
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign and synthesis of a clickable, photoreactive amino acid p-(4-(but-3-yn-1-yl)benzoyl)-L-phenylalanine for peptide photoaffinity labeling†
Penggang Han ,
Fuli Wang,
Shaoheng Bao,
Ge Yao,
Xiukun Wan,
JiaJia Liu and
Hui Jiang*
,
Fuli Wang,
Shaoheng Bao,
Ge Yao,
Xiukun Wan,
JiaJia Liu and
Hui Jiang*
State Key Laboratory of NBC Protection for Civilian, Beijing 102205, China. E-mail: synbiojiang@126.com
First published on 4th January 2023
Abstract
Photoaffinity labeling is a powerful technique to investigate the interactions between bioactive peptides and their targets. To construct a peptide-derived photoaffinity probe, at least two amino acids need to be modified or replaced, increasing experimental difficulties and negatively affecting activity. Herein, we report the synthesis of a clickable, photoreactive amino acid p-(4-(but-3-yn-1-yl)benzoyl)-L-phenylalanine (Abpa) and its Fmoc-protected version from 3-(4-bromophenyl)-1-propanol in 11 steps with an overall 12.5% yield. The amino acid contains both a photoreactive benzophenone and a clickable terminal alkyne which acts like a reporter tag by fast attachment to other functional groups via ‘click’ reaction, and a photoaffinity probe could be created by one single amino acid substitution during peptide synthesis. And its small size helps to retain bioactivity. The efficiency of Abpa was demonstrated by photoaffinity labeling experiments using photoactivatable probes of α-conotoxin MI.
1. Introduction
With outstanding potency, selectivity and low toxicity, bioactive amino acid-containing compounds and peptides have been important sources of novel drug candidates.1–4 The identification of their specific targets is the starting point for drug discovery and development. Photoaffinity labeling (PAL) is widely employed to identify the targets of bioactive molecules and to investigate ligand–receptor interactions within biological systems.5–7 Many photoreactive probes of peptide ligands have also been created for PAL studies. For example, the pre-S1 domain of the large envelope protein human hepatitis B virus is a key determinant for receptor binding and its receptor is unknown. Utilizing PAL, sodium taurocholate cotransporting polypeptide has been characterized as its functional receptor.8 Masashi Harada et al. developed a photoaffinity probe of nocistatin, a neuropeptide involved in various central functions, and identified two binding proteins.9 A series of photoreactive probes based on the potent spider toxin Huwentoxin-IV were synthesized by Tzakoniati Foteini et al. to determine its binding interface on voltage-sensor domains of NaV1.7.10 Iain A. Stepek and colleagues designed a peptide probe of a novel β-peptide antibiotic and identified some bacterial membrane proteins as prospective targets.11In general, a typical photoaffinity probe for PAL studies consists of three functional groups: a bioactive pharmacophore, a photoreactive group and a bioorthogonal reporter tag. To gain maximal and accurate information between ligands and receptors, it is required that photoaffinity probes should cause minimal alteration to the original structure of the ligands. Upon photoirradiation, the photoreactive group is capable of forming covalent bonds with the corresponding target proteins, and the reporter tag such as biotin, radioactive isotopes and fluorophores facilitates isolation and analysis of the labeled protein. The most commonly used photoreactive groups in PAL are aryl azide,9 benzophenone12,13 and diazirine.8,14 For peptide ligands, these photoreactive molecules are usually introduced into peptide ligands during peptide synthesis through the incorporation of amino acid derivatives or unnatural amino acids (UAAs). Some photoreactive amino acids such as aryl azide-containing p-azido-L-phenylalanine,15 benzophenone-containing p-benzoyl-L-phenylalanine,16 diazirine-containing photoleucine and photomethionine17 have been developed to create photoaffinity probes. In general, when creating a photoaffinity probe, two positions in the peptide need to be modified or substituted to introduce the photoreactive group and reporter tag. However, the incorporation of these functional groups may negatively affect biological activity by sterically disrupting key interactions between the cognate target and the peptide ligand. Minimizing the number and size of introduced groups helps to retain the binding affinity of the peptide ligands.18
In the present study, we describe the synthesis of a clickable, photoreactive amino acid p-(4-(but-3-yn-1-yl)benzoyl)-L-phenylalanine (Abpa) (Fig. 1), which contains both benzophenone and a terminal alkyne, allowing fast attach to a reporter tag by ‘click’ reaction. NH2-Fmoc protected derivative of the Abpa is synthesized and can be directly subjected to peptide synthesis by Fmoc chemistry. The apparent advantage of Abpa is that the photoreactive group and reporter group can be introduced together into the probe by one amino acid substitution when constructing peptide-derived photoaffinity probes. We also developed photoaffinity probes of α-conotoxin MI and performed PAL experiments to evaluate Abpa.
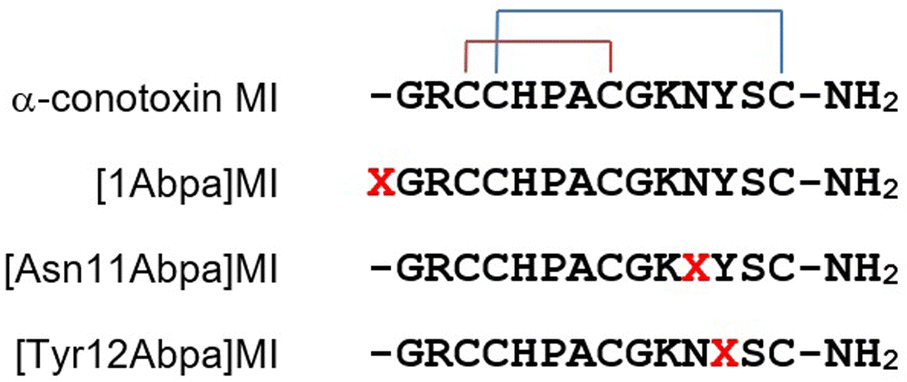 | ||
| Fig. 1 Sequences of MI and its photoactivatable probes. X presents Abpa. | ||
2. Results and discussion
Photoreactive molecules can be introduced into peptide ligands either by post-synthetic derivatizations through enzyme-catalyzed reactions and chemical modifications or during peptide synthesis through the incorporation of photoactivatable amino acids.5 The latter strategy allows the incorporation of photoreactive groups at any position in the peptide, whereas the former depends on the location of an available functional group.However, extra steps are also required to introduce reporter groups into the probes for labeling protein analysis. For more convenient and faster creation of peptide photoaffinity probes, a bi-functional amino acid that contains both a photoreactive group and a reporter group is necessary. Herein, We designed and synthesized a new amino acid, p-(4-(but-3-yn-1-yl)benzoyl)-L-phenylalanine (Abpa) that resembles phenylalanine but contains a photoreactive benzophenone and a terminal alkyne. Alkyne was introduced as a ‘clickable’ handle reporter through which many functional groups such as biotin and fluorescence could attach to the probe by click reaction. To prevent the interference of alkyne and benzophenone, two methylene groups were employed as a spacer arm.
Our plan for the synthesis of Fmoc-protected Abpa is outlined in Scheme 1. The synthesis starts from compound 1 (3-(4-bromophenyl)-1-propanol). Its hydroxyl group must be protected. Otherwise, the active hydrogen would quench the phenyl anion during the latter n-BuLi-mediated reaction. The free hydroxyl group of 1 was protected by TBDPSCl to afford compound 2 in 93.6% yield. Intermediate 2a was prepared from 2-1 in two steps (Scheme 2). Acyl chlorination of 2-1 by oxalyl chloride with DMF as catalyst afforded 2-2, which was transformed to Weinreb amide 2a by reacting with N,O-Dimethyl hydroxylamine hydrochloride. Subsequent nucleophilic substitution reaction between 2a and 2 in the presence of n-BuLi produced desired benzophenone-containing intermediate 3 in a yield of 68.8%.
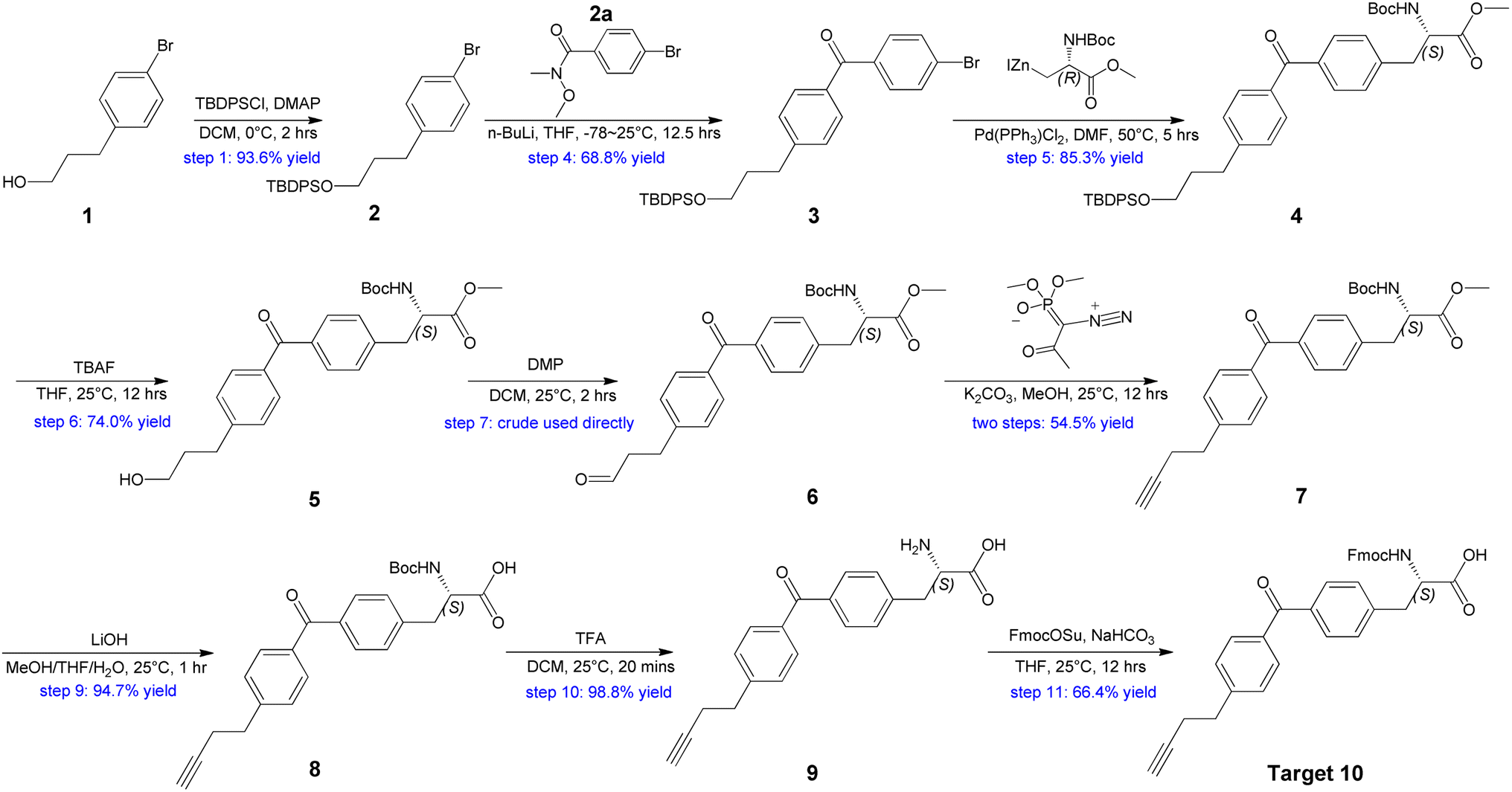 | ||
| Scheme 1 Synthesis of Abpa from 3-(4-bromophenyl)-1-propanol. | ||
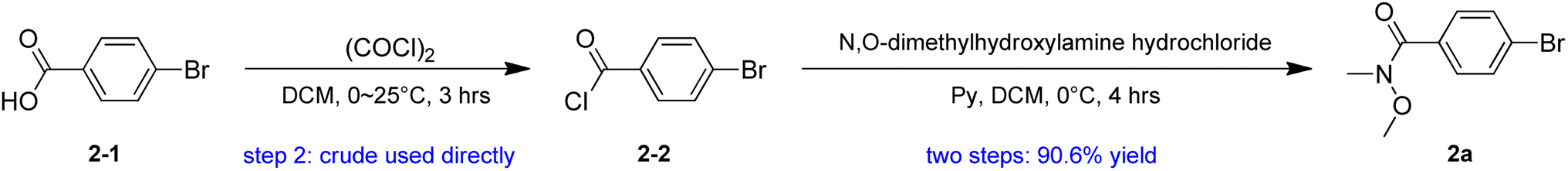 | ||
| Scheme 2 Synthesis of compound 2a. | ||
Intermediate 4 was provided by Negishi coupling between 3 and organozinc halides. We screened several palladium catalysts and ligands for their efficiency and found Pd(PPh3)Cl2 could achieve an ideal yield (Table 1).
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a
a
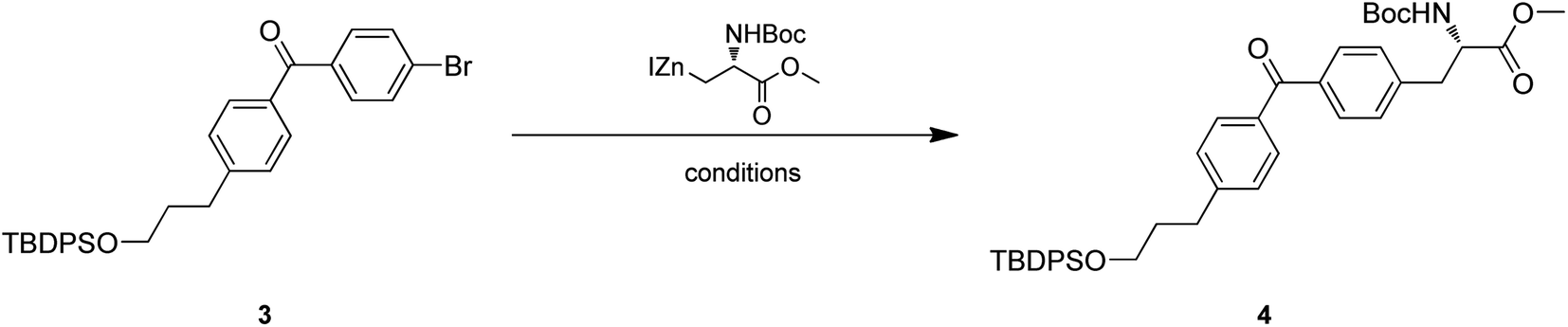
Treatment of 4 with TBAF led to the O-TBDPS deprotection, giving compound 5 in 73.9% yield. The hydroxyl group was further oxidized to carbonyl via Dess–Martin oxidation to afford aldehyde 6. As unstable, the crude mixture of 6 was directly used in the next step. Conversion of aldehyde to terminal alkyne was accomplished via Seyferth–Gilbert Homologation.19 To improve the reaction efficiency, we screened three bases including K2CO3, NaOMe and LDA/TMSCHN2, and found K2CO3/MeOH combination at room temperature could achieve a relatively high yield (Table 2).
a
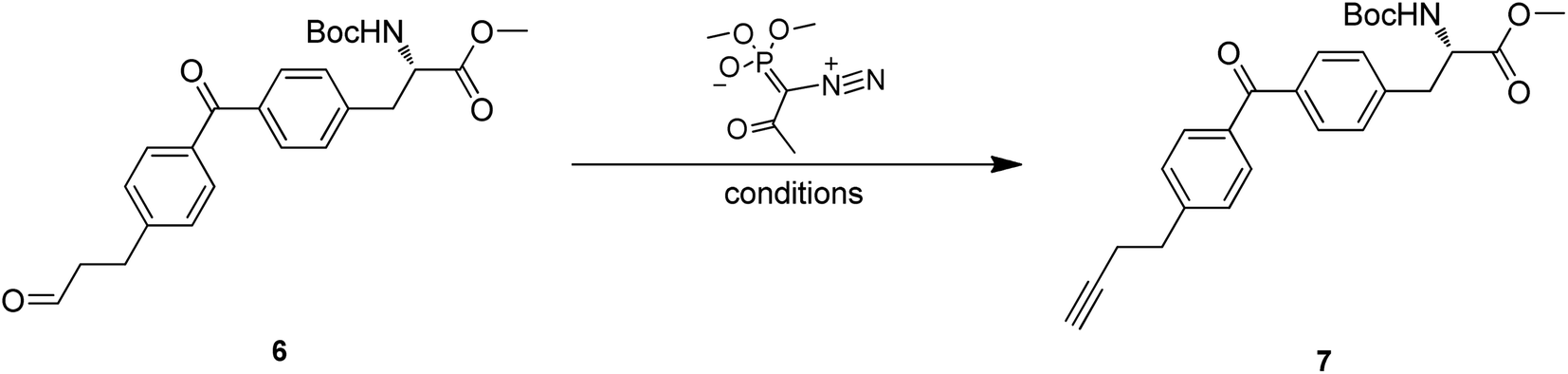
Hydrolysis of methyl ester 7 by LiOH in a mixture of MeOH, THF and H2O gave carboxylic acid 8 in desirable yield. The tert-butyloxycarbonyl (Boc) of 8 was removed with TFA in DCM to produce amino acid 9 in 98.8% yield. Finally, the NH2 protection was also accomplished by Fmoc-OSu with the aid of NaHCO3 to give the target compound 10. 1H NMR and 13C NMR spectra of 10 were measured and the purity was confirmed (ee >99%) by chiral SFC and RP-HPLC (see ESI†). Thus, Abpa, a Fmoc-protected amino acid containing both alkyne and benzophenone, was synthesized in 11 steps and 12.5% overall yield from commercially available 3-(4-bromophenyl)-1-propanol.
To test the efficiency of Abpa in peptide photoaffinity labeling, three photoactivatable probes of α-conotoxin MI were synthesized. The sequences of MI and its derivatives [1Abpa]MI, [Asn11Abpa]MI and [Tyr12Abpa]MI are shown in Fig. 1. MI is a 14-residue neuropeptide from the venom of Conus magus with the sequence GRCCHPACGKNYSC containing two disulfide bonds arranged Cys3–Cys8, Cys4–Cys14. MI shows a high level of potency and selectivity for muscle-type α1β1γδ nicotinic acetylcholine receptor (nAChR) with an IC50 value of 400 pM.20
Linear peptide probes were assembled by solid-phase peptide synthesis using Fmoc chemistry, followed by acidic cleavage of the peptides from the resin. Disulfide bonds were formed by air oxidation in 0.1 M NH4HCO3 buffer as previously described.21 The oxidized products were confirmed by mass spectrometry (see ESI†) and purified by RP-HPLC.
Using these probes, we performed photoaffinity labeling in skeletal muscle cells. Primary gastrocnemius muscle cells isolated from C57 mice were used as target cells. The three probes dissolved in DMSO-containing PBS were incubated with ∼3 × 107 cells plated on 35 mm dishes for 1 h at room temperature, respectively. Cells were exposed to 365 nm UV light for 45 min at 4 °C to induce covalent cross-linking and were then washed to remove residual free probes. Labeled proteins were then modified with biotin-azide by copper-catalyzed azide–alkyne cycloaddition (CuAAC) reaction on the surfaces of living cells.22 After cell lysis by denaturing buffer, biotinylated proteins were captured with streptavidin T1 magnetic beads and eluted by a special PBS buffer containing 2% SDS, 30 mM biotin, 6 M urea and 2 M thiourea (pH 12).23 The eluent was subjected to 4–20% SDS-PAGE, followed by Western blotting using muscle nAChR α1 subunit antibody. As shown in Fig. 2, nAChR α1 was obviously tested in [1Abpa]MI and [Asn11Abpa]MI eluted proteins, and weakly tested in the [Tyr12Abpa]MI group. The less potent of [Tyr12Abpa]MI may be explained by the key role of Tyr12 in MI–nAChR interaction20 and the substitution by Abpa affected its affinity. The results suggest that Abpa could be effectively introduced into the peptide ligands to create photoaffinity probes for the study of ligand–target interactions.
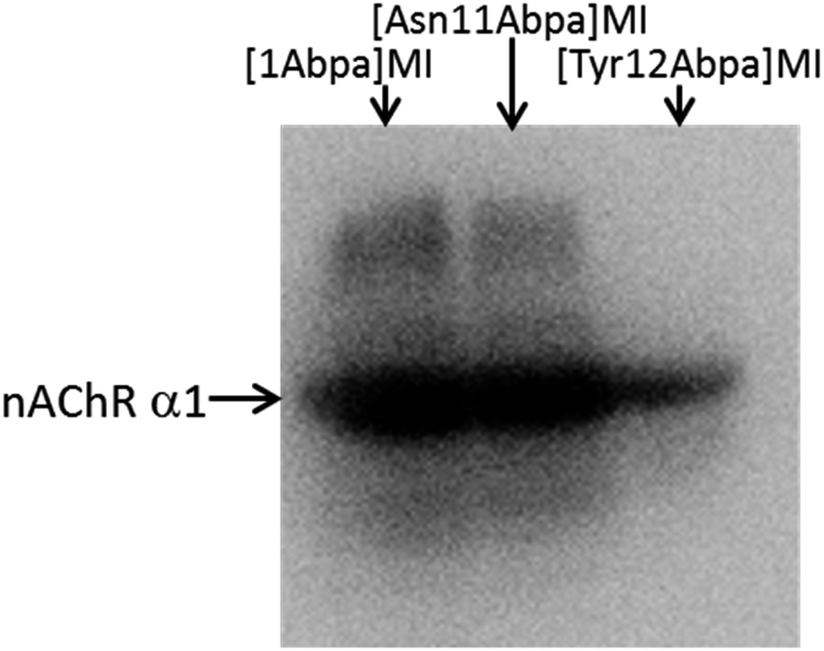 | ||
| Fig. 2 Western blot analyses of the labeled proteins using CHRNA1 antibody, which recognizes muscle nAChR α1 subunit. | ||
3. Conclusion
In summary, we have developed an efficient synthesis of Abpa and its Fmoc-protected version. The amino acid contains both photoreactive benzophenone and clickable terminal alkyne, facilitating faster and easier construction of peptide-derived photoaffinity probes through one single amino acid substitution during peptide synthesis. Using Abpa, three photoaffinity probes of α-conotoxin MI were synthesized and successfully labeled muscle nAChR from skeletal cells. Further applications of Abpa to identify target receptors of bioactive peptides are in progress and will be reported in due course.4. Material and methods
4.1 General conditions
Starting material 3-(4-bromophenyl)-1-propanol, TBDPSCl, N,O-dimethyl hydroxylamine hydrochloride, Pd2(dba)3, Pd(OAc)2, Pd(PPh3)Cl2, PdCl2, Sphos and Xphos were purchased from Bide Pharmatech, Shanghai, China. DMAP, LiOH, n-BuLi and oxalyl chloride were obtained from Sigma-Aldrich, MO, USA. DMP, K2CO3, LDA and TMSCHN2 were purchased from Energy Chemical, Shanghai, China. Rink amide resins, Fmoc-protected amino acids, Fmoc-OSu and other reagents for peptide synthesis were purchased from GL Biochem, Shanghai, China. BTTAA-based CuAAC Cell Reaction Buffer Kit was provided by Jena Bioscience GmbH, Jena, German. Cell lysis buffer SD-001 was bought from Invent Biotechnologies, Plymouth, MN, USA. Streptavidin T1 magnetic beads were purchased from Thermo Fisher Scientific, Waltham, MA, USA. CHRNA1 antibody was obtained from Signalway Antibody, Maryland, USA. HRP-conjugated goat anti-rabbit secondary antibody was purchased from Cell Signaling Technology, Massachusetts, USA. ECL kit was obtained from Millipore, Burlington, MA, USA. All other solvents and reagents were commercially available and used directly without further purification.Thin-layer chromatography (TLC) was conducted with Merck Silica Gel 60 F254 pre-coated plates and spots were visualized by ultraviolet UV and potassium permanganate stain. Liquid chromatography-mass spectra (LC-MS) analyses were performed on Shimadzu LC-MS 2020 230V (Shimadzu Scientific Instruments, Columbia, MD). Chromatographic separations were performed on a Merck Silica Gel 60 (230–400 mesh) column by flash chromatography. NMR spectra were recorded on an AVANCE III spectrometer at 400 MHz (Bruker Daltonics, Billerica, MA, USA) for proton resonance. Reversed-phase HPLC was performed on an Agilent 1100 system with a dual wavelength UV detector (Agilent, Santa Clara, CA, USA). 365 nm UV light for photoaffinity labeling was from Kylin-Bell Lab Instruments Co., China. Cell concentration was quantified by a Countess II Automated Cell Counter (Thermo Fisher Scientific, Waltham, MA, USA). Chemiluminescence was detected by an Alliance Micro Q9 Automated Chemiluminescence System (Uvitec Cambridge, UK).
4.2 Synthesis of Abpa
To a suspension of N,O-dimethylhydroxylamine hydrochloride (23.2 g, 238 mmol, 1.2 eq.) and pyridine (58.8 g, 743.36 mmol, 60 mL, 3.74 eq.) in DCM (150 mL) was added drop-wise a solution of compound 2-2 (43.6 g, 198 mmol, 1 eq.) in DCM (100 mL) at 0 °C and the mixture was stirred at 0 °C for 4 h. TLC (petroleum ether/ethyl acetate = 10/1) showed starting material (Rf = 0.8) was consumed completely and a new spot (Rf = 0.1) formed. The mixture was concentrated under vacuum to remove DCM, diluted with ethyl acetate (500 mL), washed with 1 M HCl (500 mL) and brine (500 mL). The organic layer was dried over anhydrous sodium sulfate, filtered and evaporated to give compound 2a (44.0 g, 180 mmol, 90.5% yield) as a yellow oil.
4.3 Preparation of photoactivatable α-conotoxin MI probes
Probes were manually synthesized using standard Fmoc chemistry on Rink amide resin (substitution value 1 mmol g−1) with HBTU as the coupling reagent. The following side chain protecting amino acids were used: Arg (Pbf), Cys (Trt), His (Trt), Lys (Boc), Asn (Trt), Tyr (tBu), Ser (tBu). Following chain assembly, the resin was washed 5 times with DMF. Linear peptides were cleaved from the resin with a mixture of TFA:TIS:H2O (92.5%:2.5%:2.5%) for 1 h stirring at room temperature. The peptides were precipitated with cold ethyl ether, centrifuged, and washed 4 times with cold ethyl ether. The crude peptides were purified by RP-HPLC on a semi-preparative C18 Vydac column (218TP510, 10 mm × 250 mm) eluted with a gradient of 0–50% ACN in 0.1% TFA over 50 min to give the linear peptides. Purified reduced peptides were dissolved in 0.1 M NH4HCO3 (pH 7.8–8.0) to a concentration of 0.2 mg mL−1 and stirred at room temperature. Oxidation was monitored by analytical RP-HPLC. The folded products were then purified by semi-preparative RP-HPLC and confirmed by LC-MS.
4.4 Isolations of mouse skeletal muscle cells
All animal studies were in accordance with the NIH guide for the care and use of laboratory animals and were approved by the Institutional Animal Research and Ethics Committee of State Key Laboratory of NBC Protection for Civilian (LAE-2022-07-002).Twenty healthy male/female C57 mice, aged 3 months, were sacrificed after anesthesia with massive diethyl ether, and the gastrocnemius muscle of the lower extremity was isolated. After washed with precooled PBS, muscle tissue was triturated to 1 mm3-size in PBS on ice, followed by digestion at 37 °C for 5 min with 5 mL enzyme cocktail containing 0.125% trypsin, 0.0625 mg mL−1 collagenase IV and 0.01 mg mL−1 deoxyribonuclease II. After 5 min rest, the digestion solution was removed. The digestion procedure was repeated 3–4 times until there was no obvious tissue. The cell suspension was centrifuged at 300 g at 4 °C for 10 min. The pellet was then resuspended in DMEM containing 10% FBS and filtered by cell mesh.
4.5 Photoaffinity labeling experiments
[1Abpa]MI, [Asn11Abpa]MI and [Tyr12Abpa]MI were dissolved in 20 μL DMSO and then applied to ∼3 × 107 skeletal muscle cells suspended in 4 mL PBS in 35 mm dishes, respectively. After incubation for 1 h with gentle agitation at room temperature, cells were photolabeled by irradiation by 365 nm UV at 4 °C for 40 min. The mixture was then centrifuged at 300g for 6 min and the supernatant was discarded. The cells were washed twice with PBS. Next, a click reaction mixture containing excessive biotin-PEG4-azide, BTTAA, CuSO4 and Na-ascorbate was added. After reaction for 1 h in the dark at room temperature, cells were collected by centrifugation and lysed by 3 mL denaturing lysis buffer containing 1× protease inhibitor cocktail. The cell lysates were incubated with 200 μL streptavidin T1 magnetic beads overnight at 4 °C. After removal of the supernatant, the labeled proteins were eluted with 250 μL PBS containing 2% SDS, 30 mM biotin, 6 M urea and 2 M thiourea (pH 12) by 1 h incubation at room temperature and 30 min boiling. The eluent was then subjected to SDS-PAGE on 4–20% tris-glycine gel, transferred to PVDF membrane, and immunoblotted with CHRNA1 antibody (recognizing muscle nAChR α subunit). After further incubation with horseradish peroxidase-conjugated secondary antibody, the membrane was treated with ECL reagent and visualized.Author contributions
Conceptualization, Penggang Han, Hui Jiang and Jisheng Chen; methodology, Penggang Han and Ge Yao; validation, Xiukun Wan; investigation, Shaoheng Bao and JiaJia Liu; data curation, Fuli Wang; writing – original draft preparation, Penggang Han; writing – review and editing, Hui Jiang.Conflicts of interest
The authors declare no conflict of interest.References
- Z. Yin, W. Hu, W. Zhang, H. Konno and H. Moriwaki, et al., Tailor-made amino acid-derived pharmaceuticals approved by the FDA in 2019, Amino Acids, 2020, 52, 1227–1261 CrossRef.
- J. Han, H. Konno, T. Sato, V. A. Soloshonok and K. Izawa, Tailor-made amino acids in the design of small-molecule blockbuster drugs, Eur. J. Med. Chem., 2021, 220, 113448 CrossRef.
- J. Han, H. Konno, T. Sato, K. Izawa and V. A. Soloshonok, Peptidomimetics and Peptide-Based Blockbuster Drugs, Curr. Org. Chem., 2021, 25, 1627–1658 CrossRef.
- Q. Wang, J. Han, A. Sorochinsky, A. Landa and G. Butler, et al., The Latest FDA-Approved Pharmaceuticals Containing Fragments of Tailor-Made Amino Acids and Fluorine, Pharmaceuticals, 2022, 15, 999 CrossRef PubMed.
- A. Grunbeck and T. P. Sakmar, Probing G protein-coupled receptor-ligand interactions with targeted photoactivatable cross-linkers, Biochemistry, 2013, 52, 8625–8632 CrossRef PubMed.
- E. Smith and I. Collins, Photoaffinity labeling in target- and binding-site identification, Future Med. Chem., 2015, 7, 159–183 CrossRef.
- E. Karaj, S. H. Sindi and L. M. Viranga Tillekeratne, Photoaffinity labeling and bioorthogonal ligation: Two critical tools for designing “Fish Hooks” to scout for target proteins, Bioorg. Med. Chem., 2022, 62, 116721 CrossRef CAS.
- H. Yan, G. Zhong, G. Xu, W. He and Z. Jing, et al., Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus, eLife, 2012, 1, e00049 CrossRef.
- H. Masashi, M. Toshiaki, I. Seiji and O. A. Emiko, Development of a novel photoaffinity probe for labeling nocistatin receptor, Biochem. Biophys. Res. Commun., 2018, 501, 514–519 CrossRef PubMed.
- F. Tzakoniati, H. Xu, T. Li, N. Garcia and E. W. Tate, Development of Photocrosslinking Probes Based on Huwentoxin-IV to Map the Site of Interaction on Nav1.7, Cell Chem. Biol., 2019, 27, 306–313 CrossRef.
- I. Stepek, T. Cao and A. Koetemann, et al., Antibiotic Discovery with Synthetic Fermentation: Library Assembly, Phenotypic Screening, and Mechanism of Action of β-Peptides Targeting Penicillin-Binding Proteins, ACS Chem. Biol., 2019, 1030–1040 CrossRef CAS.
- G. Wang, T. Han, D. Nijhawan, P. Theodoropoulos and J. Naidoo, et al., P7C3 neuroprotective chemicals function by activating the rate-limiting enzyme in NAD salvage, Cell, 2014, 158, 1324–1334 CrossRef CAS.
- J. Park, S. Oh and S. B. Park, Discovery and target identification of an antiproliferative agent in live cells using fluorescence difference in two-dimensional gel electrophoresis, Angew. Chem., Int. Ed. Engl., 2012, 51, 5447–5451 CrossRef CAS PubMed.
- L. Dubinsky, B. P. Krom and M. M. Meijler, Diazirine based photoaffinity labeling, Bioorg. Med. Chem., 2012, 20, 554–570 CrossRef CAS PubMed.
- R. Schwyzer and M. Caviezel, p-Azido-L-phenylalanine: a photo-affinity 'probe' related to tyrosine, Helv. Chim. Acta, 1971, 54, 1395–1400 CrossRef CAS.
- J. C. Kauer, S. Erickson-Viitanen, H. R. Wolfe and W. F. DeGrado Jr, p-Benzoyl-L-phenylalanine, a new photoreactive amino acid. Photolabeling of calmodulin with a synthetic calmodulin-binding peptide, J. Biol. Chem., 1986, 261, 10695–10700 CrossRef CAS.
- M. Suchanek, A. Radzikowska and C. Thiele, Photo-leucine and photo-methionine allow identification of protein-protein interactions in living cells, Nat. Methods, 2005, 2, 261–267 CrossRef CAS PubMed.
- D. J. Lapinsky, Tandem photoaffinity labeling-bioorthogonal conjugation in medicinal chemistry, Bioorg. Med. Chem., 2012, 20, 6237–6247 CrossRef CAS PubMed.
- A. Sapegin and M. Krasavin, One-Pot Conversion of Aldehydes and Aryl Halides to Disubstituted Alkynes via Tandem Seyferth-Gilbert Homologation/Copper-Free Sonogashira Coupling, J. Org. Chem., 2019, 84, 8788–8795 CrossRef CAS PubMed.
- R. B. Jacobsen, R. G. DelaCruz, J. H. Grose, J. M. McIntosh and D. Yoshikami, et al., Critical residues influence the affinity and selectivity of alpha-conotoxin MI for nicotinic acetylcholine receptors, Biochemistry, 1999, 38, 13310–13315 CrossRef CAS.
- P. Han, K. Wang, X. Dai, Y. Cao and S. Liu, et al., the role of individual disulfide bonds of -conotoxin giiia in the inhibition of Nav 1.4, Mar. Drugs, 2016, 14, 1–9 CrossRef PubMed.
- S. Li, L. Wang, F. Yu, Z. Zhu and D. Shobaki, et al., Copper-Catalyzed Click Reaction on/in Live Cells, Chem. Sci., 2017, 8, 2107–2114 RSC.
- J. N. Rybak, S. B. Scheurer, D. Neri and G. Elia, Purification of biotinylated proteins on streptavidin resin: a protocol for quantitative elution, Proteomics, 2004, 4, 2296–2299 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ra07248c |
| This journal is © The Royal Society of Chemistry 2023 |