
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn environment-friendly approach using deep eutectic solvent combined with liquid–liquid microextraction based on solidification of floating organic droplets for simultaneous determination of preservatives in beverages†
Jing Zhao abc,
Lingjun Houc,
Longshan Zhaoc,
Liqing Liub,
Jianhua Qia and
Longhu Wang*a
abc,
Lingjun Houc,
Longshan Zhaoc,
Liqing Liub,
Jianhua Qia and
Longhu Wang*a
aSchool of Pharmacy, Zhejiang University, 866 Yuhangtang Road, 310058, Hangzhou, Zhejiang Province, P.R. China. E-mail: wang2000@zju.edu.cn
bZhejiang Weikang Pharmaceutical Company, 15 Xinggong North Road, 324100, Jiangshan, Zhejiang Province, P.R. China
cSchool of Pharmacy, Shenyang Pharmaceutical University, 103 Wenhua Road, 110016, Shenyang, Liaoning Province, P.R. China
First published on 3rd March 2023
Abstract
With the increase in environmental protection awareness, the development of strategies to reduce the use of organic solvent used during the extraction process has attracted wide attention. A simple and green ultrasound-assisted deep eutectic solvent extraction combined with liquid–liquid microextraction based on solidification of floating organic droplets method was developed and validated for the simultaneous determination of five preservatives (methyl paraben, ethyl paraben, propyl paraben, isopropyl paraben, isobutyl paraben) in beverages. Extraction conditions including the volume of DES, value of pH, and concentration of salt were statistically optimized through response surface methodology using a Box–Behnken design. Complex Green Analytical Procedure Index (ComplexGAPI) was successfully used to estimate the greenness of the developed method and compare with the previous methods. As a result, the established method was linear, precise, and accurate over the range of 0.5–20 μg mL−1. Limits of detection and limits of quantification were in the range of 0.15–0.20 μg mL−1 and 0.40–0.45 μg mL−1, respectively. The recoveries of all five preservatives ranged from 85.96% to 110.25%, with relative standard deviation less than 6.88% (intra-day) and 4.93% (inter-day). The greenness of the present method is significantly better compared with the previous reported methods. Additionally, the proposed method was successfully applied to analysis of preservatives in beverages and is a potentially promising technique for drink matrices.
1. Introduction
Preservatives are almost indispensable ingredients in food for maintaining the color and flavor of food and extending the food shelf life. However, excessive addition of preservatives results in the high accumulation in the body, which leads to endocrine disorders and causes harm to human health. Parabens are alkyl esters of p-hydroxybenzoic acid, which are commonly used as chemical preservatives in food, cosmetics and pharmaceuticals.1,2 Shinshi Oishi has demonstrated that butyl paraben (BP) has a harmful effect on the reproductive system of male mice by damaging spermatogenesis.3 Methyl paraben (MP), ethyl paraben (EP), propyl paraben (PP), and BP are estrogenic-like substances4 and high exposure to estrogenic-like substances might lead to breast cancer.5 In addition, BP, PP, isobutyl paraben (IBP) and isopropyl paraben (IPP) showed highly genotoxic and cytotoxic effects on human lymphocyte cells in vitro.6–8 Therefore, it is crucial to propose a simple and effective method for determining and monitoring the preservatives in food and beverages.A variety of analytical methods for paraben determination in different matrices have been developed by high-performance liquid chromatograph (HPLC),9–11 gas chromatography (GC),12,13 HPLC-MS14–16 GC-MS,17,18 and supercritical fluid chromatography-tandem mass spectrometry (SFC-MS).19 However, conventional preconcentration approaches are time-consuming and often require large volumes of volatile organic solvents, which are detrimental to human health and the environment.18 As the new generation of pretreatment technology, microextraction technology, such as solid phase microextraction (SPME) and liquid–liquid microextraction (LLME) have already been utilized for analysis of preservatives.20–24 However, the organic solvents commonly used in LLME are halogenated hydrocarbons that are highly toxic. With the properties of high enrichment factor, low cost, high recovery and high efficiency, liquid–liquid microextraction-solidification of floating organic droplets (LLME-SFO) possesses highly potential for analyte preconcentration and is widely utilized for sample pretreatment. LLME-SFO has the advantages of a high extraction efficiency, increased enrichment factors (EF), a low required volume of the extraction solvent compared to other LLE techniques.25 However, LLME-SFO has some limitations. Most importantly, the number of organic solvents that fulfill the extraction solvent criteria is limited. Therefore, the search for new solvents that are applicable for LLME-SFO has grown. As an attempt to solve these problems, deep eutectic solvents (DESs) have attracted attention as being a promising new generation of green solvents.26. Subsequently, plenty of studies demonstrated the mixture of two or more components of a certain molar ratio, which were hydrogen-bond donor (HBD) and hydrogen-bond acceptor (HBA), could remain in liquid state under the room temperature, due to its lower melting point than that of either individual component.27–29 On account of the properties, such as low volatility, wide liquid range, non-toxicity, thermal stability, tunability and biocompatibility,30,31 DESs showed its potential capacity of application for alternative extraction solvents in the LLME-SFO method.32,33
In order to develop a green, rapid and sensitive method to determination of preservatives, LLME was employed combined with solidification of floating organic droplets as a sample pretreatment method using DES as a green extraction solvent. Response surface methodology (RSM) using a Box–Behnken design was used to confirm the optimal value of effective factors. Additionally, the developed method was comprehensively evaluated for its green properties by ComplexGAPI and successfully applied for the simultaneous determination of five paraben preservatives (MP, EP, PP, IPP, IBP) in different beverages. To the best of our knowledge, such a green and effective method based on UAE-DES combination with LLME and SFO quantification of preservatives in drink products has never been reported. Our work provides insight to contribute the urgent need of green chemistry for preservatives in drink matrices.
2. Materials and methods
2.1 Chemicals and reagents
Methanol was supplied from Concord Chemical Reagent Factory (Tianjin, China) and glacial acetic acid was provided from Yuwang Industry Co., Ltd., (Shandong, China) which were of chromatographic grade reagent. Wahaha pure water was obtained from Wahaha Group Co., Ltd. (Hangzhou, China). Tetrabutylammonium bromide, bitter, ammonium acetate, sodium chloride, sodium hydroxide and hydrochloric acid, which were of analytical reagent grade, were purchased from Yuwang Industry Co., Ltd., (Shandong, China). The standards, including methyl p-hydroxybenzoate (purity ≥98.5%), ethyl p-hydroxybenzoate (purity ≥98.0%), propyl p-hydroxybenzoate (purity ≥98.0%), isopropyl p-hydroxybenzoate (purity ≥98.0%) and isobutyl p-hydroxybenzoate (purity ≥98.0%), were purchased from Desite Biotechnology Co., Ltd. (Chengdu, China).2.2 Analytical instruments and operation conditions
Chromatographic analysis of the referred drugs was performed by using an Agilent 1100 liquid chromatograph (Agilent Technologies, USA). BT25S balance was used from Satorius (Germany). 78HW-1constant temperature magnetic stirrer was provided by hannuo Instrument Co., Ltd. (Shanghai, China). Ultra-sonication was performed on KQ-5200 Ultrasonic cleaner (Ultrasonic Instrument Co., Ltd. Kunshan, China). XK80-A quick mixer was used to blend uniformity, provided by Xinkang Medical Equipment Co., Ltd. (Jiangsu, China). Low speed centrifuge was purchased from Zhongjia branch of Keda Innovation Co., Ltd. Thermo Hypersil C18 (250 mm × 4.6 mm, 5 μm) was used in HPLC system and kept at 25 °C. The mobile phase consists of methanol (A) and 0.02 M ammonium acetate aqueous water (B). The gradient elution program was as follows: 0–8 min, 65–75% A; 8–15 min, 75–65% A; 15–20 min, 65% A. The flow rate was set at 1.0 mL min−1 and the injection volume was 10 μL. Detection wavelength was set at 254 nm.2.3 Preparation of DES
In the current study, six types of DESs were prepared by using the heating method at different ratios of hydrogen bond acceptors with hydrogen bond donors, as shown in Table 1. Each component was accurately weighed and mixed in a 50 mL beaker, the mixture was stirred at the proper temperature (50–80 °C) until a homogeneous liquid was formed.27 The synthesized DESs were characterized by using FT-IR to verify the formation of hydrogen bonds during the synthesis of DES.| Abbreviation | Component 1 | Component 2 | Mole ratio |
|---|---|---|---|
| DES-1 | Tetrabutylammonium bromide | Octanoic acid | 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 :2 |
| DES-2 | Tetrabutylammonium bromide | Octanoic acid | 1:3 |
| DES-3 | Tetrabutylammonium bromide | Octanoic acid | 1:4 |
| DES-4 | Tetrabutylammonium bromide | 2-Octanol | 1:2 |
| DES-5 | Tetrabutylammonium bromide | 2-Octanol | 1:3 |
| DES-6 | Tetrabutylammonium bromide | 2-Octanol | 1:4 |
2.4 Sample preparation
A single stock solution of the proposed standards was prepared in methanol with a concentration of 1.00 mg L−1 and stored at 4 °C in 10 mL brown volumetric flask. Working solution was obtained by diluting the stock solution with methanol. Three different drinks, containing fruit drink, carbonated drink and functional drink, were purchased from local market. All samples were filtered by 0.22 μm microporous membrane and stored in refrigerator at 4 °C.2.5 LLME-SFO procedure
2.0 mL of sample solution was diluted by distilled water up to 10 mL in a 50 mL centrifuge tube. The pH of the solution was adjusted to 10.0 using 0.1 M NaOH. Then, 1.8 g of NaCl was added and the mixture was vortexed for 30 s to dissolve completely.34 300 μL of DES was injected into the solution, followed by vortexing for 1 min and centrifuging for 10 min at 4200 rpm. The solution in the centrifuge tube was frozen for 10 min at −20 °C. The aim of this operation was to separate the DES phase from the solution after the upper layer was completely solidified. The collected DES extract was diluted by water was added into the DES phase up to 1.0 mL, which was filtered by 0.22 μm microporous membrane and injected into the HPLC system.2.6 RSM for the optimization of extraction conditions
RSM based on a Box–Behnken design was performed using Design-Expert.v8.0.6.1. (Stat-Ease Inc., Minneapolis, MN, USA). The following three independent variables were selected as the independent variables: salt concentration (X1, 10–50%), pH (X2, 2–10) and DES amount (X3, 100–500 μL), while the average extraction rate of target analytes was set as response variable (Y). Whole experiments were composed of seventeen experimental points that included five replicates of the center points. It was randomly performed to minimize the effects of non-controlled variables.2.7 Evaluation the greenness of the extraction process
Considering the generated waste, it was decided to use the E-factor parameter which takes into account not only waste byproducts and leftover reactants, but also spent catalysts and catalyst supports, solvent losses, and anything else that can be regarded as a waste.35 The higher the E-factor of a chemical process, the greater is the waste generated, the greater its negative environmental impact, and the less sustainable it is.36 The E factor was calculated as follows: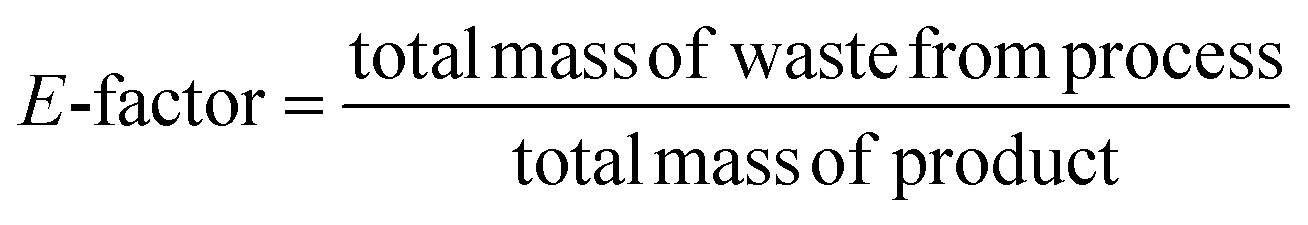
3. Results and discussion
In this study, a DES based LLME-SFO method followed by HPLC analysis was established for determination of MP, EP, IPP, PP, IBP in beverages. Under optimal conditions, the calibration curves of five preservatives were well in the range of 0.5–20 μg mL−1, the recoveries of the analytes were between 85.96% and 110.25%. Compared with traditional organic solvents, the application of DES not only reduces the impact on the environment, but also improves the extraction efficiency of preservatives.37 The method was successfully applied for determination of five preservatives in three beverage samples, including soft drink, fruity drink, and functional drinks with satisfactory precision, repeatability and accuracy.3.1 Screening of DES
The inherent characteristics of DESs such as physicochemical interaction, polarity, solubility and viscosity affect the extraction of target analytes from the sample matrixes.27,38 DESs have excellent hydrophobic properties and higher melting point and lower density than beverage samples.39 Therefore, target analytes can be easily separated from sample solutions by cryogenic freezing. Due to the hydrogen bond interaction, the deep eutectic solvent will obtain a lower freezing point than all or either of the individual constituents.28 On the other hand, the freezing point of the extractant will also affect the extraction efficiency in the SFO procedure.40 The dissolution of the analytes in DES is affected by the types and molar ratio of the HBD and HBA.41 This study investigated DES (Table 1) composed of tetrabutylammonium bromide as HBA and caprylic or secondary octanoic acid as HBD. In addition, different HBA/HBD molar ratios (1:2, 1:3, 1:4) were also evaluated. As shown in Fig. 1, the extraction efficiencies of DESs synthesized with tetrabutylammonium bromide and caprylic acid at a molar ratio of 1:2 were nearly 100% for PP, IPP and IBP and significantly higher for the other two preservatives than the other DESs. Therefore, the DES with molar ratio of 1:2 using tetrabutylammonium bromide and octanoic acid as raw materials was selected for this further investigation.
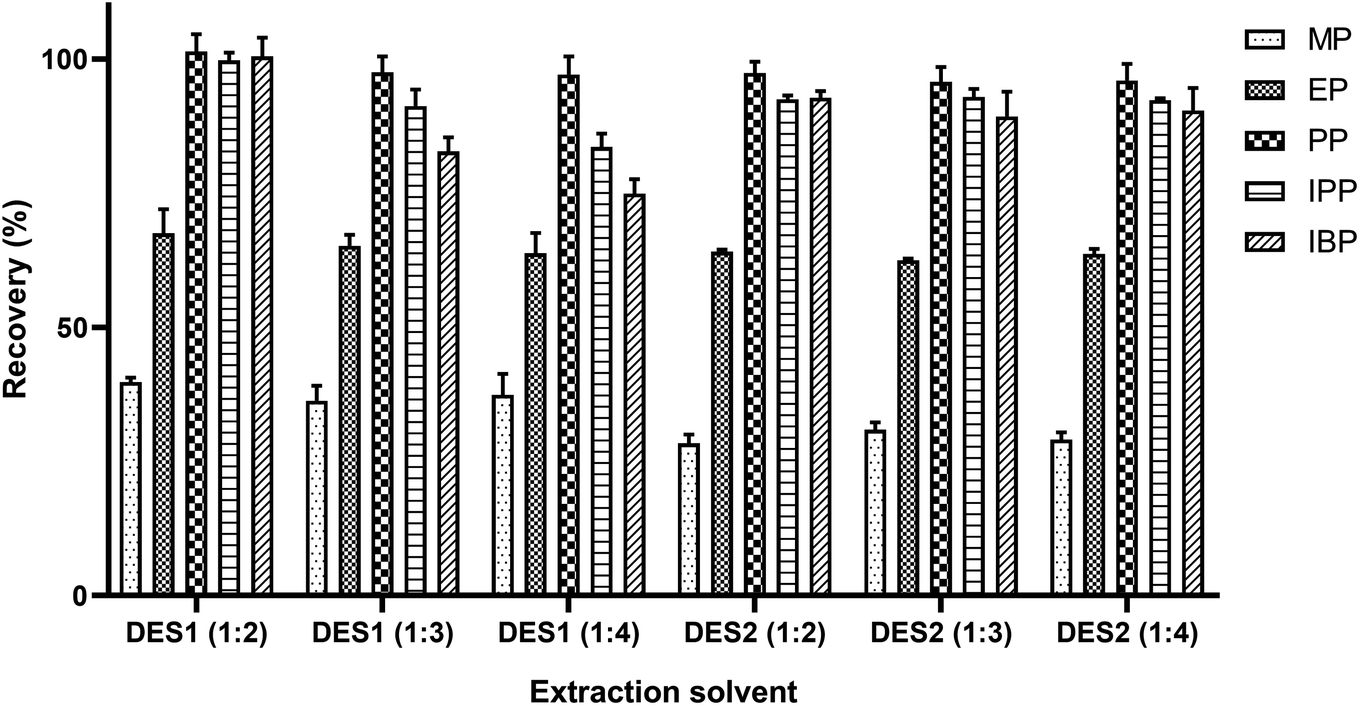 | ||
| Fig. 1 The effect of DESs at different molar ratios and species. | ||
3.2 Optimization of extraction conditions based on RSM
To optimize extraction conditions, response surface methodology (RSM) was applied to design the experiment. RSM is a commonly used experimental design method, which includes mathematical and statistical techniques.42 RSM used to determine the optimal values of the independent variables and to enable to effectively investigate the effects of multiple factors and their interactions, was employed to produce the highest extraction efficiency in this study.Salts in this method worked as a demulsier to decrease the stability of the emulsion solution generated by the adding of DES. The addition of the salts will increase the ionic strength of the emulsion, leading to the occulation and coalescence of the extractant.43 The pH of the sample solution affects the ionization of the target analyte and the SFO procedure, affecting the extraction efficiency. A symmetrical three-factor-three-level Box–Beknhen design (BBD) was investigated (ESI Table S1†) and the data was analyzed with Design-Expert. Based on the multiple regression analysis, an appropriate model of the second-order polynomial regression was established as follows: Y = 92.48 + 8.73X1 + 4.91X2 + 4.06X3 − 4.05X1X2 − 2.50X1X3 − 6.62X2X3 − 6.38X12 − 5.85X22 − 6.80X32.
Statistical analysis was carried out by analysis of variance (ANOVA) test to evaluate the statistical significance of the variables and their interactions. ANOVA, goodness-of-fit and adequacy of selected models were listed in the ESI Table S2.† The goodness-of-fit of the model was demonstrated by the determination coefficient (R2), adjusted determination coefficient (RAdj2) and coefficient of variance (C. V.%). The determination coefficient (R2) for the response was 0.9633, indicated that 93.68% of total variation could be explained by the model. The adjusted determination coefficient (RAdj2 = 0.9162) demonstrated that most variation of extraction yield could be well predicted by the model, and the coefficient variation value (C. V.% = 3.71) confirmed the reliability of the experimental values of regression model.
The significance of each coefficient was determined by using F-test and p-value. The p-value was used to check the significance of the coefficients, indicating the interaction strength between the variables. It could be known from the table that the p-value (0.0003) of the regression model was less than 0.05, which demonstrated and the result demonstrated that the proposed quadratic polynomial regression model was significant. Furthermore, the F-values for lack of fit (0.1136) was not significant (p > 0.05), which indicated that the real value was fit well with the predicted value, the model could accurately simulate the real situation, and accurately represented the experimental data.
The 3D surface diagram was shown in ESI Fig. S1.† According to statistical analysis and the 3D response plots, variables X1, X2 and X3 showed a significant influence on the extraction yields in three models on the extraction efficiencies. The aim of multiple regression model was to determine the best values for the three extraction conditions. Finally, the optimal experimental conditions were achieved with salt concentration of 18%, a pH of 10.0 and 300 μL of DES amount.
3.3 Characterization of DES
In order to elucidate the structures and main functional groups of tetrabutylammonium bromide, octanoic acid and DES (synthesized from tetrabutylammonium bromide and octanoic acid), were characterized by Fourier transform infrared spectroscopy (FT-IR). The results were shown in Fig. 2. The absorption peaks at 2959.4 cm−1 and 2873.9 cm−1 were attributable to the stretching vibration of C–H bond in the spectrum of tetrabutylammonium bromide. Moreover, the peaks at 1381.2 cm−1and 1473.1 cm−1 were belonged to the deformation vibrations of methyl (CH3), and methylene (CH2), respectively. The peak at 1166.7 cm−1 were attributable to the stretching vibration of C–N. In the spectrum of octanoic acid, the peaks at 2857.3 cm−1 and 2928.2 cm−1, 3034.7 cm−1, 1276.7 cm−1 were attributable to the stretching vibration of C–H, O–H, C–O, respectively. The absorption peak at 1711.2 cm−1 was attributable to the stretching vibration of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, and the absorption peaks at 1413.3 cm−1 and 937.0 cm−1 were belonged to the deformation vibration of O–H. In the FT-IR spectrum of DES, the peak at 2926.3−1 was attributable to the stretching vibration of C–H, and the broad peak at 3418.1 cm−1 was belonged to the stretching vibration of O–H. Furthermore, the stretching vibration of CO migrated to 1729.1 cm−1 in FT-IR spectra of deep eutectic solvent. It can be considered that there is an intermolecular hydrogen bond in DES synthesized from tetrabutylammonium bromide and octanoic acid.
O, and the absorption peaks at 1413.3 cm−1 and 937.0 cm−1 were belonged to the deformation vibration of O–H. In the FT-IR spectrum of DES, the peak at 2926.3−1 was attributable to the stretching vibration of C–H, and the broad peak at 3418.1 cm−1 was belonged to the stretching vibration of O–H. Furthermore, the stretching vibration of CO migrated to 1729.1 cm−1 in FT-IR spectra of deep eutectic solvent. It can be considered that there is an intermolecular hydrogen bond in DES synthesized from tetrabutylammonium bromide and octanoic acid.
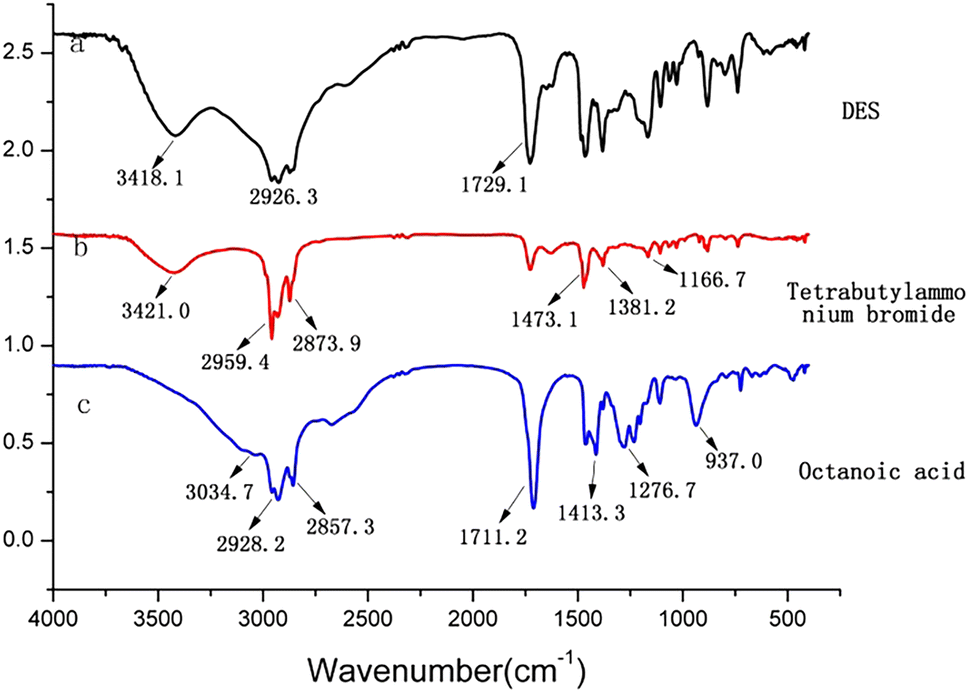 | ||
| Fig. 2 The IR spectra of DES (a), tetrabutylammonium bromide (b) and octanoic acid (c). | ||
3.4 Method evaluation
| Analytes | Linear range (μg mL−1) | Linear equation | r2 | LOD (μg mL−1) | LOQ (μg mL−1) |
|---|---|---|---|---|---|
| a MP, methyl paraben. EP, ethyl paraben. IPP, isopropyl paraben. PP, propyl paraben. IBP, isobutyl paraben. | |||||
| MP | 0.5–20.0 | y = 58.197× | 0.9986 | 0.15 | 0.40 |
| EP | 0.5–20.0 | y = 50.961× | 0.9999 | 0.15 | 0.40 |
| IPP | 0.5–20.0 | y = 49.947× | 0.9999 | 0.20 | 0.45 |
| PP | 0.5–20.0 | y = 51.418× | 0.9998 | 0.20 | 0.45 |
| IBP | 0.5–20.0 | y = 45.871× | 0.9999 | 0.20 | 0.45 |
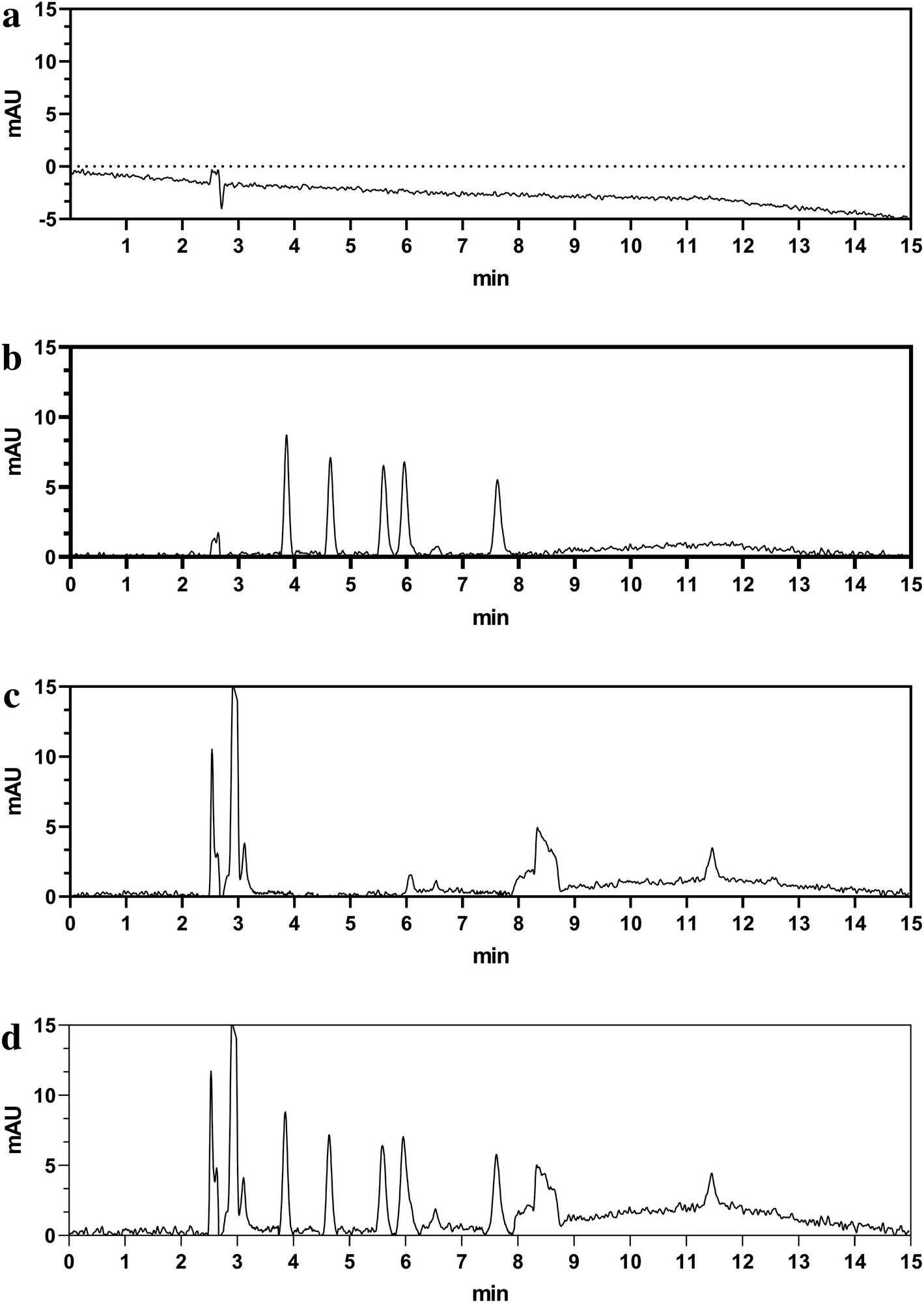 | ||
| Fig. 3 The typical chromatograms of blank (a), standards (b), beverage blank (c), and preservative spiked to beverage (d). | ||
| Analytes | Matrix | Functional drink | Fruity drink | Carbonated drink | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Spiked (μg ml−1) | Accuracy (%) | Precision (RSD%) | Accuracy (%) | Precision (RSD%) | Accuracy (%) | Precision (RSD%) | ||||
| Intra-day | Inter-day | Intra-day | Inter-day | Intra-day | Inter-day | |||||
| MP | 1.0 | 98.73 | 6.88 | 4.03 | 86.07 | 3.14 | 1.89 | 88.51 | 1.76 | 3.15 |
| 2.0 | 95.77 | 1.87 | 3.05 | 85.96 | 1.59 | 1.75 | 87.60 | 1.91 | 3.50 | |
| 5.0 | 93.21 | 1.45 | 3.41 | 88.59 | 0.65 | 1.59 | 86.76 | 1.18 | 2.56 | |
| EP | 1.0 | 94.15 | 3.10 | 1.98 | 90.57 | 6.17 | 3.33 | 90.40 | 2.95 | 3.60 |
| 2.0 | 97.18 | 1.62 | 2.54 | 91.61 | 4.14 | 3.60 | 91.81 | 3.00 | 2.90 | |
| 5.0 | 96.03 | 2.56 | 2.20 | 92.40 | 0.77 | 2.32 | 91.64 | 1.16 | 2.13 | |
| IPP | 1.0 | 95.28 | 2.88 | 2.45 | 95.92 | 3.36 | 2.41 | 99.51 | 2.69 | 2.72 |
| 2.0 | 99.03 | 1.15 | 2.54 | 96.16 | 2.44 | 2.10 | 98.70 | 1.93 | 3.46 | |
| 5.0 | 97.06 | 4.22 | 2.03 | 95.73 | 1.28 | 2.40 | 97.53 | 1.59 | 3.14 | |
| PP | 1.0 | 107.57 | 2.87 | 2.72 | 109.17 | 3.25 | 2.92 | 110.25 | 1.91 | 3.13 |
| 2.0 | 105.91 | 1.73 | 2.50 | 101.79 | 2.24 | 5.28 | 106.99 | 2.56 | 4.93 | |
| 5.0 | 100.03 | 3.80 | 1.93 | 98.19 | 1.17 | 3.41 | 105.18 | 1.15 | 4.22 | |
| IBP | 1.0 | 109.87 | 1.50 | 1.16 | 97.35 | 1.87 | 2.61 | 97.88 | 3.98 | 2.68 |
| 2.0 | 104.94 | 6.04 | 4.38 | 96.30 | 2.91 | 2.91 | 98.16 | 3.88 | 3.30 | |
| 5.0 | 99.90 | 4.14 | 4.14 | 96.61 | 0.92 | 2.88 | 97.54 | 1.40 | 3.43 | |
3.5 Application to the actual beverage samples
The validated method was applied to extract the preservatives in three actual beverage samples (fruity drink, carbonated drinks, and functional drinks). The detection results of 5 kinds of parabens in actual beverage samples were shown in Table 4. It was found that preservatives were detected in some beverage samples. Fortunately, their content did not exceed the maximum residue set by national standards. It can be seen that PP can be detected in both functional and fruit drinks, and IBP can be detected only in carbonated beverages. In addition, under the optimal conditions, and the recoveries of the various target analytes were between 85.96% and 110.25%. The results showed that the method could be applied for the detection of 5 parabens in beverage samples.| Analyte | MP (μg mL−1) | EP (μg mL−1) | IBP (μg mL−1) | PP (μg mL−1) | IBP (μg mL−1) |
|---|---|---|---|---|---|
| a ND: not detected. | |||||
| Functional drink | ND | ND | ND | 0.19 ± 0.03 | ND |
| Fruity drink | ND | ND | ND | 0.18 ± 0.02 | ND |
| Carbonated drink | ND | ND | ND | ND | 0.22 ± 0.02 |
3.6 Comparison with other methods
In order to evaluate the superiority of the developed method, the extraction method, matrix, analytes, solvent consumption extraction time and recovery were compared with the other published methods for the determination of preservatives, and the results are shown in ESI Table S3.† The results show that the proposed method demanded low amounts of extraction solvent. More importantly, the extraction solvent used in current study was green and environmentally friendly. Compared with other published methods, there is no organic solvent consumption during the extraction process in proposed method. The most significant advantage of the developed method is the use of a green and easily available deep eutectic solvent as the carrier, which is consistent with the principles of green chemistry. In brief, the highlights of this method are simple treatment process, less time-consuming and environmental friendly.3.7 Greenness assessment according to ComplexGAPI
The Complex GAPI tool uses a pictogram to classify the greenness of each stage of an entire analytical methodology, using a color scale, with two or three evaluation levels for each step.36 The colored pictogram is composed of five pentagrams involving five evaluation parameters such as sample preparation, type of method, reagents and solvents, and instrumentation. The visual presentation of GAPI allows for an at-a-glance comparison of several methods. ComplexGAPI provides a more comprehensive evaluation program for the entire analysis process. ComplexGAPI was successfully used to estimate the greenness of the developed method and compare with the previous methods using Complex GAPI v.0.2. (Gdansk University, Poland). As can be seen from ESI Fig. S3,† the greenness of the present method is significantly better compared with the other five methods, which is mainly due to the simplicity of the extraction process and the low consumption of organic solvents. The current study based on LLME, which in turn is based on the solidification of DES droplets and HPLC seems to be greener than the other methodologies. This is mainly because the processes related to the synthesis of DES as well as the micro-extraction procedure are based on non-hazardous reagents. In fact, DES synthesis is a very simple process. The synthesis occurs in 100% yield and no wastes are generated during this part (E-factor = 0), which indicates the current study is greener and environmentally friendly.4. Conclusions
In this study, a DES-LLME based on solidification of floating organic droplets method followed by high-performance liquid chromatography was established for MP, EP, IPP, PP, IBP in beverages. The beverage samples were pretreated by using deep eutectic solvent and combined with LLME-SFO technology for the purification and enrichment. Finally, target analytes were separated by using freeze solidification and simultaneously determined by using HPLC. Moreover, greenness assessment using ComplexGAPI shows the superiority of greenness of the proposed method over other published methods. DES is a green and designable solvent that is suitable for the future analysis of preservatives in complex matrices. For the study of drug quality and analysis of trace level of preservatives in drink matrices, the method had the advantages of greenness, simple operation, high accuracy and good sensitivity which contributes to the urgent need of research on green analytical chemistry of preservatives in drink products.Author contributions
J. Z.: investigation, formal analysis, methodology, writing–original draft. L. H.: formal analysis, data curation, validation. L. Z.: resources, supervision, investigation. L. L.: software, visualization, conceptualization. J. Q.: conceptualization, project administration, supervision. L. W: visualization, supervision, writing – review & editing.Conflicts of interest
The authors have declared no conflict of interest.Acknowledgements
The present research was financially supported by the Zhejiang Provincial Postdoctoral Science Foundation (ZJ2020114).References
- I. A. Wiraagni, M. A. Mohd, R. A. Rashid and D. Haron, BioMed Res. Int., 2020, 2020, 2581287 CrossRef PubMed
.
- J. Park, S. Choi, D. Oh and J. H. Mah, Food Sci. Biotechnol., 2018, 27, 291–298 CrossRef CAS PubMed
- S. Oishi., Arch. Toxicol., 2002, 76, 423–429 CrossRef PubMed
- D. K. Evanthia, B. Jean-Pierre, L. C. Giudice, H. Russ, G. S. Prins, A. M. Soto, Z. R. Thomas and A. C. Gore, Endocr. Rev., 2009, 30, 293–342 CrossRef PubMed
- P. D. Darbre, J. Appl. Toxicol., 2003, 23, 89–95 CrossRef CAS PubMed
- D. G. Bayülken and B. A. Tüylü, Drug Chem. Toxicol., 2019, 42, 386–393 CrossRef PubMed
- T. Sai, J. Maru, S. Obara, S. Endoh, H. Kajihara and K. Fujita, J. Occup. Health, 2020, 62, e12176 CAS
- S. P. Epstein, M. Ahdoot, E. Marcus and P. A. Asbell, J. Ocul. Pharmacol. Ther., 2009, 25, 113–119 CrossRef CAS PubMed
- Y. Elif and A. Hasan, J. Sep. Sci., 2018, 41, 2750–2758 CrossRef PubMed
- F. Zamani Mazdeh, S. Sasanfar, A. Chalipour, E. Pirhadi, G. Yahyapour, A. Mohammadi, A. Rostami, M. Amini and M. Hajimahmoodi, Int. J. Anal. Chem., 2017, 2017, 1–8 CrossRef PubMed
- A. Parla, E. Zormpa, N. Paloumpis, A. Kabir, K. G. Furton, E. Roje, V. Samanidou, I. V. Vrek and I. Panderi, Molecules, 2021, 26, 1526 CrossRef CAS PubMed
- M. Rajabi, A. Sarhadi, M. Bazregar, A. Asghari and E. Mirparizi, Anal. Methods, 2017, 9, 5963–5969 RSC
- B. Rda, A. Cch, A. Jmc and A. J. Whd, J. Chromatogr. B, 2016, 1036–1037, 93–99 Search PubMed
- C. Han, B. Xia, X. Chen, J. Shen, Q. Miao and Y. Shen, Food Chem., 2016, 194, 1199–1207 CrossRef CAS PubMed
- C. J. Yang, W. H. Chung and W. H. Ding, RSC Adv., 2020, 10, 35557–35564 RSC
- L. W. Jinling Lv, X. Hu, Z. Tai and Y. Yang, Liq. Chromatogr., 2012, 45, 1960–1970 Search PubMed
- R. Djatmika, C.-C. Hsieh, J.-M. Chen and W.-H. Ding, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2016, 1036–1037, 93–99 CrossRef CAS PubMed
- M. Ding, W. Liu, J. Peng, X. Liu and Y. Tang, Food Chem., 2018, 269, 187–192 CrossRef CAS PubMed
- F. Yang, H. Dong, F. Zhang, J. Shao, Y. Wang, H. Deng, G. Tang, X. Zhang, B. Li and S. Liu, J. Sep. Sci., 2022, 45, 3043–3053 CrossRef CAS PubMed
- N. Alizadeh, N. Saburi and S. E. Hosseini, Food Control, 2012, 28, 315–320 CrossRef CAS
- C. Dong, Y. Mei and L. Chen, J. Chromatogr. A, 2006, 1117, 109–114 CrossRef CAS PubMed
- T. A. Kokya, K. Farhadi and A. A. Kalhori, Food Anal. Methods, 2012, 5, 351–358 CrossRef
- T.-J. Yang, F.-J. Tsai, C.-Y. Chen, T. C.-C. Yang and M.-R. Lee, Anal. Chim. Acta, 2010, 668, 188–194 CrossRef CAS PubMed
- M. Ding, W. Liu, J. Peng, X. Liu and Y. Tang, Food Chem., 2018, 269, 187–192 CrossRef CAS PubMed
- R. E. Kannouma, M. A. Hammad, A. H. Kamal and F. R. Mansour, Microchem. J., 2021, 171, 106790 CrossRef CAS
- M. Torbati, M. A. Farajzadeh, M. R. Afshar Mogaddam and M. Torbati, Microchem. J., 2019, 147, 1103–1108 CrossRef CAS
- C. Ortega-Zamora, J. Gonzalez-Salamo and J. Hernandez-Borges, Molecules, 2021, 26, 6846 CrossRef CAS PubMed
- Y. Liu, J. B. Friesen, J. B. McAlpine, D. C. Lankin, S. N. Chen and G. F. Pauli, J. Nat. Prod., 2018, 81, 679–690 CrossRef CAS PubMed
- K. Radosevic, N. Curko, V. G. Srcek, M. C. Bubalo, M. Tomasevic, K. K. Ganic and I. R. Redovnikovic, LWT-Food Sci. Technol., 2016, 73, 45–51 CrossRef CAS
- M. Francisco, A. van den Bruinhorst and M. C. Kroon, Green Chem., 2012, 14, 2153–2157 RSC
- Z. Xu, Y. Cai, Q. Ma, Z. Zhao, D. Yang and X. Xu, Molecules, 2021, 26(6), 1729 CrossRef CAS PubMed
- D. Ge, Y. Wang, Q. Jiang and E. Dai, A Deep Eutectic Solvent as an Extraction Solvent to Separate and Preconcentrate Parabens in Water Samples Using in situ Liquid-Liquid Microextraction, Sociedade Brasileira de Quimica, 2019, vol. 30, pp. 1203–1210 Search PubMed
- W. Liu, B. Zong, X. Wang, J. Cai and J. Yu, RSC Adv., 2019, 9, 17432–17439 RSC
- L. Xu, X. Miao, Z. Yang, H. Li and B. Qiu, Food Anal. Methods, 2019, 12, 1871–1885 CrossRef
- R. A. Sheldon, Green Chem., 2017, 19, 18–43 RSC
- J. Płotka-Wasylka and W. Wojnowski, Green Chem., 2021, 23, 8657–8665 RSC
- N. A. N. M. Yusoff, N. Y. Rahim, R. E. A. Mohammad, N. Yahaya and M. Miskam, R. Soc. Open Sci., 2021, 8, 202061 CrossRef CAS PubMed
- I. Wazeer, M. K. Hadj-Kali and I. M. Al-Nashef, Molecules, 2020, 26, 75 CrossRef PubMed
- O. E. Plastiras, E. Andreasidou and V. Samanidou, Molecules, 2020, 25, 6026 CrossRef CAS PubMed
- D. N. Danielson and M. R. Fotouh, Talanta, 2017, 170, 22–35 CrossRef PubMed
- W. W. Oomen, P. Begines, N. R. Mustafa, E. G. Wilson and Y. H. Choi, Molecules, 2020, 25, 617 CrossRef CAS PubMed
- W. Huang, M. Pei, X. Liu, C. Yan and Y. Wei, Research, 2020, 2020, 1303672 CAS
- K. Z. Mousavi, Y. Yamini and S. Seidi, New J. Chem., 2018, 42, 9735–9743 RSC
- M. Mei, J. Pang and X. Huang, Anal. Methods, 2018, 10, 1977–1985 RSC
- M. M. Ariffin, N. M. Sohaimi, B. S. Yih and N. M. Saleh, Anal. Methods, 2019, 11, 4126–4136 RSC
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ra07145b |
| This journal is © The Royal Society of Chemistry 2023 |