
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ultrasound-assisted bromination of indazoles at the C3 position with dibromohydantoin†
Shengneng Ying‡
,
Xingru Liu‡,
Tao Guo,
Xuan Li,
Min Zhou,
Xia Wang*,
Mengxue Zhu,
Hongmei Jiang and
Qing-Wen Gui *
*
College of Chemistry and Materials Science, Hunan Agricultural University, Changsha 410082, Hunan, P. R. China. E-mail: gqw1216@hunau.edu.cn; wangxia@hunau.edu.cn
First published on 22nd December 2022
Abstract
Bromoaryl compounds have attracted great attention in organic chemistry, especially for the synthesis of pharmaceutical intermediates. Herein, we demonstrated a novel and efficient bromination protocol of indazoles via C–H bond cleavage to give site-specific 3-bromide products that could be further employed as synthetic blocks to prepare drugs. The reaction used DBDMH as a bromine source, tolerated a wide range of indazoles, and finished in 30 min under mild, ultrasound-assisted conditions. Besides, preliminary mechanistic studies revealed that this approach was not a radical process.
Introduction
Heterocyclics frequently constitute the core moiety of various pharmaceuticals.1 Among them, indazoles, possessing a wide range of biological activities such as antitumor, antimicrobial, and anti-inflammatory, are a critical class of N-heterocyclic molecules.2 For example, MK-4827 (ref. 3) as a PARP1 and PARP2 inhibitor has a 1H-indazole skeleton, and Indisetron4 and Granisetron,5 as antiemetics that can be used to treat nausea and emesis following chemotherapy, have an indazole construction (Fig. 1a). However, the limited methods for the functionalization of indazoles cannot meet the demands for structural diversity.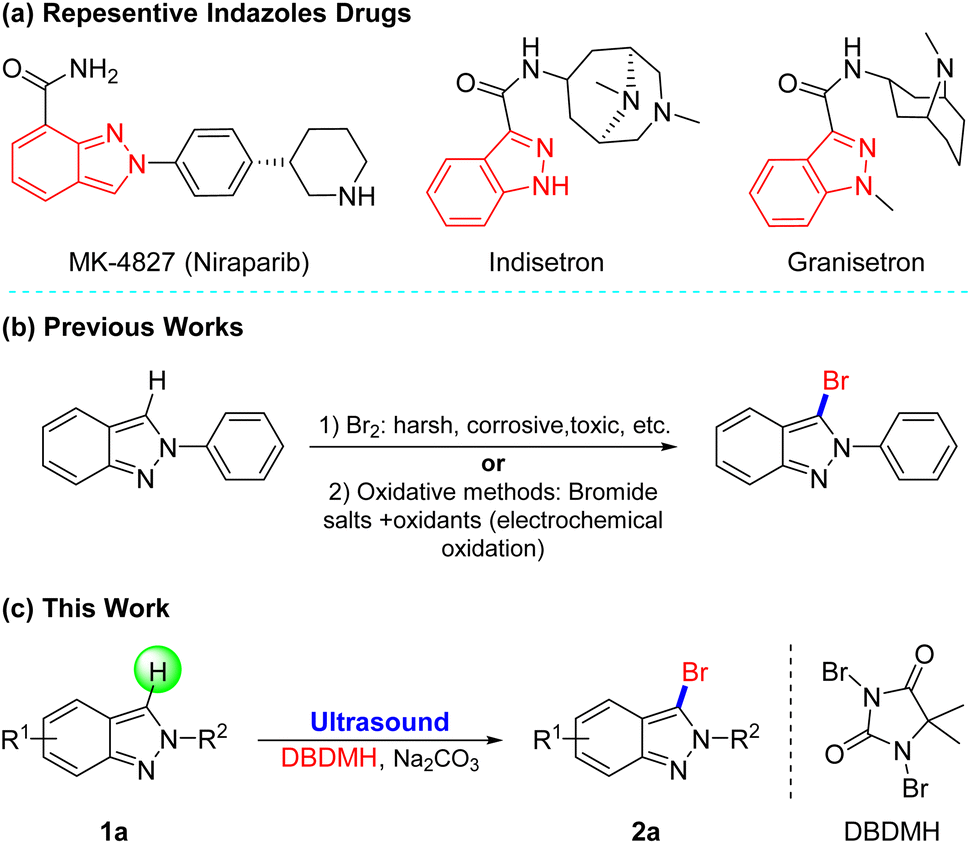 | ||
| Fig. 1 (a) Representative drugs containing indazoles. Previous works (b) and this work (c) for bromination of indazole derivatives. | ||
Due to the general application of halogenated indazoles as building blocks of the synthesis of drugs, dyes, and functional materials,6 it's significant to develop novel and efficient halogenation approaches, especially bromination which is flexible for post-group-transformation,7 Conventionally, 3-halogenated-2H-indazoles are prepared from 2H-indazoles with Br2 in acetic acid. However, these halogenated approaches often need high temperature (ca 120 °C) for activating reaction, plus Br2 is toxic to humans and difficult to operate owing to its high volatility, and byproducts are unavoidable (Fig. 1b).8 To avoid the use of the liquid bromine, a variety of oxidative protocols have then been disclosed through in situ oxidation of Br− to Br+.9 For instance, the group of Li and Shen disclosed the electrochemical oxidative halogenation of 2H-indazoles under mild conditions, recently.9a Despite the fruitful stoichiometric methods, the more attractive and novel halogenation technology is still in high demand.
Ultrasonic waves have gradually developed as a potent tool in organic synthesis, owing to their merits such as short reaction time, mild reaction conditions, good selectivity, and highly efficient.10 It can accelerate the thermal motion of molecules, further speeding up the mass and heat transfer between chemicals. Dating back to 1950, Renaud first published a paper about using ultrasound to prepare organometallic reagents.11 In 1998, Luche and Bianchi concretely described the application and potential of ultrasound to organic synthesis in “Synthetic Organic Sonochemistry”,12 and then ultrasound was gradually accepted and developed. Presently, ultrasound is a helpful technique for activating and accelerating chemical processes.13
1,3-Dibromo-5,5-dimethyl hydantoin (DBDMH) belongs to the group of cost-effective N-halamine disinfectants, which is becoming increasingly popular due to its long-term stability in dry storage or in a wide pH range of aqueous solutions, it's safety for humans and the environment, and their ability to rapidly kill microorganisms.14 Meanwhile, relative to other bromine sources, DBDMH is less corrosive, more stable and cheaper.15 Furthermore, DBDMH is among the more established, commercially available bromine carry that is gradually attracting attention for being safe, stable, easily-handled solids that can be utilized under mild conditions for highly selective organic transformations.16 Thus, in the context of our group's continued interest in improving and developing environmentally acceptable synthetic methods,17 we now describe the first practical and simple design for selective 3-bromination of 2H-indazoles with DBDMH in a green solvent accelerated by ultrasonic irradiation, which produced 3-brominated products in very high yields within a short time (Fig. 1c).
Results and discussion
Inspired by seminal works in bromination we started our investigation by designing bromination of 1H-indazole with an appropriate brominated reagent that would represent an efficient and mild strategy that could overcome many limitations of current methods. As shown in Table 1, we first screened different solvents such as DMF, MeCN, THF, DCM, EA, and EtOH with Na2CO3 as base and to afford 2a at 80 °C for 12 h (entry 1–6), while EtOH was considered to be the competent solvent among them. To decrease reaction time and temperature, ultrasound was introduced to replace conditional stir,18 excitedly, which greatly increased yields and transformation efficiency (entry 7–9). Bases were also included in the scope of optimization, like Et3N (entry 10), NaOAc (entry 11) and K2CO3(entry 12), all of which can promote the reaction process, and K2CO3 obtained similar results to Na2CO3. Therefore, the optimized experimental conditions were determined as follows: Na2CO3 as base, EtOH as solvent, and ultrasound for 0.5 h at 40 °C. The ultrasound-assisted reaction was screened with various bromine sources, such as NBS, NaBr and HBr (entries 13–15). There bromine sources did not provide any satisfactory result; rather, a very sluggish reaction rate or no reaction was observed in each case.
|
|||||
|---|---|---|---|---|---|
| Entry | Base | Solvent | Temp.(°C) | Time (h) | Yieldb (%) |
| a Reaction conditions: 1a (0.2 mmol), DBDMH (0.2 mmol), base (0.4 mmol).b Isolated yield.c Ultrasound instead of stirring.d NaBr instead of DBDMH.e NBSr instead of DBDMH.f HBr instead of DBDMH. | |||||
| 1 | Na2CO3 | DMF | 80 | 12 | 76 |
| 2 | Na2CO3 | CH3CN | 80 | 12 | 72 |
| 3 | Na2CO3 | THF | 80 | 12 | 88 |
| 4 | Na2CO3 | DCM | 80 | 12 | 68 |
| 5 | Na2CO3 | EtOAc | 80 | 12 | 80 |
| 6 | Na2CO3 | Toluene | 40 | 12 | 75 |
| 7c | Na2CO3 | EtoH | 40 | 0.5 | 92 |
| 8c | Na2CO3 | EtoH | 40 | 0.15 | 88 |
| 9c | Na2CO3 | EtoH | 30 | 0.5 | 87 |
| 10c | Et3N | EtoH | 40 | 0.5 | 23 |
| 11c | NaOAc | EtoH | 40 | 0.5 | 29 |
| 12c | K2CO3 | EtoH | 40 | 0.5 | 91 |
| 13d | Na2CO3 | EtoH | 30 | 0.5 | Trace |
| 14e | Na2CO3 | EtoH | 30 | 0.5 | 35 |
| 15f | Na2CO3 | EtoH | 30 | 0.5 | NR |

With the optimal conditions in hand, the generality of our newly developed bromination protocol was next investigated. Firstly, a series of electron-deficient 2H-indazoles were examined, including halides such as fluorine (Scheme 1, 2b, 2d), bromine (2c) and chlorine (2e), all of which proceeded smoothly to obtain target products in mild to favorable yield under standard conditions. For electron-rich groups, 2f was prepared with 81% yield that was not affected by –OMe. Notably, the bromination of 2g afforded brominated 2H-indazoles, even which included a strong electron-withdrawing group CF3.
 | ||
| Scheme 1 The Bromination of 2H-indazoles. aReaction conditions: 1 (0.2 mmol), DBDMH (0.2 mmol), Na2CO3 (0.4 mmol), 40 °C, EtOH (2.0 mL), ultrasonic (40 kHz/50 W) 30 min. | ||
Encouraged by the exciting results, we then evaluated the activities of 2-substituted indazole derivatives that reacted with DBDMH in standard conditions. As shown in Scheme 2, the bromination, of 2H-indazoles substituted by electron-donating groups such as –Me and –OMe, succeeded to afford the 3-brominated 2h–2l in mild to favorable yield, which also reported that the substitution position of groups had less effect on bromination reactivity. Subsequently, the electron-withdrawing group OCF3 was found to proceed smoothly in this transformation (2l), and acquired 73% yield. Then, we estimated the bromination of halogen-substituted 2H-indazoles derivatives. Under the standard conditions, fluorine, chlorine, bromine, and iodine replaced the hydrogen of the benzene, which brominated successfully to obtain the corresponding products with favorable yields (2n–2s). It is worth noting that the bromination of ester group substituted derivatives performed favorably to access brominated products 2t–2u with good yields and excellent selectivity. With regard to strong electron withdrawing group CF3, 2v was prepared with a slightly lower yield compared without substituents. Surprisingly, when we used cyclohexyl substituted benzene coupled with 2H-indazole, a good bromination effect was achieved (2w). Gratifyingly, methylated 1H-indazole was also carried out smoothly to generate an objective product (2x), and the transformation was unaffected by halogen iodine. Moreover, 1H-indazole substituted by benzyl also presented excellent activity to acquire 2y (93%). In addition, we tried to employ DCDMH to replace DBDMH in this reaction, as expected, chlorination could proceed smoothly under the traditional conditions (2z). All of these experiment results illustrated that this method possessed broad applicability and group tolerance.
 | ||
| Scheme 2 The Halogenation of Indazoles. aReaction conditions: 1 (0.2 mmol), DBDMH (0.2 mmol), Na2CO3 (0.4 mmol), 40 °C, EtOH (2.0 mL), ultrasonic 30 min bDCDMH (0.2 mmol). | ||
Based on the experiment results, we next investigated the mechanistic information of this transformation with some control experiments in Scheme 3. These results depicted that 2-phenyl-2H-indazole (1a) could be brominated in the presence of radical scavengers such as 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO) and 2,6-di-tert-butyl-4-methyl phenol (BHT), and gave 86% and 65% yield, respectively (Scheme 3a and b). In order to show the cavitation effect of ultrasonic irradiation, the employment of other energies (40 kHz/40 W or 40 kHz/60 W) produced 2a in slightly lower yields (Scheme 3c). In view of the primary mechanism data and previous reports, we speculate this method is not a radical process, and the proposed reaction pathway is presented in Scheme 3d. Firstly, the cleaving of DBDMH by ultrasonic irradiation generated a bromo ion and I,19 which added to 1a to form intermediate II. Finally, product 2a was obtained from II, followed by the abstraction of a hydrogen atom by base.
 | ||
| Scheme 3 Control Experiments and Mechanism study. | ||
To demonstrate the practicality of this approach, we conducted the gram-scale iodination of 2H-indazole under the optimized conditions, which gave the desired product 3-bromo-2H-indazole (2c) in a 70% yield (Scheme 4a). Considering the good selectivity of this method and the importance of 2H-indazoles in pharmaceuticals, we take advantage of products for further transformation to extend the application scope. It is noteworthy to mention that indazole can provide side products due to competitive reactive sites, but generated product 3a without having any impact on yield (75%). As reported that Br is an excellent group for coupling reaction, we chose 2c and aryl boronic acid 3 as substrates for cross-coupling reaction, expectedly, which was conducted successfully under Pd catalysis (Scheme 4c, 4, 80% isolated yield). The application extension results suggest the broad applicability potentiality of our method.
 | ||
| Scheme 4 Further Application. | ||
Conclusions
In conclusion, we have successfully developed an ultrasound-assisted, efficient, and rapid bromination approach via DBDMH as a bromine source for the synthesis of 3-Br-indazoles. This reaction underwent an ultrasound-assisted C–H bond cleavage and C–Br bond formation process, representing one of the few C–Br bond construction reactions under ultrasound waves. Furthermore, a wide range of functionalized indazoles is compatible with mild reaction conditions. The mechanism investigation revealed that the reaction is not a complete radical process. More importantly, the brominated products can be applied for further application, providing a potential strategy for the synthesis of pharmaceutical intermediates. Further investigation of related halogenation reactions is carried on studying in our group.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Science and Technology Innovation Program of Hunan Province (2021RC2079), China Postdoctoral Science Foundation (2022M720541 and 2022T150075), and Foundation of National Entrepreneurship Training Program (202110537005X, 202210537024).References
- E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS PubMed.
- (a) X. Wang, Q.-X. Yang, C.-Y. Long, Y. Tan, Y.-X. Qu, M.-H. Su, S.-J. Huang, W. Tan and X.-Q. Wang, Org. Lett., 2019, 21, 5111–5115 CrossRef CAS PubMed; (b) Y. Q. Xia Wang, C. Long and X.-Q. Wang, Chi. J. Org. Chem., 2021, 41, 795–805 CrossRef; (c) J. W. Beatty, E. A. Lindsey, R. Thomas-Tran, L. Debien, D. Mandal, J. L. Jeffrey, A. T. Tran, J. Fournier, S. D. Jacob, X. Yan, S. L. Drew, E. Ginn, A. Chen, A. T. Pham, S. Zhao, L. Jin, S. W. Young, N. P. Walker, M. R. Leleti, S. Moschütz, N. Sträter, J. P. Powers and K. V. Lawson, J. Med. Chem., 2020, 63, 3935–3955 CrossRef CAS PubMed.
- (a) P. Jones, S. Altamura, J. Boueres, F. Ferrigno, M. Fonsi, C. Giomini, S. Lamartina, E. Monteagudo, J. M. Ontoria, M. V. Orsale, M. C. Palumbi, S. Pesci, G. Roscilli, R. Scarpelli, C. Schultz-Fademrecht, C. Toniatti and M. Rowley, J. Med. Chem., 2009, 52, 7170–7185 CrossRef CAS PubMed; (b) K. A. Bridges, C. Toniatti, C. A. Buser, H. Liu, T. A. Buchholz and R. E. Meyn, Oncotarget, 2014, 5, 5076–5086 CrossRef PubMed.
- (a) E. Iritani, K. Isono, T. Izumo, N. Takeda, T. Kanemura, J. Tamaoki and A. Nagai, Gan to Kagaku Ryoho, 2009, 36, 1489–1492 Search PubMed; (b) K. Ushijima, N. Wake, H. Kobayashi, T. Hachisuga, N. Toki, H. Masuzaki, K. Kotera, T. Kawarabayashi, M. Emoto and T. Kamura, Gan to Kagaku Ryoho, 2008, 35, 1169–1173 Search PubMed.
- (a) G. J. Sanger and D. R. Nelson, Eur. J. Pharmacol., 1989, 159, 113–124 CrossRef CAS PubMed; (b) N. Maleki-Dizaji, T. Eteraf-Oskouei, A. Fakhrjou, S. H. Maljaie and A. Garjani, Int. Immunopharmacol., 2010, 10, 1010–1016 CrossRef CAS PubMed.
- (a) S. H. Henderson, R. A. West, S. E. Ward and M. A. Honey, R. Soc. Open Sci., 2018, 5, 180333 CrossRef PubMed; (b) Z. Yang, J.-T. Yu and C. Pan, Org. Biomol. Chem., 2022, 20, 7746 RSC; (c) S.-G. Zhang, C.-G. Liang and W.-H. Zhang, Molecules, 2017, 23, 2783 CrossRef PubMed.
- F. Sabuzi, G. Pomarico, B. Floris, F. Valentini, P. Galloni and V. Conte, Coord. Chem. Rev., 2019, 385, 100–136 CrossRef CAS.
- (a) M. De Angelis, F. Stossi, K. A. Carlson, B. S. Katzenellenbogen and J. A. Katzenellenbogen, J. Med. Chem., 2005, 48, 1132–1144 CrossRef CAS PubMed; (b) E. Akguen, M. B. Glinski, K. L. Dhawan and T. Durst, J. Org. Chem., 1981, 46, 2730–2734 CrossRef CAS; (c) K. Xu, Y. Zheng, Y. Ye, D. Liu and W. Zhang, Org. Lett., 2020, 22, 8836–8841 CrossRef CAS PubMed; (d) D. Dagoneau, Q. Wang and J. Zhu, Chem.–Eur. J., 2020, 26, 4866–4873 CrossRef CAS PubMed.
- (a) X. Liu, Z. Wu, C. Feng, W. Liu, M. Li and Z. Shen, Eur. J. Org. Chem., 2022, 2022, DOI:10.1002/ejoc.202200262; (b) G. Zhao, E. Wang and R. Tong, ACS Sustainable Chem. Eng., 2021, 9, 6118–6125 CrossRef CAS; (c) S. Song, X. Li, X. Sun, Y. Yuan and N. Jiao, Green Chem., 2015, 17, 3285–3289 RSC; (d) G.-W. Wang and J. Gao, Green Chem., 2012, 14, 1125–1131 RSC.
- (a) S. Puri, B. Kaur, A. Parmar and H. Kumar, Curr. Org. Chem., 2013, 17, 1790–1828 CrossRef CAS; (b) G. Kaur, A. Sharma and B. Banerjee, ChemistrySelect, 2018, 3, 5283–5295 CrossRef CAS; (c) W. Liju, K. Ablajan and F. Jun, Ultrason. Sonochem., 2015, 22, 113–118 CrossRef PubMed; (d) S. J. T. Rezaei, Y. Bide and M. R. Nabid, Tetrahedron Lett., 2012, 53, 5123–5126 CrossRef CAS.
- P. Renaud, Bull. Soc. Chim. Fr., 1950, 17, 1044–1045 Search PubMed.
- P. Cintas and J.-L. Luche, Synthetic Organic Sonochemistry. Organometallic Sonochemistry, Springer Science&Business, New York, 1998, pp. 167–234 Search PubMed.
- (a) N. Kaur, Synth. Commun., 2018, 48, 1235–1258 CrossRef CAS; (b) N. Azizi, A. Rahimzadeh-Oskooee, Z. Yadollahy and A. G. Ourimi, Monatsh. Chem., 2014, 145, 1675–1680 CrossRef CAS; (c) A. Duarte, W. Cunico, C. M. P. Pereira, A. F. C. Flores, R. A. Freitag and G. M. Siqueira, Ultrason. Sonochem., 2010, 17, 281–283 CrossRef CAS PubMed; (d) T. Kiranmye, M. Vadivelu, S. Sampath, K. Muthu and K. Karthikeyan, Sustainable Chem. Pharm., 2021, 19, 100358 CrossRef CAS; (e) S. K. Maury, D. Kumar, A. Kamal, H. K. Singh, S. Kumari and S. Singh, Mol. Diversity, 2021, 25, 131–142 CrossRef CAS PubMed.
- A. Dong, Y.-J. Wang, Y. Gao, T. Gao and G. Gao, Chem. Rev., 2017, 117, 4806–4862 CrossRef CAS PubMed.
- F. Hui and C. Debiemme-Chouvy, Biomacromolecules, 2013, 14, 585–601 CrossRef CAS PubMed.
- (a) S. I. aikia, A. J. Borah and P. Phukan, Chem. Rev., 2016, 116, 6837–7042 CrossRef PubMed; (b) H. Veisi, R. Ghorbani-Vaghei and M. A. Zolfigol, Org. Prep. Proced. Int., 2011, 43, 489–540 CrossRef CAS; (c) S. Minakata, Acc. Chem. Res., 2009, 42, 1172–1182 CrossRef CAS PubMed; (d) E. Kolvari, A. Ghorbani-Choghamarani, P. Salehi, F. Shirini and M. A. Zolfigol, J. Iran. Chem. Soc., 2007, 4, 126–174 CrossRef CAS.
- (a) H. Yang, H. Ning, N. Wang, H. Shen, F. Teng, X. Liu, H. Jiang, M.-C. Tan and Q.-W. Gui, ACS Omega, 2021, 6, 25940–25949 CrossRef CAS PubMed; (b) X. Liu, T.-C. Cai, D. Guo, Z. Nong, Y. Yang, Q. Li, H. Jiang, X. Liu and Q.-W. Gui, Chem. Commun., 2022, 58, 657–660 RSC; (c) Q.-W. Gui, Z. Xiong, F. Teng, T.-C. Cai, Q. Li, W. Hu, X. Wang, J. Yu and X. Liu, Org. Biomol. Chem., 2021, 19, 8254–8258 RSC; (d) F. Teng, J. Du, C. Xun, M. Zhu, Q. Lu, H. Jiang, Y. Chen, Y. Li and Q.-W. Gui, Org. Biomol. Chem., 2021, 19, 8929–8933 RSC; (e) H. Jiang, D. Guo, Y. Zhang, Q.-P. Shen, S. Tang, J. You, Y. Huo, H. Wang and Q.-W. Gui, Synthesis, 2020, 52, 2713–2720 CrossRef CAS.
- (a) M. Legay, N. Gondrexon, S. L. Person, P. Boldo and A. Bontemps, Int. J. Chem. Eng., 2011, 670108 Search PubMed; (b) M. A. U. Martines, M. R. Dabolos and M. Jafelicci, Quim. Nova, 2000, 2, 251–256 CrossRef; (c) J. T. Li, S. X. Wang, G. F. Chen and T. S. Li, Curr. Org. Chem., 2005, 3, 415–436 Search PubMed.
- (a) Z. Chen, W. Xia, D. Liu, M. Du and C. Cao, J. Chin. Chem. Soc., 2016, 63, 158–164 CrossRef CAS; (b) B. Han, Z. Zheng, F. Wu and A. Wang, Synth. Commun., 2017, 47, 2387–2394 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ra06867b |
| ‡ The authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |