DOI:
10.1039/D2RA07063D
(Paper)
RSC Adv., 2023,
13, 2458-2466
Experimental evidence for CH⋯π interaction-mediated stabilization of the square form in phenylglycine-incorporated ascidiacyclamide†
Received
7th November 2022
, Accepted 4th January 2023
First published on 20th January 2023
Abstract
Ascidiacyclamide [cyclo(-Ile-oxazoline-D-Val-thiazole-)2] is a cytotoxic cyclic peptide from ascidian. We examined the potential of the CH⋯π interaction at the diagonal position of ascidiacyclamide by comparing the interactions of Ile, Val, Abu (2-aminobutyric acid) or Ala with Ile, Chg (cyclohexylglycine) or Phg (phenylglycine). In solution, ascidiacyclamides are in a conformational equilibrium between square and folded forms. The CH⋯π interaction is expected to contribute to stabilization of the square form, which enhances the peptides' cytotoxicity. The distances between the alkyl side chain of Xaa and the π-plane of Phg were estimated from the crystal structures. The conformational free energies (ΔG°) determined through NMR-based quantitation indicated remarkable stabilization of the square form upon incorporation of Phg. These observations were consistent with the circular dichroism (CD) spectral measurements. Chemical shift perturbation studies suggested that stabilization of the square form of Phg-incorporated peptides was due to the CH⋯π interaction with the alkyl side chain of Xaa. Greater enthalpic losses were caused during the folding process of Phg-incorporated peptides than Ile- or Chg-incorporated peptides. It is suggested that these enthalpic losses are relevant to the CH⋯π interaction energies, which must be disrupted during folding. In addition, the CH⋯π interactions in the Phg-incorporated peptides increased cytotoxicity.
Introduction
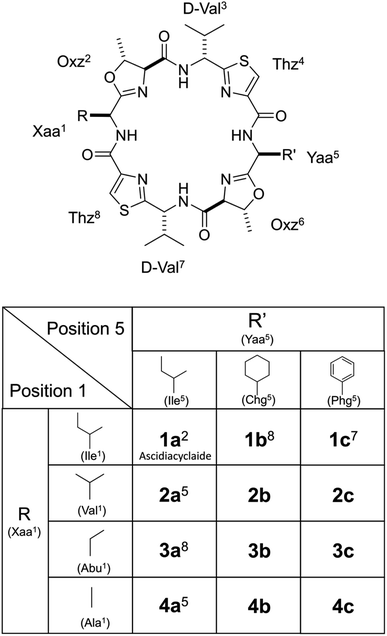
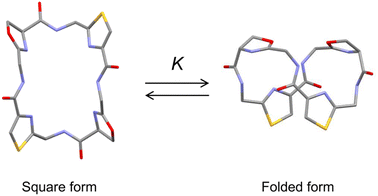
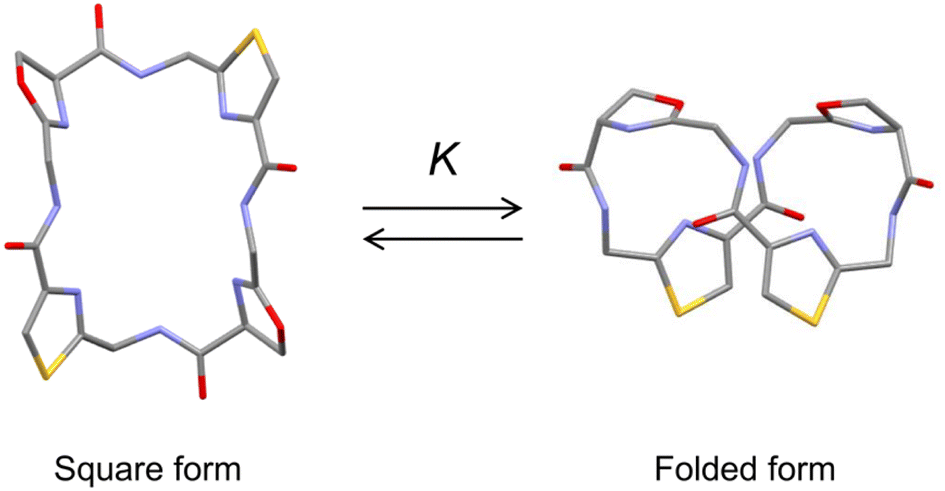
Ascidiacycalmide (1a) is a cytotoxic cyclic peptide isolated from the marine invertebrate ascidian.1 The chemical sequence of 1a, cyclo(-Ile1-Oxz2-D-Val3-Thz4-Ile5-Oxz6-D-Val7-Thz8-), contains the five-membered heterocycles oxazoline (Oxz) and thiazole (Thz) and is characterized by C2-symmetry (Fig. 1). Previous structural analyses showed that 1a assumes two major conformations, a “square form”, which is the potentially cytotoxic structure, and a “folded form”, and that the two conformations are in equilibrium in solution (Fig. 2).2–9 The conformational free energies (ΔG°) of peptides bearing various substituents at the 1-position (or 5-position) were determined in NMR-based quantitative studies.8 The 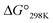 value for a peptide bearing an n-propyl group was nearly zero, while those for peptides bearing a substituent smaller than an n-propyl group (3a and 4a) were negative. On the other hand, the values for peptides bearing a bulkier substituent than an n-propyl group (1a, 2a, 1b and 1c) were positive, which means that these peptides do not spontaneously fold at 298 K. This suggests that when 1a is in the square form, the bulky side chain of the substituted amino acid residue and the sec-butyl group of the Ile residue at the diagonal position are close to each other, and the dispersion force between these two functional groups prevents spontaneous folding. In particular, a dispersion force like a CH⋯π interaction may be acting in peptides bearing a phenyl group (1c). While it is apparent that the van der Waals space occupied by a cyclohexyl group is larger than that of a planar phenyl group, the
value for a peptide bearing an n-propyl group was nearly zero, while those for peptides bearing a substituent smaller than an n-propyl group (3a and 4a) were negative. On the other hand, the values for peptides bearing a bulkier substituent than an n-propyl group (1a, 2a, 1b and 1c) were positive, which means that these peptides do not spontaneously fold at 298 K. This suggests that when 1a is in the square form, the bulky side chain of the substituted amino acid residue and the sec-butyl group of the Ile residue at the diagonal position are close to each other, and the dispersion force between these two functional groups prevents spontaneous folding. In particular, a dispersion force like a CH⋯π interaction may be acting in peptides bearing a phenyl group (1c). While it is apparent that the van der Waals space occupied by a cyclohexyl group is larger than that of a planar phenyl group, the 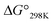 value of 1c is three times that of 1b.
value of 1c is three times that of 1b.
 |
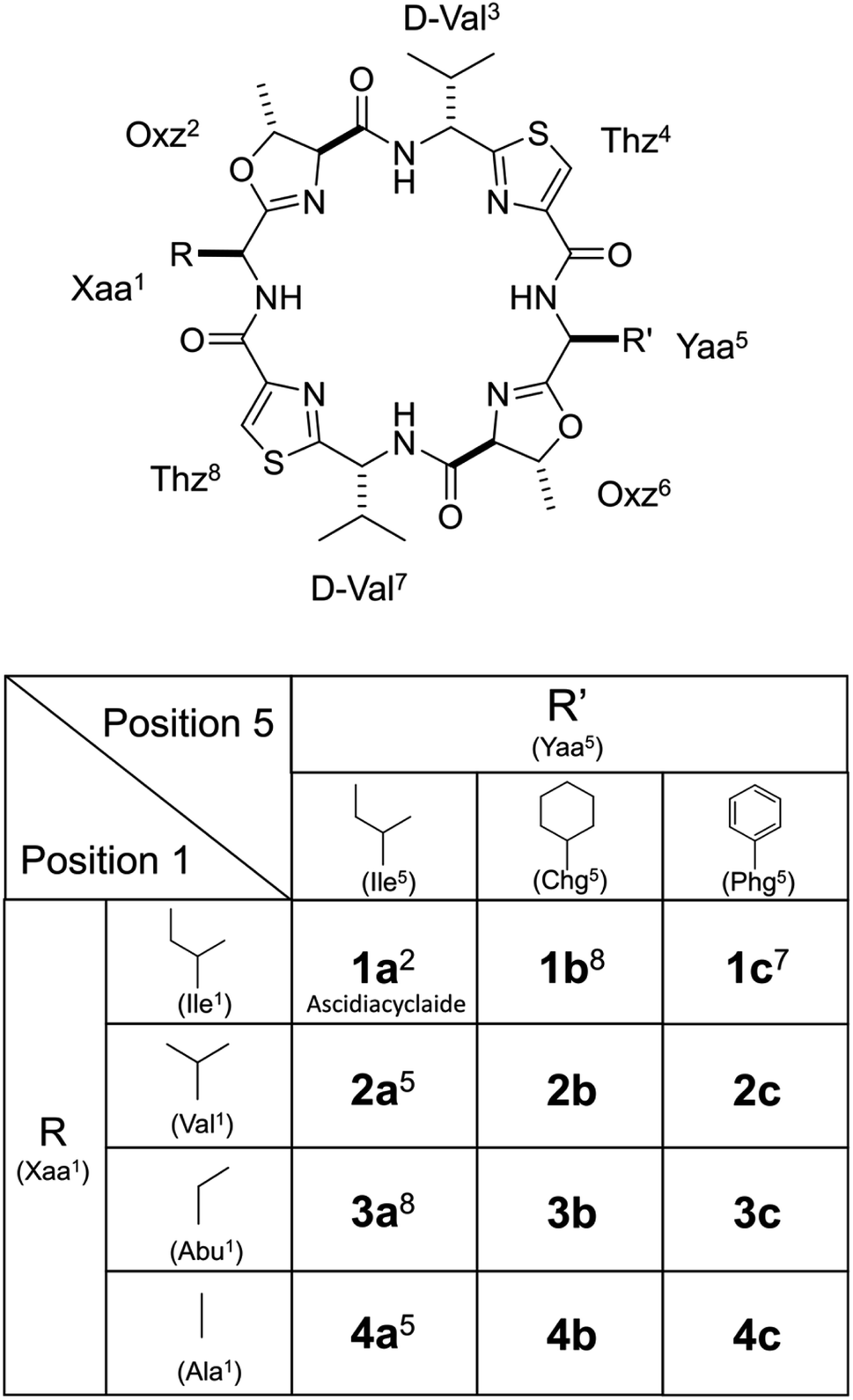
| | Fig. 1 Chemical structures of ascidiacyclamide and the side chains (R) of the Xaa1 residues and the side chains (R′) of Yaa5 residues. Peptides 1a–4a, 1b and 1c were previously synthesized.2,5,7,8 Peptides Xb (X = 2–4) and Xc (X = 2–4) are newly synthesized asymmetric analogues. | |
 |
| | Fig. 2 Conformational equilibrium between the square and folded forms of ascidiacyclamide backbone. Carbon, nitrogen, oxygen and sulfur atoms are represented in gray, blue, red and yellow, respectively. | |
Phenylglycine (Phg) is a non-proteinogenic amino acid that is, nonetheless, a reported constituent of various natural peptidic products.10–15 Phg can be regarded as a truncated version of Phe that lacks the methylene group. In other words, Phg has a bulky aromatic side chain directly attached to the α-carbon. It is therefore assumed that the degree of freedom of the aromatic ring side chain of Phg is strongly restricted, and the backbone conformation of a Phg-containing peptide is constrained accordingly. Furthermore, the electronic effects of aromatic moieties produce a weak noncovalent interaction, termed CH⋯π, which can contribute significantly to the proper folding and function of a protein.16–22 For instance, many saccharide units possessing codirected CH groups can bind into π clouds of aromatic moieties in proteins. Thus, the β-D-galactose bound to lectin makes contact with Phe131,23 and the β-D-glucose bound to an E. coli chemoreceptor protein makes contact with Trp183.24 The contact distances for these interactions were all within 3.5 Å within the X-ray structures. Given these steric and electronic features, it is expected that Phg would be a valuable pharmaceutical building block.
To gain additional details into the role of Phg in modulating the stability of square form, we investigated three series of peptides (Xa, Xb and Xc) in the present study (Fig. 1). The 5-position residues of Xa, Xb and Xc peptides were Ile5, cyclohexylglycine (Chg5) and Phg5, respectively, and the 1-position residues were replaced with amino acid residues with substituents of various bulkiness [Ile (X = 1), Val (X = 2), 2-aminobutyric acid (Abu) (X = 3) and Ala (X = 4)]. Peptides 1a–4a, 1b and 1c were previously synthesized, while peptides 2b–4b, 2c, 3c and 4c were newly synthesized. Here we describe their structural characterization based on X-ray diffraction, circular dichroism (CD) spectroscopy and variable temperature (VT) 1H NMR measurements. We then discuss the impact of Phg on stabilization of the square form, which is the potentially cytotoxic structure, by making comparisons among the Xa, Xb and Xc peptides.
Results and discussion
Crystal structures
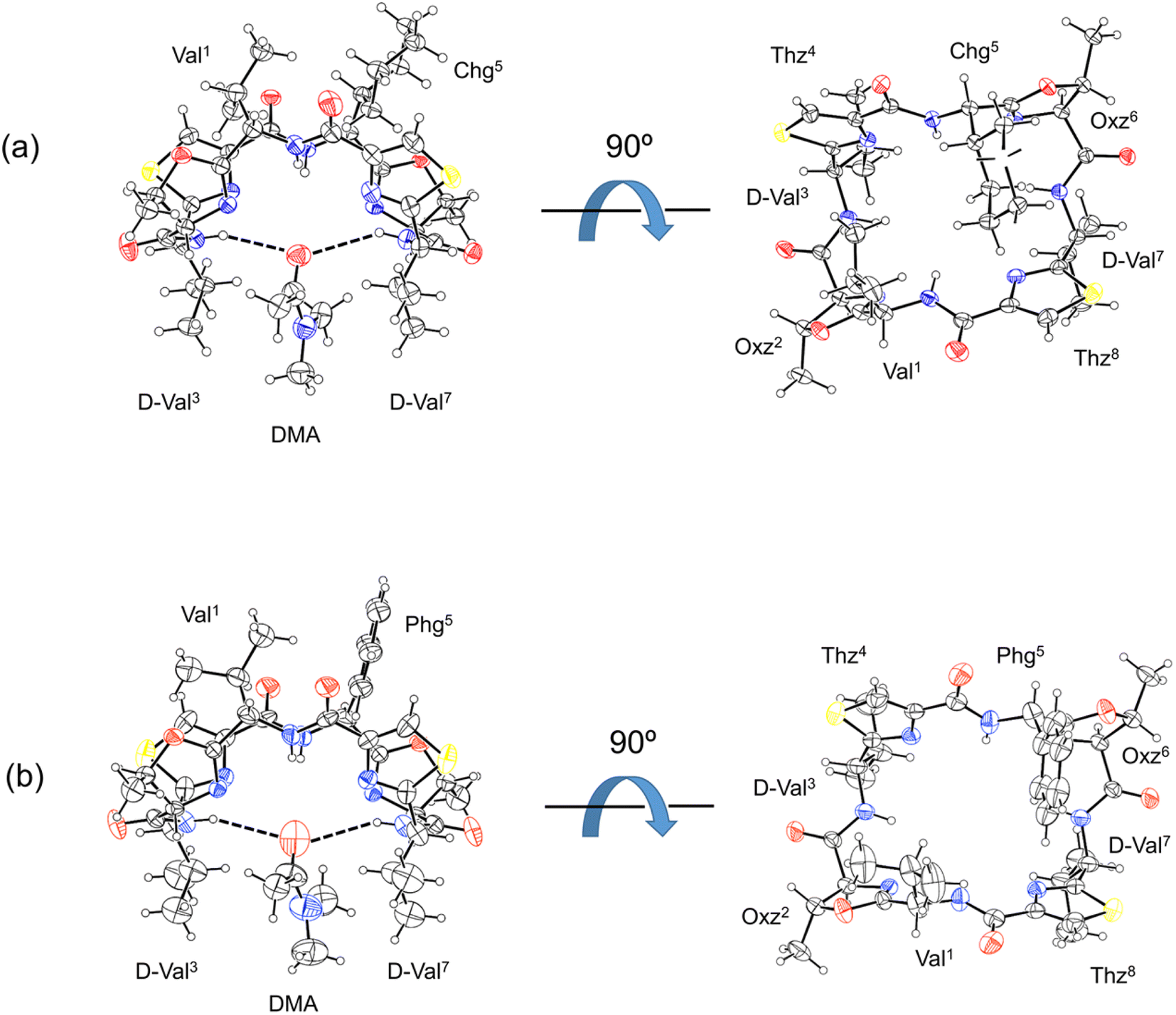
The X-ray structures of 2b and 2c are shown in Fig. 3. For both 2b and 2c, each asymmetric unit contained the peptide molecule and one N,N-dimethylacetamide (DMA) molecule. Each peptide backbone was open, and the DMA molecule was located at the center of the peptide backbone, where it formed two hydrogen bonds with the peptide: N(D-Val3)–H⋯O(DMA) and N(D-Val7)–H⋯O(DMA) (Table 1). The previously reported crystal structure of 2a was also a square form,5 and there was no significant difference in the crystal structures among 2a, 2b and 2c, whose 1-position is a Val residue. The crystal structures of 1a, 1b and 1c, whose 1-position is an Ile residue, were also open.2,7,8 On the other hand, the crystal structure of 4a was folded.5 In the Xa peptides, the crystal structures were folded when the Xaa1 residue side chain was small. However, the crystal structures of 3a, 3b, 3c, 4b and 4c have not yet been determined.
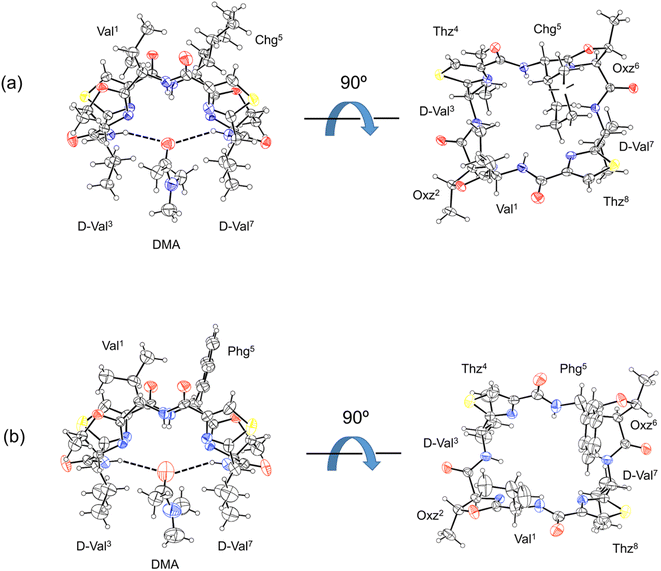 |
| | Fig. 3 Crystal structures of peptides 2b and 2c are presented in (a) and (b), respectively. Shown are the side (left) and top (right) views of the peptide rings. The dashed lines represent hydrogen bonds. | |
Table 1 Hydrogen bonds within the crystal structures of 2b and 2c
| Peptide |
Donor |
Acceptor |
Distance (Å) |
Angle (°) |
| D–H |
A |
D⋯A |
∠D–H⋯A |
| 2b |
N(D-Val3)–H |
O(DMA) |
3.223 |
150.45 |
| N(D-Val7)–H |
O(DMA) |
3.381 |
143.04 |
| 2c |
N(D-Val3)–H |
O(DMA) |
3.387 |
150.63 |
| N(D-Val7)–H |
O(DMA) |
3.387 |
150.63 |
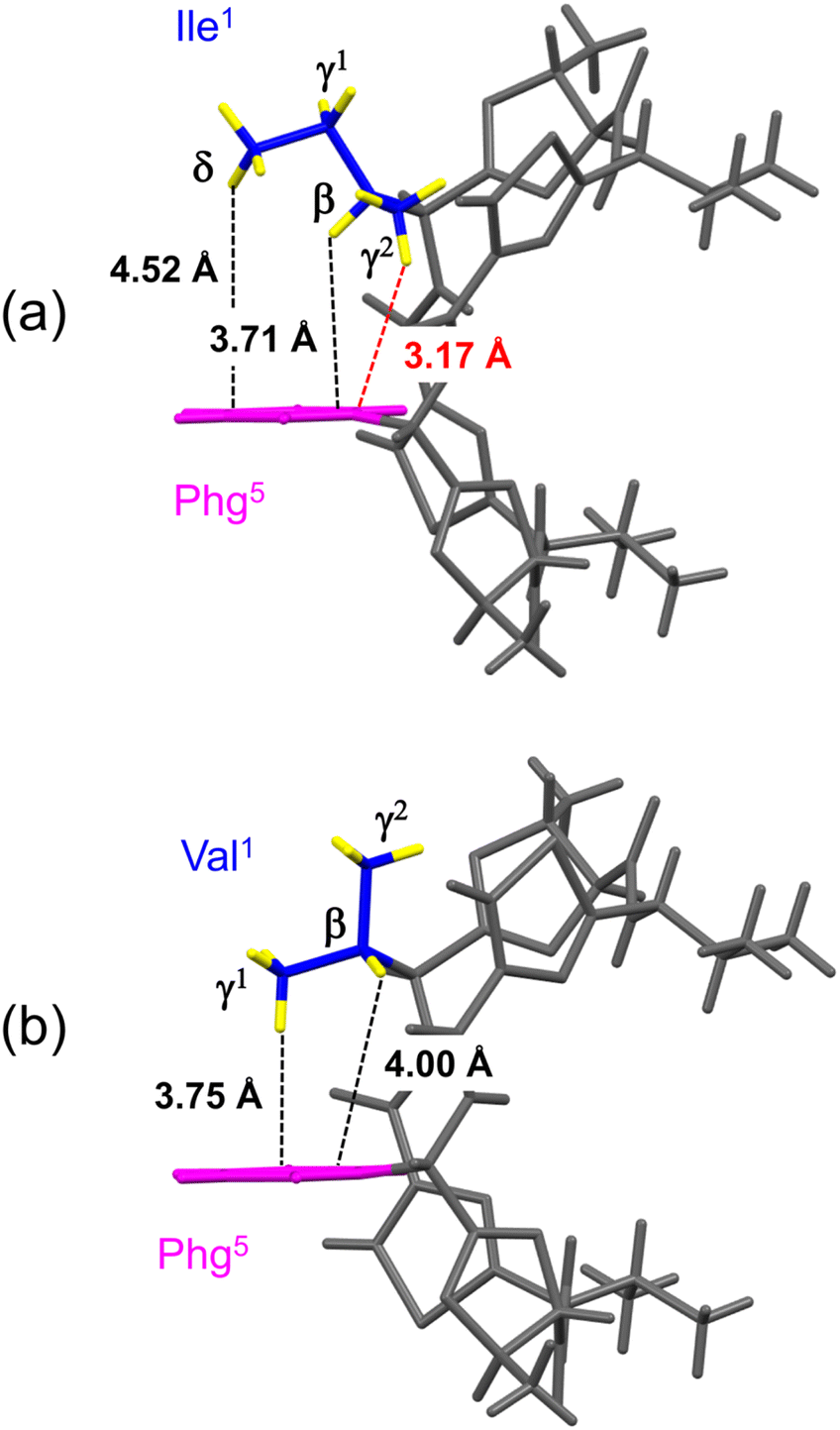
The distances between the side chains of Xaa1 and Phg5 were estimated by surveying the CH⋯π contacts for the six-membered π-system, as described by Umezawa et al.25–27 The distances from the H atoms of the Xaa1 alkyl side chain to the π-orbital of the Phg5 residue in the crystal structures of 1c (ref. 7) and 2c are shown in Fig. 4. The γ1H of Ile1 and the γ2H of Val1 were oriented in the direction opposite to the π-plane of Phg5, and CH⋯π contacts were not observed. Within the crystal structure of 1c, the δH of Ile1 was located above the π-plane, but 4.52 Å away. The CH/π distance of the βH was 3.71 Å, while that of the γ2H was 3.17 Å, which was the closest to the π-orbital. Within the crystal structure of 2c, the CH/π distances of the βH and γ1H of Val1 were 4.00 Å and 3.75 Å, respectively. The sum of the van der Waals radii of the H and Csp2 was 2.90 Å (1.20 Å for C–H plus 1.70 Å for a half thickness of the aromatic molecule28,29), and more than 77% of organic crystals have been found to have CH/π distances shorter than 3.05 Å in database studies.25–27 Although the CH/π distance between the γ2H of Ile1 and the π-orbital of Phg5 in the crystal structure of 1c was slightly longer than 3.05 Å, this distance makes it worth considering the presence of CH⋯π interactions.
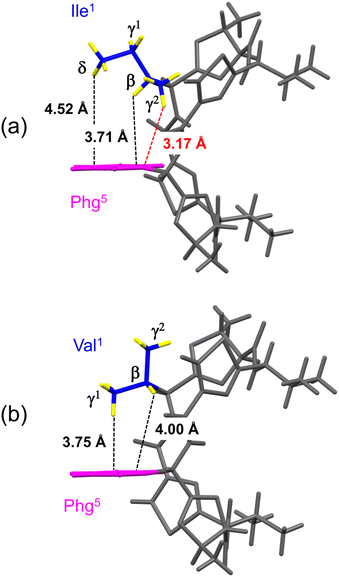 |
| | Fig. 4 Distances between the hydrogen atoms in the alkyl side chain of Xaa1 and the π-orbital of Phg5 within the crystal structures of 1c (a) and 2c (b). The alkyl carbon and hydrogen atoms of the Xaa1 side chain and the aromatic moiety of the Phg5 side chain are shown in blue, yellow and pink, respectively. | |
CD spectra
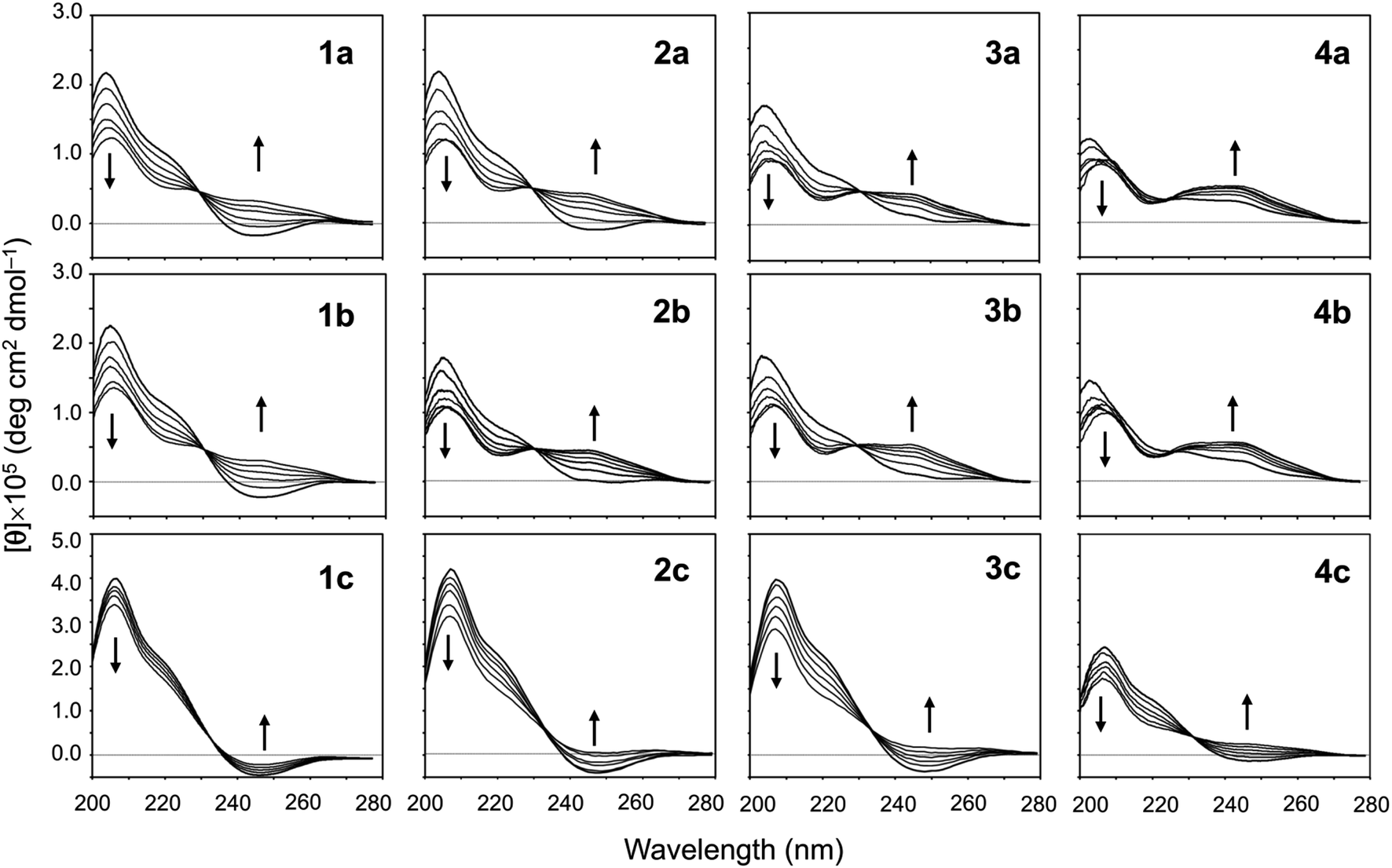
The conformational equilibrium between the square and folded forms can be measured based on the CD spectral changes that occur while titrating 2,2,2-trifluoroethanol (TFE) into acetonitrile (CH3CN) solution of the peptide.5–9 For instance, the spectrum of 1a in CH3CN solution shows a positive band at around 205 nm and a negative band at around 245 nm, which is indicative of the square form. TFE titration led to decreases in [θ]205 and increases in [θ]245, which indicate the conformation of the peptide was folding.5 By contrast, the spectrum of 4a in CH3CN solution shows a moderate positive band in the range of 230–260 nm, which is characteristic of the folded form. In other words, the conformational equilibrium position of 4a in CH3CN solution is significantly shifted in the forward direction, and there is almost no CD spectral change with TFE titration.5 The conformational equilibria for Xb and Xc (X = 2–4) were measured using the same protocol. Their spectral changes are shown in Fig. 5 along with the previously recorded spectra of 1a–4a,5,8 1b8 and 1c7 for comparison.
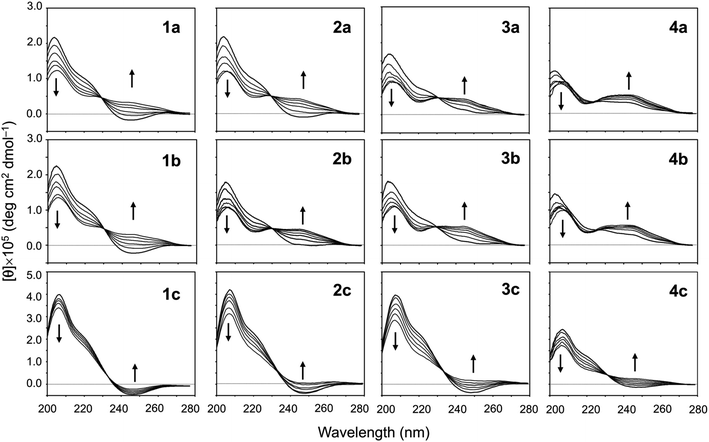 |
| | Fig. 5 CD spectral changes elicited by titration of TFE for peptides Xa, Xb and Xc (X = 1–4). The CD spectra for Xa, 1b and 1c were taken from previous reports.5,7,8 The spectra were measured in CH3CN solution while changing the TFE concentration (from 10% to 50% in increments of 10%). | |
When the spectral changes among Xa, Xb and Xc bearing the same Xaa1 were compared, very similar spectral changes were seen in Xa and Xb, but those in Xc indicated a large difference from the others. The spectra of all Xc peptides in CH3CN solution showed clearer negative bands around 245 nm than those of the corresponding Xa and Xb peptides. In particular, the spectrum of 1c in CH3CN solution was not significantly affected by TFE, suggesting the conformational equilibrium position of 1c is significantly shifted in the opposite direction. The incorporation of Chg5 did not affect the conformational equilibrium, but incorporation of Phg5 led to an apparent stabilization of the square form. For all Xa, Xb and Xc peptides, folding became easier as the bulkiness of the Xaa1 side chain declined (Ile1 > Val1 > Abu1 > Ala1).
VT-1H NMR
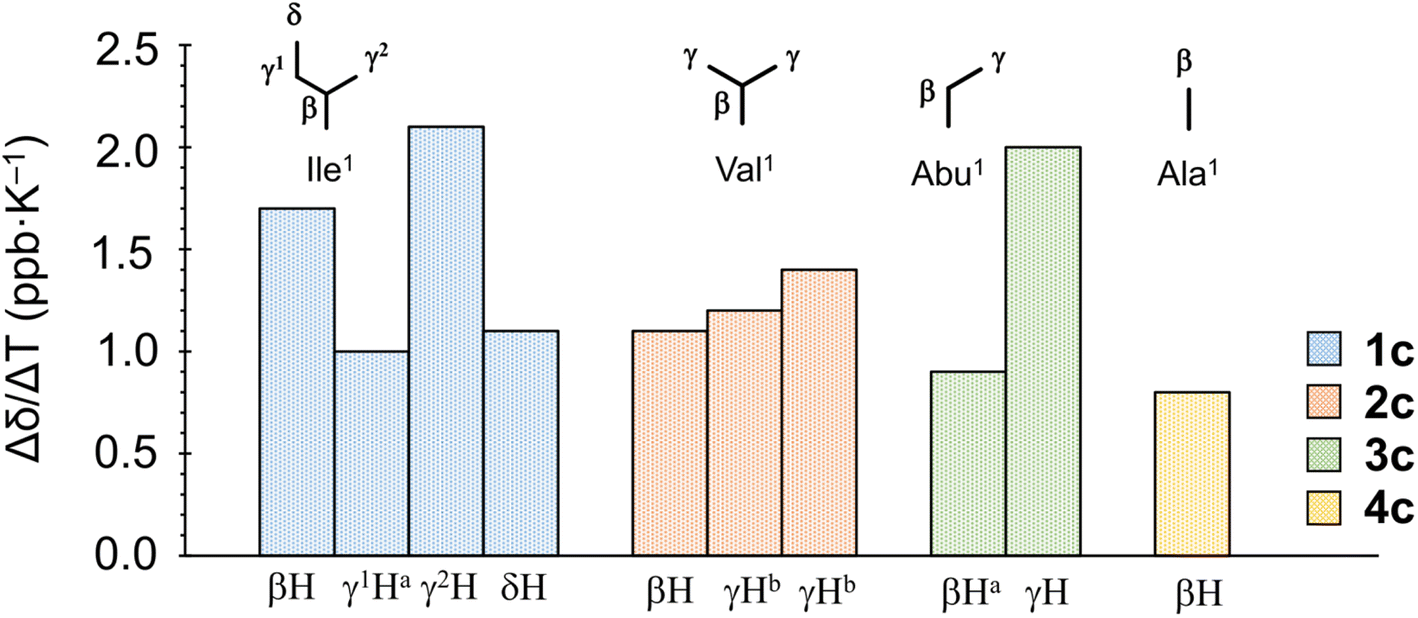
When the alkyl protons are close to the aromatic moiety, the result is a high-field shift of the alkyl proton chemical shift based on the ring-current effects of the aromatic ring. These high-field shifts can be probed to detect CH–π interactions.30,31 The chemical shifts [δ (ppm)] for protons in the Xaa1 alkyl side chain at 298 K and the chemical shift perturbations Δδ(Xb–X) (ppm) and Δδ(Xc–Xa) (ppm) are listed in Table 2. The values of Δδ(Xb–Xa) and Δδ(Xc–Xa) represent the magnitudes of the chemical shifts of alkyl protons in Xaa1 caused by incorporation of Chg5 and Phg5, respectively. All absolute values of Δδ(Xb–Xa) were very small, indicating that incorporation of Chg5 had little effect on the chemical shift of the alkyl side chain of the Xaa1 residue. By contrast, the high-field shifts of the alkyl side chain of the Xaa1 residue caused by incorporation of Phg5 were observed in all Xc peptides and resulted in negative Δδ(Xc–Xa) values for all Xaa1 side chains in Xc peptides. In addition, variable temperature 1H NMR spectroscopy (VT-1H NMR) was applied to examine the peptides every 10 K from 273 K to 333 K in CH3CN-d3 solution. The temperature coefficients [Δδ/ΔT (ppb K−1)] for the alkyl protons in the Xaa1 side chains that were high-field shifted by the ring-current effects of the π-orbital of the Phg5 residue are shown in Fig. 6. In 1c, the temperature coefficient for the γ2H and βH in the Ile1 residue were 2.1 and 1.7 ppb K−1, respectively, which are significantly elevated values. Within the crystal structure of 1c, the γ2H was located closest to the π-plane of the Phg5 residue, and the βH was second closest among the alkyl protons of the Ile1 residue. These observations suggest that the high temperature coefficient for the γ2H in the Ile1 residue is due to a decrease in the CH⋯π interaction with increasing temperature. The temperature coefficient of the γH in the Abu1 residue in 3c also had a high temperature coefficient (2.0 ppb K−1). Although the crystal structure of 3c has not yet been determined, the position of the γH in the Abu1 residue with respect to the π-plane of the Phg5 residue may be similar to that of the γ2H in the Ile1 residue within the crystal structure of 1c. Within the crystal structure of 2c, one γH of the Val1 residue was oriented toward the π-plane of the Phg5 residue while the other γH was oriented toward the opposite side. However, the temperature coefficients of these two γ protons (1.2 and 1.4 ppb K−1, respectively) did not significantly differ.
Table 2 Chemical shifts (δ, ppm) for protons in the Xaa1 alkyl side chain and chemical shift perturbations, Δδ(Xb–Xa) (ppm) and Δδ(Xc–Xa) (ppm), in CH3CN-d3 solution at 298 K
| |
Ile1 |
Val1 |
Abu1 |
Ala1 |
| βH |
γ1H |
γ2H |
δH |
βH |
γHa |
γHa |
βH |
γH |
βH |
| The chemical shifts of two γ protons in the Val1 residue are indistinguishable. These data are taken from a previous report.5 These data are taken from a previous report.8 These data are taken from a previous report.7 |
| Xa |
1.91b |
1.12b, 1.23b |
0.76b |
0.68b |
2.15b |
0.85b |
0.75b |
1.95c, 1.79c |
0.78c |
1.48b |
| Xb |
1.94c |
1.19c, 1.35c |
0.72c |
0.76c |
2.18 |
0.84 |
0.84 |
1.95, 1.82 |
0.73 |
1.44 |
| Xc |
1.45d |
0.68d, 0.98d |
0.18d |
0.37d |
1.78 |
0.32 |
0.29 |
1.41, 1.41 |
−0.02 |
0.86 |
| Δδ(Xb–Xa) |
0.03 |
0.07, 0.12 |
−0.04 |
0.08 |
0.03 |
−0.01 |
0.09 |
0.00, 0.03 |
−0.05 |
−0.04 |
| Δδ(Xc–Xa) |
−0.46 |
−0.44, −0.25 |
−0.58 |
−0.31 |
−0.37 |
−0.53 |
−0.46 |
−0.54, −0.38 |
−0.83 |
−0.62 |
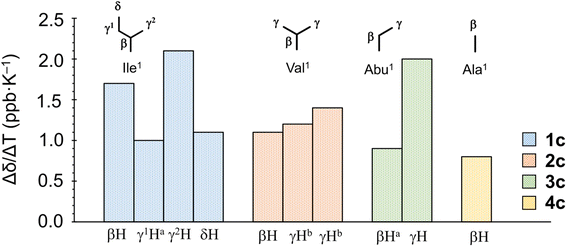 |
| | Fig. 6 Temperature coefficients [Δδ/ΔT (ppb K−1)] of protons in the Xaa1 alkyl side chain in the Xc peptides. aEstimated from the average chemical shift of two protons of methylene. bThe chemical shifts of the two γ protons in the Val1 residue are indistinguishable. | |
NMR-based quantification of the conformational equilibrium
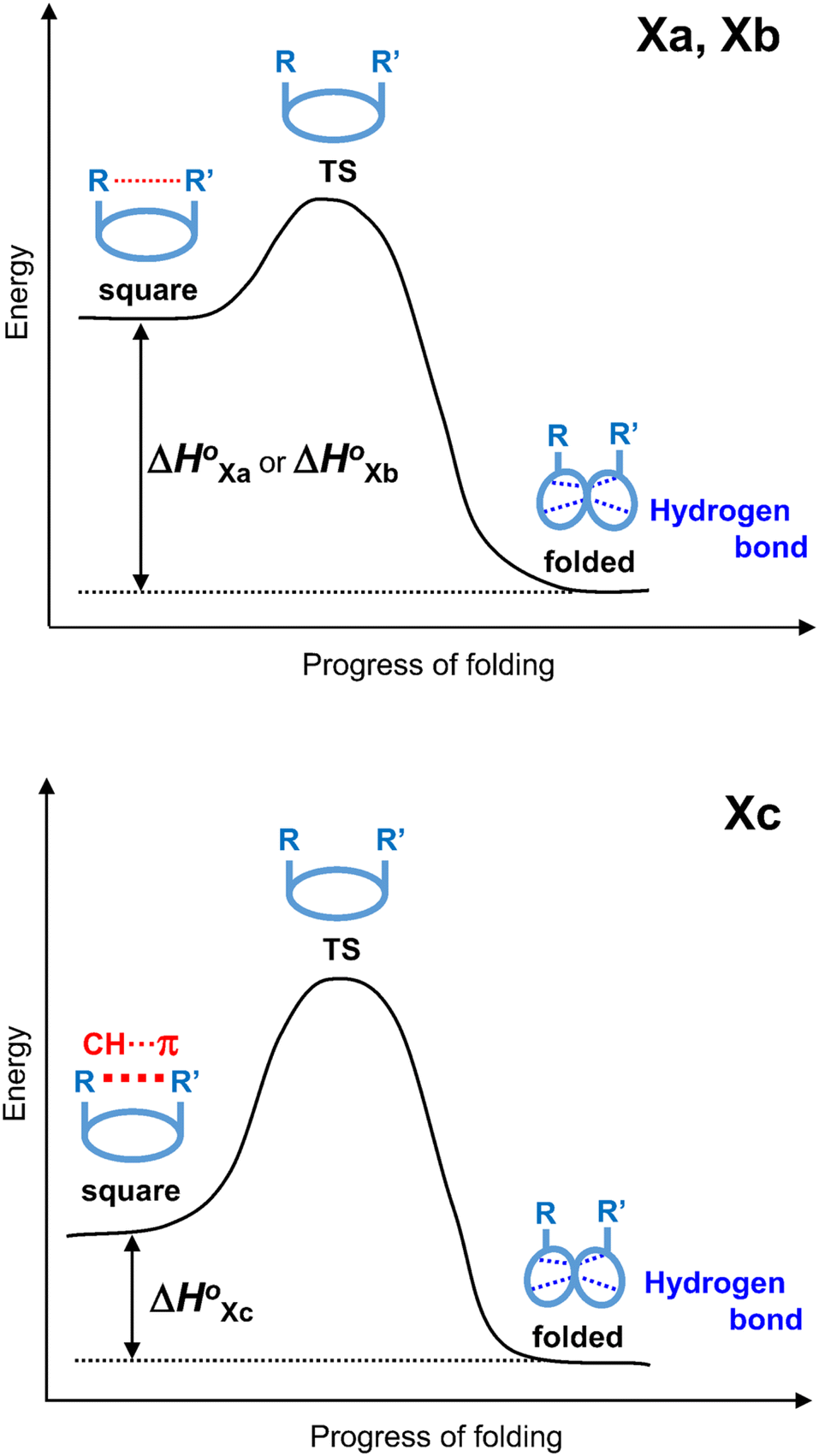
The conformational equilibrium constants (K) of the peptides were determined based on the VT-H1 NMR measurements. The thermodynamic parameters (ΔH°, ΔS° and ΔG°) were obtained from linear van't Hoff plots. These values are listed in Table 3. The ΔH° and ΔS° values for 1c were not obtained because their K values exhibited no temperature dependency.8 The folding of all peptides is enthalpically favorable (ΔH° < 0) and entropically unfavorable (ΔS° < 0). This thermodynamic profile results from four hydrogen bonds [N(Xaa1)H⋯Oγ(Oxz6), N(D-Val3)H⋯O(Thz8), N(Yaa5)H⋯Oγ(Oxz2) and N(D-Val7)H⋯O(Thz4)] formed within the folded form. The Xa and Xb peptides have similar thermodynamic profiles, whereas the thermodynamic profiles of Xc peptides differ from those of Xa and Xb. For Xc peptides, the enthalpic terms were more unfavorable than those for either the Xa or Xb peptides. This suggests that the nature of the interactions between Xaa1 and Ile5 or Chg5 are similar, but the interaction of Xaa1 with Phg5 is distinct. For the conformational change from the square to the folded form, it is necessary to through the transition state (TS) without the interaction between Xaa1 and Yaa5 (Fig. 7). It is considered that the folding of Xc peptides causes an enthalpic loss 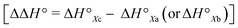 by an amount related to the energy of the CH⋯π interaction.
by an amount related to the energy of the CH⋯π interaction.
Table 3 Thermodynamic parameters of peptides
| Peptide |
ΔH° (kJ mol−1) |
ΔS° (J mol−1) |
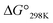
(kJ mol−1) |
| Thermodynamic parameters of Xa (X = 1–4), 1b and 1c are taken from a previous report.8 ΔH° and ΔS° values of 1c were not obtained from the van't Hoff equation because their K values showed no temperature dependencies. |
| 1aa |
−9.55 |
−39.40 |
2.19 |
| 1ba |
−9.85 |
−39.55 |
1.93 |
| 1ca |
—b |
—b |
7.09 |
| 2aa |
−13.01 |
−46.71 |
0.91 |
| 2b |
−12.39 |
−43.16 |
0.47 |
| 2c |
−5.38 |
−36.12 |
5.38 |
| 3aa |
−13.83 |
−45.23 |
−0.35 |
| 3b |
−11.78 |
−39.35 |
−0.06 |
| 3c |
−5.79 |
−34.88 |
4.60 |
| 4aa |
−16.76 |
−52.79 |
−1.03 |
| 4b |
−15.72 |
−48.91 |
−1.15 |
| 4c |
−10.58 |
−47.97 |
3.72 |
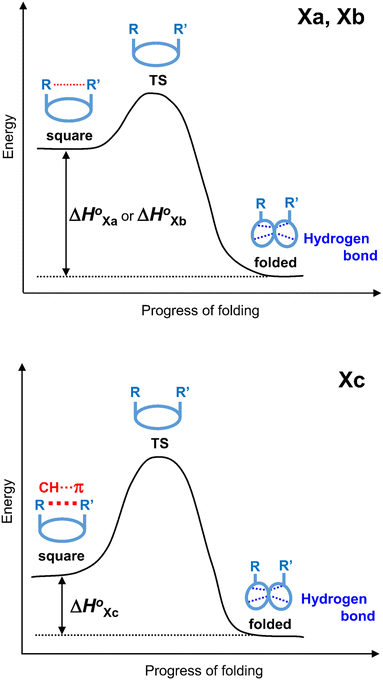 |
| | Fig. 7 Energy diagrams depicting the transition state (TS) between two conformers of Xa and Xb (top), and Xc (bottom). | |
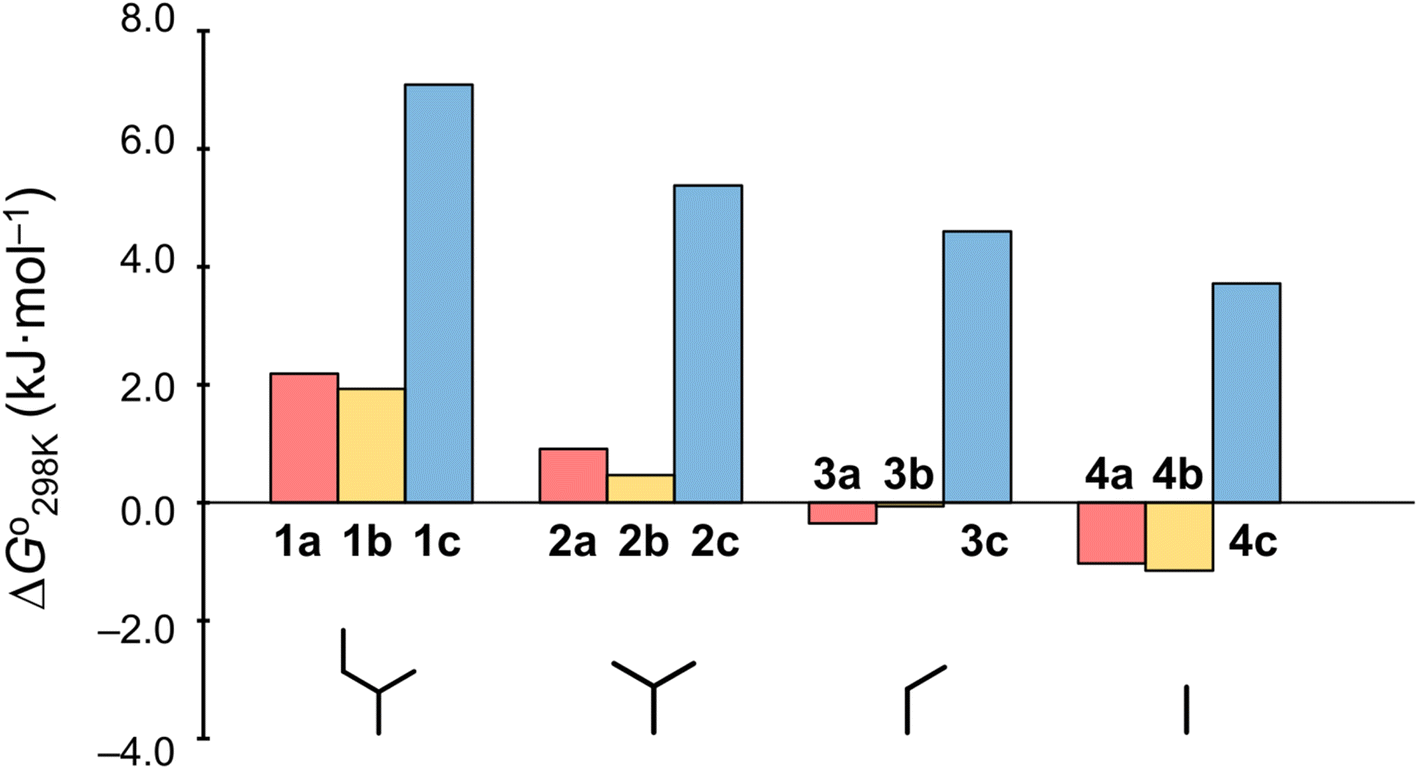
Plots of the temperature versus ΔG° values for all peptides are shown in Fig. 8. The ΔG° values for 1a, 1b and all Xc peptides were positive at every measured temperature. Comparing the results with the Xa and Xb peptides, the temperatures at which the ΔG° value of Xb peptides was zero were about 5 K higher than those for the corresponding Xa peptides (excluding 1a and 1b). This suggests that difficulty of spontaneous folding – i.e., stability of the square form – among the peptides is in the order: Xc, Xb, Xa. The ΔG° values at 298 K for all peptides are shown in Fig. 9. The 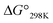 value for Xa peptides increases with increases in the bulkiness of the substituent of the Xaa1 residue [methyl (X = 4) < ethyl (X = 3) < isopropyl (X = 2) < sec-butyl (X = 1)]. The same relationship between the bulkiness of the Xaa1 side chain and the
value for Xa peptides increases with increases in the bulkiness of the substituent of the Xaa1 residue [methyl (X = 4) < ethyl (X = 3) < isopropyl (X = 2) < sec-butyl (X = 1)]. The same relationship between the bulkiness of the Xaa1 side chain and the 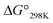 value was also seen with the Xb and Xc peptides. Among the Xa, Xb and Xc peptides, the
value was also seen with the Xb and Xc peptides. Among the Xa, Xb and Xc peptides, the 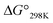 values of the Xc peptides were clearly higher than those of the corresponding Xa and Xb peptides. These results suggest that in the square form of Xc, the CH⋯π interaction was added to the dispersion force between the side chains of Xaa1 and Yaa5.
values of the Xc peptides were clearly higher than those of the corresponding Xa and Xb peptides. These results suggest that in the square form of Xc, the CH⋯π interaction was added to the dispersion force between the side chains of Xaa1 and Yaa5.
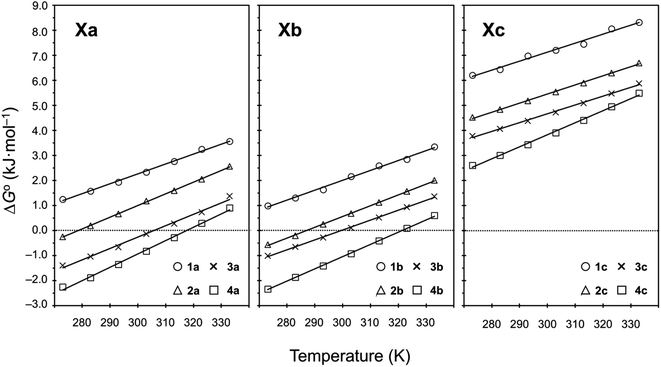 |
| | Fig. 8 Plots showing the temperature dependences of the free energies of the peptides. The data for Xa (X = 1–4), 1b and 1c were taken from a previous report.8 | |
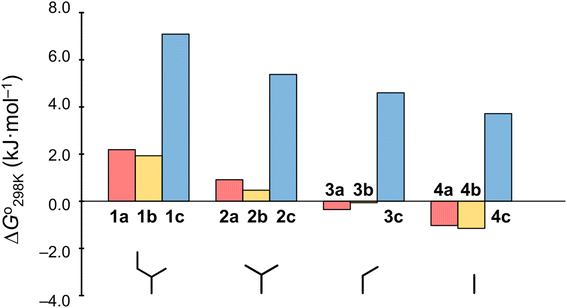 |
| | Fig. 9 Plots of the free energies of the peptides at 298 K. The 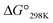 values for Xa, Xb and Xc are represented by red, yellow and blue bars, respectively. The data for Xa (X = 1–4), 1b and 1c were taken from a previous report.8 values for Xa, Xb and Xc are represented by red, yellow and blue bars, respectively. The data for Xa (X = 1–4), 1b and 1c were taken from a previous report.8 | |
Cytotoxicity
The cytotoxicities of all the peptides were assessed by determining the ED50 against HL-60 human myeloblastic leukemia cells, as described previously with some modification.31,32 These results are listed in Table 4 along with previously obtained data for 1a and 1c. From the earlier results, it was concluded that the square form exhibits cytotoxicity and that the cytotoxicity is lessened by folding.4–8 Among Xa peptides, 3a and 4a, which have negative 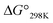 values, exhibited little or no cytotoxicity. Among Xb peptides, 1b, 3b and 4b exhibited stronger cytotoxicity than the corresponding Xa peptides. The
values, exhibited little or no cytotoxicity. Among Xb peptides, 1b, 3b and 4b exhibited stronger cytotoxicity than the corresponding Xa peptides. The 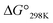 value of 1b was equal to that of 1a, but the cytotoxicity of 1b was 7 times that of 1a. Furthermore, despite having a negative
value of 1b was equal to that of 1a, but the cytotoxicity of 1b was 7 times that of 1a. Furthermore, despite having a negative 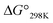 value, 3b and 4b exhibited about 2–3 times stronger cytotoxicity than the parent peptide 1a. Although the conformation and stability were very similar between the Xa and Xb peptides, there were significant differences in cytotoxicity. Xc peptides, which assume the most stable square forms, exhibited stronger cytotoxicity than the corresponding Xa peptides. However, the cytotoxicities of the Xb peptides were equal to or greater than those of the Xc peptides, except for 2b.
value, 3b and 4b exhibited about 2–3 times stronger cytotoxicity than the parent peptide 1a. Although the conformation and stability were very similar between the Xa and Xb peptides, there were significant differences in cytotoxicity. Xc peptides, which assume the most stable square forms, exhibited stronger cytotoxicity than the corresponding Xa peptides. However, the cytotoxicities of the Xb peptides were equal to or greater than those of the Xc peptides, except for 2b.
Table 4 Cytotoxicities of peptides toward HL-60 cell
| Xa peptides |
ED50 (μg mL−1) |
Xb peptides |
ED50 (μg mL−1) |
Xc peptides |
ED50 (μg mL−1) |
| These data are taken from a previous report.7 |
| 1aa |
32.9 ± 0.9 |
1b |
4.7 ± 0.03 |
1ca |
12.7 ± 0.33 |
| 2a |
21.1 ± 0.3 |
2b |
35.3 ± 2.1 |
2c |
4.6 ± 0.2 |
| 3a |
>100 |
3b |
10.7 ± 1.9 |
3c |
12.2 ± 1.5 |
| 4a |
54.9 ± 1.1 |
4b |
19.0 ± 0.3 |
4c |
16.5 ± 4.3 |
Conclusion
Our conformational studies carried out both in crystal and in solution, showed that incorporation of a Chg5 residue does not affect the conformational equilibrium but incorporation of Phg5 residue strongly promotes stabilization of the square form. It is considered that a CH⋯π interaction between the side chains of the Xaa1 and Phg5 residues is a key factor stabilizing the square form of Xc peptides. The thermodynamic parameters estimated through NMR-based quantification provided strong evidences for this suggestion. The highly positive ΔG° values in the conformational equilibrium of Xc peptides mean no spontaneous folding. The ΔH° values for the folding process of Xc peptides were estimated to be 5–8 kJ mol−1 higher than those for the corresponding Xa and Xb peptides. These enthalpic losses are relevant to the CH⋯π interaction energies, which must be disrupted during the folding of Xc peptides. Stabilization of the square form by the CH⋯π interaction also resulted in increased cytotoxicity.
Experimental
Peptide syntheses
The Thz unit (Boc-D-Val(Thz)-OMe) was synthesized as previously described.32,33 The linear peptide Boc-Xaa-allo-Thr-D-Val-Thz-Yaa-allo-Thr-D-Val-Thz-OMe was synthesized using 1-hydroxybenzotriazole (Watanabe Chemical lnd. Ltd, Hiroshima, Japan) and 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (Watanabe Chemical lnd. Ltd, Hiroshima, Japan) in a liquid phase. Macrocyclization was accomplished using benzotriazolyloxy-tris(pyrrolidino)phosphonium hexafluorophosphate (Watanabe Chemical lnd. Ltd, Hiroshima, Japan) in the presence of 4-dimethylaminopyridine (Nacalai Tesque, Kyoto, Japan). The Oxz rings were formed by reacting the Xaa-allo-Thr and Yaa-allo-Thr moieties with bis(2-methoxyethyl)aminosulfur trifluoride (Deoxo-Fluor) (Fujifilm Wako Pure Chemical Corp., Osaka, Japan).34 The synthesis and characterization of Xb (X = 2–4) and Xc (X = 2–4) are detailed in the ESI.†
X-ray diffraction
Peptides 2b and 2c were crystallized from N,N-dimethylacetamide (DMA). X-ray diffraction data for 2b was collected with a Rigaku CrysAlis Pro (Rigaku Corp., Tokyo, Japan). X-ray diffraction data for 2c was collected with a Bruker Smart APEXII (Bruker Corp., Massachusetts, USA). The structures of 2b and 2c were solved using SHELXS-97 (ref. 35) and refined using SHELXL-97.35 The crystal data for both have been deposited with the Cambridge Crystallographic Data Center under deposition numbers 2191404 and 2191405, respectively. The crystal data for the two peptides are given in ESI.†
Circular dichroism
CD spectra were obtained using a JASCO spectra-polarimeter (model J-820, JASCO, Tokyo, Japan) with a 1 cm quartz cell at room temperature. The spectra were scanned in the range of 200–280 nm at a speed of 5 nm min−1 with a 0.1 nm interval for uptake to a computer. Data were averaged over each 1 nm and plotted. The spectra were measured in acetonitrile (CH3CN) solution while changing the 2,2,2-trifluoroethanol (TFE) concentration (10%, 20%, 30%, 40% and 50%). Peptide concentrations were about 0.04 mM.
1H NMR
1H NMR spectra were recorded on an Agilent DD2 600 MHz NMR spectrometer (Agilent Technologies, California, USA). Peptide concentrations were about 5.0 mM in CH3CN-d3. Chemical shifts were measured relative to internal tetramethylsilane at 0.00 ppm. The protons were assigned using two-dimensional correlated spectroscopy (2D-COSY) and rotating-frame Overhauser effect spectroscopy (ROESY; mixing time = 500 ms). VT-1H NMR measurements were made every 10 K from 273 K to 333 K. The assignment lists and 1D, 2D-COSY and ROESY spectra for Xb (X = 2–4) and Xc (X = 2–4) are given in ESI.†
NMR-based quantitative studies
The conformational equilibrium constants (K) (Fig. 2) of the peptides were determined as previously described.8 The behaviors of the chemical shifts of conformationally sensitive Thz protons are the focus of this method. When the Thz rings face each other in the folded form, the chemical shifts for the Thz4 and Thz8 protons appear in a higher magnetic field than those in the square form due to the ring-current effects of the Thz8 and Thz4 rings, respectively. The K values of the peptides were estimated using eqn (1),| | |
K = (δS − δobs)/(δobs − δF)
| (1) |
where δobs is the chemical shift of the Thz protons in the equilibrating peptide, δS (=8.09 ppm) is the chemical shift of the Thz protons in T3ASC,36 and δF (=7.35 ppm) is the chemical shift of the Thz protons in dASC.37 Two peptides, T3ASC and dASC, were used as reference peptides to provide reference chemical shifts for the fully square and folded forms, respectively. The chemical structures of T3ASC and dASC are given in ESI.†
The enthalpy (ΔH°) and entropy (ΔS°) were determined through a linear van't Hoff plot (ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) K versus 1/T), after which the Gibbs free energy (ΔG°) at 298 K was calculated using eqn (2).
K versus 1/T), after which the Gibbs free energy (ΔG°) at 298 K was calculated using eqn (2).
| | |
ΔG° = ΔH° − TΔS° = −RT lnK
| (2) |
Assay for cytotoxicity
The cytotoxicity of the peptides was assessed using the 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) method as described previously with some modifications.38,39 HL-60 human myeloblastic leukemia cells were cultured in RPMI 1640 medium (10% fetal calf serum) at 37 °C under 5% CO2. The test materials were dissolved in dimethyl sulfoxide to a concentration of 10 mM, after which that stock solution was diluted with essential medium to concentrations of 200, 20 and 2 μM. Each solution was combined with cells suspended (1 × 105 cells per mL) in the same medium. After incubating at 37 °C for 72 h under 5% CO2, the cells were labeled with 5 mg mL−1 MTT in phosphate-buffered saline (PBS), and the absorbance of formazan dissolved in 20% sodium dodecyl sulfate in 0.1 N HCl was measured at 540 nm with a microplate reader (MTP-310, Corona electric Co. Ltd, Hitachinaka, Japan). The absorbance values are expressed as percentages relative to that of a control cell suspension prepared using the same procedure described above but without a test substance. All assays were performed three times, semilogarithmic plots were constructed from the averaged data, and the effective dose of a substance required to inhibit cell growth by 50% (ED50) was determined.
Conflicts of interest
There are no conflicts to declare.
References
- Y. Hamamoto, M. Endo, M. Nakagawa, T. Nakanishi and K. Mizukawa, Chem. Commun., 1983, 6, 323–324 RSC.
- T. Ishida, M. Inoue, Y. Hamada, S. Kato and T. Shioiri, Chem. Commun., 1987, 5, 370–371 RSC.
- T. Ishida, M. Tanaka, M. Nabae, M. Inoue, S. Kato, Y. Hamada and T. Shioiri, J. Org. Chem., 1988, 53, 107–112 CrossRef CAS.
- M. Doi, F. Shinozaki, Y. In, T. Ishida, D. Yamamoto, M. Kamigauchi, M. Sugiura, Y. Hamad, K. Kohda and T. Shioiri, Biopolymers, 1999, 49, 459–469 CrossRef CAS PubMed.
- A. Asano, K. Minoura, T. Yamada, A. Numata, T. Ishida, Y. Katsuya, Y. Mezaki, M. Sasaki, T. Taniguchi, M. Nakai, H. Hasegawa, A. Terashima and M. Doi, J. Pept. Res., 2002, 60, 10–22 CrossRef CAS PubMed.
- A. Asano, T. Yamada, A. Numata and M. Doi, Acta Crystallogr., Sect. A: Found. Crystallogr., 2003, 59, o488–o490 Search PubMed.
- A. Asano, T. Yamada and M. Doi, Bioorg. Med. Chem., 2011, 19, 3372–3377 CrossRef CAS PubMed.
- A. Asano, K. Minoura, Y. Kojima, T. Yoshii, R. Ito, T. Yamada, T. Kato and M. Doi, RSC Adv., 2020, 10, 33317–33326 RSC.
- A. Asano, M. Nakagawa, C. Miyajima, M. Yasui, K. Minoura, T. Yamada and M. Doi, J. Pept. Sci., 2021, 27, e3363 CrossRef CAS PubMed.
- C. Cocito, Microbiol. Rev., 1979, 43, 145–192 CrossRef CAS PubMed.
- J. C. Barriere, D. H. Bouanchaud, J. F. Desnottes and J. M. Paris, Expert Opin. Invest. Drugs, 1994, 3, 115–131 CrossRef CAS.
- D. Vazquez, J. Gen. Microbiol., 1966, 42, 93–106 CrossRef CAS PubMed.
- Y. Mast and W. Wohlleben, Int. J. Med. Microbiol., 2014, 304, 44–50 CrossRef CAS PubMed.
- T. Teshima, M. Nishikawa, I. Kubota, T. Shiba, Y. Iwai and S. Omura, Tetrahedron Lett., 1988, 29, 1963–1966 CrossRef CAS.
- L. Brandi, S. Maffioli, S. Donadio, F. Quaglia, M. Sette, P. Milon, C. O. Gualerzi and A. Fabbretti, FEBS Lett., 2012, 586, 3373–3378 CrossRef CAS PubMed.
- M. Nishio, M. Hirota and Y. Umezawa, The CH⋯π Interaction: Evidence, Nature and Consequences, Wiley-VCH, New York, 1998 Search PubMed.
- E. A. Meyer, R. K. Castellano and F. Diederich, Angew. Chem., Int. Ed., 2003, 42, 1210–1250 CrossRef CAS PubMed.
- M. L. Waters, Biopolymers, 2004, 76, 435–445 CrossRef CAS PubMed.
- C. D. Tatko and M. L. Waters, J. Am. Chem. Soc., 2004, 126, 2028–2034 CrossRef CAS PubMed.
- M. Harigai, M. Kataoka and Y. Imamoto, J. Am. Chem. Soc., 2006, 128, 10646–10647 CrossRef CAS PubMed.
- N. P. Brawell and A. P. Davis, J. Org. Chem., 2011, 76, 6548–6557 CrossRef PubMed.
- C. J. Pace, D. Kim and J. Gao, Chem. –Eur. J., 2012, 18, 5832–5836 CrossRef CAS PubMed.
- N. K. Vyas, M. N. Vyas and F. A. Quiocho, Science, 1988, 242, 1290–1295 CrossRef CAS PubMed.
- S. Elgavish and B. Shaana, J. Mol. Biol., 1998, 277, 917–932 CrossRef CAS PubMed.
- Y. Umezawa, S. Tsuboyama, K. Honda, J. Uzawa and M. Nishio, Bull. Chem. Soc. Jpn., 1998, 71, 1207–1213 CrossRef CAS.
- Y. Umezawa, S. Tsuboyama, H. Takahashi, J. Uzawa and M. Nishio, Bioorg. Med. Chem., 1999, 7, 2021–2026 CrossRef CAS PubMed.
- H. Suezawa, T. Yoshida, M. Hirota, H. Takahashi, Y. Umezawa, K. Honda, S. Tsuboyama and M. Nishio, J. Chem. Soc., Perkin Trans. 1, 2001, 2, 2053–2058 RSC.
- L. Pauling, The Nature of the Chemical Bond, Cornell University Press, Ithaca, New York, 1960, p. 260 Search PubMed.
- A. Bondi, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS.
- K. Kobayashi, Y. Asakawa, Y. Kikuchi, H. Toi and Y. Aoyama, J. Am. Chem. Soc., 1993, 115, 2648–2654 CrossRef CAS.
- I. Maeda, Y. Shimohigashi, I. Nakamura, H. Sakamoto, K. Kawano and M. Ohno, Biochem. Biophys. Res. Commun., 1993, 193, 428–433 CrossRef CAS PubMed.
- Y. Hamada, S. Kato and T. Shioiri, Tetrahedron Lett., 1985, 26, 3223–3226 CrossRef CAS.
- Y. Hamada, M. Shibata, T. Sugiura, S. Kato and T. Shioiri, J. Org. Chem., 1987, 52, 1252–1255 CrossRef CAS.
- A. J. Phillips, Y. Uto, P. Wipf, M. J. Reno and D. R. Williams, Org. Lett., 2000, 2, 1165–1168 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- A. Asano, T. Yamada, T. Taniguchi, N. Sasaki, K. Yoza and M. Dio, J. Pept. Sci., 2018, e3120 CrossRef PubMed.
- A. Asano, M. Doi, K. Kobayashi, M. Arimoto, T. Ishida, Y. Katsuya, Y. Mezaki, H. Hasegawa, M. Nakai, M. Sasaki, T. Taniguchi and A. Terashima, Biopolymers, 2001, 58, 295–304 CrossRef CAS PubMed.
- K. Kohda, Y. Ohta, Y. Yokoyama, T. Kato, Y. Suzumura, Y. Hamada and T. Shioiri, Biochem. Pharmacol., 1989, 38, 4497–4500 CrossRef CAS PubMed.
- K. Kohda, Y. Ohta, Y. Kawazoe, T. Kato, Y. Suzumura, Y. Hamada and T. Shioiri, Biochem. Pharmacol., 1989, 38, 4500–4502 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *,
Katsuhiko Minoura
*,
Katsuhiko Minoura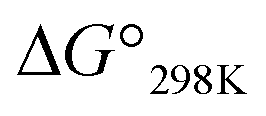 value for a peptide bearing an n-propyl group was nearly zero, while those for peptides bearing a substituent smaller than an n-propyl group (3a and 4a) were negative. On the other hand, the values for peptides bearing a bulkier substituent than an n-propyl group (1a, 2a, 1b and 1c) were positive, which means that these peptides do not spontaneously fold at 298 K. This suggests that when 1a is in the square form, the bulky side chain of the substituted amino acid residue and the sec-butyl group of the Ile residue at the diagonal position are close to each other, and the dispersion force between these two functional groups prevents spontaneous folding. In particular, a dispersion force like a CH⋯π interaction may be acting in peptides bearing a phenyl group (1c). While it is apparent that the van der Waals space occupied by a cyclohexyl group is larger than that of a planar phenyl group, the
value for a peptide bearing an n-propyl group was nearly zero, while those for peptides bearing a substituent smaller than an n-propyl group (3a and 4a) were negative. On the other hand, the values for peptides bearing a bulkier substituent than an n-propyl group (1a, 2a, 1b and 1c) were positive, which means that these peptides do not spontaneously fold at 298 K. This suggests that when 1a is in the square form, the bulky side chain of the substituted amino acid residue and the sec-butyl group of the Ile residue at the diagonal position are close to each other, and the dispersion force between these two functional groups prevents spontaneous folding. In particular, a dispersion force like a CH⋯π interaction may be acting in peptides bearing a phenyl group (1c). While it is apparent that the van der Waals space occupied by a cyclohexyl group is larger than that of a planar phenyl group, the 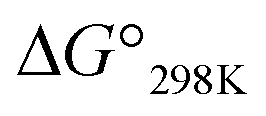 value of 1c is three times that of 1b.
value of 1c is three times that of 1b.
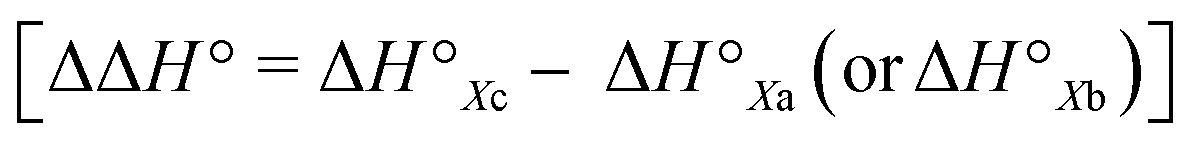 by an amount related to the energy of the CH⋯π interaction.
by an amount related to the energy of the CH⋯π interaction.
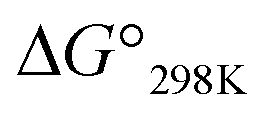
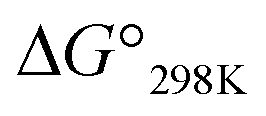 value for Xa peptides increases with increases in the bulkiness of the substituent of the Xaa1 residue [methyl (X = 4) < ethyl (X = 3) < isopropyl (X = 2) < sec-butyl (X = 1)]. The same relationship between the bulkiness of the Xaa1 side chain and the
value for Xa peptides increases with increases in the bulkiness of the substituent of the Xaa1 residue [methyl (X = 4) < ethyl (X = 3) < isopropyl (X = 2) < sec-butyl (X = 1)]. The same relationship between the bulkiness of the Xaa1 side chain and the 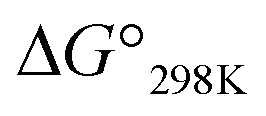 value was also seen with the Xb and Xc peptides. Among the Xa, Xb and Xc peptides, the
value was also seen with the Xb and Xc peptides. Among the Xa, Xb and Xc peptides, the 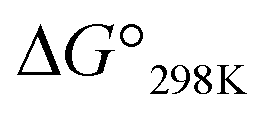 values of the Xc peptides were clearly higher than those of the corresponding Xa and Xb peptides. These results suggest that in the square form of Xc, the CH⋯π interaction was added to the dispersion force between the side chains of Xaa1 and Yaa5.
values of the Xc peptides were clearly higher than those of the corresponding Xa and Xb peptides. These results suggest that in the square form of Xc, the CH⋯π interaction was added to the dispersion force between the side chains of Xaa1 and Yaa5.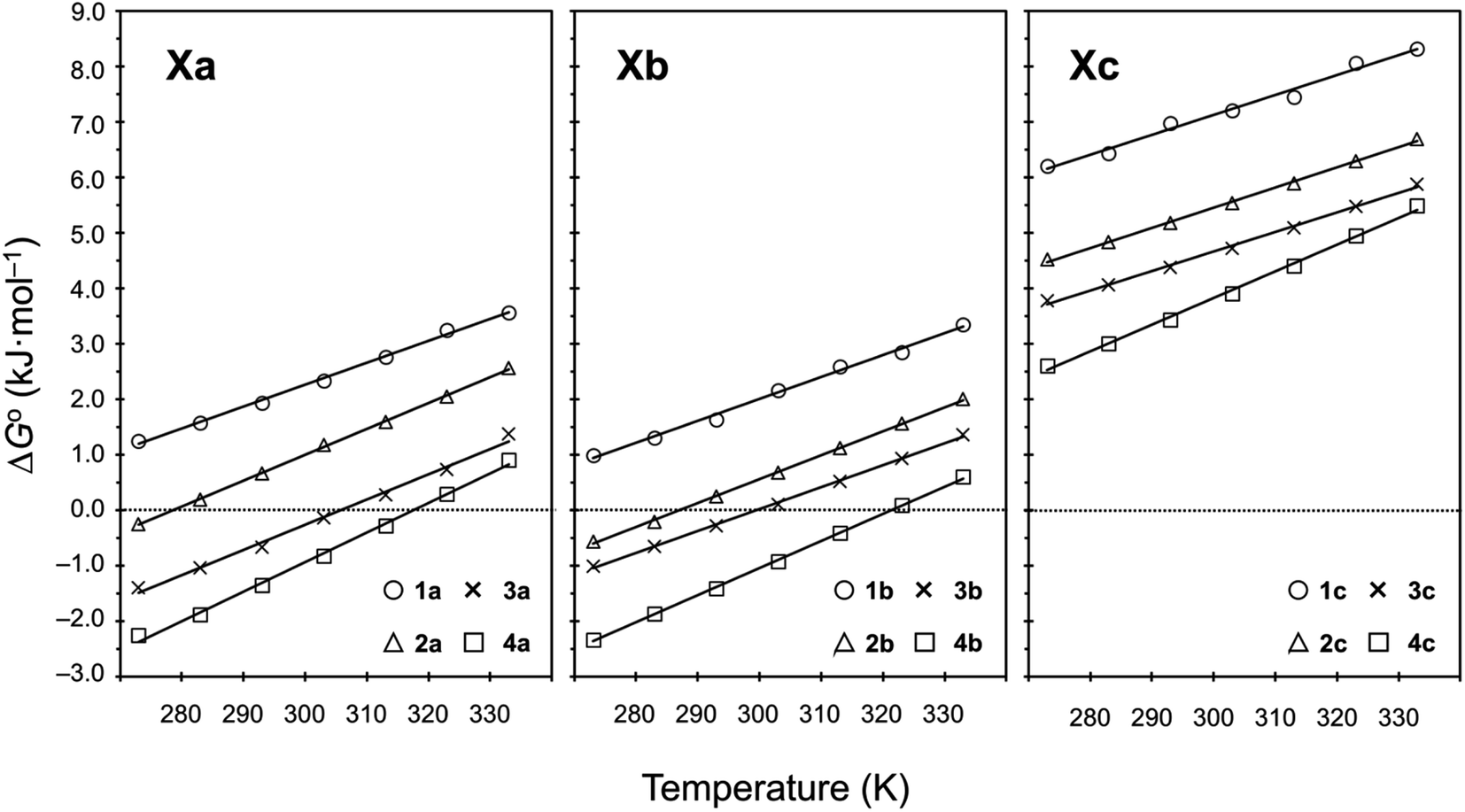
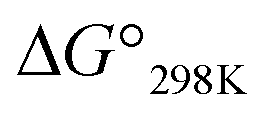 values for Xa, Xb and Xc are represented by red, yellow and blue bars, respectively. The data for Xa (X = 1–4), 1b and 1c were taken from a previous report.8
values for Xa, Xb and Xc are represented by red, yellow and blue bars, respectively. The data for Xa (X = 1–4), 1b and 1c were taken from a previous report.8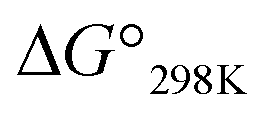 values, exhibited little or no cytotoxicity. Among Xb peptides, 1b, 3b and 4b exhibited stronger cytotoxicity than the corresponding Xa peptides. The
values, exhibited little or no cytotoxicity. Among Xb peptides, 1b, 3b and 4b exhibited stronger cytotoxicity than the corresponding Xa peptides. The 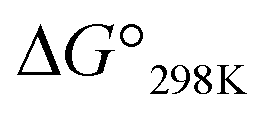 value of 1b was equal to that of 1a, but the cytotoxicity of 1b was 7 times that of 1a. Furthermore, despite having a negative
value of 1b was equal to that of 1a, but the cytotoxicity of 1b was 7 times that of 1a. Furthermore, despite having a negative 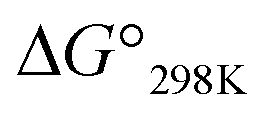 value, 3b and 4b exhibited about 2–3 times stronger cytotoxicity than the parent peptide 1a. Although the conformation and stability were very similar between the Xa and Xb peptides, there were significant differences in cytotoxicity. Xc peptides, which assume the most stable square forms, exhibited stronger cytotoxicity than the corresponding Xa peptides. However, the cytotoxicities of the Xb peptides were equal to or greater than those of the Xc peptides, except for 2b.
value, 3b and 4b exhibited about 2–3 times stronger cytotoxicity than the parent peptide 1a. Although the conformation and stability were very similar between the Xa and Xb peptides, there were significant differences in cytotoxicity. Xc peptides, which assume the most stable square forms, exhibited stronger cytotoxicity than the corresponding Xa peptides. However, the cytotoxicities of the Xb peptides were equal to or greater than those of the Xc peptides, except for 2b.