- and (2R,3S,4S)-2,3,4,5-tetrahydroxypentanal (C5-1RRR, C5-1RRS, C5-1RSR and C5-1RSS), (3R,4R)- and (3R,4S)-1,3,4,5-tetrahydroxy-2-pentanone (C5-2RR and C5-2RS), (2R,4R)- and (2R,4S)-1,2,4,5-tetrahydroxy-3-pentanone (C5-3RR and C5-3RS), (2R,3R)- and (2R,3S)-2,3,4-trihydroxy-2-hydroxymethylbutanal (C5-1b2RR and C5-1b2RS), 2,3,4-trihydroxy-3-hydroxymethylbutanal (C5-1b3) and 1,3,4-trihydroxy-3-hydroxymethyl-2-butanone (C5-2b3)). The isomers are denoted by codes Cn-mXXX and Cn-mblXXX for linear and branched isomers, respectively, where n is the carbon number, m is the position number of carbonyl, l is the position number of branch and X denotes R or S configuration if necessary (see also Fig. S6 in ESI†). The transition states should be considered to discuss the plausible reaction pathway. However, the present system was too large to be simulated by DFT calculation because the transition states should contain not only sugar and formaldehyde molecules but also water molecules and Ca(OH)2. Unavoidably, we assume that thermodynamically stable isomers were preferably formed, because the present formose reaction was carried out without adding any additive, which stabilizes specific sugars or sugar alcohols. Thus, the structures of all isomers of the trioses, tetroses and pentoses in Fig. S6 in ESI† were optimized by DFT calculations using a Gaussian 09 software44 with B3LYP functional and 6-311++G(2d,p) basis set, and the total energies were compared, as it can be seen in Fig. S7A (see also Table S2 in ESI†). Here, we assume that enantiomers possess the same total energies. Comparing the total energies of trioses, C3-2 is more stable than C3-1 by 24.1 kJ mol−1. Among the four tetroses and twelve pentoses, C4-2 and C5-3RR are the most stable, respectively. On the basis of these data, we propose a plausible reaction pathway for the preferential formation of C6* and C7* in the formose reaction under MW irradiation at a high set temperature (150 °C) for a short time (1 min), as it can be seen in Fig. S7B.† After the formation of C2 by acyloin condensation of two C1 molecules, C3-1 is formed by aldol condensation, followed by isomerization via enediol to form C3-2. C3-2 may be also formed directly from C2 and C1. Once C3-2 is formed, consequent aldol reactions favorably occur to form C4-2, C5-3RR, C6* and C7*. It should be noted here that Fig. 1C indicates that the main products were C6* and C7* even at lower conversions. These observations indicate that aldol reaction proceeds much faster than does acyloin condensation to form C2. It is also noteworthy that the HPLC and LC-MS data indicated that Cannizzaro reaction preferably occurred at a lower temperature for a longer time to form sugar alcohols.
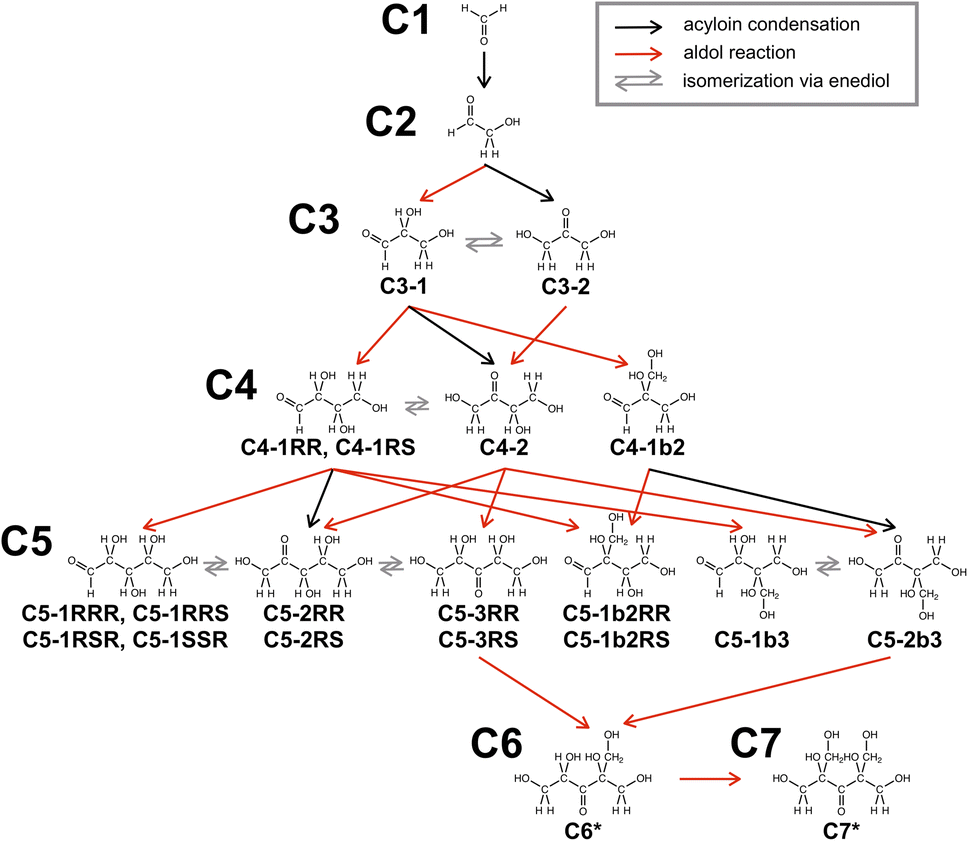 | ||
| Fig. 3 Plausible reaction pathways for the preferential formation of C6* and C7* in the formose reaction under MW irradiation. | ||
In this study, formose reaction was carried out under MW irradiation for a heterogeneous mixture containing Ca(OH)2 (275 μmol) and an aqueous solution of formaldehyde (5 mL). Fig. S8 in ESI† shows the temperature dependence of the saturation concentration of Ca(OH)2 in water ([Ca(OH)2]sat), indicating that [Ca(OH)2]sat decreases with increasing temperature.49 Since [Ca(OH)2]sat ≈ 6 mmol kg−1 (<55 mmol kg−1) at 150 °C, the present formose reaction under MW irradiation proceeded in a heterogeneous state. As can be seen in Fig. 1A, the conversions were lower than 10% at [Ca(OH)2] ≤ 20 mmol kg−1, which is larger than the [Ca(OH)2]sat value. Fig. 1C also indicates that the decomposition of products occurred markedly at [Ca(OH)2] > 57 mmol kg−1. These observations indicate that the present formose reaction was catalyzed by Ca(OH)2 which was not dissolved rather than Ca2+ ions in solution. Recently, some research groups have reported formose reaction on the surface of minerals,50,51 titania52 or meteorites.53,54 In the present system, it is thus likely that the formose reaction considerably proceeded on the surface of Ca(OH)2 crystallites. As it can be seen in Fig. S9 in ESI,† SEM images display porous morphologies of the surface of Ca(OH)2. The effect of surface of Ca(OH)2 crystallites on the formose reaction will be investigated in detail in the future.
4 Conclusions
This study demonstrated that formose reaction proceeded very fast using 1.0 mol per kg formaldehyde and 55 mmol per kg Ca(OH)2 as a catalyst under MW irradiation at a high set temperature (150 °C) for a short time (1 min) to form preferentially specific hexose and heptose. The major products (i.e., C6* and C7*), isolated by TLC, were characterized by MS and NMR. On the basis of DFT calculations, a plausible reaction pathway was proposed.Author contributions
A. H. supervised the project; A. H., T. Imai and T. M. designed the project; T. Imai, N. D., T. T., T. Ikeda, T. M. and S. K. performed the formose reaction experiments; Y. T. performed the NMR experiments; M. N. performed the DFT calculations and SEM measurements; A. H., T. Imai, N. D., T. T., T. M. and Y. K. analyzed data; A. H., M. N. and Y. K. discussed the results; A. H. Y. K. and M. N. wrote the paper.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was partly supported by JSPS Kakenhi Grant Number JP26288061.Notes and references
- Handbook of Petrochemicals Production Processes, ed. R. A. Meyers, McGraw-Hill, New York, 2019 Search PubMed.
- A. J. Ragauskas, C. K. Williams, B. H. Davison, G. Britovsek, J. Cairney, C. A. Eckert, W. J. Frederick, J. P. Hallett, D. J. Leak, C. L. Liotta, J. R. Mielenz, R. Murphy, R. Templer and T. Tschaplinski, Science, 2006, 311, 484–489 CrossRef PubMed.
- M. E. Himmel, S.-Y. Ding, D. K. Johnson, W. S. Adney, M. R. Nimlos, J. W. Brady and T. D. Foust, Science, 2007, 315, 804–807 CrossRef CAS PubMed.
- L. Hu, G. Zhao, W. Hao, X. Tang, Y. Sun, L. Lin and S. Liu, RSC Adv., 2012, 2, 11184–11206 RSC.
- M. Singhvi and D. Gokhale, RSC Adv., 2013, 3, 13558–13568 RSC.
- M. Asadieraghi, W. M. Ashri Wan Daud and H. F. Abbas, RSC Adv., 2015, 5, 22234–22255 RSC.
- J. Gao, Q. Liu, F. Gu, B. Liu, Z. Zhong and F. Su, RSC Adv., 2015, 5, 22759–22776 RSC.
- L. Korzen, I. N. Pulidindi, A. Israel, A. Abelson and A. Gedanken, RSC Adv., 2015, 5, 16223–16229 RSC.
- H. Xia, S. Xu, H. Hu, J. An and C. Li, RSC Adv., 2018, 8, 30875–30886 RSC.
- Z.-Z. Yang, Y.-N. Zhao and L.-N. He, RSC Adv., 2011, 1, 545–567 RSC.
- S. Das and W. M. A. Wan Daud, RSC Adv., 2014, 4, 20856–20893 RSC.
- T. W. van Deelen, C. Hernández Mejía and K. P. de Jong, Nat. Cat., 2019, 2, 955–970 CrossRef CAS.
- W. Zhou, K. Cheng, J. Kang, C. Zhou, V. Subramanian, Q. Zhang and Y. Wang, Chem. Soc. Rev., 2019, 48, 3193–3228 RSC.
- G. Chen, G. I. N. Waterhouse, R. Shi, J. Zhao, Z. Li, L.-Z. Wu, C.-H. Tung and T. Zhang, Angew. Chem., Int. Ed., 2019, 58, 17528–17551 CrossRef CAS PubMed.
- Y. Liu, D. Deng and X. Bao, Chem, 2020, 6, 2497–2514 CAS.
- Q. Zhang, J. Yu and A. Corma, Adv. Mater., 2020, 32, 2002927 CrossRef PubMed.
- L. Kang, B. Wang, A. T. Güntner, S. Xu, X. Wan, Y. Liu, S. Marlow, Y. Ren, D. Gianolio, C. C. Tang, V. Murzin, H. Asakura, Q. He, S. Guan, J. J. Velasco-Vélez, S. E. Pratsinis, Y. Guo and F. R. Wang, Angew. Chem., Int. Ed., 2021, 60, 14420–14428 CrossRef CAS PubMed.
- A. Butlerow, Ann. Chem., 1861, 120, 295–298 CrossRef.
- R. Breslow, Tetrahedron Lett., 1959, 1, 22–26 CrossRef.
- T. Mizuno and A. H. Weiss, Adv. Carbohydr. Chem. Biochem., 1974, 29, 173–227 CrossRef CAS.
- A. H. Weiss, R. F. Socha, V. A. Likholobov and M. M. Sakharov, Appl. Catal., 1981, 1, 237–246 CrossRef CAS.
- R. F. Socha, A. H. Weiss and M. M. Sakharov, J. Catal., 1981, 67, 207–217 CrossRef CAS.
- Y. Shigemasa and H. Saimoto, Trends Glycosci. Glycotechnol., 1990, 2, 119–123 CrossRef CAS.
- I. V. Delidovich, A. N. Simonov, O. P. Taran and V. N. Parmon, ChemSusChem, 2014, 7, 1833–1846 CrossRef CAS PubMed.
- C. Appayee and R. Breslow, J. Am. Chem. Soc., 2014, 136, 3720–3723 CrossRef CAS PubMed.
- L. Cheng, C. Doubleday and R. Breslow, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 4218–4220 CrossRef CAS PubMed.
- Y. Shigemasa, O. Nagae, C. Sakazawa, R. Nakashima and T. Matsuura, J. Am. Chem. Soc., 1978, 100, 1309–1310 CrossRef CAS.
- T. Matsumoto, H. Yamamoto and S. Inoue, J. Am. Chem. Soc., 1984, 106, 4829–4832 CrossRef CAS.
- A. Ricardo, M. A. Carrigan, A. N. Olcott and S. A. Benner, Science, 2004, 303, 196 CrossRef CAS PubMed.
- J. B. Lambert, S. A. Gurusamy-Thangavelu and K. Ma, Science, 2010, 327, 984–986 CrossRef CAS PubMed.
- K. Usami and A. Okamoto, Org. Biomol. Chem., 2017, 15, 8888–8893 RSC.
- M. Masaoka, T. Michitaka and A. Hashidzume, Beilstein J. Org. Chem., 2016, 12, 2663–2667 CrossRef CAS PubMed.
- T. Imai, T. Michitaka and A. Hashidzume, Beilstein J. Org. Chem., 2016, 12, 2668–2672 CrossRef CAS PubMed.
- T. Michitaka, T. Imai and A. Hashidzume, Polymers, 2017, 9, 549 CrossRef PubMed.
- P. Lidström, J. Tierney, B. Wathey and J. Westman, Tetrahedron, 2001, 57, 9225–9283 CrossRef.
- A. de la Hoz, Á. Díaz-Ortiz and A. Moreno, Chem. Soc. Rev., 2005, 34, 164–178 RSC.
- C. O. Kappe and D. Dallinger, Nat. Rev. Drug Discovery, 2006, 5, 51–63 CrossRef CAS PubMed.
- C. O. Kappe, Chem. Soc. Rev., 2008, 37, 1127–1139 RSC.
- Microwaves in Organic Synthesis, A. de la Hoz and A. Loupy, Wiley-VCH, Weinheim, Germany, 2012 Search PubMed.
- R. T. McBurney, F. Portela-Cubillo and J. C. Walton, RSC Adv., 2012, 2, 1264–1274 RSC.
- M. Henary, C. Kananda, L. Rotolo, B. Savino, E. A. Owens and G. Cravotto, RSC Adv., 2020, 10, 14170–14197 RSC.
- D. Kopetzki and M. Antonietti, New J. Chem., 2011, 35, 1787–1794 RSC.
- Y. Y. Maruo, J. Nakamura and M. Uchiyama, Talanta, 2008, 74, 1141–1147 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision, B.01, Gaussian Inc., Wallingford CT, 2009 Search PubMed.
- The yields were approximate values because some impurities were included in both the fractions. But the main impurities for C7* and C6* are C6* and C7*, respectively, as can be seen in Fig. 2. Since the purity and yield are trade-off, we chosen a reasonable balance in this study. The yields of C6* and C7* were quite low in this study presumably because of the treatment of larger amounts of ion-exchange resins. This is because inorganic salts had to be removed prior to the HPLC analysis..
- Y. Shigemasa, S. Akagi, R. Nakashima and S. Saito, Carbohydr. Res., 1980, 80, C1–C3 CrossRef CAS.
- Y. Shigemasa, T. Ueda and H. Saimoto, Bull. Chem. Soc. Jpn., 1990, 63, 389–394 CrossRef CAS.
- S. A. Benner, H.-J. Kim, M.-J. Kim and A. Ricardo, Cold Spring Harbor Perspect. Biol., 2010, 2, a003467 Search PubMed.
- J. Vanderdeelen, J. Phys. Chem. Ref. Data, 2012, 41, 023105–023137 CrossRef.
- S. Lamour, S. Pallmann, M. Haas and O. Trapp, Life, 2019, 9, 52 CrossRef CAS PubMed.
- M. Haas, S. Lamour, S. B. Christ and O. Trapp, Commun. Chem., 2020, 3, 140 CrossRef CAS PubMed.
- S. Civiš, R. Szabla, B. M. Szyja, D. Smykowski, O. Ivanek, A. Knížek, P. Kubelík, J. Šponer, M. Ferus and J. E. Šponer, Sci. Rep., 2016, 6, 23199 CrossRef PubMed.
- Y. Furukawa, Y. Chikaraishi, N. Ohkouchi, N. O. Ogawa, D. P. Glavin, J. P. Dworkin, C. Abe and T. Nakamura, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 24440–24445 CrossRef CAS PubMed.
- Y. Furukawa, Y. Iwasa and Y. Chikaraishi, Sci. Adv., 2021, 7, eabd3575 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ra07249a |
| This journal is © The Royal Society of Chemistry 2023 |