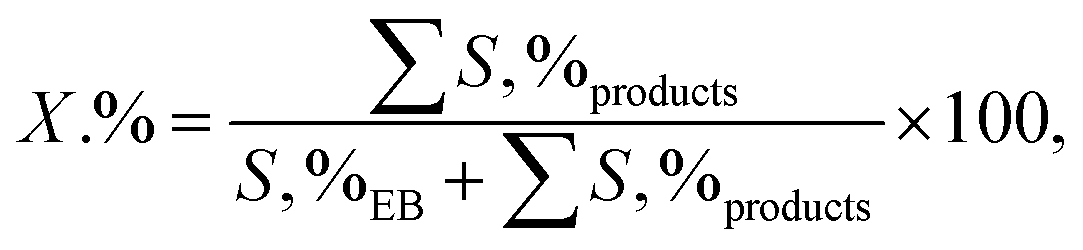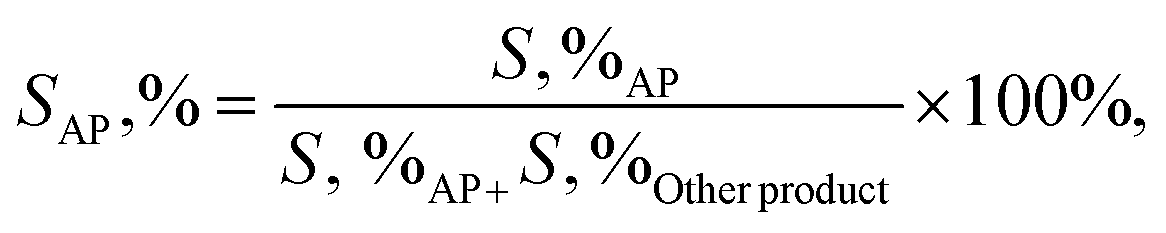DOI:
10.1039/D2RA07102A
(Paper)
RSC Adv., 2023,
13, 9846-9863
Fabrication of a magnetic Mn(II) cross-linked chitosan-amine/glutaraldehyde nanocomposite for the rapid degradation of dyes and aerobic selective oxidation of ethylbenzene†
Received
8th November 2022
, Accepted 11th March 2023
First published on 28th March 2023
Abstract
Owing to the great demand for using sustainable, renewable, and widely available materials in catalytic systems for the conversion of waste/toxic material to high value-added and harmless products, biopolymers derived from natural sources have demonstrated great promise as an alternative to state-of-the-art materials that suffer from high costs and limitations. These have encouraged us to design and fabricate a new super magnetization of Mn–Fe3O4–SiO2/amine-glutaraldehyde/chitosan bio-composite (MIOSC-N-et-NH2@CS-Mn) for advanced/aerobic oxidation process. The morphological and chemical characterization of the as-prepared magnetic bio-composite was assessed using ICP-OES, DR UV-vis, BET, FT-IR, XRD, FE-SEM, HR-TEM, EDS, and XPS techniques. The PMS + MIOSC-N-et-NH2@CS-Mn system was capable of degrading methylene orange (98.9% of removal efficiency) and selectively oxidizing ethylbenzene to acetophenone (conversion 93.70%, selectivity 95.10% and TOF 214.1 (103 h−1) within 8.0 min and 5.0 h, respectively. Moreover, MO was efficiently mineralized (TOC removal of ∼56.61) by MIOSC-N-et-NH2@CS-Mn with 60.4%, 5.20, 0.03 and 86.02% of the synergistic index, reaction stoichiometric efficiency, specific oxidant efficiency, and oxidant utilization ratio in wide pH ranges, respectively. An understanding of its vital parameters and relationship of catalytic activity with structural, environmental factors, leaching/heterogenicity test, long-term stability, inhibitory effect of anions in water matrix, economic study and response surface method (RSM) were evaluated in detail. Overall, the prepared catalyst could be employed as an environmentally friendly and low-cost candidate for the enhanced activation of PMS/O2 as an oxidant. Additionally, MIOSC-N-et-NH2@CS-Mn exhibited great stability, high recovery efficiency, and low metal leaching, which eliminated the harsh condition reaction and supplied practical application performance for water purification and selective aerobic oxidation of organic compounds.
1. Introduction
The application of biopolymers derived from natural sources (with polypeptide- and protein-based, polynucleotide-based and polysaccharide-based materials) has expanded into many applied scientific fields (such as biology, biomedical, medicine, agricultural, food, environmental and chemical) and industrial areas. High attention to polysaccharides (such as carrageenan, alginic acid, hyaluronic acid & chondroitin sulfate with acidic nature, chitin, chitosan, cellulose, polyline with basic nature and agarose, pullulan, dextran & glycosaminoglycans with neutral nature) can be due to their unique properties, such as biodegradability, biocompatibility, non-toxicity, economical, renewability and availability.1 Chitin, the second most abundant marine polymer (after cellulose) on earth, is an essential biomass resource made in plants and lower animals. It is utilized as a raw material for the generation of chitosan through the partial deacetylation mechanism, in which the main steps include demineralization, deproteination of the amino group at the C![[2 with combining macron]](https://www.rsc.org/images/entities/char_0032_0304.gif) position of glucosamine, and decolorization. These processes are conducted via chemical or biological methods (fermentation or enzyme-based treatment).2–5
position of glucosamine, and decolorization. These processes are conducted via chemical or biological methods (fermentation or enzyme-based treatment).2–5
Assessing molecular weight and/or solubility is crucial to comprehending the possible applications of polymers, particularly in biological and chemical systems. Chitosan viscosity depends on many factors, such as molecular weight, degree of deacetylation (DD), degree of polymerization (DP), ionization, and pH value.4,5 Water cannot dissolve chitosan in itself; however, chitosan is dissolved by certain organic acids, such as lactic, acetic, formic, glutamic, hydrochloric acid solutions and diluted nitric acid (pH up to 6.5). Moreover, the abundant presence of –OH and –NH2 groups on the chitosan chain can facilitate its efficient graft to heavy metal ions via chelation and H-bonding interactions with water molecules. Thus, chitosan has great potency as a versatile platform for supporting active species and has been produced into various forms of powders (such as gels, spheres and fibers) for the immobilization/adsorption of transition metal and then the fabrication of a magnetic bio-catalyst. Briefly, magnetic core shell composite fabrication mainly involves three steps: first is the synthesis of magnetic nanomaterials, the second is the coating of magnetic beads by a thin shell of the desired material, such as amorphous silica,6 metal,7 metal–organic-framework,8 polymer,9,10 bio-polymers,11,12 hybrid shells13 and etc.), and after that, loading of the active site. The selection of shell materials in core–shell NPs is largely determined by their end application and utility. Furthermore, the magnetic-core shell is separated from the reaction medium, thus increasing its efficiency.14 Owing to their remarkable physicochemical properties, magnetic bio-polymers as core–shell NPs are used for diverse practical biomedical, environmental, catalytic synthesis and industrial biotechnological applications.3–5,11–14 Two very important aspects in reducing environmental hazards are pollution degradation, such as the mineralization of organic dyes, and the conversion of toxic waste to high-value-added material, such as the oxidation of ethylbenzene (EB) to acetophenone (AP). Non-biodegradable dyes (such as methylene blue, methyl orange, congo red rhodamine B and etc.) are applied extensively in textile, paint, printing, leather, paper, plastic, cosmetics, food and pharmaceutical industries, and they are improperly discharged into the environment via wastewater. Dye contaminants are often toxic, carcinogenic and mutagenic; consequently, exposure to them causes many sicknesses, such as skin irritation, dermatitis and red skin rash with headache, liver and kidney damage, diarrhea, nausea, joint and/or muscle pain, irregular heartbeat, seizures and central nervous system infections, in both humans and animals.15 For instance, methyl orange (MO, dimethylaminoazobenzenesulfonate), a common anionic azo dye, can cause digestion and heart problems in individuals exposed to it and can even decrease the oxygen level of water, which can directly threaten the aquatic life of creatures and ecosystems. Furthermore, MO contains aromatic ring and azo groups in their structures, which are highly toxic, carcinogenic and teratogenic, with negative effects on the environment and organisms.16 Thus, owing to recalcitrant to biodegradation degradation, which may accumulate in the environment and threaten ecology and human health, it is inevitable to develop an eco-friendly way to degrade dyes in wastewater. Many strategies, such as adsorption, biological digestion, coagulation/flocculation, membrane separation, ion exchange, ultrafiltration/nanofiltration, and UV/metal-mediated activation of oxidants (i.e., proxymonosulfate (PMS), proxydisulfate (PDS), proxysulfate (PS) and hydrogen peroxide), have been proposed or demonstrated for the treatment of dye-containing wastewaters. Amid them, advanced oxidation processes (AOPs) are known as one of the most efficient degradation techniques for completely mineralizing harmful dyes into harmless minerals.15–18 Compared to homogeneous activation, heterogeneous catalysts exhibit excellent PMS activation capability, chemical/mechanical stability, low leachability, rapid and efficient separation and reuse. Therefore, the evaluation of efficient heterogeneous catalysts (e.g., CoFe2O4 (ref. 19 and 20) CoMn2O4,21 Cox Mn3xO4,22 CoNi2O4, CoCu2O4, M–Co2Ox (M is transition metals),23,24 CuCo2O4,25 Fenton-like catalyst,26,27 magnetic nano-fibers,28 phosphate doping of Nb2O5,29 Ag/AgCl/3D-rGO,30 and Mn–Fe2O3 (ref. 31)) for PMS activation has become a precedency in decolorization of methylene blue and methyl orange.
The selective oxidation of EB (as a cheap and toxic waste in petroleum industries) to value-added AP is a great industrially remarkable process. AP is widely used in the industrial production of polyester, pharmaceuticals, drug, resin, tear gas flavoring agent, chewing gum, alcohols, aldehydes, fiber, and perfumes; it can also be utilized as a solvent for cellulose ether and ester preservatives, and dyestuff for our daily life.9 Industrially, AP is produced using the Hock process, Fridel–Crafts acylation, and Co(OAc)2 as a homogenous catalyst, which is a harsh condition, impurity with irreversible deactivation, self-aggregation, by-products, costly, low selectivity, difficult to separate and not an environmentally friendly process.9,32,33 Numerous applications of AP and high industry demand for high-purity AP, as well as numerous environmental problems of the accumulation of dye pollutants, confirm that the fabrication of highly efficient catalytic systems is one of the important challenges in industries and academic research.
This study aims to evaluate the decontamination and aerobic oxidation performance of the developed doubly shell magnetic composite (MIOSC-N-et-NH2@CS-Mn) in a pilot scale and to evaluate possible structural, morphological, and surface changes in the activator during the long-term operation, economic study and response surface method. We combined the unique properties of magnetic beads as ferromagnetic Fe3O4 having magnetic sensitivity, environmental compatibility, low production cost, and easy separation after use; amine groups-as cross linker agent for effective, stable and compact coating of chitosan onto magnetic-silica beads; SiO2/GT-AEAPS-CT multi-layers as an insulator, passive and anti-sintering shell for improving the solubility, thermal and chemical stability of magnetic core against oxidation, rapid mass transfer, surface area, and increase efficient interaction with the aqueous environment; calcination process as a compact stage for improve purity and stability; and Mn(II) NPs as a green and highly active site at a favorable oxidation state for organic dye discoloration and selective conversion of EB to AP.
2. Experimental
2.1. Materials and method
All chemicals were of reagent grade and used without further purification. FeCl2·4H2O, FeCl3·6H2O, tetraethyl orthosilicate (TEOS), N-(2-aminoethyl)-3-aminopropyltrimethoxysilane (AEAPS), and manganese acetate (II) were purchased from Sigma-Aldrich and Merck companies. Methyl orange (SRL India) was used to study the catalytic efficiency, while PMS (Merck) was used to evaluate the catalytic efficiency of MIOSC-N-et-NH2@CS-Mn.
2.2 Preparation of MIOSC-N-et-NH2@CS-Mn
Chemical co-precipitation and Stöber sol–gel methods were applied as well-known and efficient methods for the synthesis of MIO NPs and MIO NPs coated with monodispersed silica,6,9,34 which are described in Text 1S.† Then, AEAPS was added to the mixture and vigorously stirred for 4 h. The functionalized magnetic nanoparticles were separated by a bare magnet, washed using H2O (3 × 5), EtOH (3 × 5), and diethyl ether (2 × 5), and then dried at 60 °C. The chitosan solution was prepared by dissolving 0.5 g low molecular weight chitosan in 30 mL AcOH (2%). Then, 20 mL drops of chitosan solution were added to the NH2-silica-coated Fe3O4 NPs (1.0 g) mixture and 50 mL H2O and vigorously stirred for 5 min. The process was continued by introducing 20 mL of glutaraldehyde (GT, 5% wt) to the solution drop by drop and stirred for 4 h. The prepared MIOSC-NH2@CS was washed as a preliminary phase and dried at 80 °C to obtain brown sediment. After calcination under an N2 atmosphere at 400 °C for 1.5 h, the final step of the fabrication of the heterogeneous catalyst was dispersed 0.5 g MIOSC-N-et-NH2@CS in 100 mL of H2O by ultrasonication. Finally, 0.5 g Mn(OAc)2 was dissolved in 20 mL H2O and introduced to the reaction solution. The mixture was stirred for a day under reflux. The ultimate heterogeneous catalyst was collected using a super magnet and washed with H2O, acetone, diethyl ether, and ethanol, which allowed the catalyst to dry at 60 °C for 7 h (Fig. 1).
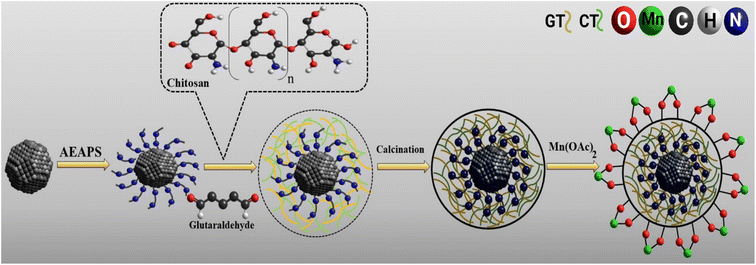 |
| | Fig. 1 Synthesis route of MIOSC-N-et-NH2@CS nanoparticles. | |
2.3. Catalytic performance
2.3.1. General procedure for the decolorization of methyl orange. All experiments were carried out using a laboratory scale and at ambient temperature (25 °C) and pressure (1 atm) in 500 mL conical flasks. In a typical experiment, the desired amount of PMS and pollutant (MO stock solutions) was added to the reactor, and pH adjustment was performed using 1.0 M H2SO4 and 1.0 M NaOH. Then, a certain amount of MIOSC-N-et-NH2@CS-Mn was added to the solution containing PMS and pollutants. During the degradation process, the solution was magnetically stirred to ensure a complete mixing state. At the given reaction time intervals, approximately 2 mL of the sample was withdrawn, and the pollutant content was determined immediately using a UV spectrophotometer. The degradation of the MO was monitored immediately by maximum absorbance at λmax = 465 nm. The removal efficiency (%) and constant rate were used to illustrate the degradation capability (eqn (1)):| | |
ln(Ct/C0) = ln(At/A0) = kobst
| (1) |
where the ratio of the absorbance of MO at the desired removal time to the initial time is denoted by At/A0, the concentration ratio of MO at the desired removal time to the initial time is denoted by Ct/C0, the observation rate constant is denoted by kobs, and time is denoted by t (Table 1S†). Afterward, the SF (synergistic factor), η (reaction stoichiometric efficiency), SOE (specific oxidant efficiency) and the UR (oxidant utilization ratio) of PMS/MIOSC-N-et-NH2@CS-Mn in MO degradation were measured based on eqn (2)–(5):35| |
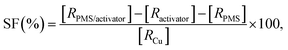 | (2) |
| |
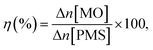 | (3) |
| |
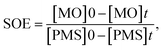 | (4) |
| |
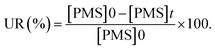 | (5) |
To evaluate the efficiency of the proposed system, other pollutants, such as methylene blue (MB) and bisphenol A (BPA), were tested (Table 1S†).
2.3.2. General procedure for aerobic oxidation of ethylbenzene. A suspension of the combined catalyst system (0.09 g catalyst and 0.4 g NDHPI), 6 mL AcH–H2O (60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :40 v/v), and EB (0.20 g, 2 mmol) was added to a three-necked flask, which was fitted using an equipped water condenser through a balloon filled with O2 at 80 °C with vigorous stirring. After isolating by magnet stick and washing with EtOH/MeOH and H2O, the quantity (conversion (X, %), selectivity (S, %), TON (turn over number) and TOF (turn over frequency, h−1)) and type of the resulting products from oxidation were measured (eqn (6)–(8)), identified by GC and GC-MS analyses and isolated (Text 2S†):
:40 v/v), and EB (0.20 g, 2 mmol) was added to a three-necked flask, which was fitted using an equipped water condenser through a balloon filled with O2 at 80 °C with vigorous stirring. After isolating by magnet stick and washing with EtOH/MeOH and H2O, the quantity (conversion (X, %), selectivity (S, %), TON (turn over number) and TOF (turn over frequency, h−1)) and type of the resulting products from oxidation were measured (eqn (6)–(8)), identified by GC and GC-MS analyses and isolated (Text 2S†):| |
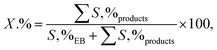 | (6) |
| |
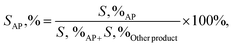 | (7) |
| |
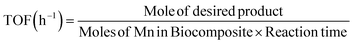 | (8) |
3. Results and discussion
Calcination temperature is a significant parameter for preparing catalysts, which can affect the physicochemical properties of heterogeneous samples, such as the distribution of active site and surface properties, thus impacting catalytic activity. By increasing the calcination temperature from 400 to 500 °C under nitrogen atmosphere, the average particle size increased to 75 ± 3.0 nm, and SBET diminished to 45.3 m2 g−1 because of the compact agglomeration of MIOSC-N-et-NH2@CS-Mn nanoparticles. Based on these results, the calcination temperature and dwelling time are 400 °C and 1.5 h, respectively. The nature and number of peripheral groups are identified using various techniques, as mentioned in Text 3S.† Furthermore, the elemental analysis (CHN) and ICP-OES results with the summary of the measured values for carbon, hydrogen, nitrogen and manganese particle elements are shown in Table 1.
Table 1 Chemical composition and morphological properties of magnetic catalysts
| Samples |
aEA (wt%) |
bMn (mmol g−1) |
Structural parametersc |
| C |
N |
SBET (m2 g−1) |
VBJH (cm2 g−1) |
MPD (nm) |
| Elemental analysis was estimated from the EDS analyses. Mn content by ICP-OES. Pore size calculated using the BJH method. Spent catalyst after 5 runs. Spent catalyst after 9 runs. |
| MIO NPs |
— |
— |
— |
38.82 |
0.174 |
6.56 |
| MIOSC NPs |
— |
— |
— |
54.63 |
0.121 |
8.31 |
| MIOSC-N-et-NH2@CS-Mn (fresh) |
31.2 |
5.87 |
1.13 |
61.43 |
0.902 |
25.3 |
| MIOSC-N-et-NH2@CS-Mnd |
32.5 |
5.91 |
1.13 |
61.76 |
0.904 |
26.2 |
| MIOSC-N-et-NH2@CS-Mne |
37.7 |
6.10 |
0.92 |
59.22 |
0.843 |
32.6 |
3.1. Characterization
3.1.1. Chemical and surface area analyses. The CHN results with a summary of measured values for C, H and N elements are shown in Table 1. These data confirm the modification and encapsulation processes during the fabrication of the magnetic activator. Furthermore, the amount of Mn NPs at MIOSC-N-et-NH2@CS-Mn was estimated to be 1.13 ± 0.001 mmol g−1 by applying ICP-OES analyses. This result indicates that Mn NPs within the synthesized biopolymer-coated magnetic activator are stabilized by the CS-GT-AEPS skeleton, providing catalytic active sites. These results illustrate MIOSC-N-et-NH2@CS-Mn formation and demonstrate that CHN content increased with adding AEPS, GT, CS and Mn to the surface of MIOSC (Table 1). The N2 adsorption–desorption isotherm curves of the MIO nanoparticles and MIOSC-N-et-NH2@CS-Mn nanocatalysts are depicted in Fig. 2. The calculated BET surface areas (SBET) of the MIO nanoparticles and MIOSC-N-et-NH2@CS-Mn were 38.8 and 61.43 m2 g−1, respectively. Thus, the microporous MIOSC-N-et-NH2@CS-Mn indicated an SBET due, principally, to chitosan-glutaraldehyde-AEPTS modification, total pore volume (VBJH, cm2 g−1) and mean pore size (MPD, nm) compared to less porous MIO (Table 1).
 |
| | Fig. 2 BET analysis of (A) MIONPs and (B) MIOSC-N-et-NH2@CS-Mn. | |
MIOSC-N-et-NH2@CS-Mn with a high surface area and uneven structure usually indicates an increased removal capacity for the target pollutants.35 The SBET, VBJH and MPD showed that the existence of mesoporous MIOSC-N-et-NH2@CS-Mn is less than 25.3 nm. Studies in the literature have shown that MPD declines as SBET and VBJH increase.35 Fig. 2B depicts the N2 adsorption–desorption isotherms of MIOSC-N-et-NH2@CS-Mn. The BET isotherms and the hysteresis loop of MIOSC-N-et-NH2@CS-Mn were fitted to type IV and type H3 based on IUPAC classifications, illustrating the presence of typical microspores and hysteresis loops at high relative pressure. Compared with the magnetic core, the enhancement in SBET observed for MIOSC-N-et-NH2@CS-Mn could mainly be related to the introduction of the CS-GT-AEPTS skeleton.
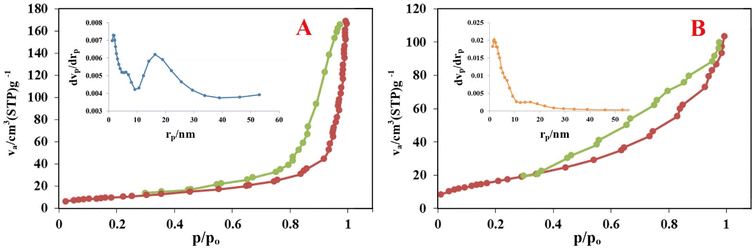
3.1.2. Spectroscopic studies (FT-IR). The FT-IR spectra analysis was conducted to determine the chemical structure of the MIO, MIOSC-N-et-NH2, MIOSC-N-et-NH@CS and MIOSC-N-et-NH2@CS-Mn (ν represents stretching, σ bending, s symmetric, and as asymmetric vibrations). As shown in Fig. 3A, the ν (OH) and ν (NH2) in samples appeared as a broad band in the range of 3200–3600 cm−1.9,35 Additionally, the related peaks to νs (O–Si–O), νs (Si–O–Si), νs (Fe–O–Fe) and νas (Si–O–Fe) can be observed at 462792, 542 and 1072 cm−1, respectively.35,36 The νas (CH3) and σ (NH2) vibrations became visible at around 2854 and 1615 cm−1, respectively, indicating the AEAPS attachment with the surface of the magnetic beads, which verifies the formation of a silica shell on the MIO NPs.35,37 Moreover, the spectrum of IOSC-N-et-NH2@CS shows characteristic peaks at 1384 cm−1, which is ascribed to polymeric chitosan, explicitly confirming the formation of chitosan multi-layers on the core–shell nanocomposite.38 As can be observed, the slight transition in the vibration band from 554 cm−1 to 634 cm−1 confirmed the coordination of Mn with the chitosan-coated nanocomposite.9,35
 |
| | Fig. 3 (A) FT-IR, (B) XRD pattern, (C) Uv-vis spectra, and (D) TGA profile of MIONPs, CS and MIOSC-N-et-NH2@CS-Mn . | |
3.1.3. Crystal structure analysis (XRD). Powder X-ray diffraction was conducted as an excellent analysis for detecting the phase behavior and crystallinity of the heterogeneous catalyst. Fig. 3B shows the diffraction patterns of the samples, including MIO, MIOSC-N-et-NH2, MIOSC-N-et-NH2@CS and MIOSC-N-et-NH2@CS-Mn. Evidence of phase separation in the diffractogram was not detected, which confirmed a single-phase catalyst.6,35 Diffraction peaks at (220), (311), (400), (422), (511), and (440) at around 2θ of 31, 36, 44, 54, 58, and 63, respectively, are the characteristic peaks of the MIO crystal having an inverse cubic spinel structure compared with standard MIO nanoparticles (JCPDS 65-3107).8,9,35 Regarding MIOSC, similar characteristic peaks were also observed, which indicated that the crystalline structure of MIO was stable after coating the silica shell around its surface.37 Additionally, the broad hump in the range of 15–30 in the diffraction angle scale is ascribed to the amorphous carbon and SiO2 structures, which confirmed that MIO nanoparticles were coated by silica.37 XRD examinations of pure chitosan show extremely expansive peaks in the range of 10–20.25–27 No detectable XRD peaks arising from impurities were seen, demonstrating no evidence that foreign materials pollute MIOSC-N-et-NH2@CS-Mn.38–41
3.1.4. Optical properties study (UV-visible). The UV-vis absorption spectra of MIOSC-N-et-NH2@CS & MIOSC-N-et-NH2@CS-Mn are shown in Fig. 3C. MIO NPs show thermally induced electron delocalization between Fe2+ and Fe3+ ions. From the data obtained, the peak in the NIR (near infrared region, 800–2500 nm) and 407 nm illustrates the presence of MIO NPs.42,43 Surface plasmon resonances (SPRs) dominated the optical spectra of the metal nanoparticles, thereby transferring them to longer wavelengths with an increase in particle size. A broad peak at 490 nm (d → d*) in the spectrum of MIOSC-N-et-NH2@CS-Mn is due to excitation in the surface plasmon vibrations of manganese particles.6
3.1.5. Thermal stability (TGA). The thermal behavior and durabilities of the MIOSC-N-et-NH2@CS-Mn were examined by performing TG-DTG analysis. As can be observed, the TG-DTA curves demonstrated that the weight loss of MIOSC-N-et-NH2@CS-Mn nanoparticles is a multistage process, which appears in two stages. Initially, the heterogeneous catalyst indicated a slight weight loss of almost 9% when the temperature increased from 50 to 150 °C, concerning the loss of physically and/or chemically adsorbed water.6,32 The second weight-loss stage (300–600 °C) is ascribed to the loss of chitosan and other compounds.42 Ultimately, the total weight change of the nanocatalyst was almost 15.37%. The secondary and primary amino groups in AEPS and CS can act as nucleophilic groups and attack the carbonyl in GT to construct a stable and compact cross-linked polymeric coating on the magnetic core to strengthen the stability of CS multi-layers (Fig. 3D). Therefore, it is concluded that the MIOSC-N-et-NH2@CS is an excellent scaffold for the construction of a magnetically recoverable biocomposite with expected durability and chemical stability.
3.1.6. M![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) −H curve analysis (VSM). The magnetic properties of the MIO, MIOSC-N-et-NH2, MIOSC-N-et-NH2@CS and MIOSC-N-et-NH2@CS-Mn were studied using a vibrating-sample magnetometer (VSM) as a function of magnetic field (H), temperature, and time, respectively (Fig. 1S†). The hysteresis loops are associated with superparamagnetic behavior, which can be seen for all magnetic beads.5 By plotting M against 1/H2, the Ms (saturation magnetization) samples were determined by the simple extrapolation of the M value to 1/H2 when the numerical value of 1/H2 is zero. The Ms value of MIO reduced after the formation of the core–shell structure and coating by CS and organic compounds from almost 47 to 41 and 30 emu g−1, respectively.42 This decrease in Ms value confirmed that MIO NPs are surrounded by non-magnetic materials. However, the following can be concluded from the decrease in Ms values: cover the magnetic support by the non-magnetic shell, and lower percentage (17.4%) of MIO NPs in the MIOSC-N-et-NH2@CS-Mn and surface order/disorder interaction of the magnetic spin moment.6,38,42
−H curve analysis (VSM). The magnetic properties of the MIO, MIOSC-N-et-NH2, MIOSC-N-et-NH2@CS and MIOSC-N-et-NH2@CS-Mn were studied using a vibrating-sample magnetometer (VSM) as a function of magnetic field (H), temperature, and time, respectively (Fig. 1S†). The hysteresis loops are associated with superparamagnetic behavior, which can be seen for all magnetic beads.5 By plotting M against 1/H2, the Ms (saturation magnetization) samples were determined by the simple extrapolation of the M value to 1/H2 when the numerical value of 1/H2 is zero. The Ms value of MIO reduced after the formation of the core–shell structure and coating by CS and organic compounds from almost 47 to 41 and 30 emu g−1, respectively.42 This decrease in Ms value confirmed that MIO NPs are surrounded by non-magnetic materials. However, the following can be concluded from the decrease in Ms values: cover the magnetic support by the non-magnetic shell, and lower percentage (17.4%) of MIO NPs in the MIOSC-N-et-NH2@CS-Mn and surface order/disorder interaction of the magnetic spin moment.6,38,42
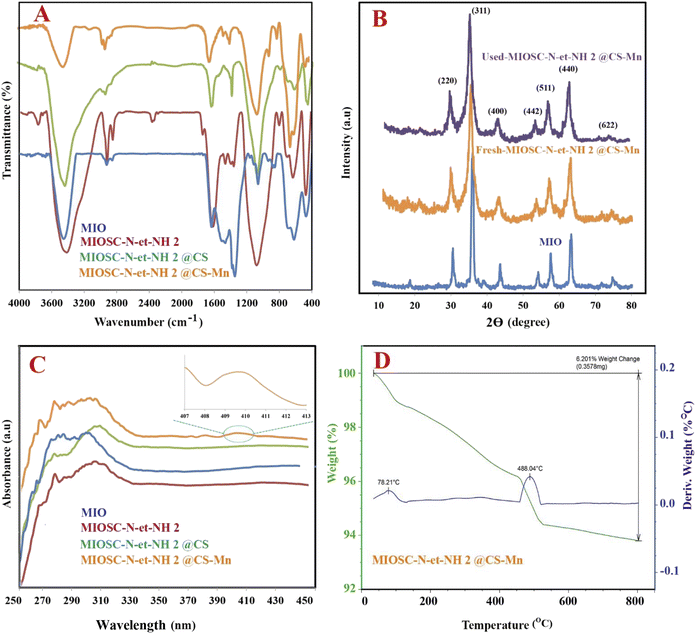
3.1.7. Study of energy state (XPS). As shown in the survey scan spectrum of MIOSC-N-et-NH2@CS-Mn core–shell biocomposite (Fig. 4), the characteristic peaks, such as Fe, C, N, O and Mn, with the binding energies (BE) 723.5 eV (2p1/2) and 710.8 eV (2p3/2) for Fe2+, 725.2 eV (2p1/2) and 711.7 eV (2p3/2) for Fe3+, 287.4 for C(1s), 401 for O(1s), 530.78 for N(1s) and 641.3 eV for Mn(2p3/2) and 654.3 eV for Mn(2p1/2).6 The existence of Fe(2p) peaks with a ratio of 2.701:1.0 (Fe3+/Fe2+) and the absence of Si(2P) illustrated the existence of the MIO NP phase in the MIO NPs and the complete coating of them by SiO2 shell with thin thickness, and no other peaks were detected, which illustrates the good purity of the biocomposite (Fig. 4A). Additionally, the 2p3/2 to 2p1/2 splitting in the catalyst is around 11.13 eV, confirming oxidation sate 2+ for Mn in MIOSC-N-et-NH2@CS Mn. In the case of N(1s) and O(1s), the shift N(1s) to a higher BE (from 399 eV in pure chitosan to 400.0 in MIOSC-N-et-NH2@CS Mn and the shift O(1s) to lower BE (from 530.04 and 531.34 eV to 531.49 and 534.88 eV, respectively) in MIOSC-N-et-NH2@CS-Mn confirms the capping of MnNPs with core shell scaffold (Fig. 4B).
 |
| | Fig. 4 (A) Expand XPS analysis, (B) O1s, (C) Fe2p and (D) Mn2p of MIOSC-N-et-NH2@CS-Mn. | |
3.1.8. Microscopic analysis (SEM and HRTEM). Electron microscopy (SEM, TEM) analysis was conducted to characterize the morphology and particle size of the nanocomposite. The selected SEM images indicate that the morphology of MIO NPs is spherical with an average size of approximately 21 nm. Fig. 5 shows that the core–shell nanocomposite still maintains the morphological properties of MIO except for the negligibly larger particle size and uneven surface. It is concluded that the intensity of the thickness of the external layer has been enhanced with an increase in the layer thickness of the nano-composite. Furthermore, the SEM image demonstrates that the heterogeneous catalyst surface is rough and porous.44 Furthermore, the existence of Mn, Fe, Si, C, O, and N elements was examined using the EDAX spectrum and FE-SEM image of MIOSC-et-NH2@CS-Mn. TEM analysis was performed for an in-depth investigation of morphology and structure. As can be seen in Fig. 5, MIO nanoparticles are not separated effectively during the modification process owing to the tendency to agglomerate.44 A selected TEM image illustrated in Fig. 5 reveals that MIOSC-NH2 is of the uniform microsphere with good dispersibility; in contrast, MIOSC-et-NH2@CS exhibit small aggregation, which means that MIOSC-et-NH2 is coated entirely by biopolymer. These functionalized core–shell nanoparticles have a global shape with an 8–22 nm diameter. Additionally, the size of these microspheres remarkably increased, which can be ascribed to the subsequent deposition of CS multi-layers. Fig. 5G shows the FESEM image of the MIOSC-et-NH2@CS-Mn, which reveals that the metal loading was done successfully, and Fe and Mn nanosized are dispersed regularly over the surface of the catalyst.38,44,45 Furthermore, the HRTEM image of the catalysts illustrates that the nanoparticles are dispersed in an organic matrix (Fig. 5D). The selected area electron diffraction (SAED) exhibited concentric blurred rings, which corresponded with the XRD results (Fig. 5F).
 |
| | Fig. 5 . Microscopic analyses of MIOSC-N-et-NH2@CS-Mn (A–C) SEM, (D, E) HR-TEM images, (F) SAED pattern, and (G) EDAX and SEM-map analysis. | |
3.2. Catalytic performance
3.2.1. Degradation of methyl orange dye. The temporal UV-vis spectral changes in target pollutants in the course of catalytic degradation under different conditions are evaluated in detail. The decrease in absorbance (at 465 nm) illustrates the efficient decolorization of MO in an optimum condition. Different influential operating factors, such as reaction temperature (5 °C < T < 35 °C), time (0 < t < 12 min), activator loading (0.1< [Cat]0< 0.6 g L−1), PMS dosage (0.1 < [PMS] < 0.5 g L−1), the MO dosage (5.0 < [MO]0< 15 g L−1), and pH (3.0 < pH < 9.0), coexisting anions (Cl−, CO32−, SO42− and NO3−) and natural organic matters (NOMs) on the discoloration efficiency of MO were thoroughly assessed (Fig. 6). The results show that the adsorption capacity of the compact activator for MO was only 8.3%. This low adsorption capacity may be attributed to a high dosage of loaded Mn and the compacted skeleton after the calcination process. Furthermore, these results indicate that the self-degradation rate of MO without the PMS was traced (<13.5% with kobs = 0.0113 min−1). The introduction of MIOSC-N-et-NH2@CS-Mn increased the discoloration efficiency (and constant rate) ca. 98.3% (0.5237 min-1) within 8.0 min. In the preliminary experiment, where MO and PMS were mixed without MIOSC-N-et-NH2@CS-Mn, the discoloration efficiency (and constant rate) ca. 39.4% (0.0389 min−1) of dye was achieved within 8.0 min (Fig. 6A). In contrast to the homogenous Mn2+ activation process, the activation of Mn2+/Mn3+ occurs on the surface of the activator, thereby operatively avoiding full contact, the generation of undesirable intermediates, and the rapid consumption of active sites.
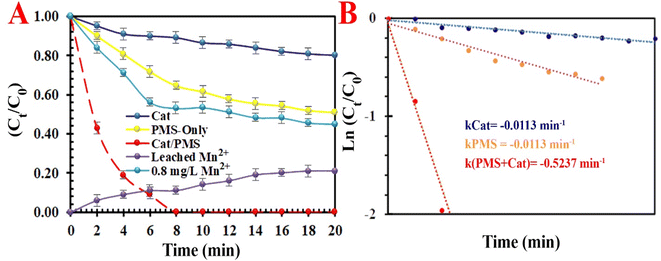 |
| | Fig. 6 (A) Degradation efficiency under various systems and (B) reaction rates of TC degradation by various systems. | |
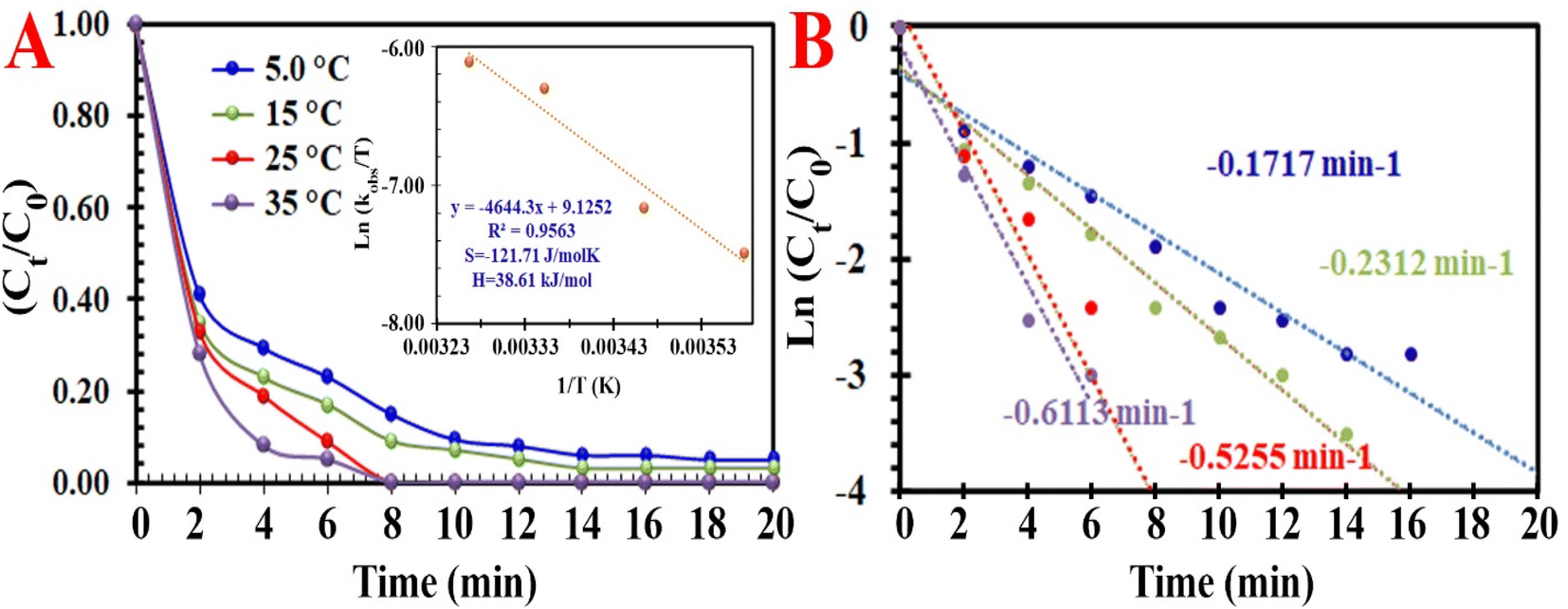
Exploring the two kinds of pivotal factors (time and temperature) on the RE of MO demonstrated that both of these factors influence MO discoloration reactions. This is due to the easier transfer of electrons from the MIOSC-N-et-NH2@CS-Mn to the PMS at higher reaction temperatures during the time. Higher temperature results in a higher RE (and kobs) of MO, and the RE at 278, 288, 298 and 308 K was measured to be 79.8% (0.097 min−1), 89.2% (0.1653 min−1), 98.3% (0.5255 min−1) and 99.2% (0.8574 min−1), respectively. The RE (%) was enhanced as the reaction temperature increased from 278 K to 298 K, which demonstrated that the discoloration process was an endothermic reaction; consequently, the kinetic energy of the PMS and activator increased at higher temperatures. These results can be related to the enhancement of the intensity of the collision/transfer of the reactants at high temperatures. In the following, Ea (activation energy), ΔS# (activation entropy), and ΔH# (activation enthalpy) were measured using the Arrhenius and Eyring equation (eqn (9) and (10)):
| | |
ln|kobs| = ln(A0) − Ea(1/RT)
| (9) |
| | |
ln|kobs/T| = ln(kB/ħ) + ΔS#/R − ΔH#(1/RT)
| (10) |
where
A0 (pre-exponential factor, min
−1),
T (temperature, kelvin),
R (ideal gas constant, 8.314 J K
−1 mol),
kB (Boltzmann constant, 1.381 × 10
−23 J K
−1) and
ħ (Planck constant, 6.626 × 10
−34 J K
−1 mol
−1) are constants. The
A0,
Ea, Δ
S# and Δ
H# values of the degradation process of MO were measured to be 78.2 × 10
4 min
−1, 35.62 kJ mol
−1, −121.71 J mol
−1 K
−1 and 38.61 kJ mol
−1, respectively (
Fig. 7).
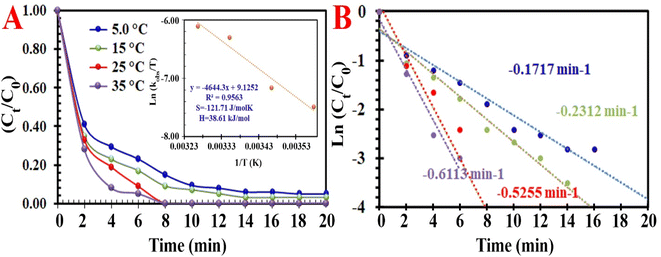 |
| | Fig. 7 (A) Influence of temperature on the degradation of MO [inset: energy activation of process], and (B) constant rate of degradation at different temperatures. | |
Based on the low value of Ea and the desirable thermodynamic parameter values (ΔS# and ΔH#), MIOSC-N-et-NH2@CS-Mn can be considered an efficient activator for MO degradation (Table 2S†). The performance of PMS-based AOPs strongly depended on the PMS dosage. Fig. 7 illustrates that the activator dosage significantly influences the activation of PMS and, consequently, the RE of MO. As expected, the RE (and kobs) value of MO significantly increased from 78.4% (0.1455 min−1) to 83.0% (0.1969 min−1) and 99.8% (0.5219 min−1) within 8 min with the enhancement of the activator dosage from 1.0 mM to 1.5 and 2.0 mM, respectively. Additionally, at a dosage higher than the optimum dosage (>0.20 g L−1), the RE (%) decreases because of the radical-quenching ability of excessive PMS.46 A high dosage of PMS can induce the reverse reaction and recombination of active radicals, and the scavenging reaction of reactive radicals and the reaction of unreacted residual PMS with active radicals produce inactive radicals and secondary pollution (eqn (11)–(16)).47 Moreover, the UR (%) and SOE were 86.02% and 0.03, respectively, confirming that most of the PMS was activated and utilized.
| | |
SO4˙− + SO4˙− → S2O82−, (k = 4.0 × 108 L mol−1 s−1)
| (11) |
| | |
˙OH + ˙OH → H2O2, (k = 5.3 × 109 L mol−1 s−1)
| (12) |
| | |
HSO5−+ SO4˙− → HSO42− + SO5˙−
| (13) |
| | |
SO5˙− + SO5˙− → S2O82− + O2
| (14) |
| | |
HSO5− + ˙OH → H2O + SO5˙−
| (15) |
| | |
SO4˙− + ˙OH → HSO5−, (k = 0.95 × 1010 L mol−1 s−1)
| (16) |
The initial pH value, as a key factor, controlled the RE of MO by influencing the degree of the ionization of hydrogen ions, hydroxide ions in solutions, the proportion of PMS, species of MO molecules, and structural stability (Fig. 8A). The MIOSC-N-et-NH2@CS-Mn showed high RE with pH values ranging from 3.0 to 7.0, and RE (and kobs) of MO gradually increased from 76.3% (0.1188 min−1) to 98.3% (0.5239 min−1) as the pH value increased. Although, under acidic conditions, the amino/hydroxyl groups and MnNPs on the chitosan shell were protonated, resulting in desirable electrostatic attraction with MO dye but a decrease in RE under strongly acidic conditions occurred. This could be described as follows: (i) the H-bond construction between H+ and the O–O group of PMS, which exerted as a scavenger by attaching H+ to the HSO5− and possibly obstructed the effective interaction between PMS and the MIOSC-N-et-NH2@CS-Mn and (ii) production of Mn–OH+ and prevent the generation of active free radicals (eqn(17)–(19)):49–51
| | |
˙OH/SO4˙− + H+ + e− → HSO4˙−/H2O
| (17) |
| | |
Mn2+ + H2O → [Mn–OH]+ + H+
| (18) |
| | |
[Mn–OH]+ + HSO5˙− → Mn–O + SO4˙− + H2O
| (19) |
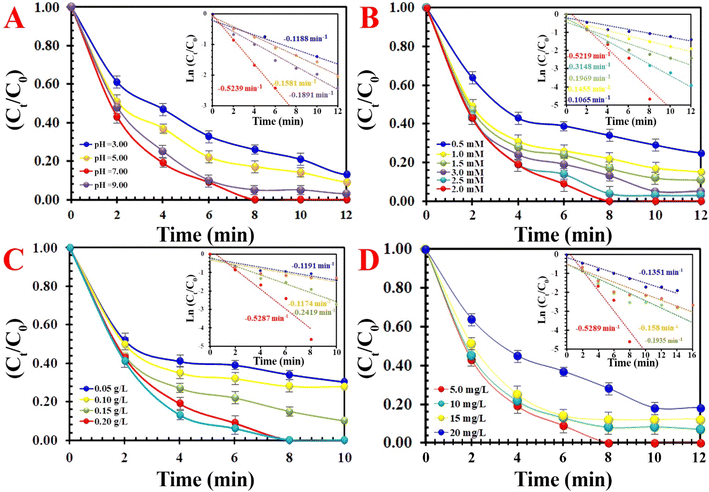 |
| | Fig. 8 The effects of various parameters on the degradation of MO: (A) effect of pH, (B) effect of PMS concentration, (C) effect of activator dosage, and (D) effect of MO concentration. | |
It seems that under alkaline conditions (>9.0), the amino groups on the CS molecule were deprotonated, and an electrostatic repulsion appeared between MIOSC-N-et-NH2@CS-Mn and MO, resulting in the decrease in RE. However, based on the pKa value of PMS (9.40), the dominant species at pH 9.40 and pH > 9.40 is HSO5− and S2O52−, respectively. Therefore, under harsh alkaline conditions, PMS self-decomposes to non-radical specie (S2O82−). The outstanding RE of MO in the PMS + MIOSC-N-et-NH2@CS-Mn system under certain pH ranges confirms the promising application potential for dye removal in various wastewater.
As expected, the RE (and kobs) value of MO significantly increased from 72.2% (0.1174 min−1) to 85.1% (0.2419 min−1) and 99.8% (0.5287 min−1) within 10 min with an enhancement of activator dosage from 0.10 g L−1 to 0.15 and 0.20 g L−1, respectively. This can be attributed to the increase in surface area and active center content.
The catalytic activity of MIOSC-N-et-NH2@CS-Mn towards the discoloration of MO with various concentrations of MO are illustrated in Fig. 8D. These results demonstrate that the RE (%) and constant rate value of MO decreased as the initial MO dosage increased. As shown in Fig. 8D, a higher dosage of MO molecules (>5.0 mg) blocks the active sites on the surface of MIOSC-N-et-NH2@CS-Mn; subsequently, it reduces the RE and kobs from 98.3% and 0.5289 min−1 to 83.0% and 0.1351 min−1 at 20.0 mg, respectively. Consequently, the pivotal parameters, such as a higher reaction of PMS, with dye molecules instead of the interaction surface of the MIOSC-N-et-NH2@CS-Mn and oxidant block the active center on MIOSC-N-et-NH2@CS-Mn with dye and by-product, and the reduction in the generation of free radicals owing to the decrease in the PMS/MO ratio can decrease the REs at higher initial MO dosages.47
3.2.2. Potential activation mechanism using MIOSC-N-et-NH2@CS-Mn. To identify the reactive oxygen species for MO decolorization, scavenger agents (i.e., tert-butyl alcohol (TBA) for ˙OH (k = 3.8–7.6 × 108 M−1 s −1), EtOH for SO4˙−(k = 1.6–7.7 × 108 M−1 s−1) and ˙OH (k = 1.2–2.8 × 109 M−1 s−1), p-benzoquinone (p-BQ) for O2˙− (k = 0.9–1.0 × 109 M−1 s−1)), and sodium azide for ˙OH (k = 1.2 × 1010 M−1 s−1), SO4 ˙− (k = 2.51 × 109 M−1 s−1) and 1O2 (k = 2 × 109 M−1 s−1)) were added to quench free radicals in aqueous medium.52,53 The RE (%) of MO at optimum conditions decreased to 54.8% in the presence of EtOH (50.0 mM) and 23.2% in the presence of NaN3 (15.0 mM). The results indicated that TBA and p-BQ had a lower effect on MO degradation compared to EtOH and NaN3. Based on radical scavenge experiments, we conclude that the ˙OH, 1O2, O2˙− and SO4˙− participated in the PMS-AOPs system, but 1O2 and SO4˙− play a dominant role in PMS/MIOSC-N-et-NH2@CS-Mn system (Fig. 9). Additionally, the MO degradation rate in D2O (∼100%) was higher than that in H2O (98.3%), illustrating the existence of 1O2 as the dominant species (Fig. 2S†). Furthermore, the experiments under anoxic conditions with argon bubbling illustrated that dissolved O2 had a negligible effect on MO degradation. Therefore, the apparent quenching effect of p-BQ was related to the elimination of O2˙− and consequently hindered the generation of 1O2. Moreover, the role of MIOSC-N-et-NH2@CS-Mn and surface-bond radicals was further confirmed by the addition of KI (10.0 mM) to the aqueous medium. In this condition, the reactivity of the surface of MIOSC-N-et-NH2@CS-Mn is determined by capturing surface-bound radicals with KI.54 This comparison results illustrated that the RE (%) significantly decreased, proving that the main contribution to the degradation process was 1O2 generated at the surface of MIOSC-N-et-NH2@CS-Mn. In addition, low REs (18.7%) were obtained in the presence of 0.8 mM Mn(II) ions, confirming that Mn residuals had an ignorable influence on MO degradation.
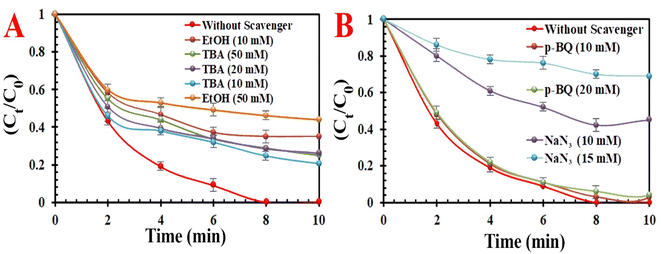 |
| | Fig. 9 . Effect of quenching scavengers (A) EtOH and TBA and (B) p-BQ & NaN3. on degradation efficiency(%) for MO degradation. | |
Furthermore, at the fresh MIOSC-N-et-NH2@CS-Mn, the relative content of Mn(II) was approximately two times higher than that of Mn(III). Based on the literature, PMS activation by transitional metal approximately followed the order Co(II) > Ru(III) > Fe(II) > Mn(II) > Fe(III) > Ni(II), in which a higher oxidation state generates SO5˙− and with a lower oxidation state generates SO4˙−. Therefore, the degradation of pollutants through homogenous/heterogonous activators containing Mn(II) has attracted significant attention owing to the results that have been reported recently,54–56 which illustrated that Mn(II) has a more activation effect on PMS compared to Fe(III). For the MIOSC-N-et-NH2@CS-Mn(II), after reaction (Fig. 4), the element composition remained unchanged; however, the content of Mn(II) decreased, whereas the content of Mn(III) increased. After the removal process, the percentage of Mn(II) exhibited a significant decrease from 77.4% to 64.8%, whereas the relative content of Mn(III) increased from 22.6% to 35.2%. Based on the result of the Mn 2p high-resolution spectrum, Mn(II) is the main form in MIOSC-N-et-NH2@CS-Mn surface partially converted into Mn(III), and the electron transfer of Mn(III)/Mn(II) (E0 = 1.51 V) on the solid surface is a significant contribution to the activation of oxidants (Fig. 4D). Therefore, the Mn(II) on the surface of the catalyst could directly react with the peroxy bond of PMS to generate SO4˙− and ˙OH (eqn (20)–(24)). Then, the 1O2 and O2˙− could be generated in a degradation system via a self-interaction of SO5−/SO4˙− and the interaction of Mn(III) and generated H2O2, respectively (eqn (25)–(34)).54–56 Finally, MO can be degraded into organic intermediates and finally mineralized with the participation of SO4˙−, ˙OH, O2˙− and 1O2 (eqn (35)).
| | |
Mn(II) + HSO5− → Mn(III) + SO4˙− + OH−, E0 ((Mn(III)/Mn(II)) = 1.51 V
| (20) |
| | |
Mn(III) + HSO5− → Mn(II) + SO5˙− + H+, E0 (HSO5−/ SO5˙−) = 1.10 V
| (21) |
| | |
HSO5− + SO4˙− → SO4˙− + SO5˙− + H+
| (22) |
| | |
H2O + SO4˙− → SO42− + ˙OH + H+
| (23) |
| | |
−OH + SO4˙− → SO42− + ˙OH
| (24) |
| | |
HSO5− + H2O → HSO4− + H2O2
| (25) |
| | |
H2O2 + ˙OH → −OH + HO2˙
| (26) |
| | |
2O2˙− + 2 ˙OH → H2 + 21O2
| (28) |
| | |
2 O2˙− + H2O → H2O2+ −OH + 1O2
| (29) |
| | |
SO5˙− + SO5−˙ → S2O82− + 1O2
| (30) |
| | |
SO5˙− + SO5˙− → 2SO42− + 1O2
| (31) |
| | |
SO5˙− + HSO5− → HSO4− + SO42− + 1O2
| (32) |
| | |
SO4˙− + SO4−˙ → S2O82−
| (33) |
| | |
O2˙− + S2O82− → SO4˙− + SO42− + O2
| (34) |
| | |
SO4˙−/˙OH/O2˙− /1O2 + MO → [intermediates]→ CO2 + H2O +, etc.
| (35) |
Additionally, no difference in the FT-IR illustrates that the reaction is not performed by the adsorption mechanism and is a catalytic process.
3.2.3. Effect of naturally occurring ions and NOMs on catalytic activity. Various wastewaters, such as dyestuff wastewater, medical sewage and river water, contain several ions, i.e., Cl−, SO42−, CO32−, HCO32−, NO3− and many other anions, as well as natural organic matters (NOMs), which usually act as scavengers and in combination with active free radicals. Therefore, the interference of various anions and NOM on the degradation of MO in the PMS/MIOSC-N-et-NH2@CS Mn system was investigated. The inhibition results (98.3% for 0.0 M, 94.2% for 5.0 cM and 92.3% for 10 cM) were concluded when the concentration of Cl− ranged from 0.0 to 10 cM. Based on the literature,57 the Cl− would scavenge the active radicals SO4˙− and somewhat ˙OH to produce Cl˙, Cl2˙− and Cl–OH˙− (eqn (36)–(45)). The redox potential of Cl˙, Cl2˙− and Cl–OH˙− radicals is lower than that of SO4˙− and ˙OH, resulting in lower degradation efficiency.57| | |
2Cl− + HSO5˙ − + H+ → SO42− + Cl2 + H2O
| (36) |
| | |
Cl− + HSO5˙ − → HOCl + SO42−
| (37) |
| | |
Cl− + HO˙ → HOCl˙− (k = 2.1 × 1010 L mol−1 s−1)
| (38) |
| | |
Cl− + SO4˙− → SO42− + Cl˙ (k = 3.1 × 108 L mol−1 s−1)
| (39) |
| | |
ClHO˙− + H+ → Cl˙ + H2O
| (42) |
| | |
Cl2˙− + Cl2˙− →2Cl− + Cl2
| (43) |
| | |
Cl2˙− + Cl˙ → Cl−+Cl2
| (45) |
The degradation efficiency of MO had a negligible effect when the concentration of SO42− was enhanced (0.0 to 10.0 cM) in PMS/MIOSC-N-et-NH2@CS Mn system. It seems that SO42− was constantly refilled throughout the decontamination system (eqn (46) and (47)). Thus, SO42− had little effect (<1.20%) on the degradation of MO.58
| | |
SO42− + SO4˙− → S2O82− + e−
| (46) |
| | |
SO42− + ˙OH → SO4˙− + OH−
| (47) |
In the case of NO
3−, CO
32− and HCO
32−, the quenching effect on the degradation efficiency of MO was observed because of the rapid reaction of these ions with ˙OH and SO
4˙
− to generate NO
3˙ (
eqn (48) and
(49)), CO
3˙
− (
eqn (50) and
(51)), and HCO
3˙ (
eqn (52)–(54)) with lower redox potential radicals, respectively. Furthermore, the degradation of MO was insignificantly affected (<1.0%) when 20 cM CO
32− in PMS + MIOSC-
N-et-NH
2@CS system was added; this is because most of the CO
32− in aqueous media can be converted to HCO
32− with negligible effect on the degradation of MO.
59| | |
NO3− + ˙OH → OH− + NO3˙
| (48) |
| | |
NO3− + SO4˙− → SO42− + NO3˙
| (49) |
| | |
CO32− + ˙OH → OH− + CO3˙−
| (50) |
| | |
CO32− + SO4˙− → SO42− + CO3˙−
| (51) |
| | |
CO32− + H2O → OH− + HCO3−
| (52) |
| | |
HCO3− + ˙OH → H2O + CO3˙−
| (53) |
| | |
HCO3− + SO4˙− → SO42− + HCO3˙
| (54) |
Additionally, in the presence of 50 mg L−1 of humic acid, 79.60% of MO could be degraded. The decreased degradation efficiency by humic acid may be due to the competition process on the surface of MIOSC-N-et-NH2@CS, including the adsorption/oxidation competition between humic acid and MO as well as the adsorption competition between humic acid and PMS.
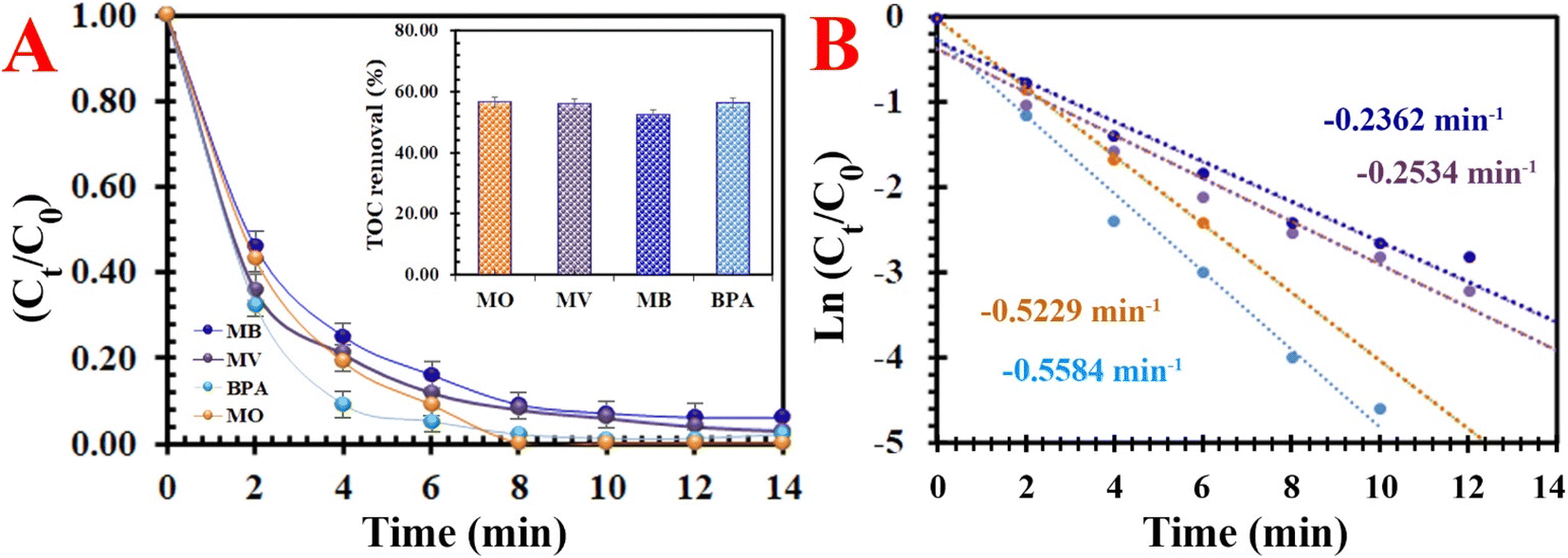
3.2.4. Universality. The universality of the PMS/MIOSC-N-et-NH2@CS-Mn system was evaluated based on its removal efficiency of various organic pollutants (i.e. methyl blue (MB), methyl violet (MV) and bisphenol A (BPA)). Fig. 10 illustrates that the removal efficiency (and TOC removal) of MO, MB, MV and BPA degradation reached 98.8% (∼56.61), 93.6% (∼52.4), 97.5% (∼55.8) and 98.4% (∼56.1) after 14 min at ambient temperature. The obvious difference in RE (%) is apparently owing to their various chemical structures and molecular weights. These observations indicate that the PMS/MIOSC-N-et-NH2@CS-Mn can be applied as an efficient catalyst for the high-performance degradation of various organic pollutants.
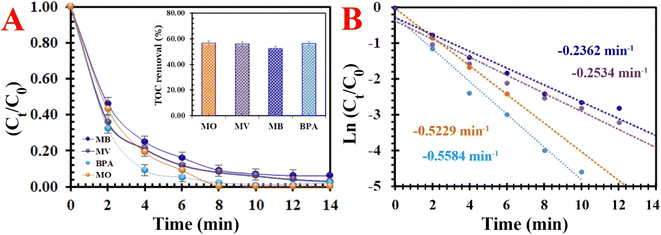 |
| | Fig. 10 Degradation of various typical organics: (A) removal efficiency [inset the TOC removal efficiency] and (B) kinetics rate [[MIOSC-N-et-NH2@CS-Mn]0 = 1.0 g L−1, [pollutant]0 = 10 mg L−1, pH = 7.0, T = 25 °C, [PMS]0 = 1.5 mM)]. | |
3.3. Aerobic oxidation of ethylbenzene
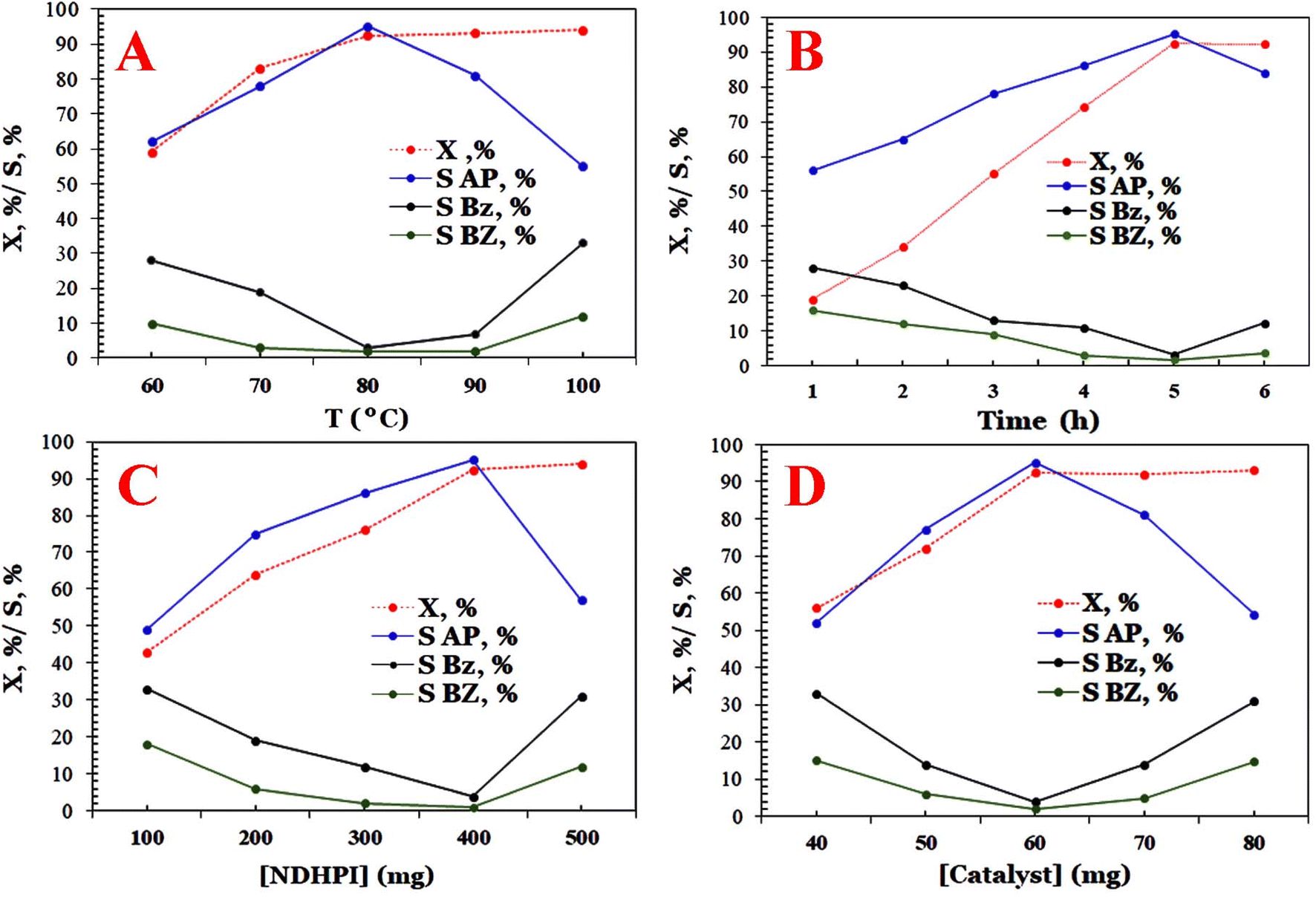
The selective aerobic oxidation of EB is commonly laborious with AP as its predominant product, accompanied by a minor amount of detectable benzaldehyde (BZ), benzoic acid (Bz) and phenyl ethyl alcohol (PEA) as side products. Therefore, the influence of catalyst dosage, temperature, time, amount of benzaldehyde and type of N-hydroxyimide (i.e., N-hydroxysuccinimide (NHSI), 4-do-decyloxycarbonyl-N-hydroxyphthalimide (C13O2)–NHPI), N,N-di-hydroxypyromellitimide (NDHPI), N-hydroxypyromellitimide (NHPI) and N-acetoxyphthalimide (NAPI)) and the nature of solvents on the conversion (X, %) and selectivity (S, %) of aerobic oxidation of EB were studied using MIOSC-N-et-NH2@CS-Mn in detail. First, the aerobic oxidation process used MIOSC-N-et-NH2@CS-Mn and NDHPI alone as catalysts, and when the processes were performed under harsh conditions (80 °C and 48 h), very low efficiency was achieved (X (<10.40%) & SAP (27.80%)). Only MIOSC-N-et-NH2@CS-Mn was applied in combination with NDHPI; AP as the main product was obtained.
From Fig. 11, the X (%) and SAP (%) of EB were 93.7% and 95.1% when the amount of MIOSC-N-et-NH2@CS-Mn was 40.0 mg, respectively. Based on these results, it is clear that the S (%) decreases for values higher than 40.0 mg as the optimum catalyst dosage due, principally, to the increase in the available oxidant and the particle–particle interaction and subsequently deep oxidation and generation of by-products. The gathering of magnetic particles blocked the active centers on the catalyst and then reduced the surface area.
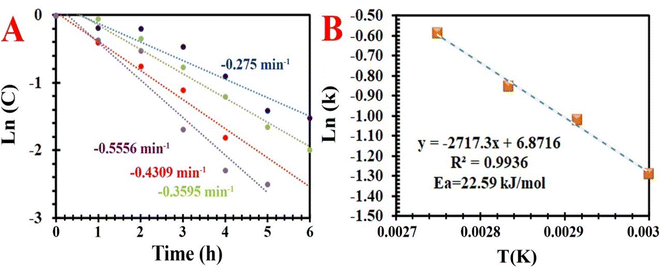 |
| | Fig. 11 . (A) First order kinetics fit of aerobic oxidation of EB under various temperatures, and (B) Arrhenius plot for the aerobic oxidation of EB over MIOSC-N-et-NH2@CS-Mn. | |
As shown in Fig. 11, increasing the time (>5.0 h) and temperature (>80.0 °C) above the optimum condition increased the X (%) and decreased the SAP (%). The SAP (%) diminishes as the deep oxidation of both AP and PEA to BZ/Bz occurs. Therefore, the reaction time of 5.0 h and temperature of 80.0 °C are the most appropriate conditions by which X (%) of 93.7% and SAP (%) of 95.1% were obtained. Further increasing the temperature process is not conducive to the stability of NDHPI. After excluding the effects of internal/external diffusion, aerobic oxidation was evaluated at different temperatures to investigate the kinetic aspects of the process. The fitting results illustrated that oxidation is a first order reaction (Fig. 11). The slope and the intercept of the Arrhenius equation can be obtained as −2717.32 and 6.8716, respectively. The values of the apparent Ea and A0 of MIOSC-N-et-NH2@CS-Mn can be measured using eqn (1), and they are 22.59 kJ mol−1 and 9.64 × 103 min−1, respectively. Low Ea indicates the high catalytic activity of MIOSC-N-et-NH2@CS-Mn, which should be related to its unique structure (Table 3S†).
The data shown in Fig. 12 illustrate that catalytic performances are influenced by the type and number of N-hydroxyimide derivatives in the aerobic process. Among the catalysts examined, it is found that the X (%) and SAP (%) on N-hydroxyimide derivatives decreased in the following order: NHDPI (93.7% and 95.1%) > NHPI (86.2% and 95.9%) > NAPI (23.7% and 10.2%)> NHSI (17.6% and 11.9%) > (C13O2)–NHPI (10.0% and 32.3%).6 It seems that the EB-O˙ generated with NHPI is remarkably greater than that generated with NHSI. Moreover, NAPI is a more stable precursor of N-oxyl radical than NHPI, because NHPI is produced owing to the hydrolysis of the (N–CO2CH3) group in HAc/H2O.6,48 It appears that the HAc–H2O as a mixed solvent diminishes the SAP (%) using NAPI. In the case of NDHPI, two N-hydroxyl groups in its structure and the electronic effect of each imide ring on each other are beneficial for the generation of N-oxyl radicals. Additionally, utilizing the (C13O2)–NHPI yields a lower X (%) than NHPI, which is related to the effect of lipophilic and/or electron donating group on solubility and/or hydrogen-atom-transfer process. The results shown in Fig. 12 indicate that the concentration of NDHPI from 100 to 500 mg also significantly affects the catalytic performance of the fabricated activator. Interestingly, when increasing the NDHPI (>400.0 g), the SAP (%) decreased, but the X (%) remained approximately unchanged.6
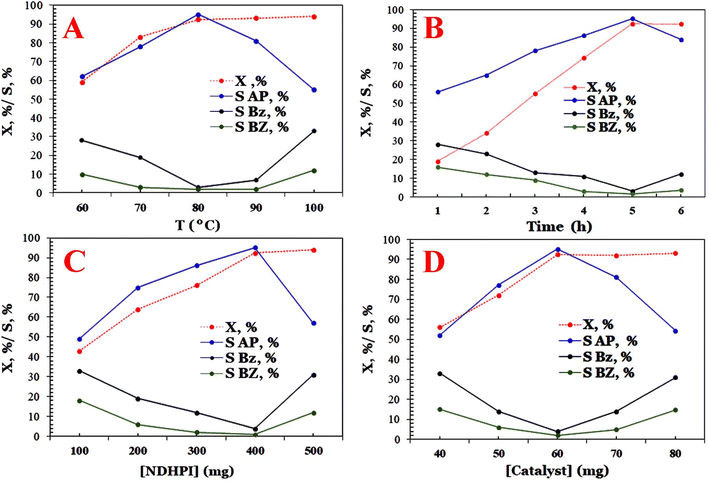 |
| | Fig. 12 Effect of pivotal parameters in catalytic oxidation of EB on X [%] and S [%], (A) effect of temperature, (B) effect of time, (C) effect of the amount of bio-composite, and (D) effect of NDHPI dosage. | |
Table 4S† depicts the influence of various solvents (i.e., cyclohexane (CY), acid acetic (HAc), acetonitrile (ACN), methanol (MeOH), dichloromethan (DCM), 1,2-dichloromethan (1,2-DCM), TCM (trichloromethan) benzaldehyde (Bz), benzoeic acid (BZ), ethanol (EtOH), toluene (TOL)) and solvent mixture (HAc–H2O) on the catalytic oxidation of EB. The experimental results suggest that HAc–H2O (1.5/1.0 v/v) is a suitable solvent for the sustainable production of AP (X (93.7%) and SAP (95.1%)). It must be considered that NDHPI is insufficiently soluble in EB and nonpolar solvents, particularly at or near temperature reactions. Therefore, a polar medium, such as a single solvent (HAc & ACN) or solvent mixture (HAc/H2O), is suitable for dispersing the quantities of NDHPI necessary to achieve excellent catalytic performance. Furthermore, a high amount of pure HAc (>25.0 mL) or water probably blocked the metallic sites on the surface of MIOSC-N-et-NH2@CS-Mn or had a diffusion competition with O2 and EB˙ molecules, resulting in reduced catalytic activity ((X = 63.2%) & (SAP = 78.2%)). This process was carried out without any additional oxidation of the solvent and only with selective aerobic oxidation of EB.
Based on previous studies,9 a possible mechanism (Scheme 1) is proposed. As demonstrated in Fig. 3D, the redox value of Mn(II)/Mn(III) indicated that the Mn(II) on the surface of MIOSC-N-et-NH2@CS-Mn performed catalytic functions to EB oxidation.54,55 The electron transfer around the surface of MIOSC-N-et-NH2@CS-Mn is an important contribution to increasing catalytic efficiency. First, NDHPI abstracted hydrogen to generate free radical in the presence of Mn–OO˙, which showed that the N-oxyl radical could abstract a benzylic hydrogen atom from EB to generate NDHPI. In the next step, the generated benzylic radical was trapped by O2 to generate EB-O2˙, which was further converted to EB-O2H in the presence of NDHPI (steps I and II). EB-O2H could be further decomposed and oxidized to AP, BZ and Bz using a radical process (Path I-III). For more clarity, the radical nature of the benzylic oxidation mechanism using the MIOSC-N-et-NH2@CS-Mn was evaluated by the interruption of an ongoing reaction after adding 5.0 mol% BHT (butylated hydroxytoluene) as a radical scavenger. It was concluded that the addition of BHT scavenged the oxidative process.54,55
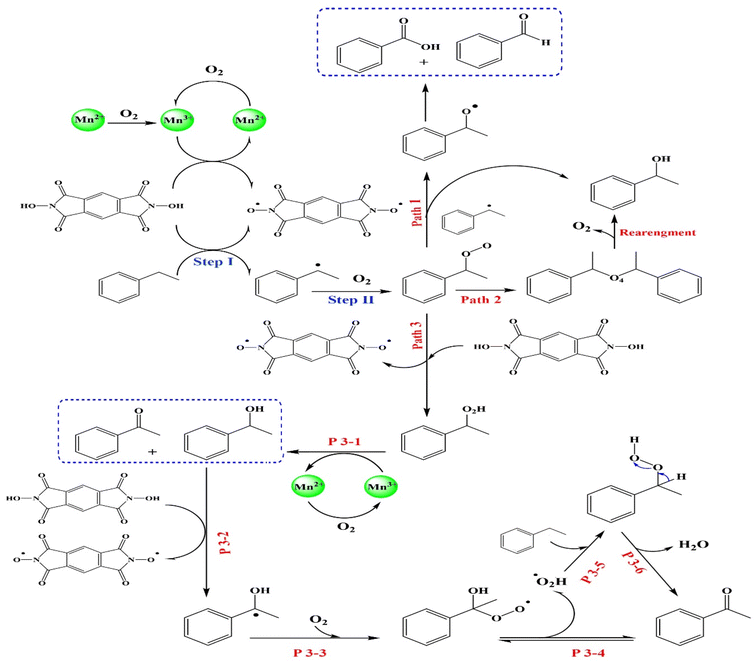 |
| | Scheme 1 Plausible pathway of aerobic oxidation of ethylbenzene in the presence of NDHPI/MIOSC-N-et-NH2@CS-Mn. | |
3.4. Recycle tests, metal leaching tests and long-term stability
One of the key issues for degradation and oxidation systems is maintaining high catalytic efficiency and good stability during the recovery process. The results indicated that MIOSC-N-et-NH2@CS-Mn has excellent reproducibility, achieving 95.10% removal efficiency and 89.31% conversion after 6 and 5 runs of degradation of MO and aerobic oxidation of EB, respectively. After the stability testing, the MIOSC-N-et-NH2@CS-Mn were characterized by ICP-OES, XRD, FT-IR, SEM and TEM analyses to further evaluate the structural and chemical stability of MIOSC-N-et-NH2@CS-Mn (Fig. 3S and 4S†). Furthermore, the TOC, SF (%) and η (%) and kobs decreased from 56.61, 60.4 (%), 5.20 (%) and 0.5219 min−1 to 44.2, 48.2 (%), 3.03 (%) and 0.1021 min−1 with the increase in reuse times (run 6) in the degradation process, respectively. Based on the leaching test and ICP-OES detection limit (Text 4S†), we were impotent to find Mn-particles in the reaction medium, which confirms the mechanical stability of MIOSC-N-et-NH2@CS-Mn and the encapsulated Mn NPs in the 3D-organic framework. In this study, a magnetic biocomposite with a high SBET area and extremely low density can facilitate the recovery process and simultaneously exclude high catalytic efficiency. For more clarity, the leaching concentrations of Mn(II) ions from MIOSC-N-et-NH2@CS-Mn were detected. The maximum concentration values of 0.217 mg L−1 were achieved for Mn ions in the first run. In the following cycles (from run 2 to run 6), the leaching concentrations of Mn ions decreased to 0.11, 0.081, 0.076, 0.045, and 0.041 mg L−1, respectively.
Additionally, the XRD spectra of MIOSC-N-et-NH2@CS-Mn before and after catalytic degradation are used to further investigate the structural stability of the catalyst during the degradation reaction. As depicted in Fig. 2B, the structure of MIOSC-N-et-NH2@CS-Mn remained unchanged before and after degradation. However, the relative solubility of chitosan in aqueous media, MIOSC-N-et-NH2@CS-Mn might exhibit even better removal efficiency during the recovery process. The decrease in catalytic performance after several runs can be because of absorbed intermediates and undegraded MO on the surface of the catalyst, leaching of the active site and oxidation of the MIOSC-N-et-NH2@CS-Mn surface by PMS during the consecutive recovery process.
Overall, the magnetic core–shell structure improved the essential properties (such as size and shape) of the prepared catalyst, in which their extremely small size produced a tremendous surface area-to-volume ratio (Fig. 2 and 5). The structural and textural properties of the prepared catalyst, such as favorable SBET and specific morphology, play an important role in the excellent catalytic activity of MIOSC-N-et-NH2@CS-Mn. Typically, the higher the SBET, the larger the number of exposed active centers. Furthermore, a porous property permits the diffusion of the PMS/O2 inside the pores, which leads to easier attainability of the Mn(II) centers (Table 1). However, Mn(II) as a main active center has a high oxygen storage capacity along with faster oxygen/PMS adsorption, which can be used to produce ROS (Fig. 4).
3.5. Response surface method (RSM)
In this study, the relationship between response (removal efficiency) and six independent parameters (temperature, time, catalyst loading, pH, methyl orange concentration and PMS dosage) on removal efficiency were evaluated. The experimental design involved the use of the Box–Behnken design and response surface statistical method. The RSM method was applied for the decolorization of MO using PMS + MIOSC-N-et-NH2@CS-Mn because this combination resulted in high RE (%). Optimization of treatments to maximize the RE (%) was carried out using RSM (MIOSC-N-et-NH2@CS-Mn = 0.2 mg L−1, PMS = 2.0 mM, t = 8 min, and T = 25 °C). Additionally, the response model equations and ANOVA (analysis of variance) for the response surface quadratic model were done (as described in Text 5S†). These results showed that R2 (coefficient of determination) and adjusted-R2 were 0.9641 and 0.9134, respectively, illustrating that the model was suitable to represent the factors for MIOSC-N-et-NH2@CS-Mn. Moreover, a comparison of the RSM models and the experimental results showed that the RSM has good predictions and is a powerful tool for modeling and optimizing MB degradation (p < 0.05). Fig. 13 illustrates the combined effect of temperature and MIOSC-N-et-NH2@CS-Mn loading on RE (%). Canonical analysis of the six independent factors determined that the most critical factor was MO dosage, with the amount of catalyst being the second most influential factor on RE (%).
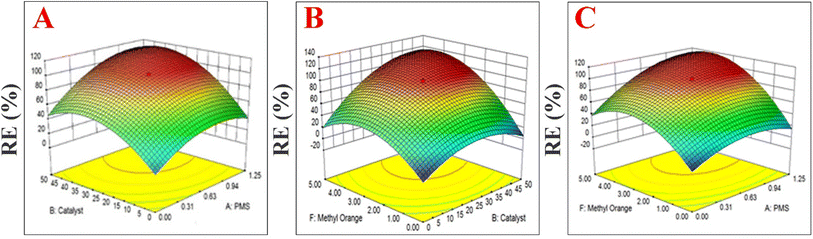 |
| | Fig. 13 The combined effect of (A) PMS and Cat concentration, (B) MO dosage and amount of MIOSC-N-et-NH2@CS-Mn (Cat), and (C) PMS and MO concentration on RE (%) of MO dye. | |
3.6. Uniqueness of this protocol
To confirm the distinctiveness of our developed method, we compared our results with those of other studies (especially in the past three years, Tables 4S and 5S†).
3.6.1. Degradation system. A catalytic comparison of several catalytic systems reported 3 years ago is summarized in Table 5S.† These comparisons with our system for remediation and refractory azo-dye confirmed that the PMS + MIOSC-N-et-NH2@CS-Mn system has certain advantages in terms of ecofriendly, biocompatible shell and solubility in polar/nonpolar media, effective coating of chitosan onto the core with amino groups, simple recovery due to super magnetic property, simple operational procedure and use of commercially accessible compounds for fabrication, stability against leaching, thermal and chemical destruction, longevity, mild condition reaction and rapid degradation process, applicable in extensive ranges of temperature and pH, utilization of PMS as a green and waste avoiding oxidant, highly efficient in MO decolorization. Thus, this catalytic system is very appropriate for the degradation process.
3.6.2. Aerobic oxidation system. A catalytic comparison of several studies (Table 6S†) with our system indicated that the MIOSC-N-et-NH2@CS-Mn as an eco-friendly bio-composite has remarkable merits in terms of its heterogeneous nature based on bio-magnetic cores and complete separation, low-cost active sites as an alternative to costly and toxic metals, simple in fabrication and application, mild condition reaction (low temperature and short time), high stability and longevity, use of O2 as greener and waste-avoiding oxidant, greener and ordinary solvent, excellent catalytic conversion, and high selectivity. Thus, this procedure is very appropriate for practical applications.
3.6.3. Economic feasibility study. The economic feasibility was assessed by comparing the cost of MIOSC-N-et-NH2@CS-Mn with other activators (i.e., ultraviolet, thermal and chemical) to activate the oxidant (energy consumption is ignored). It is clear that the calculation of the processing charge highly depends on the cost of producing the MIOSC-N-et-NH2@CS-Mn. Because the cost of the process is the main factor in the choice of the method, the charge for discoloration of MO in the PMS + MIOSC-N-et-NH2@CS-Mn system should be compared to other systems.49,50 For single-use, the charge of our degradation system was much more than those of the PS + thermal (44.41 $ per m3), PS + ultraviolet (0.176 $ per m3) and chemical systems (PS+γ-Fe2O3–CeO2 = 0.106 $ per m3 and PS + Fe2+ = 0.106 $ per m3) (see Table 7S and 8S†). However, the MIOSC-N-et-NH2@CS-Mn had excellent reusability (Fig. 3S†), and the charge for discoloration of a single-use was estimated to be approximately 26.46 $ per m3. Universality for aerobic oxidation and AOPs, excellent catalytic performance, and rapid decontamination with reasonable cost and recyclable capability can introduce this system as an effective system.
4. Conclusion
In this study, the new MIOSC-N-et-NH2@CS-Mn bio-composite was successfully prepared by simple chemical co-precipitation and Stöber sol–gel methods; its ability to rapidly decolorize azo-dye molecules from water/wastewater and catalyze the selective oxidation of cheap ethylbenzene to its valuable derivative with molecular oxygen in the absence of promoters and/or reducing agents was demonstrated. To the best of our knowledge, a high TOF of 214.1 (103 h−1) was observed within 8 h and with excellent selectivity (95.1%) for the aerobic oxidation of ethylbenzene in HAc/H2O. The removal efficiency (and TOC removal) of methyl orange, methylene blue, methylene violet and BPA degradation reached 98.8% (∼56.61), 93.6% (∼52.41), 97.5% (∼55.83) and 98.4% (∼56.12) after 14 min at room temperature, respectively. In addition, MO degradation is affected by high levels in reaction temperature, wide pH application range, coexisting anions and various dosages of natural organic matters. The RSM was successfully applied to optimize the degrading conditions regarding the removal efficiency of dye, temperature, time and concentration of activator and oxidant as dependent variables or responses. More importantly, the of MIOSC-N-et-NH2@CS-Mn nanocomposite was magnetically recovered from the reaction mixture through 6 and 5 catalytic cycles with only 11.5% and 18.2% deactivation for the degradation of MO dye and aerobic oxidation process, respectively. The catalyst showed good efficiency, and no significant change in the crystal structure or morphology even after several cycles, which confirmed the highly efficient, great reusability and heterogeneous nature of MIOSC-N-et-NH2@CS-Mn bio-composite. Finally, the economic feasibility study confirms that PMS + MIOSC-N-et-NH2@CS-Mn is an economically efficient system for the decolorization of MO dye. Therefore, the bio-composite can benefit from the facile route to synthesize the magnetic characteristics of the core, the chelating activity, stability, costly and versatility of the chitosan as shell, the high catalytic activity of encapsulated-Mn NPs and the universality for degradation/oxidation capability under mild conditions. The investigation would facilitate the commercial fabrication of a new type of activator/catalyst composite, illustrating its potential usage to overcome the environmental issues related to the degradation of organic dyes and the aerobic oxidation process of raw materials.
Conflicts of interest
No conflicts of interest exists.
Acknowledgements
A. F. acknowledges research council of Tehran Medical Sciences, Islamic Azad University for the research founding of this project.
References
- M. S. Reddy, D. Ponnamma, R. Choudhary and K. K. Sadasivuni, Polymers, 2021, 13, 1105 CrossRef CAS PubMed.
- I. Younes, O. Ghorbel-Bellaaj, R. Nasri, M. Chaabouni, M. Rinaudo and M. Nasri, Process Biochem., 2012, 47, 2032–2039 CrossRef CAS.
- S. Ifuku, Molecules, 2014, 19, 18367–18380 CrossRef PubMed.
-
(a) P. R. Sivashankari and M. Prabaharan, Deacetylation modification techniques of chitin and chitosan, in Chitosan Based Biomaterials Volume 1, Woodhead Publishing, 2017 Jan 1, pp. 117–133 Search PubMed;
(b) P. R. Sivashankari and M. Prabaharan, Chitosan Based Biomaterials: Fundamentals, Elsevier, 2017, vol. 1, pp. 117–133 Search PubMed.
- C. Casadidio, D. V. Peregrina, M. R. Gigliobianco, S. Deng, R. Censi and P. Di Martino, Mar. Drugs, 2019, 17, 369 CrossRef CAS PubMed.
- A. R. Faraji, F. Ashouri, Z. Hekmatian, S. Heydari and S. Mosazadeh, Polyhedron, 2019, 157, 90–106 CrossRef CAS.
- A. P. Kumar, D. Bilehal, T. Desalegn, S. Kumar, F. Ahmed, H. A. Murthy, D. Kumar, G. Gupta, D. K. Chellappan, S. K. Singh and K. Dua, Adsorpt. Sci. Technol., 2022, 2022, 1–18 Search PubMed.
- M. Wan, F. Xiang, Z. Liu, D. Guan, Y. Shao, L. Zheng, M. Jin, Y. She, L. Cao, F. Jin and R. Chen, Food Chem., 2021, 365, 130485 CrossRef CAS PubMed.
- S. Movahedian, A. R. Farahani and A. R. Faraji, J. Alloys Compd., 2022, 908, 164585 CrossRef CAS.
- Y. Ma, C. Hou, H. Zhang, Q. Zhang, H. Liu, S. Wu and Z. Guo, Electrochim. Acta, 2019, 315, 114–123 CrossRef CAS.
- C. C. Fu, H. N. Tran, X. H. Chen and R. S. Juang, Ind. Eng. Chem. Res., 2020, 83, 235–246 CrossRef CAS.
- K. Wang, L. Li, X. Xu, L. Lu, J. Wang, S. Wang, Y. Wang, Z. Jin, J. Z. Zhang and Y. Jiang, ACS Appl. Mater. Interfaces, 2019, 11, 10452–10461 CrossRef CAS PubMed.
- Y. Wang, Y. Zhang, T. C. Zhang, G. Xiang, X. Wang and S. Yuan, ACS Appl. Nano Mater., 2020, 8, 8495–8504 CrossRef.
- A. Funes, J. de Vicente and I. de Vicente, Chemosphere, 2017, 171, 571–579 CrossRef CAS PubMed.
- A. Gholami and F. Mousavinia, Environ. Sci. Technol., 2022, 22, 3444–3461 CrossRef PubMed.
- U. E. Amjad, L. Sherin, M. F. Zafar and M. Mustafa, Arabian J. Sci. Eng., 2019, 44, 9851–9857 CrossRef CAS.
- X. Sun, D. Xu, P. Dai, X. Liu, F. Tan and Q. Guo, Chem. Eng. J., 2020, 402, 125881 CrossRef CAS.
- S. Li, Y. Wu, H. Zheng, H. Li, Y. Zheng, J. Nan, J. Ma, D. Nagarajan and J. S. Chang, Chemosphere, 2022, 311, 136977 CrossRef PubMed.
- Y. Du, W. Ma, P. Liu, B. Zou and J. Ma, J. Hazard. Mater., 2016, 308, 58–66 CrossRef CAS PubMed.
- R. Tabit, O. Amadine, Y. Essamali, K. Dânoun, A. Rhihil and M. Zahouily, RSC Adv., 2018, 8, 1351–1360 RSC.
- C. X. Li, C. B. Chen, J. Y. Lu, S. Cui, J. Li, H. Q. Liu, W. W. Li and F. Zhang, Chem. Eng. J., 2018, 337, 101–109 CrossRef CAS.
- Y. Yao, Y. Cai, G. Wu, F. Wei, X. Li, H. Chen and S. Wang, J. Hazard. Mater., 2015, 296, 128–137 CrossRef CAS PubMed.
- I. Shakir, M. Sarfraz, U. A. Rana, M. Nadeem and M. A. Al-Shaikh, RSC Adv., 2013, 3, 21386–21389 RSC.
- M. G. Bellino, J. G. Sacanell, D. G. Lamas, A. G. Leyva and N. E. Walsöe de Reca, J. Am. Chem. Soc., 2007, 120, 3066–3067 CrossRef PubMed.
- Y. Feng, J. Liu, D. Wu, Z. Zhou, Y. Deng, T. Zhang and K. Shih, J. Chem. Eng., 2015, 280, 514–524 CrossRef CAS.
- L. S. Ardakani, V. Alimardani, A. M. Tamaddon, A. M. Amani and S. Taghizadeh, Heliyon, 2021, 7, e06159 CrossRef PubMed.
- P. L. Yi, W. J. Zhang, L. H. Kong, R. F. Shen, X. J. Guo, X. Yan, Y. Chen and W. Z. Lang, Chem. Eng. J., 2022, 3, 374–384 Search PubMed.
- J. A. Fuentes-García, B. Sanz, R. Mallada, M. R. Ibarra and G. F. Goya, Mater. Des., 2023, 226, 111615 CrossRef.
- L. Wolski, K. Sobańska, G. Nowaczyk, M. Frankowski, M. Pietrowski, M. Jarek, M. Rozmyślak and P. Pietrzyk, J. Hazard. Mater., 2022, 440, 129783 CrossRef CAS PubMed.
- X. Liu, J. Xu, K. Jing, L. Lu and H. Liu, Chem. Eng. Res. Des., 2022, 186, 22–33 CrossRef CAS.
- Q. Wang, Z. Guan, S. Ding, D. Xia and D. Li, Sep. Purif. Technol., 2022, 289, 120625 CrossRef CAS.
- V. Chaudhary and S. Sharma, Chem. Eng. J., 2020, 15, e2441 CAS.
- L. Gao, W. Zhuge, X. Feng, W. Sun, X. Sun and G. Zheng, New J. Chem., 2019, 43, 8189–8194 RSC.
- Y. Deng, D. Qi, C. Deng, X. Zhang and D. Zhao, J. Am. Chem. Soc., 2008, 130, 28–29 CrossRef CAS PubMed.
- H. Veisi, T. Ozturk, B. Karmakar, T. Tamoradi and S. Hemmati, Carbohydr. Polym., 2020, 235, 115966 CrossRef CAS PubMed.
- M. Karami, A. R. Faraji, S. Saremnezhad and M. Soltani, RSC Adv., 2022, 12(50), 32280–32296 RSC.
- F. Ghorbani and S. Kamari, Environ. Technol. Innovation, 2019, 14, 100333 CrossRef.
- S. Li, S. R. Zhai, Q. D. An, M. H. Li, Y. Song and X. W. Song, Mater. Res. Bull., 2014, 60, 665–673 CrossRef CAS.
- L. Zhuang, W. Zhang, Y. Zhao, H. Shen, H. Lin and J. Liang, Sci, 2015, 5, 1–6 Search PubMed.
- G. Unsoy, S. Yalcin, R. Khodadust, G. Gunduz and U. Gunduz, J. Nanopart. Res., 2012, 14, 1–3 CrossRef.
- S. Kumar and J. Koh, Int. J. Mol. Sci., 2012, 13, 6102–6116 CrossRef CAS PubMed.
- M. A. Ghasemzadeh, M. H. Abdollahi-Basir and M. Babaei, Green Chem. Lett. Rev., 2015, 8, 40–49 CrossRef.
- V. Sureshkumar, S. C. K. Daniel, K. Ruckmani and M. Sivakumar, Appl. Nanosci., 2016, 6, 277–285 CrossRef CAS.
- M. S. Izgi, M. Ş. Ece, H. Ç. Kazici, Ö. Şah
![[i with combining dot above]](https://www.rsc.org/images/entities/char_0069_0307.gif) n and E. Onat, Int. J. Hydrogen Energy, 2020, 45, 30415–30430 CrossRef.
n and E. Onat, Int. J. Hydrogen Energy, 2020, 45, 30415–30430 CrossRef. - S. Li, S. R. Zhai, Q. D. An, M. H. Li, Y. Song and X. W. Song, Mater. Res. Bull., 2014, 60, 665–673 CrossRef CAS.
- J. Luo, S. Bo, Y. Qin, Q. An, Z. Xiao and S. Zhai, Chem. Eng. J., 2020, 395, 125063 CrossRef CAS.
- S. Zhu, Z. Wang, C. Ye, J. Deng, X. Ma, Y. Xu, L. Wang, Z. Tang, H. Luo and X. Li, Chem. Eng. J., 2022, 432, 134180 CrossRef CAS.
- G. Dobras and B. Orlińska, Appl. Catal., A, 2018, 561, 59–67 CrossRef CAS.
- M. Amasha, A. Baalbaki and A. Ghauch, Chem. Eng. J., 2018, 350, 395–410 CrossRef CAS.
- L. Niu, G. Zhang, G. Xian, Z. Ren, T. Wei, Q. Li, Y. Zhang and Z. Zou, Sep. Purif. Technol., 2021, 259, 118156 CrossRef CAS.
- M. J. Amiri, A. Faraji, M. Azizi, B. G. Nejad and M. Arshadi, Process Saf. Environ. Prot., 2021, 147, 626–641 CrossRef CAS.
- J. Wang, D. Zhi, H. Zhou, X. He and D. Zhang, Water Res., 2018, 137, 324–334 CrossRef CAS PubMed.
- R. Rezaee, A. Faraji and F. Ashouri, Inorg. Chem. Commun., 2022, 146, 110184 CrossRef CAS.
- J. Cui, T. Liu, Q. Zhang, T. Wang and X. Hou, Chem. Eng. J., 2020, 404, 126453 CrossRef.
- J. Zhang, W. Zhao, S. Wu, R. Yin and M. Zhu, J. Hazard. Mater., 2021, 410, 124623 CrossRef CAS PubMed.
- F. Ghanbari and M. Moradi, Chem. Eng. J., 2017, 310, 41–62 CrossRef CAS.
- Z. Li, S. Ning, H. Zhu, X. Wang, X. Yin, T. Fujita and Y. Wei, Chemosphere, 2022, 288, 132600 CrossRef CAS PubMed.
- B. Guo, J. Ma, Y. Shi, K. Zheng, M. Wu, G. Ren and S. Komarneni, Ceram. Int., 2021, 47, 27617–27623 CrossRef CAS.
- J. Cui, T. Liu, Q. Zhang, T. Wang and X. Hou, Chem. Eng. J., 2020, 404, 126453 CrossRef.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *bc and
Fatemeh Ashourid
*bc and
Fatemeh Ashourid