
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProfitable Fischer Tropsch realization via CO2–CH4 reforming; an overview of nickel–promoter–support interactions
M. Alhassanad,
A. A. Jalil *ab,
M. B. Baharic,
A. H. K. Owgia,
W. Nabgane,
N. S. Hassana,
T. V. Tranag,
A. A. Abdulrasheedf,
M. Y. S. Hamida,
M. Ikram*h,
M. L. Firmansyahi,
H. Holilahjk and
N. A. Sholejahl
*ab,
M. B. Baharic,
A. H. K. Owgia,
W. Nabgane,
N. S. Hassana,
T. V. Tranag,
A. A. Abdulrasheedf,
M. Y. S. Hamida,
M. Ikram*h,
M. L. Firmansyahi,
H. Holilahjk and
N. A. Sholejahl
aSchool of Chemical and Energy Engineering, Faculty of Engineering, Universiti Teknologi Malaysia, 81310, UTM Johor Bahru, Johor, Malaysia. E-mail: aishahaj@utm.my
bCentre of Hydrogen Energy, Institute of Future Energy, Universiti Teknologi Malaysia, 81310, UTM Johor Bahru, Johor, Malaysia
cFaculty of Science, Universiti Teknologi Malaysia, 81310 UTM Johor Bahru, Johor, Malaysia
dDepartment of Chemistry, Sokoto State University, PMB 2134, Airport Road, Sokoto, Nigeria
eDepartament d'Enginyeria Química, Universitat Rovira I Virgili, Av Països Catalans 26, 43007, Tarragona, Spain
fDepartment of Chemical Engineering, Abubakar Tafawa Balewa University, PMB 0248, Bauchi, Bauchi State, Nigeria
gApplied Technology and Sustainable Development, Nguyen Tat Thanh University, 300A Nguyen Tat Thanh District 4, Ho Chi Minh City 755414, Vietnam
hSolar Cell Applications Research Lab, Department of Physics, Government College University Lahore, 54000 Punjab, Pakistan. E-mail: dr.muhammadikram@gcu.edu.pk
iNanotechnology Engineering, Faculty of Advanced Technology and Multidiscipline, Airlangga University, Jl. Dr. Ir. H. Soekarno, Surabaya 60115, Indonesia
jDepartment of Chemistry, Faculty of Science and Data Analytics, Institut Teknologi Sepuluh Nopember, Sukolilo, Surabaya, 60111, Indonesia
kResearch Center for Biomass and Bioproducts, National Research and Innovation Agency of Indonesia (BRIN), Cibinong, 16911, Indonesia
lCollege of Vocational Studies, Bogor Agricultural University (IPB University), Jalan Kumbang No. 14, Bogor 16151, Indonesia
First published on 9th January 2023
Abstract
Environmental pollution, climate change, and fossil fuel extinction have aroused serious global interest in the search for alternative energy sources. The dry reforming of methane (DRM) could be a good technique to harness syngas, a starting material for the FT energy process from greenhouse gases. Noble metal DRM catalysts are effective for the syngas generation but costly. Therefore, they inevitably, must be replaced by their Ni-based contemporaries for economic reasons. However, coking remains a strong challenge that impedes the industrialization of the FT process. This article explains the secondary reactions that lead to the production of detrimental graphitic coke deposition on the surface of active nickel catalyst. The influence of nickel particle size, impact of extra surface oxygen species, interaction of Ni catalysts with metal oxide supports/promoters, and larger fraction of exposed nickel active sites were addressed in this review. Size of active metal determines the conversion, surface area, metal dispersion, surface reactions, interior diffusion effects, activity, and yield. The influence of oxygen vacancy and coke deposition on highly reported metal oxide supports/promoters (Al2O3, MgO and La2O3) was postulated after studying CIFs (crystallographic information files) obtained from the Crystallography open database (COD) on VESTA software. Thus, overcoming excessive coking by La2O3 promotion is strongly advised in light of the orientation of the crystal lattice characteristics and the metal–support interaction can be used to enhance activity and stability in hydrogen reforming systems.
1. Introduction
The depletion of fossil fuel stocks, including coal and petroleum products, has spurred extensive study into hydrogen (H2) as an environmentally beneficial energy carrier for the post-fossil fuel regime. At present, there seems to be a consensus that H2 could be the most effective strategy for combating the trifecta of resource depletion, pollution, and climate change.1–4 H2 as an energy carrier and clean fuel might be a viable replacement for fossil fuels due to its great energy efficiency and absence of greenhouse gas emissions.5,6 (i) Low-carbon pathways using fossil fuels, such as natural gas conversion and coal gasification, combined with carbon capture, utilization, and storage (CCUS); (ii) biomass through biological processes such as anaerobic digestion, which used organic matter transformation and waste to energy; and (iii) the splitting of water into hydrogen and oxygen using nuclear energy are some technologies for H2 production. Over 90% of hydrogen is created from fossil fuels including coal, natural gas, and petroleum. However, hydrocarbon reforming, notably methane, and recent discoveries are the most significant.7,8 Interstellar methane incorporated in oil reservoirs along with landfill sources supply in abundance, CH4 gas which is dangerous to the ecosystem.DRM (1) has gained much interest since it uses two carbon-containing hazardous feedstocks (CH4 and CO2) to produce syngas with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 H2/CO ratio appropriate for use in chemical processes that could harness syngas such as FT energy process for liquid fuel and chemical manufacture. As well, the reaction can store and transmit energy. Despite its significance in energy and material applications, it has not been fully deployed in the industry. The design of a DRM catalyst that is industrially acceptable and affordable is an ongoing challenge. Precious metals are obviously ideal as active catalysts for the DRM, but they are outrageously costly and scarce; therefore, substituting them with more economically affordable ones is unavoidable.9–12
:1 H2/CO ratio appropriate for use in chemical processes that could harness syngas such as FT energy process for liquid fuel and chemical manufacture. As well, the reaction can store and transmit energy. Despite its significance in energy and material applications, it has not been fully deployed in the industry. The design of a DRM catalyst that is industrially acceptable and affordable is an ongoing challenge. Precious metals are obviously ideal as active catalysts for the DRM, but they are outrageously costly and scarce; therefore, substituting them with more economically affordable ones is unavoidable.9–12
Nickel is the most commonly used active metal in CO2–CH4 reforming catalysts due to its high intrinsic activity, availability, and inexpensive cost compared to precious metals and fairly notable catalytic behaviour. However, they suffer severe coking and subsequently deactivation due to harsh tuning operating conditions, catalyst preparation techniques, poor homogenous metal–support interface interactions, inaccurate metal oxide promotion, and a low surface carbon oxidation rate among others.13–20
| CH4 + CO2 = 2CO + 2H2, ΔH0298 K = 247 kJ mol−1 | (1) |
Clear records in the literature21–27 have addressed key issues regarding impediments to FT industrialization via DRM. Table 1 gives a summary of key findings in some similar articles strategizing profitable DRM processes. Major topics addressed in the current review centred on nickel crystal size, oxygen vacancy, pore size distribution, support acidity–basicity, nature of support–promoter interactions and reducibility effects. Additionally, the interaction between the support/promoter, with active metal (Ni) was studied after analysing the crystallographic information files on VESTA, which is the novelty of the present review along with other problems impeding the coke formation in Ni catalysts.
| Year | Study focus | Ref. |
|---|---|---|
| 2013 | Low-temperature DRM | 28 |
| Particle size (1.6–7.3 nm) effect of SiO2-supported Ni | ||
| 2014 | Influence of alkaline earth promotion; MgO, BaO and CaO | 29 |
| Coking and activity of catalysts | ||
| 2014 | Effects of Pt-addition on CeZrO2 and Al2O3 supported-Ni for DRM | 30 |
| 2017 | Effect of variable nano size (2.6 nm, 5.2 nm, 9.0 nm and 17.3 nm) Ni catalysts on SiO2, Al2O3, MgO, ZrO2, and TiO2 supports | 31 |
| 2018 | Support effects on performance of Ni catalysts in DRM; SiO2, TiO2 and ZrO2 | 32 |
| Agglomeration of support and stability for DRM | ||
| 2019 | Effects of support materials such as Al2O3, ZrO2, TiO2, SBA-15, MgO and CeO2–ZrO2 | 33 |
| Tri-reforming potency of Ni catalysts | ||
| 2020 | Effect of Ni–Ta ratio on the selectivity of Ni–Ta/ZSM-5 DRM catalyst | 34 |
| Insight on coke-stimulating side reactions | ||
| 2021 | Effect of Y, Ce and La on MgO-promoted SBA-16 for DRM catalyst | 35 |
| 2022 | Influence of Ni incorporation during CeO2 crystallization in DRM | 36 |
| Catalyst durability, metal support interaction, Ni NP uniform size dispersion | ||
| Current article | Ni particle size, pore distribution and support influence | |
| Coke build-up strategies and possibilities in DRM processes | ||
| Analysis of crystallography files; MgO, La2O3 and Al2O3 on VESTA software to describe coking nature and interaction possibilities with Ni metal | ||
| Metal–support–promoter interactions | ||
| Relevance of oxygen vacancy, Lewis basicity and strategies for coke supression |
Compared to steam reforming of CH4 (SRM) (2), where coke deposition can be reduced by excessive amounts of steam, coke formation becomes more problematic in DRM since the co-reactant (CO2) also contributes to coke deposition.37,38
| CH4 + H2O = CO + 3H2, ΔH0298 K = 206 kJ mol−1 | (2) |
Although the kinetics and mechanism of reactions (1) and (2) are identical; however, the CO:H2 ratio produced by DRM (1) is more valuable (near unity) for the Fischer Tropsch's (FT) energy process39–44 although it has peculiar challenges.
1.1 Vulnerability of Ni catalysts to coking
Susceptibility for coke deposition is predicted based on O/C and H/C ratios of the feed gas. Coke production is more likely in DRM with low O/C and H/C ratios of 1 and 2 respectively, in comparison to other CH4 reforming techniques. Table 2 compares potential for coke formation in DRM, SRM and partial CH4 oxidation (POM).25,45,46| Reaction | ΔH0298 K (kJ mol−1) | Ratio | |
|---|---|---|---|
| O/C | H/C | ||
| a The least ratios obtained for both O/C and H/C are for dry reforming of methane hence, it has the highest susceptibility to coking than other reforming reactions but, the best for FT process. | |||
| CH4 + CO2 = 2CO + 2H2 | 247 | 4/4 = 1a | 4/4 = 1a |
| CH4 + H2O = CO + 3H2 | 206 | 2/2 = 1 | 12/2 = 6 |
| CH4 = C + 2H2 | 75 | 0 | 8/2 = 4 |
 |
−22.6 | 2/2 = 1 | 8/2 = 4 |
| CH4 + 2O2 = CO2 + 2H2O | −806 | 8/2 = 4 | 8/2 = 4 |
| 2CO = C + CO2 | −172 | 4/4 = 1 | 0 |
| CO + H2 = C + H2O | −131 | 2/2 = 1 | 4/2 = 2 |
| CO2 + H2 = CO + H2O | 41.1 | 4/2 = 2 | 4/2 = 2 |
Side and main reactions encountered while dealing with the feed gases in DRM processes are given in (3) to (10) in addition to those discussed elsewhere.47
| CH4 = C + 2H2, ΔH0298 K = 75 kJ mol−1 | (3) |
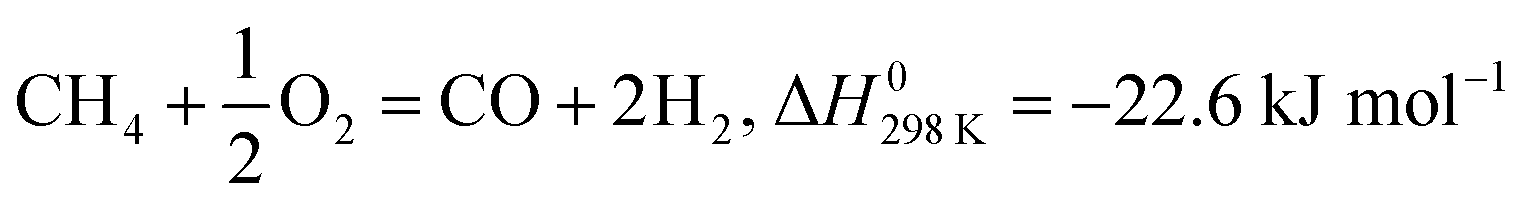 | (4) |
| CH4 + 2O2 = CO2 + 2H2O, ΔH0298 K = −806 kJ mol−1 | (5) |
| 2CO = C + CO2, ΔH0298 K = −172 kJ mol−1 | (6) |
| CO + H2 = C + H2O, ΔH0298 K = −131 kJ mol−1 | (7) |
| CO2 + H2 = CO + H2O, ΔH0298 K = 41.1 kJ mol−1 | (8) |
| CO2 + 2H2 = C + 2H2O, ΔH0298 K = −90 kJ mol−1 | (9) |
| CO + H2O = CO2 + H2, ΔH0298 K = −41.1 kJ mol−1 | (10) |
Nucleation, predominantly occurs due to reactions (3), (6), (7) and (9) in undercoordinated spots of the Ni surface by producing carbon islands, which expand spontaneously. Coke formation and gasification are dynamically essential in both DRM and SRM reactions, and coke-free operations can only be attained when coke deposition does not overrate gasification.48 However, coking phenomenon could give rise to catalytic coke, which is usually filamentous and proceeds via the metal carbide intermediate, non-catalytic coke which exhibits a spherical morphology usually formed under dehydrogenation atmosphere with a high degree of coalescence or bridging spheres, solid coke which forms via polynuclear aromatic intermediates or via the reaction of olefins, acetylenes, dienes and free radicals. Coke deposits on DRM catalysts have been classified as graphitic, filamentous, amorphous or crystalline. Unlike the second, the first is differentiated through its H/C ratio contents, where, the coke is predominantly, carbon rich with less hydrogen. Amorphous and crystalline cokes are identified with a high d-spacing broad and low d-spacing sharp XRD peaks respectively.
Reactions (4) and (5) represent the partial oxidation of methane (POM) under limited (4) and excess (5) oxygen supply; while, eqn (7) and (8) connote respectively, CO and CO2 reduction. Consequently, thermodynamic investigations15,39,49–54 of the reforming reaction have revealed that side reactions (3) and (6) are the primary drivers of carbon deposition, which occurs significantly between 633 and 700 °C; while the coke formation due to reactions (7) and (9) is affected by reactants molar ratio, and that of reaction (10) is the water gas shift reaction which is favoured at temperatures lower than 800 °C.55
2. Particle size-dependent effects of Ni catalysts
It has recently become possible to lessen the quantity of coke that is deposited on Ni catalysts by using a variety of methods. According to earlier literature,56–58 a high aggregate size of metal atoms favours CO and CH4 dissociation on metal surfaces. Subsequently, one common practice is to make the Ni particles smaller. Generating Ni clusters is required for carbon deposition on a Ni surface, and it is widely reported that larger Ni particles result in severe coke deposition.28 However, the endothermic nature of the dry reforming reaction causes the incredibly small Ni particles to get compressed and the size of the Ni domains to eventually grow quite large due to heavy coke deposition. Employing strong metal–support interactions as reported by28,32 when working at high reaction temperatures maintains particle size of the Ni. Table 3 provides a summary of Ni particle size effects on some parameters of reaction. The turnover frequency (TOF) of Ni catalysts with varying particle sizes of 2.6, 5.2, 9.0, and 17.3 nm, as well as CO2 and CH4 initial and final conversions for each particle size, and CO final and initial uptake, were calculated to better understand the coking nature of Ni catalysts and the role of particle size-effect. Despite its relatively high Ni concentration, the 2.9 nm Ni-catalyst demonstrated the highest CO uptake levels at the reaction's beginning and end. Although it was assumed that the initial and final TOFs for CH4 and CO2 would be superior in all catalysts, it became apparent that the smaller size cannot be disregarded.| Parameters(s) | Nickel particle size (nm) | Average | |||
|---|---|---|---|---|---|
| 2.6 | 5.2 | 9.0 | 17.3 | 8.525 | |
| a Appreciable.b Higher.c Much higher value compared to others. | |||||
| Ni content (wt%) | 0.21a | 0.32b | 0.16 | 0.16 | 0.2125 |
|
|||||
| CO uptake (μmol g−1) | |||||
| Initial | 0.42a | 0.81b | 0.59 | 0.46 | 0.57 |
| Final | 0.54a | 0.93b | 0.67 | 0.54 | 0.67 |
|
|||||
| CO2 conversion (%) | |||||
| Initial | 66.0b | 62.2 | 37.8 | 24.0 | 47.5 |
| Final | 62.4b | 61.6 | 35.4 | 26.6 | 46.4 |
|
|||||
| CO2 TOF (s−1) | |||||
| Initial | 104.4c | 51.6 | 42.5 | 34.8 | 58.325 |
| Final | 77.3c | 44.4 | 35.4 | 32.9 | 47.5 |
|
|||||
| CH4 conversion (%) | |||||
| Initial | 39.0b | 38.3 | 18.8 | 10.4 | 26.625 |
| Final | 36.7b | 36.8 | 18.8 | 12.6 | 26.225 |
|
|||||
| CH4 TOF (s−1) | |||||
| Initial | 61.7c | 31.8 | 21.2 | 15.1 | 32.45 |
| Final | 44.4c | 26.5 | 18.8 | 15.6 | 26.325 |
During synthesis, the size of Ni particles might be reduced if there are powerful interactions between the Ni particles and their support (for example, Al2O3). Also, the interaction could help reduce clumping since the support tends to keep Ni particles in place even when the temperature is high. At a long response time, however, particle size preservation becomes weak and most Ni particles will end up getting bigger.59
3. Coke accumulation (build-up) effects
Thermodynamically, Ni catalysts have a high potential for carbon deposition.60 Coke formation principally due to methane decomposition (3) and Boudouward reaction or CO disproportionation (6) is responsible for several issues; including the induction of a high-pressure drop and crushing of catalyst pellets, the clogging of the reactor, and the accelerated degradation of the catalyst.31,32 Another mechanism for catalyst deactivation at high temperatures (>700 °C) is the aggregation of active Ni species.33,34 The ability to eradicate or limit carbon deposition to a minimal level is likewise consistent with the findings of previous investigations.61 Finely disseminated, small particle Ni size can rescue excessive sintering and agglomeration, regulate the acidity62,63 and produce a catalytically active Ni size1,27 that withstands carbon deposition. Several studies showed a Ni nanoparticle size threshold (2 nm or 7–10 nm) below which carbon deposition is reduced.64,65 Incorporating another metal into nickel's surface structure improves performance due to its inexpensive cost and strong activity, Al2O3, MgO, TiO2, SiO2, and La2O3 are common DRM supports. In another instance, Larmier and Comas-Vives et al.66 used a multiscale modelling approach and experiments to investigate the reactivity of the Ni/Al2O3 interface towards WGS and DRM. The metal/support contact serves as a reservoir for oxygenated species in the WGS reaction, whereas all Ni surface atoms are active in the DRM reaction.Dekkar et al.67,68 reported the activity and properties of Ni/Al2O3 and Ni–Si ME catalysts (whose SEM images are shown in Fig. 1). After DRM evaluation, a variation in morphology between fresh and expended Ni/Al2O3 catalyst was noticed; minute nickel particles and carbon deposits emerged. After 16 and 66 h time on stream (TOS), the silica-coated Ni and, impregnated Ni supported on mesoporous alumina catalysts had the best conversions and long-term stability. The high activity of the Ni–Si-ME was attributed to its small Ni particles, strong metal–support interaction, and homogeneous metal dispersion on silica.
 | ||
| Fig. 1 SEM images of the (a) fresh and (b) used Ni/Al2O3 catalyst and (c) fresh and (d) used Ni–Si-ME catalyst. Reproduced from ref. 68 with permission from [Elsevier], copyright [2022]. | ||
Studies on coke gasification kinetics reported by Tingting et al.48 implied the promotional impacts of Fe in MgO supported Ni–Fe alloy catalysts in CO2 reforming of methane (depicted in Fig. 2(a)) and ascribed the delayed coke deposition and quicker surface-coke gasification to the Fe promotion without changing the pristine Ni catalyst's basic reaction mechanism or kinetic properties. The stimulating effect of Fe was found to be highly dependent on CO2 levels in the contracting environment and Fe/Ni alloy ratios. Similarly, Zahra et al.69 synthesized a series of Ni/SBA-16-MgO catalysts with various promoters (3 wt% of Y, Ce, and La) using a double-solvent technique and tested them in the CO2 reforming of CH4. The graphical representation is shown in Fig. 2(b). Compared to the other catalysts, the Y–Ni/SBA-16-MgO catalyst showed better activity due to finer and more dispersed nanoparticles size. The stability experiments revealed that further promotion with yttrium, lanthanum, and cerium made the catalyst more resistant to coking. Cerium promotion aided coke removal and prevented from forming on all catalysts. The Y-promoted catalyst had the highest NiO, which prevented the formation and spread of carbonaceous deposits.
 | ||
| Fig. 2 (a) Influence of Fe promotion on the carbon-resistant property and kinetics using Ni–Fe–MgO for DRM;48 (b) scheme of DRM over unpromoted and promoted Ni/SBA-16-MgO catalyst.35 Reproduced from ref. 35,48 with permission from [Elsevier], copyright [2022]. | ||
The exceptional surface features of TiO2, aided its resistance to carbon deposition and great dispersion of nickel nanoparticles in Ni/TiO2 catalysts, as reported by.70
Additionally, the cluster (Fig. 3) depicts stressing significance of particle size to the support, and its relation to surface area of Ni-catalysts during the CO2–CH4 reforming to value-added compounds in FT synthesis. The cluster was generated from plain text file of author keywords screening from 1000 articles on web of science. The central keywords in the construction of an appropriate DRM catalyst in Fig. 3 include coke resistance, catalyst surface area, alumina-supported Ni, MgO, and ZrO2 support or promoters. The appearance of “coke resistance” close to “Ni” implies that coke resistance is critical to Ni DRM catalysts as reported by.47 A comprehensive look into the 3 cluster shows the need for a coke-resistant Ni catalyst. The yellow centers on catalytic activity, resistance to coking, hydrogen production, silica, ceria and mixed oxides; while the blue cluster stresses on hydrogen production which is achieved through CO2 and CH4 conversion, as well as relationship between Ni-particle size, coke resistance and surface area of DRM catalysts supports.
 | ||
| Fig. 3 Word group highlighting the most-important terms. Generated from plain text file extracted from WoS using VOSviewer. No permissions required. | ||
Another appealing catalyst support, silicon dioxide (SiO2) is an amorphous substance utilized in microsystems as a dielectric in capacitors and transistors, as an insulator in various electronic devices, which is chemically inert and stable even at high temperatures. Loaiza-Gil et al.71 employed an ammonia-synthesized Ni/SiO2 catalyst to achieve a 60% methane conversion (at 700 °C). At the same temperature, Guo and co-researchers72 reported a methane conversion of 50 per cent using catalysts produced by impregnation and plasma breakdown (CH4:H2O of 1:1). At a lower reaction temperature (500 °C), Matsumura and Nakamori73 evaluated a high-loaded methane steam reforming catalyst prepared by impregnating 20 wt% Ni/SiO2 with a methane conversion of just 20%, with the catalyst deactivating completely after 4 hours. The trend testifies that methane reforming displays poorer activity at lower temperatures and high active metal loadings. Fig. 4(a) is based on the reports of Wenming et al.8 according to which, a novel core–shell Ni–ZrO2 catalyst small nickel nanoparticles as cores and microporous silica as shell (denoted as Ni–ZrO2@SiO2) was successfully developed for CO2/CH4 dry reforming to produce syngas utilizing a one-pot simple approach. Under challenging reaction circumstances (800 °C), the Ni–ZrO2@SiO2 catalyst displayed excellent stability after 240 h TOS compared to previously reported core–shell structured catalysts, with no coking after 240 h TOS.
 | ||
| Fig. 4 (a) The development of a Ni–ZrO2@SiO2 catalyst with ultra-high sintering and coking resistance depicted in a schematic diagram;8 (b) graphical illustration of boosted stability of Ni/SiO2 catalyst. Reproduced from ref. 8,74 with permission from [Elsevier], copyright [2022]. | ||
A complementary method for generating starting materials for the DRM, CO2 methanation reported by Run-Ping and colleagues74 utilized a Ni/SiO2 catalyst by ammonia-evaporation method (Fig. 4(b)). The ammonia-evaporation method (AEM) had a particle size of around 4.2 nm, substantially Ni/SiO2 smaller than Ni-based catalysts made through impregnation. The specific surface area of the Ni/SiO2-AEM (446.3 m2 g−1) was significantly larger than that of hydrotalcite or perovskites. In comparison to Ni/SiO2-impregnation method (IM), the Ni/SiO2-AEM had a longer lifespan and a higher methane output. For 100 h at 370 °C, there was no observed deactivation in the Ni/SiO2-AEM, which is better than most previously reported Ni/SiO2-IM catalysts, which deactivated after 20 h TOS. Therefore, the AEM is a useful technique that can be employed for preparing coke-recalcitrant Ni–Si-based catalysts with high nickel loading and great stability.
La2O3 has been extensively explored due to its superior catalytic qualities, such as low methane ignition temperature and long-term stability. SEM images of La2O3 are shown in Fig. 5(a)–(d) for example, superior activity in gradient temperature DRM was reported by Grabchenko et al.75 for modified Ni/La2O3 catalysts Fig. 5(e). The greater activity of the Ni La CA–NH3 sample was attributed to the production of carbon-oxidizing La2O2CO3 species and the interaction of small Ni particles with the support, which allowed for high dispersion during the reaction.
 | ||
| Fig. 5 SEM images of La2O3 (a) nanoparticles (b) nanorods (c) nanosheets and (d) nanoflowers. Reproduced from ref. 76 with permission from [Elsevier] copyright [2022]. (e) Graphical illustration of DRM over Ni/LaO3 catalysts. Reproduced from ref. 75 with permission from [Elsevier] copyright [2022]. | ||
4. Metal–support–promoter interaction and reducible metal-oxides
Active metal interaction with support or promoter materials is critical and remains a challenge in numerous important catalytic reactions including DRM. The metal–support interaction is usually significant for a good reforming catalyst. Strong metal–support contact keeps active metal dispersion minimal at high DRM temperatures.77 The greater the metal–support contact, the higher the temperature needed to break away and unite with a neighbouring metal particle, limiting agglomeration. Weaker active metal–inert support contact makes it more suitable for bimetallic catalysts because it improves metal–metal contact.78By starting the adsorption and activation stage, the support features specifically for Ni-based catalysts control the DRM reaction route. A typical bi-functional DRM reaction pathway involves CH4 activation on the active metal surface and CO2 activation on a mildly acidic (Al2O3) or basic (CeO2, La2O3) support, whereas catalyst supported on inert silica-based materials follows a mono-functional pathway where both CH4 and CO2 activation are on the active metal surface. A semi-empirical indication of the temperature at which sintering begins is the development of thermal resistance to Hüttig and Tamman temperature constraints in catalysts with significant metal–support interaction.21
The MSPI was suggested in the literature at optimum La2O3 loading (near to monolayer) since excessive La-addition stimulates Ni migration from the surface to the bulk of the catalyst, resulting in a lower Ni surface area. The strength of metal–support interaction (MSI) determines the build-up of particle sizes. A stronger interaction inhibits both CO and CH4 disproportionation and other possibilities favouring product yield and stability. Although the impact of the support is multidimensional, CO2 adsorption and decomposition on the support could have the most influence.5,77,79–81 Increased catalytic activity in DRM, depends not only on the particle size of the active metal, rather literature82–86 claims that support characteristics (textural qualities, redox attributes, surface basicity, reducibility, oxygen storage capacity and metal–support).
Active metals localization in the support skeletal structures is controlled by catalysts synthesis methods. Notable ones reported to have excellent recalcitrance to coking and sintering by immobilization of Ni particles include mesosilica shell structure confinement, immobilization in channels of polyol-assisted mesoporous silica, alloy formation with a promoter, aerogel, sublimation-deposition confinement21 and solution-combustion synthesis (SCS).
Yahia and co-authors87 adopted SCS for the synthesis of K, Na, Cs, Li, and Mg-promoted Ni/La2O3 catalysts for the DRM. At 700 °C, all the samples showed substantial catalytic activity, with excellent CO2 and CH4 conversions above the equilibrium values. Unlike the unpromoted samples, boosted catalysts showed improved catalytic activity and durability, attributed to the high metal support interaction due to increased Lewis basicity. The Mg–Ni–La2O3 exhibited outstanding anti-coking qualities. There are two main causes for this: first, it has more surface area and porosity than its competitors, giving it the benefit of more active DRM sites; and second, Mg was added, increasing the number of basic sites while maintaining high MSI and MSPI. This boosted CO2 adsorption and the availability of oxygen species on the surface, so reducing coke formation and prolonging catalyst deactivation due to high-temperature sintering.
4.1 VESTA analysis strategy and justification
Visualization for Electronic and Structural Analysis (VESTA) is a 3D (three dimensional) visualization tool that provides structural models for volumetric data, nuclear densities, and crystal morphologies for COD evaluation. It also provides information on the crystal phase, atom labels, bond sites, symmetry operations that supports around 47 data input structures format and 31 output formats. The selection of the package is influenced by the necessity to find the correlation between surface and bulk atoms of the chosen (Al2O3, MgO, and La2O3) support/promoters for Ni DRM catalysts. The choice of these oxides is based on the work of by Guojie et al.88 who reviewed supports and promoters of DRM catalyst from 2010 to 2017 and reported them as the most proven, most recommended and most utilized oxides amongst others.To propose the nature of coke formation, seizure in the activity of DRM catalyst, the potency of oxygen vacancy and interaction between active metal, support and promoter, the CIF files of Al2O3-supported, MgO, and La2O3-promoted Ni catalysts were analyzed using Vesta software; the orientation of the crystal shape, crystal structures and oxygen vacancies are presented in Fig. 6(a)–(c). A detailed examination of the crystal structures in Fig. 6 revealed a proposal on how the MSI and MSPI occur. The Ni particle size is in the order Ni/La2O3 < Ni/MgO < Ni/Al2O3; therefore, it is highly anticipated that agglomeration at high DRM reaction temperatures will be dominant in the Ni/Al2O3 and there will be a high susceptibility to metal sintering which corresponds to the reports of Tahir and colleagues.89
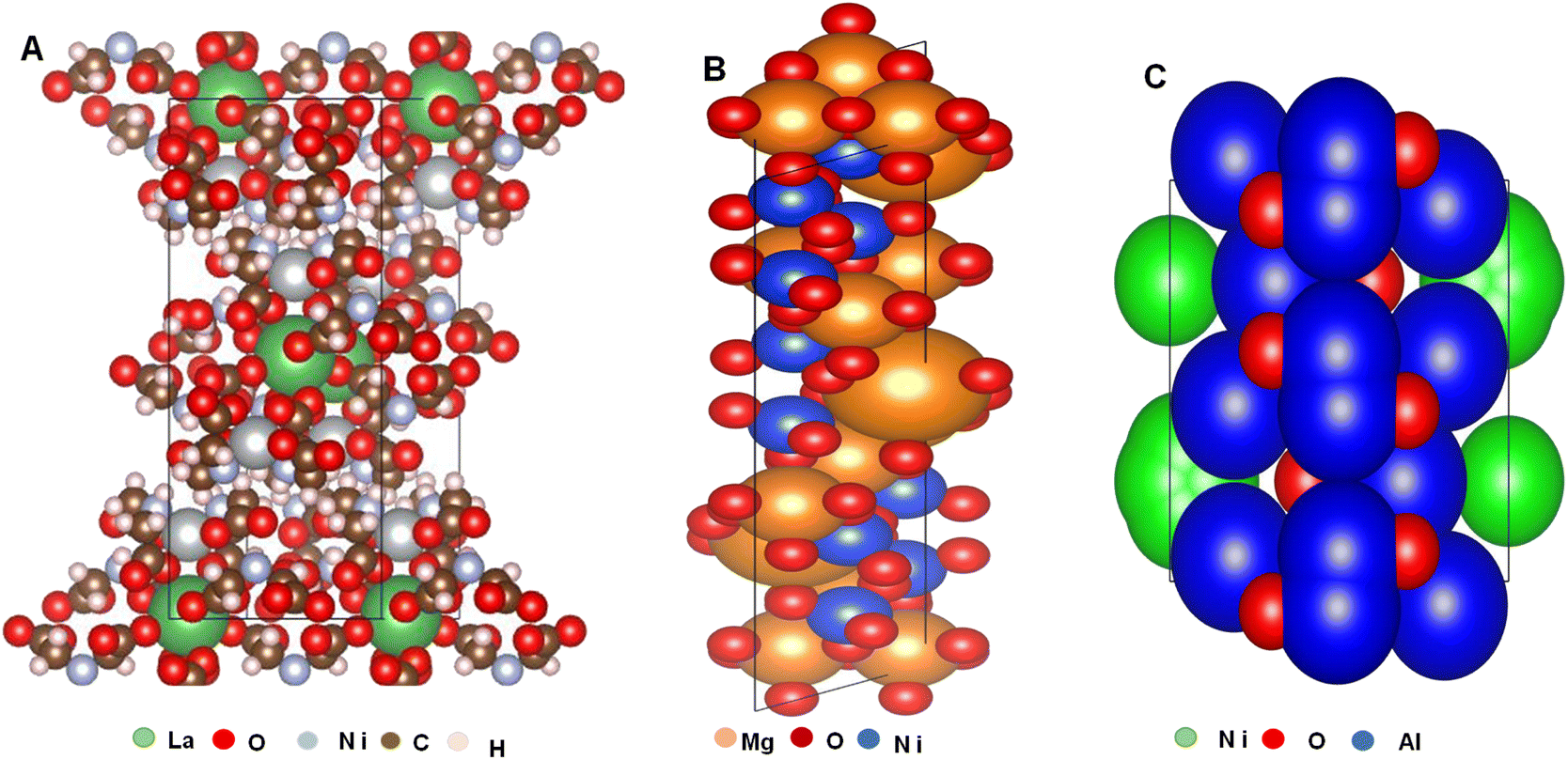 | ||
| Fig. 6 Standard orientation of the skeletal structure, crystal shape and oxygen vacancies from (A) La2O3 promoted Ni catalyst (B) MgO promoted Ni catalyst and (C) Ni/Al2O3. Figure developed from Vesta software using crystallographic information file of the compounds. No permissions required. | ||
Accordingly, scientific research software have made easier, the exploration and analysis of complicated models.81,90–95 A careful study the structural parameters, skeletal orientation, binding sites, and interaction between the support and active metal confirms that the oxygen atoms are more noticeable in the Al2O3 (Fig. 6(c)) connoting a high possible rate of reducibility. Abdulrasheed et al.21 reported that oxygen vacancies on support materials aid the oxidation of deposited coke to CO thereby reducing the coking potential and increase product yield. However, the possibility that metal-oxide overlayer effects may expose Ni particles to size shoot up at high DRM temperatures in Al2O3 being more pronounced than the other two cannot be ruled out here. In contrast, the oxidation of Ni surface in Al2O3 could inhibit the formation of coke, so for low-temperature processes, Al2O3 support may be the best alternative. However, under high DRM heating settings, it is anticipated that La2O3 will exhibit greater stability and fundamental qualities that will promote CO2 chemisorption.
At present, the de supports like, ZrO2, Fe2O3, CeO2, TiO2, Co3O4, etc. are mainly utilized in dry methane reforming since they materials possess high oxygen vacancies, which could store a vast amount of oxygen under oxidizing conditions before releasing it under reducing requirements for CO2 activation.96
This capability dramatically controls CO2 adsorption and activation and directly influences the selection of the intermediate's reaction, thus improving the reforming activity. Indeed, the high release of oxygen or mobile oxygen by this reducible metal oxide support will further assist the suppression of coke during dry methane reforming, hence inhibiting the occurrence of catalyst deactivation and leading to stable reforming activity.22 Besides the intense interaction between active metal and reducible metal oxides, supports also facilitate the suppression of metal growth, hence hindering the sintering phenomenon.
Steinhauer et al.97 compared the reforming activity of Ni–Pd (5 wt%) catalysts supported over irreducible (γ-Al2O3, SiO2) and reducible (La2O3–ZrO2, La2O3, ZrO2, TiO2) metal oxides support. The authors noticed that most reducible supports employed in reforming exhibited a superior activity than irreducible supports in the order of ZrO2–La2O3 > La2O3 > ZrO2 > SiO2 > Al2O3 > TiO2 (Fig. 7(a) and (b)). The observed trend was accredited to the strength of metal–support interaction created along with the excellent distribution of active Ni–Pd over the employed support. The authors also claimed the blockage of active sites was responsible for the inferior reforming activity recorded for TiO2 and Al2O3. The superior activity resulting from La2O3 employment as support for Ni-based catalysts in dry methane reforming agreed with the findings of Fatesh et al.98
 | ||
| Fig. 7 Role of supports towards (a) CO2 and CH4 conversion (CH4-dotted) and (b) CO and H2 yield (H2-dotted) of Ni–Pd catalyst. Reproduced from ref. 97 with permission from [Elsevier] copyright [2022]. | ||
In a different study, Naeem et al.99 investigated the role of support γ-Al2O3, CeO2, and ZrO2 toward the dry methane reforming activity of Ni (5 wt%) catalysts, synthesized through a polyol approach. By carrying out the dry methane reforming at 973 K, Ni-supported reducible ZrO2 demonstrated the highest catalytic performance (CH4 conversion = 87.2%), followed by Ni-supported γ-Al2O3 (CH4 conversion = 87.2%), and Ni supported CeO2 (CH4 conversion = 83.2%). It was justified that the superior activity by Ni/ZrO2 corresponded to unique features of ZrO2, including high surface acidity, reducibility, redox potential and thermal stability.
On the contrary, Zhang et al.32 found that the Ni supported by reducible metal oxides like ZrO2 and TiO2 demonstrated lower catalytic activity as compared to irreducible metal oxide (SiO2, Al2O3 and MgO). The authors justified that Ni/ZrO2 and Ni/TiO2 suffered poor performance due to weak MSI associated with the low Ni distribution on the small surface area of reducible metal oxides (TiO2 = 9 m2 g−1, ZrO2 = 25 m2 g−1). Moreover, Ni/ZrO2 and Ni/TiO2 exhibited the highest accumulation of coke, about 21.0 and 24.7 mg gcat−1 h−1, respectively.
A similar agreement was reported by Naeem et al.98 where the inferior activity of Ni supported with reducible CeO2 support has resulted from low Ni active metal distribution on the CeO2 surface, thus leading to an increment in Ni crystallite size. Consequently, this condition favoured the high coke accumulation (0.025 g gcat−1 h−1) on the low surface area of CeO2 compared to Ni/ZrO2 (0.02 g gcat−1 h−1) and Ni/γ-Al2O3 (0.015 g gcat−1 h−1). On the contrary, the high surface area of irreducible metal oxides such as SiO2 (239 m2 g−1) and Al2O3 (167 m2 g−1) caused excellent Ni distribution, providing a more accessible active site, hence leading to high catalytic performance.
Wang and Ruckenstein100 assessed the reducibility of metal oxide supports, including Ta2O5, ZrO2, Nb2O5, TiO2, and CeO2, through temperature-programmed reduction analysis. A significant reduction peak assigned to TiO2, CeO2, and Nb2O5 appeared around 1023 K, 1123 K, and 1163 K respectively; while there was no noticeable reduction peak revealed by Ta2O5 and ZrO2 (Fig. 8(a)). The reducibility strength of the tested metal oxide support declined in the order of CeO2 > Nb2O5 > TiO2 > ZrO2 > Ta2O5, which agrees with findings reported by Tauster and Fung.77 However, the authors highlighted that these metal oxide supports are inappropriate for dry methane reforming, which is ascribed to low conversion and yield. They reported that irreducible metal oxides, namely MgO and Al2O3, provide superior and stable performance with time compared to those tested with reducible metal oxide support.
 | ||
| Fig. 8 (A) TPR profiles for (a) CeO2, (b) Nb2O5, (c) Ta2O5, (d) TiO2, (e) ZrO2; the conversion (B) of CH4 and (C) CO2 in dry reforming over 5NiYZ catalysts. Reproduced from ref. 100 with permission from [Elsevier] copyright [2022]. | ||
Furthermore, Li and Veen5 evaluated the impact of reduction temperature on reducible CeO2-supported Ni nanoparticles and reported that the reduction of CeO2 support in H2 within 773–973 K led to an intense bonding between the CeO2 support and Ni, thus suppressing Ni particle sintering. However, when the reduction is conducted at a higher temperature, it was found that Ni nanoparticles were encapsulated by the migration of CeO2 from reduced support. Consequently, this phenomenon caused the decline in the catalyst capability for both CH4 and CO2 adsorption/activation, leading to lower catalytic performance, although low coke accumulation was recorded.
Since the reducible metal oxides mainly contributed to coke suppression compared to increased performance, some modifications towards the reducible metal oxide have been extensively reported in the literature. For instance, Bellido et al.101 successfully modified ZrO2 support on nickel catalyst by introducing CaO (8 mol%) and evaluating its activity in dry reforming methane at 800 °C for about 6 h. It was noticed that the incorporation of CaO led to the increment in oxygen vacancies in the reducible support, which facilitated the reduction of NiO to active Ni. This improvement effectively caused the increment in the number of the accessible Ni active sites on the support, thus enhancing the catalytic activity up to CH4 conversion = 66.5% and CO2 conversion = 76.3% in dry methane reforming. Nevertheless, the authors highlighted that the increment of CaO addition towards ZrO2 support (>8 mol%) caused a considerable drop-in reforming activity (reactant conversion lower than 50.0%). This negative trend has resulted from the excessive presence of oxygen vacancies on the surface, which hinders the CO2 activation. A similar trend was reported by Roh and Jun102 during the modification of support with different loading of CaO (0.04–0.06) for dry methane reforming. Furthermore, they also emphasized that the modification by CaO enhanced the basicity of the catalyst, leading to effective suppression of coke, hence improving the stability performance of the catalyst.
In other efforts, Pietraszek et al.84 studied the effect of reducible support modification over 5 wt% Ni-based catalysts for dry methane reforming. The authors modified Ce2Zr2O8 by incorporating 0.5 wt% noble metal, Rh and Ru via a pseudo-sol–gel strategy. It was found that the modification of Ce2Zr2O8 support by Rh and Ru effectively boosted the catalytic activity up to 87% compared to unmodified support (76%). The studies revealed the growing reflection peak related to Ni species in XRD analysis, which prompted the increase in the number of Ni active sites. Furthermore, they observed better catalytic stability following the change due to both noble metals' superior coke suppression capabilities.
5. Dispersion, pore distribution, oxygen vacancy and acidity–basicity effects
The evenness of active metal dispersion over the support influences performance of catalysts85,103 and properties such as agglomeration, activity and stability are dependent on the active metal dispersion. Excellent dispersion in Ni catalysts is essential for anti-carbon ability, high cross-sectional area and turn over frequencies for DRM reactions.Highly-loaded active metal catalysts experience blockage of the pores, and this defect is overcome with a moderate Ni loading. Contrary to type I hysteresis mostly reported for microporous materials, the BET adsorption–desorption isotherm for mesoporous materials (typically, type IV), implies a multilayer adsorption, followed by capillary condensation. The amount of unfilled pore space (p/p0) represents the empty pores identified after multilayer covering has been filled with condensate and isolated from the gas phase. A hierarchical or multi-modal pore structure may be inferred from the BJH based on the pore size distribution that is displayed. The occurrence of metastable states linked with the capillary condensation of probe molecules on mesopores causes hysteresis loops to occur often at high relative pressures. However, low pressure hysteresis phenomena have been reported for porous solids such as ordered mesoporous SiO2, zeolites and activated carbons.79,104
The oxygen vacancy increases the electron density of metal, stimulating a weakening of the carbonyl group (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) bond on CO2 over the metal sites. These imply that oxygen vacancies preserves the metal in the active metallic phase and hinder coke accumulation. In bifunctional catalysts, it provides labile electrons that improve the electrostatic interactions between metal and support, stabilize metal phases, and produce labile oxygen ions for carbon gasification both of which are mobilised to the nearby unpaired electron-deficient metal cations, resulting in electrostatic interactions. By using this property, the difference in relative intensities can be used to figure out how strongly the oxygen vacancies and active metal particles interact with the support.64
O) bond on CO2 over the metal sites. These imply that oxygen vacancies preserves the metal in the active metallic phase and hinder coke accumulation. In bifunctional catalysts, it provides labile electrons that improve the electrostatic interactions between metal and support, stabilize metal phases, and produce labile oxygen ions for carbon gasification both of which are mobilised to the nearby unpaired electron-deficient metal cations, resulting in electrostatic interactions. By using this property, the difference in relative intensities can be used to figure out how strongly the oxygen vacancies and active metal particles interact with the support.64
Similarly, acidity or basicity of the support that affects the coke deposition and, the catalysts life.9 For example, it has been found that increasing the Lewis basicity of the support can increase the chemisorption of CO2, in particular when using alkaline earth metals (MgO, CaO, and BaO), which are predominantly studied as promoters, modifiers, and supports. The generation of coke is reduced to an absolute bare minimum as a result of the CO2 activation, which also enhances catalyst deactivation.18,45 At excessive surface acidity, the catalyst deactivation fast occurs due to the CO2 disproportionation, which favours coking over syngas conversion, followed by metal oxide formation. RWGS reaction is usually favoured at higher CO2 than CH4 conversions and confirmed by the presence of water droplets at the reactor outlet.21
At moderate acidity and basicity, however, DRM occurs without appreciable deactivation because CO2 and CH4 show comparable activation energy. In practice, the alkalinity of La2O3 coupled with the confinement strategy of the mesoporous shell improves the overall catalytic performance of the catalyst.
Promoters play the role of improving the properties of the active phase. For example, the addition of CaO and MgO increases CO2 conversion, by forming strongly ionic oxides that attract CO on the catalyst surface;11,105–107 potassium addition improves the reducibility of metal particles; decrease the concentration of nickel ensembles leading to carbon formation by behaving as a coke gasification additive.6,108 Table 4 summarizes the properties of some common metal oxide supports and promoters.
| Catalyst system | wt%; method of preparation | Findings | Ref. |
|---|---|---|---|
| a N.R. = not reported. | |||
| Co–Ni/CeO2, Ni/CeO2 and Co/CeO2 | 7.5 wt% Co (Co/CeO2), 7.5 wt% Ni (Ni/CeO2), 3.75 wt% each Co and Ni (Co–Ni/CeO2); coprecipitation | CeO2 mitigated the coke deposition and provided a reducing atmosphere for CO2 activation attributed to its oxygen storage | 109 |
| Ni/CeO2 | 5 wt%; wet impregnation | Surface Ni content (7.1% to 15.5%) and stronger interaction between Ni and CeO2 reduced Ni particle agglomeration/sintering | 5 |
| Pt–Ni/CeO2 | 2 wt%, Pt:Ni (25:75; 50:50; 75:25); incipient wetness impregnation |
A synergistic impact between platinum and nickel enhanced metal dispersion and reduction, as well as the reducibility of cerium | 110 |
| NiCe-x/SiO2 catalysts | (x = 0.17, 0.50, 0.67, 0.84) promoted over Sm, Co, and Ce | Higher activity was noted with Sm, Co and Ce-promoted Ni-based catalysts than Ni/SiO2 | 111 |
| Ni/2.5La–ZrO2 and Ni/5La–ZrO2 | 2.5–5 wt%; impregnation | MgO, CaO, K2O, and La2O3 do improve both resistance to coke formation and catalyst activity | 112 |
| CeO2–Cu/KIT-6 | 10 wt% Cu 2 wt% Ce; impregnation | Remarkable average methanol conversion (80%) after 100 h reaction time, assigned to decrease in the catalyst particle size | 113 |
| Ni/La2O3 | 5 wt%; incipient wetness impregnation | Confinement of La2O3 suppressed coke deposition, and metal sintering | 114 |
| Ni/SBA-15, Ni–La/SBA-15, Ni–La–Ce/SBA-15 | 10 wt% Ni (Ni/SBA-15), 10 wt% Ni–5 wt% La (Ni–La/SBA-15), 10 wt% Ni–5 wt% La–5 wt% Ce (Ni–La–Ce/SBA-15); hydrothermal | La2O3 was crucial in regulating the average particle size of nickel and its interactions | 115 |
| Ni–CaO/ZrO2–La2O3 | 2 wt% Ca–5 wt% Ni; impregnation | La promotion enables an alternate and more effective method for CO2 activation by oxidizing carbonaceous deposits on metallic surfaces with oxygen species | 116 |
| Ni–La2O3/Si–C | 7 wt% Ni, 1 wt% La (Ni–La 7–1/SiC-foam); impregnation | High Ni–La2O3 interactions stop Ni from sintering and make C-elimination stronger, which stops carbon from forming | 117 |
| Ni/Zr, Ni/LaZr and Ni/CeZr | 8 wt% Ni; wet impregnation | By adding La2O3 and CeO2, the oxygen ion lability, resistance to sintering, and stability/efficiency are all greatly improved | 118 |
| Ni/La2O3–ZrO2 | 5 wt% Ni; impregnation | Carbon building on Ni particles relies on the ratio of CH4 degradation and deposit-CO2 reactions inhibited by La2O3 addition | 119 |
| Ni–MgO/ATP, Ni–ATP | 10 wt% Ni; impregnation | Mg-modified Ni/ATP prevented Ni sintering and amorphous carbon formation. Ni/10Mg/ATP catalyst showed the best activity and stability | 11 |
| NiO/MgO | N.R.; impregnation | NiO/MgO solid solution catalyst is active and selective. However, mechanical mixture calcination time increases activity and selectivity | 120 |
| Ni–MgO/SiO2 | NiO and MgO were 4 wt% and 0.4 wt%, respectively; impregnation | MgO is an electron donor that may transfer electrons to Ni particles, increasing Ni electron density. As Ni particle size decreased, MgO's electron-supplying ability increased | 121 |
| Ni/MgO–Al2O3 | 10 wt% Ni, 3, 5, and 10 wt% MgO; impregnation | 5 and 10% catalysts have identical activity and stability. The Ni/5MgOeAl2O3 catalyst showed the best results, with decreased coke formation and a high H2:CO molar ratio of the synthesis gas |
51 |
| Ni–MgO/Al2O3 | N.R., incipient wetness | The activation of CO2 might be aided by the injection of MgO, which increases the surface oxygen (i.e. hydroxyl oxygen or defect oxygen) and the number of basic sites on the catalyst surface | 122 |
| La2O2CO3-modified Ni/Al2O3 catalysts | 5 wt%; impregnation | La2O3 and Al2O3 interact strongly therefore Ni may penetrate La2O3 layers and develop a strong contact with Al2O3, reducing the Ni's surface area | 123 |
| Ni–Mg(Ca)–Al2O3 | 5, 7.5 and 10 wt%; evaporation induced self-assembly (EISA) | Given the endothermic nature of the combined steam and dry reforming reaction, CH4 and CO2 conversions declined with lowering temperature, with the influence being particularly apparent for CO2 below 700 °C | 124 |
6. Future and prospects
The success stories that have been documented in the laboratory and the pilot scales of the plant suggest that the FT is closer than ever, to being realized commercially. However, efforts can focus on important perspectives thus:(i) Given the significance of the CO2 conversion in the preceding discussion of the RWGS reaction, it is necessary to strategize such that the CH4 conversion can outweigh that of CO2, since a higher CO2 conversion indicates the occurrence of the RWGS reaction, which impedes the formation of syngas.
(ii) Smaller Ni particles are reduced at low temperatures while agglomeration of Ni particles happens at high temperatures. However, stability of catalysts can be improved by loading optimum amount of Ni, increasing the Lewis basicity of DRM catalysts supports and careful selection of good promoter and preparation method.
(iii) Low and high-temperature DRM reactions have advantages and disadvantages each, and the FT requirement prefers the DRM over the other possibilities due to its less O/H and C/H ratios. Establishing MSI and MSPI of other transition metal and inner transition metal oxides is highly encouraged.
(iv) To complement the claim raised by researchers based on data available in the literature, VESTA can be utilized for proposing the possibilities of other emerging metal oxides supports and promoters.
7. Conclusion
The induced impact of Ni surface-decoration by metal oxide supports such as MgO, La2O3, or CeO2 in suppressing coke formation in DRM is described in the current review article. The role of Ni-particle sizes; supports morphology and promoters' interactions with emphasis on critical observation of synergistic interplay among these properties for performance enhancement.Interactions between Ni catalyst and metal oxide supports, nickel particle size, and surface oxygen species is shown to provide an enhanced coke suppression. Selecting the appropriate reducible support can increase reforming activity and stability while minimizing coke accumulation.
Conflicts of interest
There are no conflicts to declare.List of abbreviations
| AEM | Ammonia evaporation method |
| CH4 | Methane |
| CIF | Crystallographic information file |
| CO | Carbon monoxide |
| CO2 | Carbon dioxide |
| COD | Crystallography open database |
| DRM | Dry reforming of methane |
| FT | Fischer Tropsch |
| H2 | Hydrogen |
| H2O | Water |
| kJ mol−1 | kilo Joule per mole |
| ME | Microemulsion method |
| MSI | Metal–support interaction |
| MSPI | Metal–support–promoter interaction |
| POM | Partial oxidation of methane |
| SCS | Solution combustion synthesis |
| SRM | Steam reforming of methane |
| TOF | Turnover frequency |
Acknowledgements
The authors are thankful to Universiti Teknologi Malaysia High Impact Research Grant (08G92).References
- N. Fajrina and M. Tahir, Int. J. Hydrogen Energy, 2019, 44, 540–577 CrossRef CAS.
- N. M. Gupta, Renewable Sustainable Energy Rev., 2017, 71, 585–601 CrossRef CAS.
- P. K. Dubey, P. Tripathi, R. S. Tiwari, A. S. K. Sinha and O. N. Srivastava, Int. J. Hydrogen Energy, 2014, 39, 16282–16292 CrossRef CAS.
- H. Ahmad, S. K. Kamarudin, L. J. Minggu and M. Kassim, Renewable Sustainable Energy Rev., 2015, 43, 599–610 CrossRef CAS.
- M. Li and A. C. van Veen, Appl. Catal., B, 2018, 237, 641–648 CrossRef CAS.
- R. Espinal, E. Taboada, E. Molins, R. J. Chimentao, F. Medina and J. Llorca, Appl. Catal., B, 2012, 127, 59–67 CrossRef CAS.
- W. J. Jang, J. O. Shim, H. M. Kim, S. Y. Yoo and H. S. Roh, Catal. Today, 2019, 324, 15–26 CrossRef CAS.
- W. Liu, L. Li, X. Zhang, Z. Wang, X. Wang and H. Peng, J. CO2 Util., 2018, 27, 297–307 CrossRef CAS.
- S. Das, M. Sengupta, J. Patel and A. Bordoloi, Appl. Catal., A, 2017, 545, 113–126 CrossRef CAS.
- P. K. Chaudhary, N. Koshta and G. Deo, Int. J. Hydrogen Energy, 2020, 45, 4490–4500 CrossRef CAS.
- X. Qingli, Z. Zhengdong, H. Kai, X. Shanzhi, M. Chuang, C. Chenge, Y. Huan, Y. Yang and Y. Yongjie, Int. J. Hydrogen Energy, 2021, 46, 27380–27393 CrossRef.
- W. S. Dong, H. S. Roh, K. W. Jun, S. E. Park and Y. S. Oh, Appl. Catal., A, 2002, 226, 63–72 CrossRef CAS.
- B. Tahir, M. Tahir and N. A. S. Amin, Energy Convers. Manage., 2018, 159, 284–298 CrossRef CAS.
- F. Touahra, A. Rabahi, R. Chebout, A. Boudjemaa and D. Lerari, Int. J. Hydrogen Energy, 2015, 41, 2477–2486 CrossRef.
- I. Hussain, A. A. Jalil, C. R. Mamat, T. J. Siang, A. F. A. Rahman, M. S. Azami and R. H. Adnan, Energy Convers. Manage., 2019, 199, 112056 CrossRef CAS.
- K. N. Papageridis, G. Siakavelas, N. D. Charisiou, D. G. Avraam, L. Tzounis, K. Kousi and M. A. Goula, Fuel Process. Technol., 2016, 152, 156–175 CrossRef CAS.
- A. Iglesias-juez, F. Fresno, J. M. Coronado, J. Highfield, A. M. Ruppert and N. Keller, Curr. Opin. Green Sustainable Chem., 2022, 37, 100652 CrossRef CAS.
- M. C. J. Bradford and M. A. Vannice, Catal. Rev. – Sci. Eng., 1999, 41, 1–42 CrossRef CAS.
- A. M. Gadalla and M. E. Sommer, Chem. Eng. Sci., 1989, 44, 2825–2829 CrossRef CAS.
- K. S. Baamran and M. Tahir, Energy Convers. Manage., 2019, 200, 112064 CrossRef CAS.
- A. Abdulrasheed, A. A. Jalil, Y. Gambo, M. Ibrahim, H. U. Hambali and M. Y. Shahul Hamid, Renewable Sustainable Energy Rev., 2019, 108, 175–193 CrossRef CAS.
- A. M. Ranjekar and G. D. Yadav, J. Indian Chem. Soc., 2021, 98, 100002 CrossRef CAS.
- A. A. Jalil, M. Alhassan, W. Nabgan, M. Y. B. S. Hamid, M. B. Bahari and M. Ikram, SSRN Electronic Journal, 2022, 328, 125240 Search PubMed.
- T. Xie, X. Zhao, J. Zhang, L. Shi and D. Zhang, Int. J. Hydrogen Energy, 2015, 40, 9685–9695 CrossRef CAS.
- J. Edwards and A. M. Maitra, Technology and undefined, Elsevier, 1995 Search PubMed.
- L. Xu, Y. Liu, Y. Li, Z. Lin, X. Ma, Y. Zhang, M. D. Argyle and M. Fan, Appl. Catal., A, 2014, 469, 387–397 CrossRef CAS.
- A. H. K. Owgi, A. A. Jalil, I. Hussain, N. S. Hassan, H. U. Hambali, T. J. Siang and D. V. N. Vo, Environ. Chem. Lett., 2021, 19, 2157–2183 CrossRef CAS.
- D. Baudouin, U. Rodemerck, F. Krumeich, A. De Mallmann, K. C. Szeto, H. Ménard, L. Veyre, J. P. Candy, P. B. Webb, C. Thieuleux and C. Copéret, J. Catal., 2013, 297, 27–34 CrossRef CAS.
- Z. Alipour, M. Rezaei and F. Meshkani, J. Ind. Eng. Chem., 2014, 20, 2858–2863 CrossRef CAS.
- E. G. Mahoney, J. M. Pusel, S. M. Stagg-Williams and S. Faraji, J. CO2 Util., 2014, 6, 40–44 CrossRef CAS.
- J. W. Han, J. S. Park, M. S. Choi and H. Lee, Appl. Catal., B, 2017, 203, 625–632 CrossRef CAS.
- J. H. Park, S. Yeo and T. S. Chang, J. CO2 Util., 2018, 26, 465–475 CrossRef CAS.
- R. Kumar, K. Kumar, N. V. Choudary and K. K. Pant, Fuel Process. Technol., 2019, 186, 40–52 CrossRef CAS.
- H. U. Hambali, A. A. Jalil, A. A. Abdulrasheed, T. J. Siang, B. B. Nyakuma, W. Nabgan and T. A. T. Abdullah, Chem. Eng. Sci., 2020, 227, 115952 CrossRef CAS.
- Z. Taherian, A. Khataee and Y. Orooji, J. Energy Inst., 2021, 97, 100–108 CrossRef CAS.
- S. B. Kim, A. A.-S. Eissa, M.-J. Kim, E. S. Goda, J.-R. Youn and K. Lee, Catalysts, 2022, 12, 423 CrossRef CAS.
- K. Song, M. Lu, S. Xu, C. Chen, Y. Zhan, D. Li, C. Au, L. Jiang and K. Tomishige, Appl. Catal., B, 2018, 239, 324–333 CrossRef CAS.
- C. C. Chong, S. N. Bukhari, Y. W. Cheng, H. D. Setiabudi, A. A. Jalil and C. Phalakornkule, Appl. Catal., A, 2019, 584, 117174 CrossRef CAS.
- T. J. Siang, A. A. Jalil, A. A. Abdulrasheed, H. U. Hambali and W. Nabgan, Energy, 2020, 198, 117394 CrossRef CAS.
- Y. Pei, Y. Ding, H. Zhu and H. Du, Chin. J. Catal., 2015, 36, 355–361 CrossRef CAS.
- M. Claeys, M. E. Dry, E. Van Steen, E. Du Plessis, P. J. Van Berge, A. M. Saib and D. J. Moodley, J. Catal., 2014, 318, 193–202 CrossRef CAS.
- H. Fazeli, M. Panahi and A. Rafiee, J. Nat. Gas Sci. Eng., 2018, 52, 549–558 CrossRef CAS.
- W. L. Luyben, J. Process Control, 2016, 39, 77–87 CrossRef CAS.
- J. G. Speight, Gasification of Unconventional Feedstocks, 2014, pp. 118–134 Search PubMed.
- D. Li, Y. Nakagawa and K. Tomishige, Appl. Catal., A, 2011, 408, 1–24 CrossRef CAS.
- M. A. Peña, J. P. Gómez and J. L. G. Fierro, Appl. Catal., A, 1996, 144, 7–57 CrossRef.
- A. A. Jalil, M. Alhassan, W. Nabgan, M. Y. B. S. Hamid, M. B. Bahari and M. Ikram, SSRN Electronic Journal, 2022, 328, 125240 Search PubMed.
- T. Zhang, Z. Liu, Y. A. Zhu, Z. Liu, Z. Sui, K. Zhu and X. Zhou, Appl. Catal., B, 2020, 264, 118497 CrossRef CAS.
- M. Jafarbegloo, A. Tarlani, A. W. Mesbah and S. Sahebdelfar, Int. J. Hydrogen Energy, 2015, 40, 2445–2451 CrossRef CAS.
- C. Jensen and M. S. Duyar, Energy Technol., 2021, 9, 1–12 Search PubMed.
- V. R. Bach, A. C. de Camargo, T. L. de Souza, L. Cardozo-Filho and H. J. Alves, Int. J. Hydrogen Energy, 2020, 45, 5252–5263 CrossRef CAS.
- N. Abdel Karim Aramouni, J. Zeaiter, W. Kwapinski and M. N. Ahmad, Energy Convers. Manage., 2017, 150, 614–622 CrossRef CAS.
- D. Xiang and S. Zhao, Energy Convers. Manage., 2018, 172, 1–8 CrossRef CAS.
- H. Xie, Q. Yu, Y. Zhang, J. Zhang, J. Liu and Q. Qin, Int. J. Hydrogen Energy, 2017, 42, 2914–2923 CrossRef CAS.
- A. Macario, P. Frontera, S. Candamano, F. Crea, P. De Luca and P. L. Antonucci, J. Nanosci. Nanotechnol., 2019, 19, 3135–3147 CrossRef CAS.
- R. A. Van Santen and M. Neurock, Catal. Rev., 1995, 37, 557–698 CrossRef CAS.
- C. Minot, M. A. Van Hove and G. A. Somorjai, Surf. Sci., 1983, 127, 441–460 CrossRef CAS.
- A. De Koster and R. A. Van Santen, J. Catal., 1991, 127, 141–166 CrossRef CAS.
- J. Kim, D. Jin, T. Park and K. Kim, Appl. Catal., A, 2000, 197, 191–200 CrossRef CAS.
- P. Frontera, A. Macario, A. Aloise, P. L. Antonucci, G. Giordano and J. B. Nagy, Catal. Today, 2013, 218–219, 18–29 CrossRef CAS.
- J. Guo, H. Lou, H. Zhao, D. Chai and X. Zheng, Appl. Catal., A, 2004, 273, 75–82 CrossRef CAS.
- B. Fidalgo, A. Domínguez, J. J. Pis and J. A. Menéndez, Int. J. Hydrogen Energy, 2008, 33, 4337–4344 CrossRef CAS.
- C. Wang, Y. Wang, M. Chen, D. Liang, Z. Yang, W. Cheng, Z. Tang, J. Wang and H. Zhang, Int. J. Hydrogen Energy, 2021, 46, 5852–5874 CrossRef CAS.
- T. J. Siang, A. A. Jalil, A. Abdulrahman and H. U. Hambali, Environ. Chem. Lett., 2021, 19, 2733–2742 CrossRef CAS.
- A. A. Ibrahim, A. H. Fakeeha, M. S. Lanre, A. S. Al-Awadi, S. B. Alreshaidan, Y. A. Albaqmaa, S. F. Adil, A. A. Al-Zahrani, A. E. Abasaeed and A. S. Al-Fatesh, Catalysts, 2022, 12, 361 CrossRef CAS.
- L. Foppa, T. Margossian, S. M. Kim, C. Müller, C. Copéret, K. Larmier and A. Comas-Vives, J. Am. Chem. Soc., 2017, 139, 17128–17139 CrossRef CAS PubMed.
- D. Li, Y. Nakagawa and K. Tomishige, Appl. Catal. A, 2011, 408, 1–24 CrossRef CAS.
- S. Dekkar, S. Tezkratt, D. Sellam, K. Ikkour, K. Parkhomenko, A. Martinez-Martin and A. C. Roger, Catal. Lett., 2020, 150, 2180–2199 CrossRef CAS.
- Z. Taherian, M. Yousefpour, M. Tajally and B. Khoshandam, Int. J. Hydrogen Energy, 2017, 42, 24811–24822 CrossRef CAS.
- D. Hu, C. Liu, L. Li, K. Le Lv, Y. H. Zhang and J. L. Li, Int. J. Hydrogen Energy, 2018, 2, 21345–21354 CrossRef.
- A. Loaiza-Gil, M. Villarroel, J. F. Balbuena, M. A. Lacruz and S. Gonzalez-Cortés, J. Mol. Catal. A: Chem., 2008, 281, 207–213 CrossRef CAS.
- X. Guo, Y. Sun, Y. Yu, X. Zhu and C. J. Liu, Catal. Commun., 2012, 19, 61–65 CrossRef CAS.
- Y. Matsumura and T. Nakamori, Appl. Catal., A, 2004, 258, 107–114 CrossRef CAS.
- R. P. Ye, W. Gong, Z. Sun, Q. Sheng, X. Shi, T. Wang, Y. Yao, J. J. Razink, L. Lin, Z. Zhou, H. Adidharma, J. Tang, M. Fan and Y. G. Yao, Energy, 2019, 188, 116059 CrossRef CAS.
- M. Grabchenko, G. Pantaleo, F. Puleo, O. Vodyankina and L. F. Liotta, Int. J. Hydrogen Energy, 2021, 46, 7939–7953 CrossRef CAS.
- T. Jiang, J. Song, M. Huo, N. T. Yang, J. Liu, J. Zhang, Y. Sun and Y. Zhu, RSC Adv., 2016, 6, 34872–34876 RSC.
- S. J. Tauster and S. C. Fung, J. Catal., 1978, 55, 29–35 CrossRef CAS.
- K. Li, C. Pei, X. Li, S. Chen, X. Zhang, R. Liu and J. Gong, Appl. Catal., B, 2020, 264, 118448 CrossRef CAS.
- W. O. Alabi, K. O. Sulaiman, H. Wang, Y. Hu and C. Patzig, J. CO2 Util., 2020, 37, 180–187 CrossRef CAS.
- L. Zhang, F. Wang, J. Zhu, B. Han, W. Fan, L. Zhao, W. Cai, Z. Li, L. Xu, H. Yu and W. Shi, Fuel, 2019, 256, 115954 CrossRef CAS.
- J. R. Carvalho and L. N. Vidal, J. Comput. Chem., 2022, 43, 1484–1494 CrossRef CAS PubMed.
- J. M. García-Vargas, J. L. Valverde, F. Dorado and P. Sánchez, J. Mol. Catal. A: Chem., 2014, 395, 108–116 CrossRef.
- O. Omoregbe, H. T. Danh, S. Z. Abidin, H. D. Setiabudi, B. Abdullah, K. B. Vu and D. V. N. Vo, Procedia Eng., 2016, 148, 1388–1395 CrossRef CAS.
- A. Pietraszek, B. Koubaissy, A. C. Roger and A. Kiennemann, Catal. Today, 2011, 176, 267–271 CrossRef CAS.
- P. Djinović, I. G. Osojnik črnivec, B. Erjavec and A. Pintar, Appl. Catal., B, 2012, 125, 259–270 CrossRef.
- T. Zhang, Z. Liu, Y. A. Zhu, Z. Liu, Z. Sui, K. Zhu and X. Zhou, Appl. Catal., B, 2020, 264, 118497 CrossRef CAS.
- Y. H. Ahmad, A. T. Mohamed, A. Kumar and S. Y. Al-Qaradawi, RSC Adv., 2021, 11, 33734–33743 RSC.
- G. Zhang, J. Liu, Y. Xu and Y. Sun, Int. J. Hydrogen Energy, 2018, 43, 15030–15054 CrossRef CAS.
- K. S. Baamran, M. Tahir, M. Mohamed and A. Hussain Khoja, J. Environ. Chem. Eng., 2020, 8, 103604 CrossRef CAS.
- E. Clavijo, J. R. Menéndez and R. Aroca, J. Raman Spectrosc., 2008, 39, 1178–1182 CrossRef CAS.
- A. R. Gargaro, L. D. Barron and L. Hecht, J. Raman Spectrosc., 1993, 24, 91–96 CrossRef CAS.
- S. Wartewig, IR and Raman Spectroscopy, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2003 Search PubMed.
- C. H. Chuang and Y. T. Chen, J. Raman Spectrosc., 2009, 40, 150–156 CrossRef CAS.
- R. Jaquet and H. Haeuseler, J. Raman Spectrosc., 2008, 39, 599–606 CrossRef CAS.
- S. Wartewig, IR and Raman Spectroscopy, 2004, pp. 27–33 Search PubMed.
- W. J. Jang, J. O. Shim, H. M. Kim, S. Y. Yoo and H. S. Roh, Catal. Today, 2019, 15–26 CrossRef CAS.
- B. Steinhauer, M. R. Kasireddy, J. Radnik and A. Martin, Appl. Catal., A, 2009, 366, 333–341 CrossRef CAS.
- A. S. Al-Fatesh, M. A. Naeem, A. H. Fakeeha and A. E. Abasaeed, Chin. J. Chem. Eng., 2014, 22, 28–37 CrossRef CAS.
- M. A. Naeem, A. S. Al-Fatesh, W. U. Khan, A. E. Abasaeed and A. H. Fakeeha, Int. J. Chem. Eng. Appl., 2013, 4, 315–320 CAS.
- H. Y. Wang and E. Ruckenstein, Appl. Catal., A, 2000, 204, 143–152 CrossRef CAS.
- J. D. A. Bellido, J. E. De Souza, J. C. M'Peko and E. M. Assaf, Appl. Catal., A, 2009, 358, 215–223 CrossRef CAS.
- H. S. Roh and K. W. Jun, Catal. Surv. Asia, 2008, 12, 239–252 CrossRef CAS.
- L. A. Schulz, L. C. S. Kahle, K. H. Delgado, S. A. Schunk, A. Jentys, O. Deutschmann and J. A. Lercher, Appl. Catal., A, 2015, 504, 599–607 CrossRef CAS.
- A. Rathi, P. D. Babu, P. K. Rout, V. P. S. Awana, V. K. Tripathi, R. Nagarajan, B. Sivaiah, R. P. Pant and G. A. Basheed, J. Magn. Magn. Mater., 2019, 474, 585–590 CrossRef CAS.
- F. Mirzaei, M. Rezaei, F. Meshkani and Z. Fattah, J. Ind. Eng. Chem., 2015, 21, 662–667 CrossRef CAS.
- C. Shi and P. Zhang, Appl. Catal., B, 2015, 170–171, 43–52 CrossRef CAS.
- S. Sengupta and G. Deo, J. CO2 Util., 2015, 10, 67–77 CrossRef CAS.
- J. Á. E. LauBomfim, J. F. S. C. Filho, T. D. Bezerra, F. C. Rangela, T. A. Simões, P. N. Romano, R. S. Cruza, Heterogeneous Catalysis. Materials and Applications, Elsevier, 2022, pp. 175–206 Search PubMed.
- I. Luisetto, S. Tuti and E. Di Bartolomeo, Int. J. Hydrogen Energy, 2012, 37, 15992–15999 CrossRef CAS.
- D. G. Araiza, D. G. Arcos, A. Gómez-Cortés and G. Díaz, Catal. Today, 2021, 360, 46–54 CrossRef CAS.
- B. Li, X. Xu and S. Zhang, Int. J. Hydrogen Energy, 2013, 38, 890–900 CrossRef CAS.
- R. A. El-salamony, A. M. A. El Naggar, D. R. A. El-hafiz and A. M. Al-sabagh, Int. J. Energy Res., 2021, 45, 3899–3912 CrossRef CAS.
- M. Taghizadeh and M. H. Abbandanak, Int. J. Hydrogen Energy, 2022, 47, 16335–16710 CrossRef.
- K. Li, X. Chang, C. Pei, X. Li, S. Chen, X. Zhang, S. Assabumrungrat, Z. J. Zhao, L. Zeng and J. Gong, Appl. Catal., B, 2019, 259, 118092 CrossRef CAS.
- S. Moogi, I. G. Lee and J. Y. Park, Int. J. Hydrogen Energy, 2019, 44, 29537–29546 CrossRef CAS.
- B. Bachiller-Baeza, C. Mateos-Pedrero, M. A. Soria, A. Guerrero-Ruiz, U. Rodemerck and I. Rodríguez-Ramos, Appl. Catal., B, 2013, 129, 450–459 CrossRef CAS.
- Z. Zhang, G. Zhao, G. Bi, Y. Guo and J. Xie, Fuel Process. Technol., 2021, 212, 106627 CrossRef CAS.
- M. A. Goula, N. D. Charisiou, G. Siakavelas, L. Tzounis, I. Tsiaoussis, P. Panagiotopoulou, G. Goula and I. V. Yentekakis, Int. J. Hydrogen Energy, 2017, 42, 13724–13740 CrossRef CAS.
- S. Sokolov, E. V. Kondratenko, M. M. Pohl and U. Rodemerck, Int. J. Hydrogen Energy, 2013, 38, 16121–16132 CrossRef CAS.
- E. Ruckenstein and Y. H. Hu, Appl. Catal., A, 1999, 183, 85–92 CrossRef CAS.
- J. y. Jing, Z. h. Wei, Y. b. Zhang, H. c. Bai and W. y. Li, Catal. Today, 2020, 356, 589–596 CrossRef CAS.
- B. Jin, S. Li and X. Liang, Fuel, 2021, 284, 119082 CrossRef CAS.
- K. Li, C. Pei, X. Li, S. Chen, X. Zhang, R. Liu and J. Gong, Appl. Catal., B, 2020, 264, 118448 CrossRef CAS.
- K. Jabbour, P. Massiani, A. Davidson, S. Casale and N. El Hassan, Appl. Catal., B, 2017, 201, 527–542 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2023 |