DOI:
10.1039/D2RA07120G
(Paper)
RSC Adv., 2023,
13, 3371-3376
Unsupported nanoporous gold catalyst for highly selective hydroamination of alkynes†
Received
9th November 2022
, Accepted 17th January 2023
First published on 23rd January 2023
Abstract
An efficient and highly selective heterogeneous catalyst system for hydroamination of alkynes was developed using unsupported gold nanopores (AuNPore) for the first time. The AuNPore-catalyzed highly regioselective hydroamination of alkynes proceeded smoothly without any additive and solvent under mild conditions (rt-50 °C) to yield Markovnikov imines in satisfactory to excellent yields. No gold leached from AuNPore during the hydroamination of alkynes. Moreover, the catalyst was easily recovered and reused without any loss of catalytic activity. A one-pot, two-step procedure using a single AuNPore catalyst has been devised to produce secondary amines derived from readily available alkynes and anilines with high atom efficiency.
Introduction
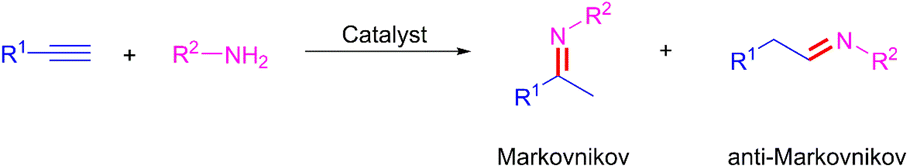
Nitrogen-based functionalities such as imines, enamines, and amines are ubiquitous structural motifs in biologically active molecules, fine chemicals, pharmaceuticals, or the agriculture industry.1 Over the past decades, many methods such as reductive amination of the carbonyl compounds,2 C–N cross-coupling,3 aminomercuration/demercuration of alkynes,4 and hydroamination of alkynes5 have emerged for their construction. Among these methods, the hydroamination of alkynes is the most convenient and promising synthetic route because of its 100% atom economy and easy access of the raw materials. However, the electronic repulsion between the electron-rich functional groups involved, namely the nitrogen lone pair and the π system of alkyne, as well as the regioselectivity towards the Markovnikov and anti-Markovnikov products, make this reaction challenging (Scheme 1).6 Therefore, many types of homogeneous7 and heterogeneous8 catalyst systems have been developed for the hydroamination of alkynes. Taking into consideration of the pharmaceutical process and potential industrial application, the use of recoverable and reusable heterogeneous catalysts with a high catalytic activity for the hydroamination of alkynes would be more desirable. However, these catalysts still have the following considerable drawbacks: (i) high reaction temperatures (80–150 °C) and additives were required, (ii) organic solvent as reaction medium, (iii) poor yield and regioselectivity, (iv) metal nanoparticles needed specific carriers to prevent aggregation-induced catalytic deactivation, (v) leaching of metal, (vi) additional apparatus (microwave and light sources) may be indispensable (Scheme 2a). From the perspective of green chemistry and other practical concerns, the development of more environmentally benign and efficient catalyst systems, without any additive and solvent, under much milder conditions, for the hydroamination of alkynes is highly desirable.
 |
| | Scheme 1 Possible imine products from hydroamination of terminal alkynes. | |
 |
| | Scheme 2 Synthesis of Markovnikov imines. | |
In recent years, unsupported nanoporous metal material has attracted much attention because of their unique three-dimensional nanoporous structure, large surface-to-volume ratio, nontoxic nature, high recyclability, and simple recovery.9 Moreover, the bulk metallic feature of nanoporous metals could prevent the aggregation-induced catalytic deactivation and nanoporous metals without any supports may help to understand the intrinsic catalytic activity more easily by elimination of the support-effect.10 We have demonstrated that nanoporous metals are promising green heterogeneous catalysts for liquid-phase organic synthesis.11 In the current study, we attempted the hydroamination of alkynes using unsupported gold nanopores (AuNPore) as the catalyst, which can be fabricated easily by selective leaching of Ag from an alloy foil of Au30Ag70 at chemical dealloying conditions. The highly regioselective hydroamination of alkynes proceeded smoothly under relatively low temperatures (rt-50 °C) to provide Markovnikov imines in good to excellent yields without any additive and solvent (Scheme 2b). Furthermore, a one-pot, two-step procedure using AuNPore catalyst has been devised to produce secondary amines derived from alkynes and anilines with high atom efficiency. The results are reported in the current work.
Experimental
Materials
Au (99.99%) and Ag (99.99%) were purchased from Tanaka Kikinzoku Hanbai K. K. and Mitsuwa's Pure Chemicals, respectively. Au/TiO2 (1% wt/wt loading; average size of AuNPs is around 2–3 nm) were purchased from Strem. The starting materials were purchased from Energy Chemicals Co., Ltd. Solvents were purified by standard techniques without special instructions. All other reagents were used as received.
Catalyst preparation
The unsupported nanoporous gold (AuNPore) catalyst was prepared according to a method described previously.12 Au (99.99%) and Ag (99.99%) were melted with electric arc-melting furnace under argon atmosphere to form Au/Ag alloy (30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :70, in at%), which was rolled down to thickness of 0.04 mm. The resulting foil was annealed at 850 °C for 20 h. The foil was cut into small pieces (5 × 2 mm square). Treatment of the resulting chips (67.1 mg) with 70 wt% nitric acid (7.5 mL) for 18 h at room temperature in a shaking apparatus resulted in the formation of the nanoporous structure by selective leaching of silver. The material was washed with a saturated aqueous solution of NaHCO3, pure water, and acetone, successively. Drying of the material under reduced pressure gave the nanoporous gold (Fig. S1†).
:70, in at%), which was rolled down to thickness of 0.04 mm. The resulting foil was annealed at 850 °C for 20 h. The foil was cut into small pieces (5 × 2 mm square). Treatment of the resulting chips (67.1 mg) with 70 wt% nitric acid (7.5 mL) for 18 h at room temperature in a shaking apparatus resulted in the formation of the nanoporous structure by selective leaching of silver. The material was washed with a saturated aqueous solution of NaHCO3, pure water, and acetone, successively. Drying of the material under reduced pressure gave the nanoporous gold (Fig. S1†).
Catalyst characterization
SEM observation was carried out using HITACHI FE-SEM S4300 operated at an accelerating voltage of 10 kV. TEM was performed on a Tecnai G2 microscope operating at 120 kV. EDX analysis was carried out using EDAX Genesis with HITACHI FE-SEM S4300 operated at an accelerating voltage of 20 kV. The XPS measurements were carried out using an ESCALAB 250Xi spectrometer (Thermo Fisher Scientific Inc., UK) employing monochromatic AlKα X-ray radiation. ICP-MS analysis was performed with Agilent ICP-MS HP 4500.
Typical procedure for hydroamination of phenylacetylenes
AuNPore (4.92 mg, 5 mol%), phenylacetylene (1a, 51.07 mg, 0.5 mmol), and phenylamine (2a, 55.55 mg, 0.6 mmol) were placed in a V-shaped vial reactor with a magnetic stir bar under N2 atmosphere. The reaction mixture was stirred at 50 °C for 24 h and cooled down to room temperature. Then, the AuNPore was recovered by filtration, followed by washing with acetone, and the residual was purified via silica gel chromatography (eluent: petroleum ether/ethyl acetate = 5/1 with 10% of triethylamine) to afford imine (3a) as a yellow oil.
Results and discussion
Characterization of AuNPore catalyst
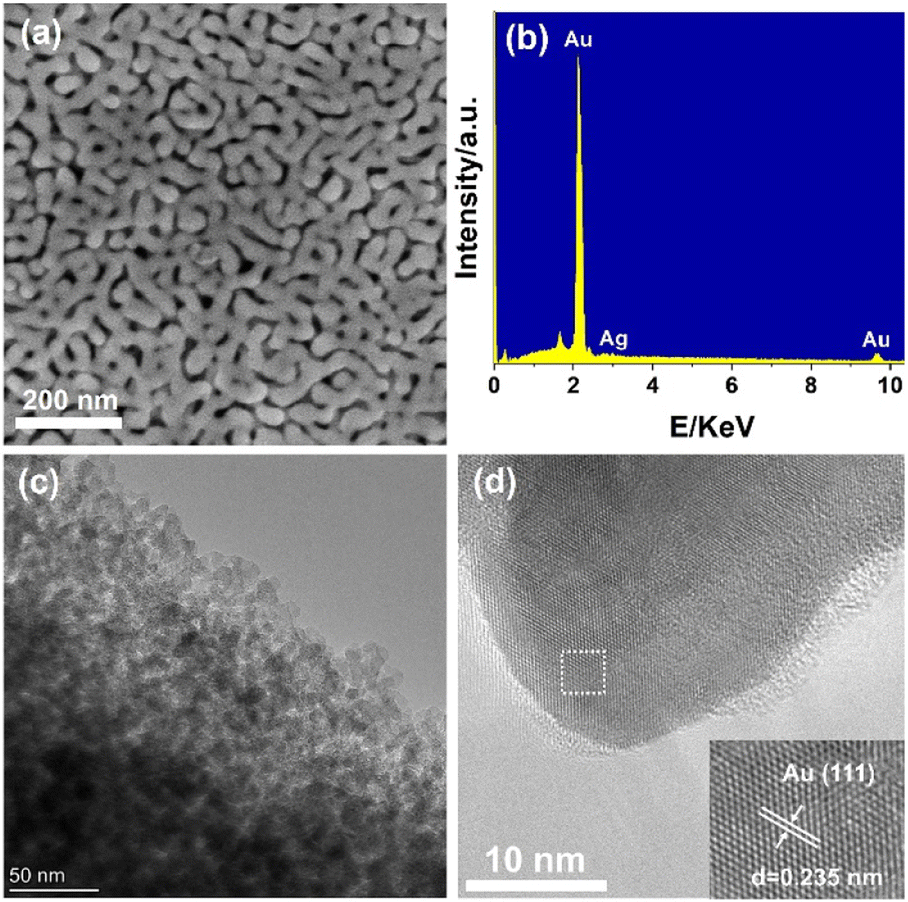
The scanning electron micrograph (SEM) in Fig. 1a clearly indicate the formation of three-dimensional (3D) nanoarchitecture constructed by hyperboloid-like ligaments. The ligament and nanopore channel were formed uniformly across the entire AuNPore with an average ligament size around 30 nm. The dealloyed material was found to comprise Au>99Ag<1 based on energy dispersive X-ray spectroscopy (EDX) analysis (Fig. 1b). The transmission electron micrograph (TEM) showed the structure of the AuNPore with bright nanopores and dark-contrast ligaments (Fig. 1c). Further, the high-resolution TEM (HRTEM) of freshly prepared AuNPore demonstrated that the topmost surface of these ligaments is not smooth but various steps and kinks are found representing low coordinated gold atoms as shown in Fig. 1d. The high density of low coordinated gold atoms on surface are proposed to be catalytically active sites in this catalyst. The lattice spacing was 0.235 nm corresponding to the (111) crystal plane of the AuNPore. The Au 4f 7/2 peak was observed at 84.0 eV from the Au 4f X-ray photoelectron spectroscopy (XPS) measurements. This observation indicated that the oxidation state of Au in the AuNPore was Au(0) (Fig. S2†).13
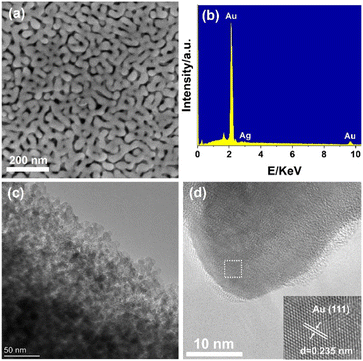 |
| | Fig. 1 (a) SEM image, (b) EDS analysis, (c) TEM image, (d) HRTEM image (inset: close-up views of the boxed regions) of fresh AuNPore. | |
Hydroamination of phenylacetylene catalyzed by AuNPore
The hydroamination of phenylacetylene (1a) in the presence of AuNPore (5 mol%) was chosen as a model in our preliminary studies to optimize the reaction conditions. The results are shown in Table 1. Toluene, ethanol (EtOH), water (H2O), acetonitrile (CH3CN), and tetrahydrofuran (THF) were initially tested at 50 °C for 24 h. Good conversion of 1a and high yield of 3a were observed (entries 1–5). Trace amounts of acetophenone (4a) were also obtained, probably due to hydrolysis of the imine 3a. Notably, the best results were obtained without solvent, leading to 99% conversion of 1a and 97% yield of Markovnikov product 3a (entry 6). The conversion and yield decreased with decreases in both the catalyst amount and the temperature (entries 7 and 8, respectively). The conversion and yield also decreased with relatively short reaction time (entry 9). No reaction was observed in the absence of catalyst AuNPore or by using the precursor of AuNPore, Au30Ag70 alloy, in the hydroamination of 1a (entries 10 and 11). The homogeneous AuCl also did not exhibit any catalytic activity (entry 12). Accordingly, we set the standard conditions as follows: AuNPore (5 mol%) and 50 °C for 24 h. Low conversion and yield were observed when the Au/TiO2 catalyst was used instead of the AuNPore catalyst (entry 13). This result clearly indicated that the activity of the AuNPore catalyst is higher than that of the commercially available catalyst. In all cases, the Markovnikov product was obtained exclusively. No isomeric anti-Markovnikov product could be detected by 1H NMR analyses of the crude reaction mixtures.
Table 1 Screening of reaction conditionsa
|
| Entry |
Solvent |
Conversionb (%) |
Yieldb (%) |
| 3a |
4a |
| Reaction conditions: phenylacetylene (1a, 0.5 mmol), phenylamine (2a, 0.6 mmol), AuNPore (5 mol%), and solvent (2 mL) at 50 °C for 24 h. Determined by 1H NMR and GC-MS analysis; the value in parenthesis indicates isolated yield. 3.0 mol% of AuNPore was used. The hydroamination reaction was conducted at room temperature. The reaction mixture was treated for 20 h. The reaction was treated in the absence of AuNPore. Au30Ag70 alloy was used. AuCl was used. Au/TiO2 was used as the catalyst instead of AuNPore. No reaction; starting material 1a and 2a were recovered. |
| 1 |
Toluene |
67 |
64 |
3 |
| 2 |
EtOH |
87 |
85 |
2 |
| 3 |
H2O |
96 |
92 |
4 |
| 4 |
CH3CN |
82 |
80 |
2 |
| 5 |
THF |
57 |
56 |
1 |
| 6 |
— |
99 |
97(94) |
2 |
| 7c |
— |
83 |
82 |
1 |
| 8d |
— |
83 |
81 |
2 |
| 9e |
— |
87 |
86 |
1 |
| 10f |
— |
NRj |
— |
— |
| 11g |
— |
NRj |
— |
— |
| 12h |
— |
NRj |
— |
— |
| 13i |
— |
63 |
61 |
2 |
Scope of the substrates
The catalytic activity of AuNPore was further examined with terminal alkynes and anilines under the aforementioned conditions (Scheme 3). The hydroamination of aromatic alkynes 1b–1f bearing an electron-donating group such as methyl, methoxy or dimethylamino group proceeded smoothly to afford the corresponding imines 3b–3f in good yields (75–84%). The sterically hindered ortho-substituted aryl alkynes 1d exhibited high reactivity, which indicated that electron-donation in the ortho-position was tolerated well despite the increased steric bulk. The product 3d contained a 2:1 ratio of E:Z isomers. This behavior was attributed to the E/Z interconversion caused by the ortho-effect.14 The substrates 1g–1j bearing an electron-withdrawing group fluoro (F), chloro (Cl), bromo (Br) or trifluoromethyl (CF3) at the para-position exhibited higher reactivity than the substrates bearing an electron-donating group in the hydroamination reaction. The products 3g–3j were obtained in high yields (88%, 85%, 84% and 90% respectively) at room temperature. The hydroamination of 2-naphthalene acetylene (1k) under the standard reaction conditions gave the corresponding imine 3k in 85% yield. Subsequently, various anilines were tested with phenylacetylene under the standard set of reaction conditions. The hydroamination of the substrates 1l–1q bearing an electron-donating group such as methyl, methoxy or hydroxy group proceeded smoothly to afford the corresponding imines 3l–3q in high yields (89–94%). Similarly, the steric hindrance did not affect the catalytic performance as the bulky aniline (1n) bearing the methyl group at the ortho-position. Aniline substrates 1r–1w bearing a strong electron-withdrawing group halogen (F, Cl, Br), acetyl (CH3CO), methoxycarbonyl (CH3OCO) or trifluoromethyl (CF3) at the para or meta position, respectively, exhibited relatively low reactivity for completion of hydroamination compared with that of anilines 1l–1q. The desired products 3r–3w were obtained in 80–85% yields. The hydroamination of 1x proceeded well under the standard reaction conditions to produce the corresponding imine 3x in 87% yield. These results described above indicate that the reactivity of phenylacetylenes and anilines were influenced by the electronic property of substituents in substrates.
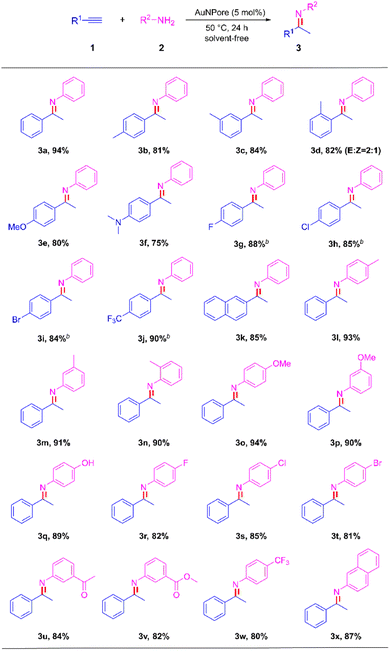 |
| | Scheme 3 AuNPore-catalyzed hydroamination of terminal alkynes and anilinesa. aReaction conditions: AuNPore (5 mol%), 1 (0.5 mmol), 2 (0.6 mmol), at 50 °C for 24 h. Isolated yield. bThe hydroamination reaction was conducted at room temperature. | |
Leaching studies of AuNPore catalyst
We next examined the leaching test to clarify whether the AuNPore-catalyzed reaction proceeds heterogeneously or not (Scheme 4). The hydroamination of 1a was performed under standard conditions for 12 h. At this stage, 3a was formed in 40% yield. Then, half of the reaction mixture was transferred to another reaction vessel, and it was continuously stirred in the absence of a solid catalyst for 12 h. As we expected, no change in the yield of 2a was observed. By contrast, 2a was obtained in 92% yield from the reaction of residual containing the AuNPore catalyst. The inductively coupled plasma-mass spectrometry (ICP-MS) measurements were also used to confirm that no Au leached from the AuNPore catalyst (detection limit of 0.01 ppm). These results clearly indicated that the catalysis proceeded through a heterogeneous process.
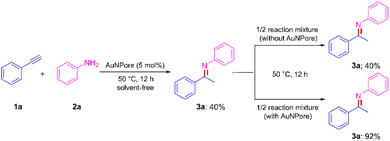 |
| | Scheme 4 Leaching test of AuNPore catalyst in the hydroamination of 1a. | |
Gram-scale reaction
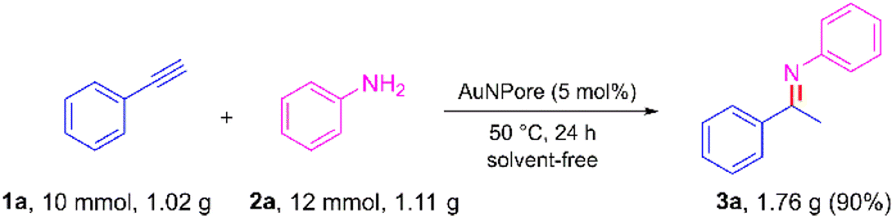
To further explore the practical applicability of our method, the hydroamination of phenylacetylene was scaled up to the gram scale, and the result is shown in Scheme 5. When 1.02 g of 1a and 1.11 g of 2a were treated under standard conditions, 1.76 g of product 3a was obtained with 90% yield. This result indicated that the gram-scale reactions also proceeded smoothly without loss of efficiency.
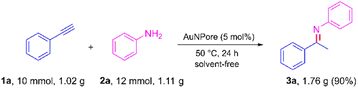 |
| | Scheme 5 AuNPore-catalyzed gram-scale hydroamination of 1a. | |
Reusability and recovery of AuNPore catalyst
Recycling tests of AuNPore catalyst were performed under the optimal conditions. The results are shown in Fig. 2. After each recycle, the AuNPore catalyst was simply washed with acetone for several times to remove the surface substances and then reused in the next run without further purification. After six recycles, high yield of 3a was still obtained. The SEM image of AuNPore after six runs is shown in Fig. S3.† The result for the fresh one and that after six runs were essentially same, indicating that AuNPore catalyst is very stable and can be used repeatedly without loss of activity.
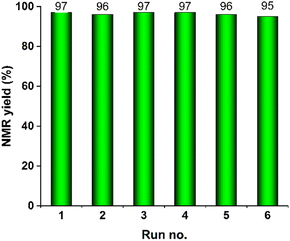 |
| | Fig. 2 Reusability of AuNPore for hydroamination of 1a. | |
Catalytic synthesis of secondary amines through one-catalyst, one-pot, two-step process
To expand this reaction, we also investigated the reductive hydroamination reaction of alkynes with amines. In a typical reductive hydroamination reaction, the second reduction (hydrogenation) step is generally achieved by using either a separate catalyst system, such as ZnCl2/NaBH3CN,15 or a strong reducing agent, such as LiAlH4.16 These procedures can create troublesome experimental procedures and require extra chemicals and solvents, and the strong reducing reagents can lead to a decrease in the chemoselectivity toward functional groups that are sensitive to reducing conditions. Therefore, the development of a one-pot method that uses a single catalyst for a reductive hydroamination reaction that occurs under mild reaction conditions would be highly worthwhile. During the investigation of the catalytic activity for the dissociation of various chemical bonds in the last decade, we have disclosed that the Si–H bond is readily activated by AuNPore, reacting with water to produce silanols and H2,17 which was successfully extended to the selective reduction of unsaturated chemical bonds.18 Herein, considering the high product yield in hydroamination of alkynes as well as the fact that AuNPore catalyst can catalyze transfer hydrogenation of unsaturated chemical bonds with hydrosilanes as reductants, we envision a one-catalyst, one-pot, two-step process in which the initial imine formed in the hydroamination of alkynes is subsequently reduced to secondary amine. If the proposed process would take place with high efficiency and selectivity under mild conditions, it would be an interesting general catalytic process for the formation of secondary amines. The process is shown in Scheme 6. As anticipated based on the catalytic activity of AuNPore, we have been able to obtain different secondary amines 4 in one-catalyst, one-pot, two-step reactions without isolation of the imine intermediates 3 at room temperature under solvent-free conditions.
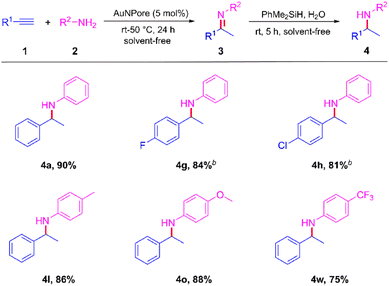 |
| | Scheme 6 AuNPore-catalyzed one-pot, two-step synthesis of secondary aminea. aReactions were performed using AuNPore (5 mol%), 1 (0.5 mmol), 2 (0.6 mmol), at 50 °C for 24 h. Then, PhMe2SiH (0.6 mmol) and H2O (0.6 mmol) were added directly and the mixture continued to react at room temperature for 5 h. Isolated yield. bThe hydroamination reaction was conducted at room temperature. | |
On the basis of our experimental outcomes, a plausible catalytic cycle is proposed to account for the present one-catalyst, one-pot, two-step reaction (Scheme 7). The initial step involves the generation of a AuNPore-activated alkyne intermediate A. The amine reagent coordinates to the electron-deficient intermediate A, forming intermediate B, which then undergoes a nucleophilic attack by the nitrogen of the amine resulting in the formation C. Intermediate C experiments a proton transfer to form a AuNPore-alkene species D, which releases the active AuNPore catalyst and the enamine E. After a tautomerization process, the imine product F is obtained. Then, PhMe2SiH and H2O were adsorbed onto the surface of the AuNPore catalyst. The hydrogen bond forms between imine F and water followed by the reaction of hydrosilane and water to give [AuNPore−H]− and protonated imine species, in which the latter reacts subsequently with hydride on AuNPore to form the corresponding secondary amine G and regenerates the AuNPore catalyst.
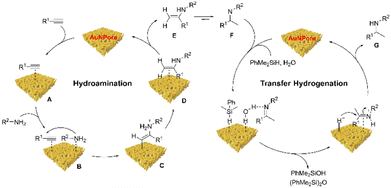 |
| | Scheme 7 Proposed mechanism for one-catalyst, one-pot, two-step synthesis of secondary amine. | |
Conclusions
In conclusion, we have demonstrated for the first time that the unsupported AuNPore is an efficient heterogeneous catalyst for the regioselective hydroamination of alkynes. Good to excellent yields of Markovnikov imines were obtained with excellent regioselectivities under low temperature without using any additive and solvent. The AuNPore catalyst could be easily recovered and reused several times without any loss of activity. Furthermore, the AuNPore is able to selectively catalyze the formation of secondary amines through a simple, one-catalyst, one-pot, two-step reaction that involves one hydroamination step followed by transfer hydrogenation. The unique features of the AuNPore catalyst system (no additive, no solvent, low temperature, high regioselectivity, good functional group tolerance, high stability, and no leaching) render this catalyst useful for laboratory and industrial applications.
Author contributions
Conceptualization, J. L., Y. L. and M. B.; methodology, Y. L.; analysis and validation, Y. W., Y. L. and H. L.; resources, J. L. and Y. L.; writing-original draft preparation, Y. L. and Y. W.; writing-review and editing, J. L., M. B. and Y. Y.; visualization, P. L. and X. F.; supervision, Y. L.; and funding acquisition, J. L., M. B. and Y. L. All authors have read and agreed to the published version of the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (21961024, 21961025 and 22261042), Natural Science Foundation of Inner Mongolia Autonomous Region of China (2018JQ05, 2020BS02009, and 2020BS02014), Inner Mongolia Science & Technology Planning Project for Applied Technology Research and Development (2019GG261) and Doctoral Scientific Research Foundation of Inner Mongolia Minzu University (BS445, BS481 and BS560).
Notes and references
-
(a) L. Huang, M. Arndt, K. Gooßen, H. Heydt and L. J. Gooßen, Chem. Rev., 2015, 115, 2596–2697 CrossRef CAS PubMed;
(b) G. Guillena, D. J. Ramón and M. Yus, Chem. Rev., 2010, 110, 1611–1641 CrossRef CAS PubMed;
(c) T. E. Muller, K. C. Hultzsch, M. Yus, F. Foubelo and M. Tada, Chem. Rev., 2008, 108, 3795–3892 CrossRef PubMed.
-
(a) S. Liang, P. Monsen, G. B. Hammond and B. Xu, Org. Chem. Front., 2016, 3, 505–509 RSC;
(b) H. Alinezhad, H. Yavari and F. Salehian, Curr. Org. Chem., 2015, 19, 1021–1049 CrossRef CAS;
(c) S. Werkmeister, K. Junge and M. Beller, Green Chem., 2012, 14, 2371–2374 RSC.
-
(a) D. S. Surry and S. L. Buchwald, Chem. Sci., 2011, 2, 27–50 RSC;
(b) J. F. Hartwig, Acc. Chem. Res., 2008, 41, 1534–1544 CrossRef CAS PubMed.
- R. C. Larock, Angew. Chem., Int. Ed., 1978, 17, 27–37 CrossRef.
-
(a) M. Sengupta, S. Das, S. Islam and A. Bordoloi, ChemCatChem, 2020, 13, 1089–1104 CrossRef;
(b) Z. Tashrifi, M. M. Khanaposhtani, B. Larijani and M. Mahdavi, Asian J. Org. Chem., 2020, 9, 969–991 CrossRef CAS;
(c) R. Severin and S. Doye, Chem. Soc. Rev., 2007, 36, 1407–1420 RSC.
- T. E. Müller, K. C. Hultzsch, M. Yus, F. Foubelo and M. Tada, Chem. Rev., 2008, 108, 3795–3892 CrossRef PubMed.
- Selected examples of homogeneous hydroamination of alkynes. For Au catalysis, see:
(a) C. Ruiz-Zambrana, M. Poyatos and E. Peris, ACS Catal., 2022, 12, 4465–4472 CrossRef CAS;
(b) T. Jia, S. Fan, F. Li, X. Ye, W. Zhang, Z. Song and X. Shi, Org. Lett., 2021, 23, 6019–6023 CrossRef CAS PubMedFor Pd catalysis, see:
(c) M. Virant, M. Mihelač, M. Gazvoda, A. E. Cotman, A. Frantar, B. Pinter and J. Košmrlj, Org. Lett., 2020, 22, 2157–2161 CrossRef CAS PubMedFor Rh catalysis, see:
(d) E. Kumaran and W. K. Leong, Organometallics, 2012, 31, 1068–1072 CrossRef CASFor Cu catalysis, see:
(e) Q. Sun, Y. Wang, D. Yuan, Y. Shen and Q. Shen, Dalton Trans., 2015, 44, 20352–20360 RSCFor Ag catalysis, see:
(f) J. Bahri, B. Jamoussi, A. van Der Lee, M. Taillefer and F. Monnier, Org. Lett., 2015, 17, 1224–1227 CrossRef CAS PubMed;
(g) X. Zhang, B. Yang, G. Li, X. Shu, D. C. Mungra and J. Zhu, Synlett, 2012, 4, 622–626 Search PubMed.
- Selected examples of heterogeneous hydroamination of alkynes. For Au catalysis, see:
(a) H. Shen, Q. Wu, S. Malola, Y.-Z. Han, Z. Xu, R. Qin, X. Tang, Y.-B. Chen, B. K. Teo, H. Häkkinen and N. Zheng, J. Am. Chem. Soc., 2022, 144, 10844–10853 CrossRef CAS PubMed;
(b) V. I. Isaeva, K. Papathanasiou, V. V. Chernyshev, L. Glukhov, G. Deyko, K. K. Bisht, O. P. Tkachenko, S. V. Savilov, N. A. Davshan and L. M. Kustov, ACS Appl. Mater. Interfaces, 2021, 13, 59803–59819 CrossRef CAS PubMed;
(c) C. Parise, B. Ballarin, D. Barreca, M. C. Cassani, P. Dambruoso, D. Nanni, I. Ragazzini and E. Boanini, Appl. Surf. Sci., 2019, 492, 45–54 CrossRef CAS;
(d) L.-C. Lee and Y. Zhao, ACS Catal., 2014, 4, 688–691 CrossRef CAS;
(e) J. Zhao, Z. Zheng, S. Bottle, A. Chou, S. Sarina and H. Zhu, Chem. Commun., 2013, 49, 2676–2678 RSCFor Pd catalysis, see:
(f) M. K. Richmond, S. L. Scott and H. Alper, J. Am. Chem. Soc., 2001, 123, 10521–10525 CrossRef CAS PubMedFor Ru catalysis, see:
(g) C. Risi, E. Cini, E. Petricci, S. Saponaro and M. Taddei, Eur. J. Inorg. Chem., 2020, 1000–1003 CrossRef CASFor Co, Ni catalysis, see:
(h) B. Davaasuren, A.-H. Emwas and A. R. Inorg, Chem, 2017, 56, 9609–9616 CASFor Cu catalysis, see:
(i) M. Sengupta, S. Das and A. Bordoloi, Mol. Catal., 2017, 440, 57–65 CrossRef CASFor Zn catalysis, see:
(j) A. K. Cook and C. Copéret, Organometallics, 2018, 37, 1342–1345 CrossRef CAS.
-
(a) R. Song, J. Han, M. Okugawa, R. Belosludov, T. Wada, J. Jiang, D. Wei, A. Kudo, Y. Tian, M. Chen and H. Kato, Nat. Commun., 2022, 13, 5157 CrossRef CAS PubMed;
(b) H.-J. Qiu, I. Johnson, L. Chen, W. Cong, Y. Ito, P. Liu, J. Han, T. Fujita, A. Hirata and M. Chen, Nanoscale, 2021, 13, 10916–10924 RSC;
(c) Z. Xiao, H. Yang, S. Yin, J. Zhang, Z. Yang, K. Yuan and Y. Ding, Nanoscale, 2020, 12, 4314–4319 RSC;
(d) H.-J. Qiu, H.-T. Xu, L. Liu and Y. Wang, Nanoscale, 2015, 7, 386–400 RSC;
(e) K. M. Kosuda, A. Wittstock, C. M. Friend and M. Bäumer, Angew. Chem., Int. Ed., 2012, 51, 1698–1701 CrossRef CAS PubMed;
(f) A. Wittstock, V. Zielasek, J. Biener, C. M. Friend and M. Bäumer, Science, 2010, 327, 319–322 CrossRef CAS PubMed.
- T. Fujita, P. Guan, K. McKenna, X. Lang, A. Hirata, L. Zhang, T. Tokunaga, S. Arai, Y. Yamamoto, N. Tanaka, Y. Ishikawa, N. Asao, Y. Yamamoto, J. Erlebacher and M. Chen, Nat. Mater., 2012, 11, 775–780 CrossRef CAS PubMed.
-
(a) Y. Lu, J. Wang, X. Feng, Y. Li, W. Zhang, Y. Yamamoto and M. Bao, Nanoscale, 2022, 14, 9341–9348 RSC;
(b) Y. Lu, Y. Yamamoto, A. I. Almansour, N. Arumugam, R. S. Kumar and M. Bao, Chin. J. Catal., 2018, 39, 1746–1752 CrossRef CAS;
(c) Y. Lu, X. Feng, B. S. Takale, Y. Yamamoto, W. Zhang and M. Bao, ACS Catal., 2017, 7, 8296–8303 CrossRef CAS;
(d) B. S. Takale, X. Feng, Y. Lu, M. Bao, T. Jin, T. Minato and Y. Yamamoto, J. Am. Chem. Soc., 2016, 138, 10356–10364 CrossRef CAS PubMed.
- M. Yan, T. Jin, Y. Ishikawa, T. Minato, T. Fujita, L. Y. Chen, M. Bao, N. Asao, M. W. Chen and Y. Yamamoto, J. Am. Chem. Soc., 2012, 134, 17536–17542 CrossRef CAS PubMed.
- K. Raiber, A. Terfort, C. Benndorf, N. Krings and H.-H. Strehblow, Surf. Sci., 2005, 595, 56–63 CrossRef CAS.
- J. Bjøgo, D. R. Boyd, C. G. Watson and W. B. Jennings, J. Chem. Soc. Perkin Trans., 1974, 2, 757–762 RSC.
-
(a) K. Alex, A. Tillack, N. Schwarz and M. Beller, ChemSusChem, 2008, 1, 333–338 CrossRef CAS PubMed;
(b) A. Heutling, F. Pohlki and S. Doye, Chem.–Eur. J., 2004, 10, 3059–3071 CrossRef CAS PubMed.
-
(a) Q. Sun, Y. Wang, D. Yuan, Y. Yao and Q. Shen, Dalton Trans., 2015, 44, 20352–20360 RSC;
(b) E. Kumaran and W. K. Leong, Organometallics, 2012, 31, 1068–1072 CrossRef CAS.
- N. Asao, Y. Ishikawa, N. Hatakeyama, Menggenbateer, Y. Yamamoto, M. Chen, W. Zhang and A. Inoue, Angew. Chem., Int. Ed., 2010, 49, 10093–10095 CrossRef CAS PubMed.
- Y. Zhao, S. Zhang, Y. Yamamoto, M. Bao, T. Jin and M. Terada, Adv. Synth. Catal., 2019, 361, 4817–4824 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Ying Wang‡a,
He Lia,
Peihe Lia,
Xiujuan Fengb,
Yoshinori Yamamoto
*a,
Ying Wang‡a,
He Lia,
Peihe Lia,
Xiujuan Fengb,
Yoshinori Yamamoto