
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAntiviral activity of amide-appended α-hydroxytropolones against herpes simplex virus-1 and -2†
Andreu Gazquez Casals‡
a,
Alex J. Berkowitz‡bc,
Alice J. Yua,
Hope E. Watersa,
Daniel V. Schiavonebc,
Diana M. Kapkayevab,
Lynda A. Morrison*a and
Ryan P. Murelli *bcd
*bcd
aDepartment of Molecular Microbiology and Immunology, Saint Louis University School of Medicine, St. Louis, MO, USA. E-mail: andreu.gazquez@health.slu.edu; alice.yu@slu.edu; hope.waters@slu.edu; lynda.morrison@health.slu.edu
bDepartment of Chemistry, Brooklyn College, The City University of New York, Brooklyn, NY, USA. E-mail: alex.berkowitz@brooklyn.cuny.edu; daniel.schiavone@brooklyn.cuny.edu; kapkayeva@gmail.com
cPhD Program in Chemistry, The Graduate Center, The City University of New York, New York, NY, USA
dPhD Program in Biochemistry, The Graduate Center, The City University of New York, New York, NY, USA
First published on 15th March 2023
Abstract
α-Hydroxytropolones (αHTs) have potent antiviral activity against herpes simplex virus-1 and -2 (HSV-1 and HSV-2) in cell culture, including against acyclovir-resistant mutants, and as a result have the potential to be developed as antiviral drugs targeting these viruses. We recently described a convenient final-step amidation strategy to their synthesis, and this was used to generate 57 amide-substituted αHTs that were tested against hepatitis B virus. The following manuscript describes the evaluation of this library against HSV-1, as well as a subset against HSV-2. The structure–function analysis obtained from these studies demonstrates the importance of lipophilicity and rigidity to αHT-based anti-HSV potency, consistent with our prior work on smaller libraries. We used this information to synthesize and test a targeted library of 4 additional amide-appended αHTs. The most potent of this new series had a 50% effective concentration (EC50) for viral inhibition of 72 nM, on par with the most potent αHT antivirals we have found to date. Given the ease of synthesis of amide-appended αHTs, this new class of antiviral compounds and the chemistry to make them should be highly valuable in future anti-HSV drug development.
Introduction
More than half of the U.S. population suffers from infection with herpes simplex virus (HSV-1),1 and two thirds of people worldwide.2 This large double-stranded DNA virus replicates in the oral or corneal epithelium, causing painful ulcerative lesions. HSV-1 is also beginning to supplant the related HSV-2 as the principal cause of sexually transmitted ulcerative disease of the genital epithelium.3 HSV enters sensory nerve endings innervating the epithelia and establishes life-long latent infection in nerve cell bodies within sensory ganglia. Periodic reactivations lead to reappearance of disease in the epithelium, which manifests as cold sores, sight-threatening keratitis or, in the rare event of virus spread into the central nervous system, potentially lethal encephalitis. In addition, babies born to mothers shedding virus from the genital epithelium are at risk for disseminated HSV infection that induces high rates of morbidity and mortality.4 Although anti-HSV drug therapies exist they are incompletely effective5 and can drive emergence of resistant viruses.6 As a result, there is substantial interest in developing new and novel antivirals for HSV-1.7 Some of the novel enzyme and protein targets for HSV drug development include helicase–primase,8 thymidine kinase,9 and glycoprotein B.10HSV-1 has a variety of divalent magnesium-based nucleases that are important for replication and infection and could be targeted by molecules possessing metal-binding fragments.11 One such family of molecules possessing a metal-binding fragment is α-tropolones,12 which have been of broad interest in drug development over the last few decades.13 α-Tropolones are known to be potent inhibitors of HSV-1 and -2,14 and this activity may be due to viral nuclease inhibition. For example, a sub-class of tropolones called α-hydroxytropolones (αHTs, e.g. 5, Scheme 1)15 are known to inhibit two magnesium-dependent HSV-1 enzymes, UL15 (ref. 16) and UL12,17 that are attractive targets for antiviral development. pUL15 is essential for viral DNA packaging,18 and disruption of its cytomegalovirus homolog pUL89 was validated clinically by letermovir prevention of infection.19 Meanwhile, HSV-1 pUL12 has alkaline nuclease activity essential for in vivo function.20
 | ||
| Scheme 1 Synthetic routes to ketone and amide-appended αHTs. | ||
One challenge associated with developing αHTs as drugs has been the scarcity of synthetic methods available to make structural variants of them,21 and up until very recently22 all synthetic chemistry-driven structure–function studies on the chemotype relied on direct modification of either 3,7-dihydroxytropolone23 or manicol.24 Of late, oxidopyrylium cycloaddition/ring-opening strategies have emerged that are highly effective for αHTs synthesis25 and the strategy has been exploited in various medicinal chemistry-based pursuits, including against HSV-1 and -2.26 One key study focused on ketone-appended αHTs, which demonstrated a strong correlation between lipophilicity and anti-HSV efficacy27 and led to the discovery of a molecule with an EC50 of 60 nM (5, Scheme 1). The synthesis of this ketone library was not ideal for generating libraries of molecules, however, as each appendage often had to be tracked back to its associated ynone, which itself would often have to be made in one or two steps (top, Scheme 1). Thus, for each new αHT that is desired, as many as 5 steps are needed from the nearest common starting material. In addition, the final steps of the synthetic route are extremely harsh (usually triflic acid followed by hydrobromic acid in acetic acid, as shown), which limits the types of groups that can be installed. Thus, in pursuit of strategies more conducive to αHT library synthesis, amide-appended αHTs have been explored, which can be made through a final-step amidation strategy (bottom, Scheme 1).28 A major advantage of this approach is that diversity is generated in the final step, allowing for larger and more diverse libraries. Consequently, a library of 57 amide-appended αHTs was recently generated and assessed against hepatitis B virus, Cryptococcus neoformans, and various bacteria.29 Given the potential of αHTs as a valuable chemotype for anti-HSV development, and the practical benefits to amide-appended αHTs for library synthesis, studies on the anti-HSV activity of amide-appended αHTs was pursued.
Results and discussion
57 amide-appended αHTs previously reported29 were assessed for capacity to suppress viral replication at 50 μM and 5 μM, and active compounds (along with several inactive compounds) were also tested at 1 μM (Table 1). For clarity, these are reported based upon class through which they are subdivided. Thus, those derived from aniline are reported in Table 1A, benzylamine in Table 1B, piperidine/piperazine in Table 1C, amino acids in Table 1D, and a series of miscellaneous compounds that do not fall within any of these categories are in Table 1E. For further assistance with analysis, these are ordered based upon lipophilicity measurement (c![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) logP, determined with ChemDraw Professional, Version 16.0),30 which is calculated in the monoanionic form that is expected at physiological pH31.
logP, determined with ChemDraw Professional, Version 16.0),30 which is calculated in the monoanionic form that is expected at physiological pH31.
| Entry | Amide (#) name | HSV-1 log suppression | Lipophilicity | ||
|---|---|---|---|---|---|
| 50 μM | 5 μM | 1 μM | clogP |
||
| (A) Aniline-based amides | |||||
| 1 |  |
+ | − | − | −3.28 |
| 2 |  |
5.98 | 0.43 | nd | −2.97 |
| 3 |  |
5.55 | 5.23 | 0 | −1.87 |
| 4 | 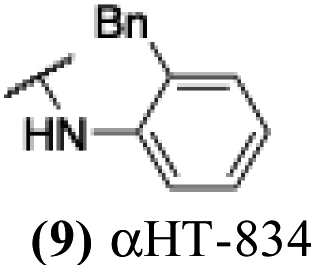 |
4.67 | 1.8 | − | −1.77 |
| 5 | 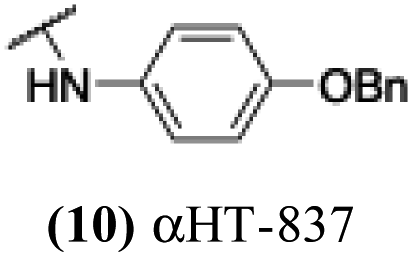 |
4.88 | 4.09 | − | −1.28 |
| 6 | 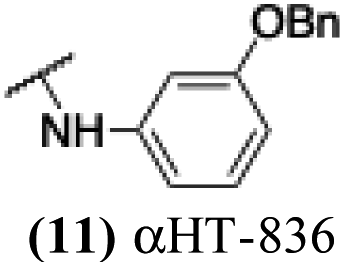 |
4.35 | 5.2 | 3.15 | −1.28 |
| 7 | 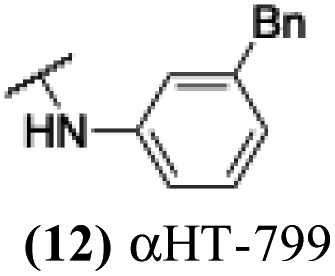 |
+ | 5.48 | 1.96 | −0.9 |
| 8 | 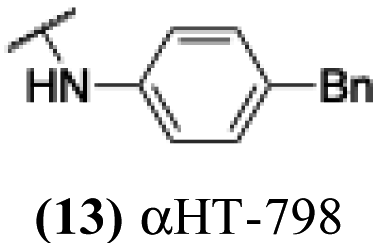 |
+ | 5.61 | 1.82 | −0.9 |
|
|||||
| (B) Benzyl amine-based amide | |||||
| 9 |  |
+ | − | − | −2.87 |
| 10 |  |
5.23 | 0.05 | nd | −2.79 |
| 11 | 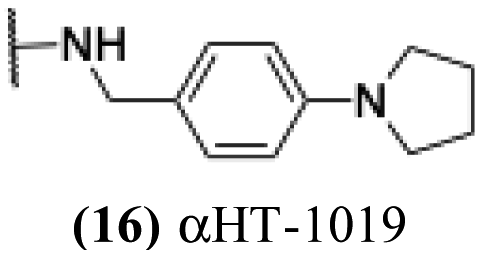 |
5.11 | 2.29 | 0.29 | −2.51 |
| 12 | 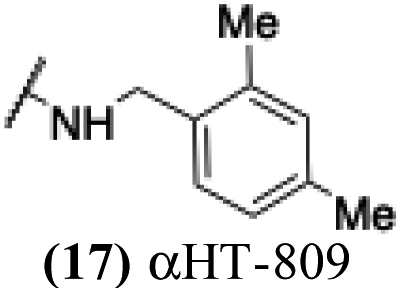 |
+ | 2.78 | 0 | −1.84 |
| 13 | 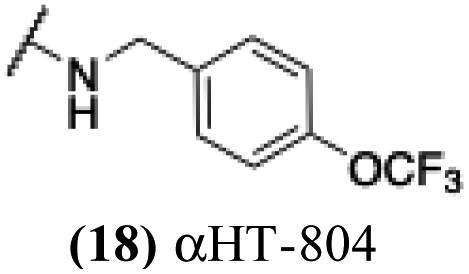 |
+ | 1.23 | −0.14 | −1.76 |
| 14 | 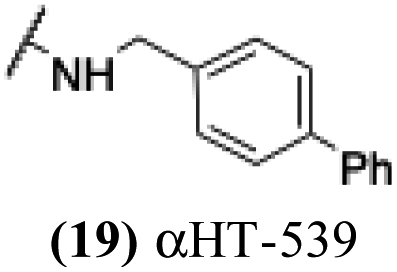 |
T | 5.29 | 4 | −0.9 |
| 15 | 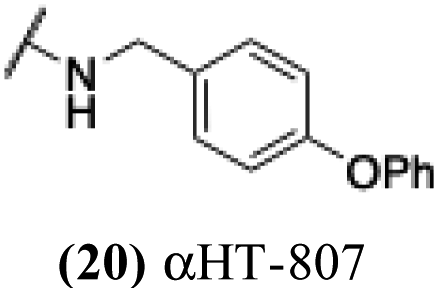 |
+ | 5.73 | 1.4 | −0.69 |
|
|||||
| (C) Piperazine and piperidine-based amide | |||||
| 16 | 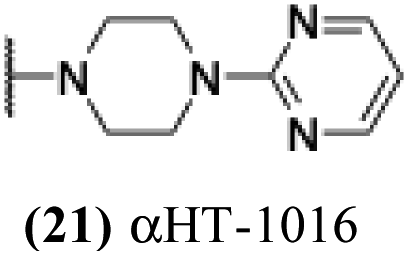 |
5.35 | 0.73 | nd | −5.51 |
| 17 | 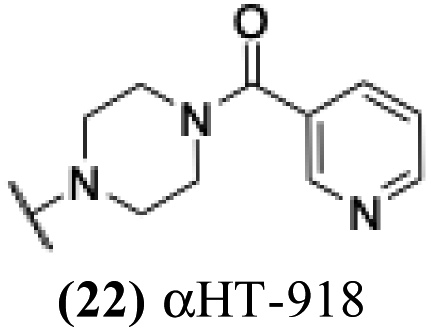 |
1.01 | 0.19 | 0.02 | −4.83 |
| 18 |  |
2.32 | 0.29 | nd | −4.2 |
| 19 |  |
+ | − | − | −4.16 |
| 20 | 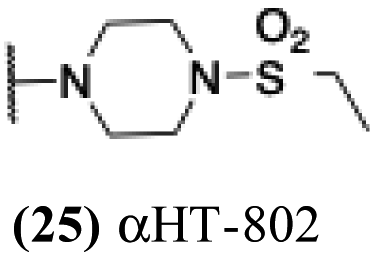 |
+ | − | − | −3.97 |
| 21 | 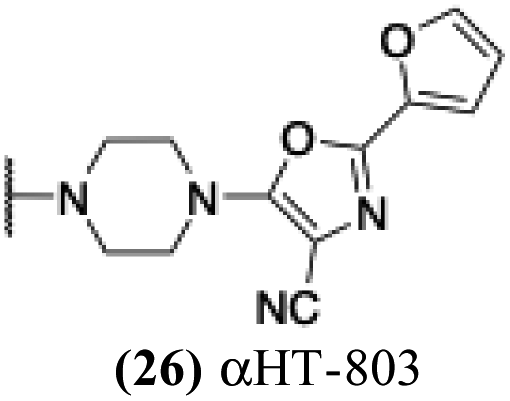 |
+ | − | − | −3.72 |
| 22 | 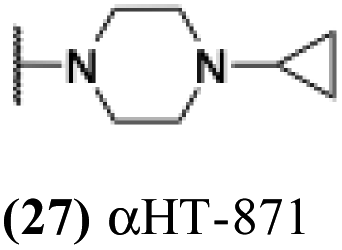 |
0.81 | −0.02 | −0.1 | −3.57 |
| 23 | 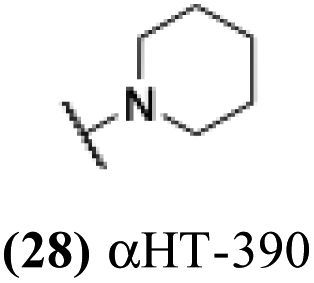 |
5.75 | 0.52 | nd | −3.48 |
| 24 | 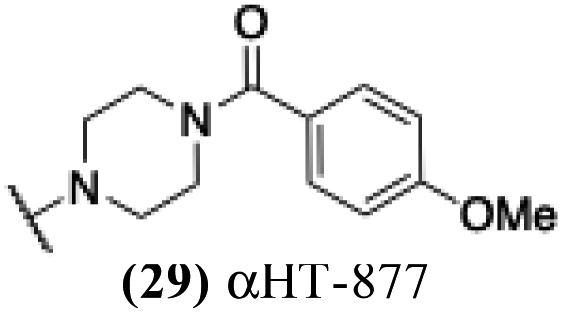 |
4.21 | 0.17 | −0.24 | −3.42 |
| 25 | 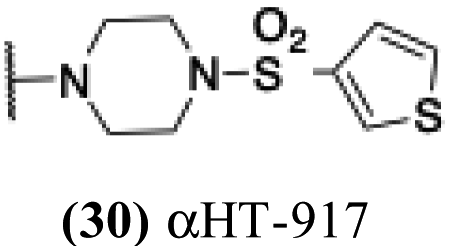 |
5.04 | 0.57 | 0.06 | −3.18 |
| 26 | 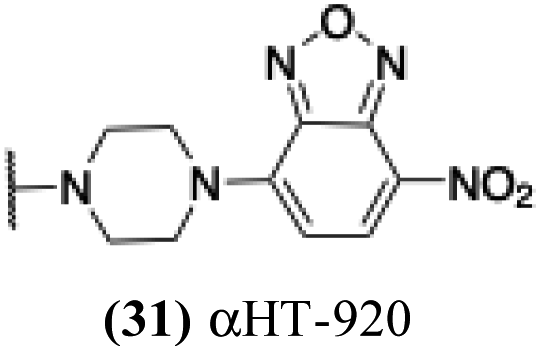 |
1.49 | −0.16 | 0.1 | 3.15 |
| 27 | 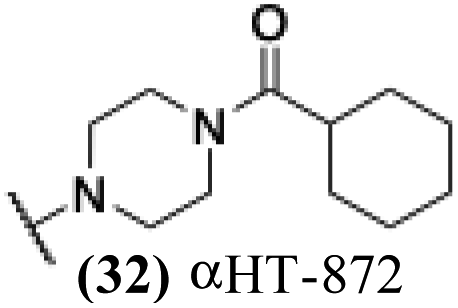 |
7.04 | 0.64 | −0.03 | 2.89 |
| 28 | 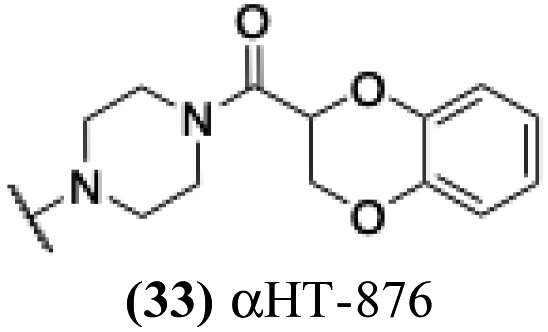 |
6.92 | 0.28 | −0.07 | 2.83 |
| 29 | 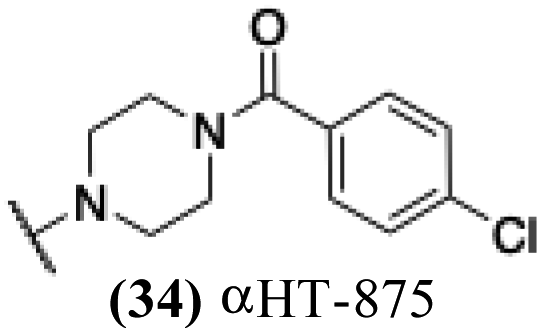 |
6.92 | 1.32 | −0.25 | −2.62 |
| 30 | 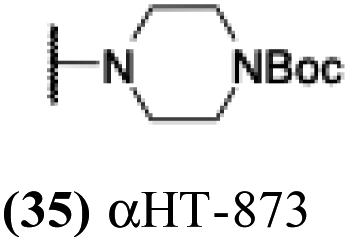 |
6.21 | 0.2 | −0.15 | −2.53 |
| 31 | 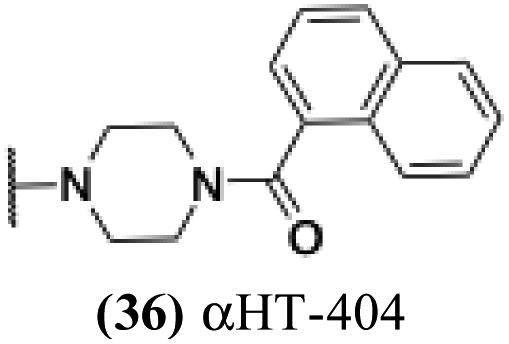 |
2.18 | − | − | −2.163 |
| 32 | 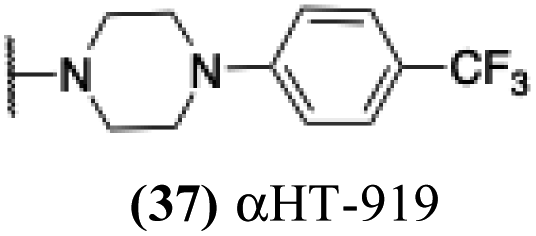 |
5.28 | 6.07 | 0.84 | −1.63 |
| 33 | 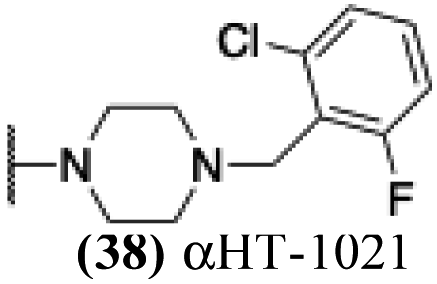 |
5.84 | 1.77 | 0.23 | −1.36 |
| 34 | 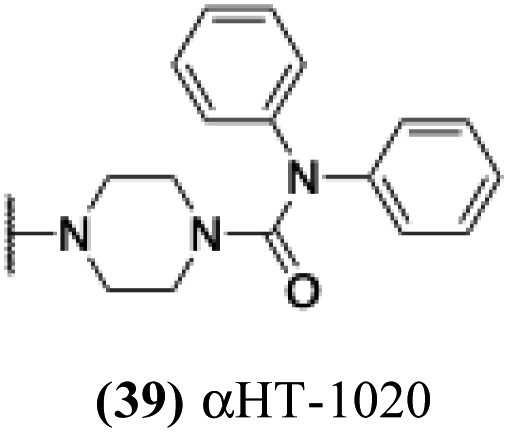 |
5.21 | 0.36 | nd | −1.17 |
| 35 | 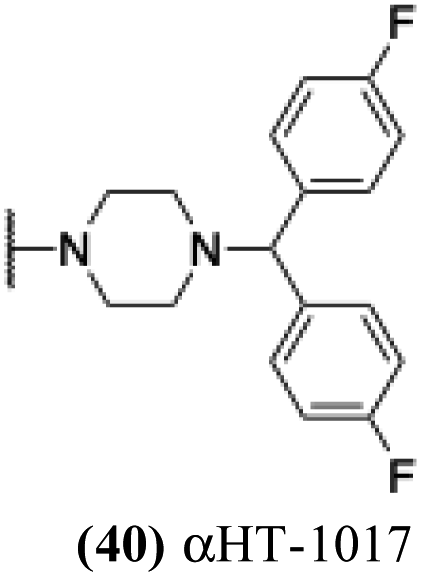 |
6.48 | 5.49 | 5.03 | −0.59 |
|
|||||
| (D) Amino acid-based amide | |||||
| 36 | 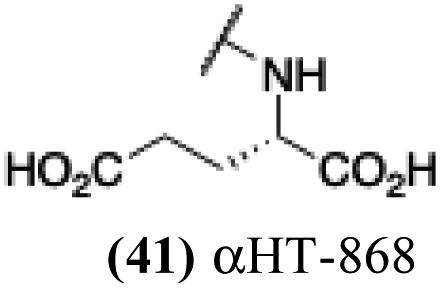 |
0.26 | 0.04 | −0.15 | −5.54 |
| 37 | 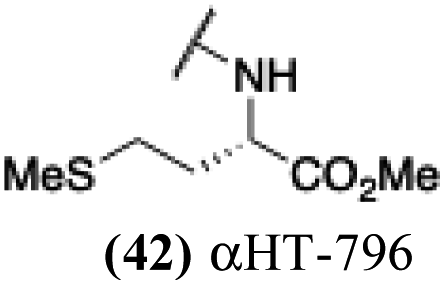 |
+ | − | − | −4.29 |
| 38 | 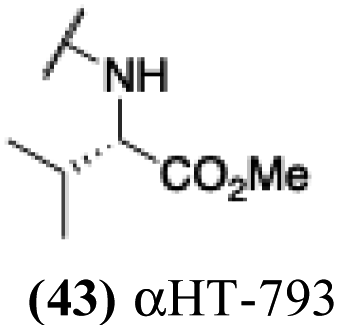 |
+ | −0.41 | − | −3.51 |
| 39 | 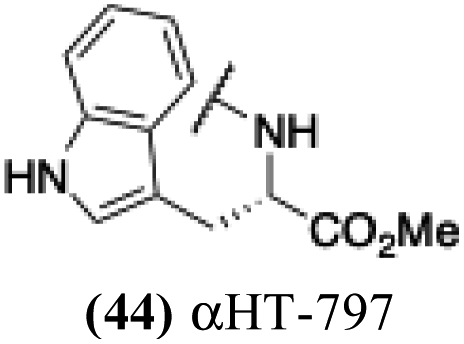 |
+ | − | − | −3.03 |
| 40 | 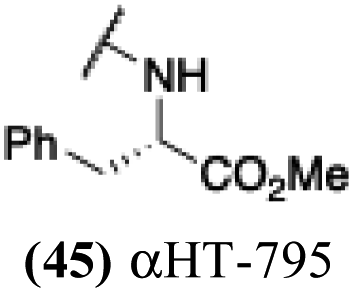 |
+ | − | − | −3.02 |
| 41 | 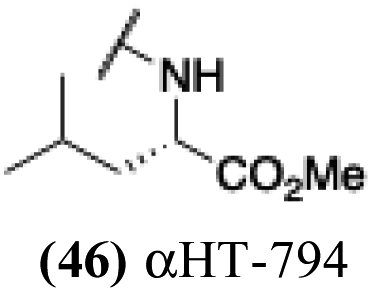 |
+ | − | − | −2.99 |
| 42 | 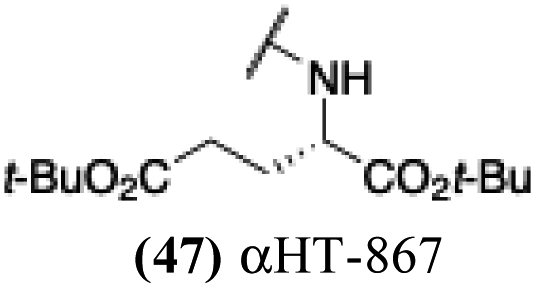 |
5.21 | 5.91 | 0.16 | −2.48 |
| 43 | 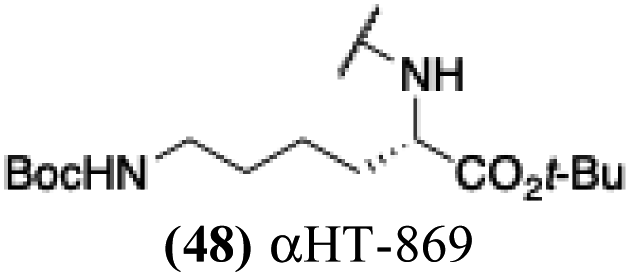 |
5.49 | −0.14 | nd | −1.76 |
| 44 | 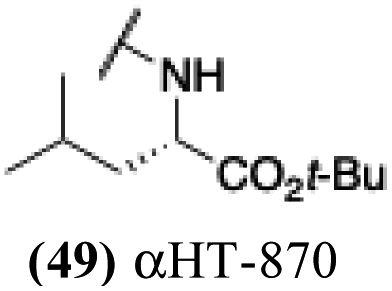 |
5.34 | −0.81 | nd | −1.75 |
|
|||||
| (E) Other amides | |||||
| 45 | 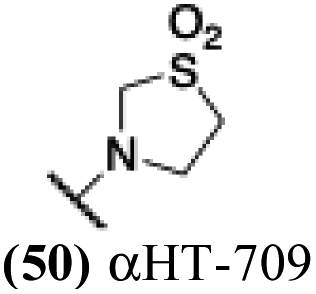 |
− | − | − | −5.09 |
| 46 | 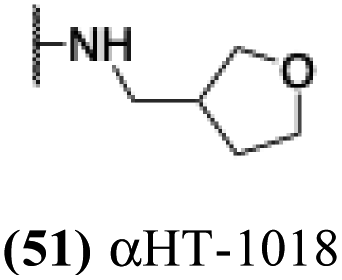 |
0.51 | −0.03 | nd | −4.93 |
| 47 | 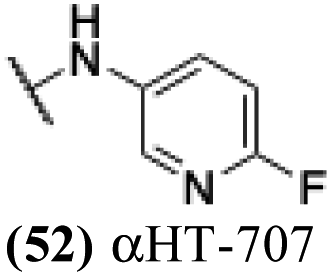 |
− | − | − | −4.32 |
| 48 | 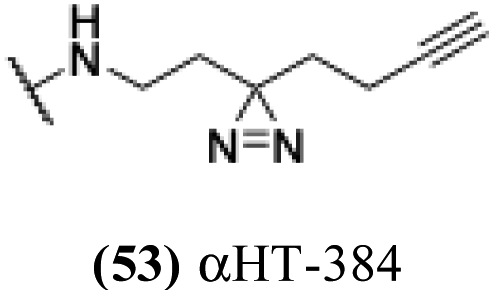 |
4.49 | 1.04 | 0.36 | −4.19 |
| 49 |  |
+ | − | − | −3.73 |
| 50 |  |
+ | − | − | −3.7 |
| 51 |  |
4.57 | 2.95 | 0.23 | −3.17 |
| 52 | 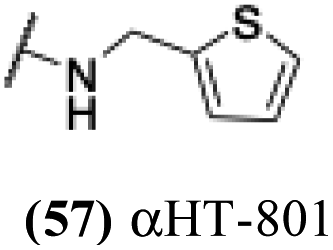 |
+ | − | − | −3.14 |
| 53 | 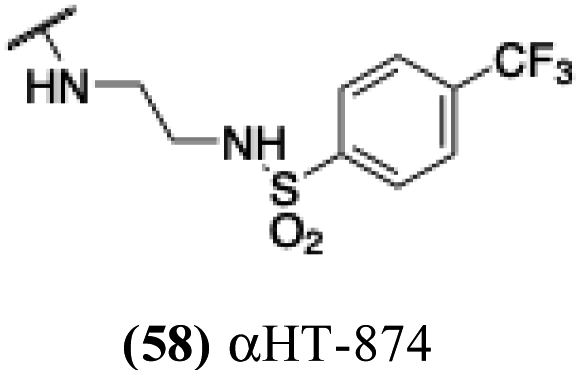 |
2.41 | −0.15 | nd | −2.72 |
| 54 | 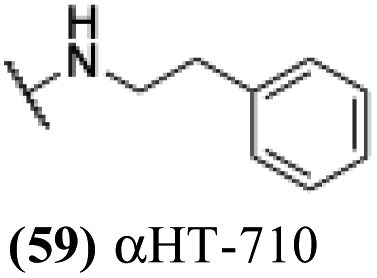 |
4.86 | 0.04 | 0.06 | −2.66 |
| 55 | 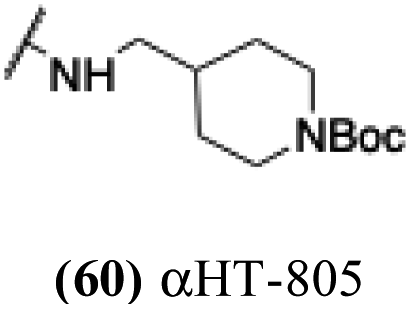 |
+ | − | − | −2.59 |
| 56 | 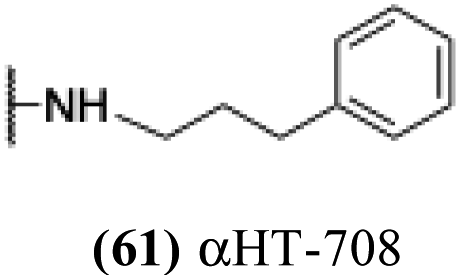 |
+ | − | − | −2.28 |
| 57 | 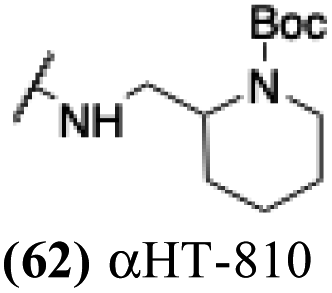 |
+ | − | − | −1.73 |
Lipophilicity was important for activity against HSV-1. The vast majority of molecules were capable of substantial inhibition of HSV-1 replication at a concentration of 50 μM; only 5 of the 57 molecules tested did not suppress the virus by at least 10-fold (i.e., log10 > 1.0) at that concentration (see entries 22, 36 and 45–47, Table 1). At the lower concentration of 5 μM, however, less than half of the library was capable of suppressing, and lipophilicity highly correlated with this activity (Fig. 1A). Specifically, only two out of the 32 molecules with clogP values under −3.0 showed any appreciable antiviral activity (log10 > 1.0) at 5 μM. Conversely, all 5 molecules with clogP values greater than −1.0 showed strong viral suppression (log10 > 3.0). When evaluating molecules at 1 μM, the lipophilicity threshold for suppression log10 > 1.0 increased to a clogP of roughly −1.3 (Fig. 1B). Specifically, all of the 32 molecules tested at this concentration that had a clogP less than −1.3 suppressed viral replication less than 1.0log10. Meanwhile, all 6 molecules with clogP values greater than −1.3 inhibited replication by at least 1.0log10. None of these 6 molecules demonstrated any significant anti-hepatitis B virus activity in prior studies,29 with EC50 values (1.7–5.6 μM) comparable to their host cell-specific cytotoxicity (CC50 = 3.3–8.7 μM). Furthermore, of the 16 αHT amides with previously reported hepatitis B antiviral EC50 values below 1 μM, only one (53, αHT-384) suppressed HSV-1 replication by 10 fold at 5 μM. Thus, the antiviral activity has some virus-based selectivity.
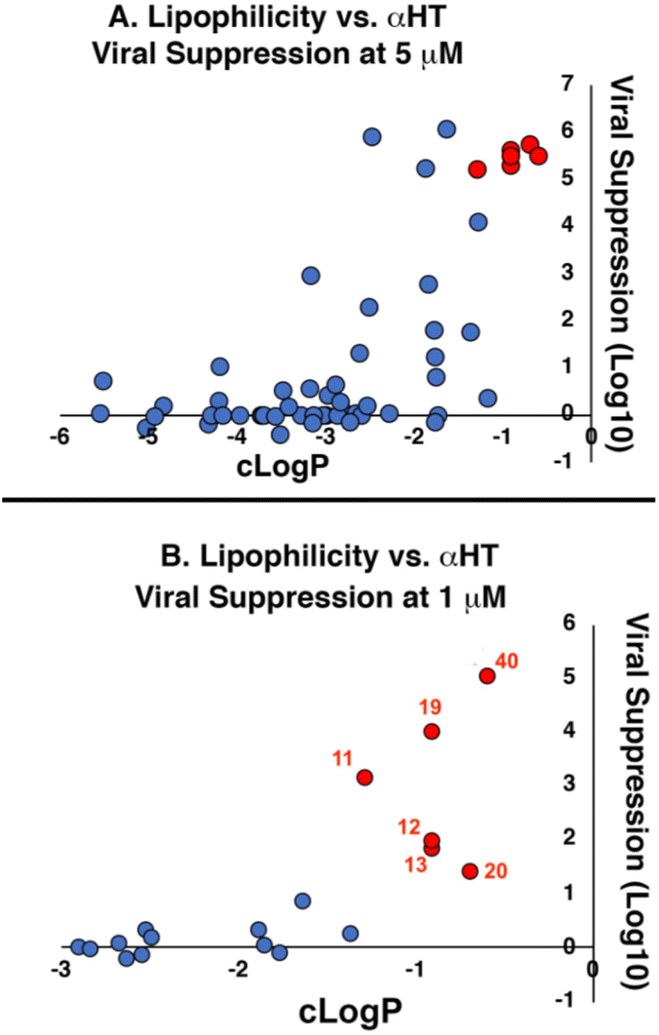 | ||
| Fig. 1 Correlation between lipophilicity and antiviral activity of amide-appended αHTs at (A) 5 μM and (B) 1 μM. Red-shaded points in (A) correspond to 6 molecules shaded and numbered red in (B). Molecules showing no viral suppression but not quantitated (‘−’ in Table 1) are denoted as ‘0’ for graphical purposes. | ||
These 6 molecules with c log P values greater than −1.3, as well as 9 other αHTs, were also tested against HSV-2. Their ability to inhibit HSV-2 replication at 5 μM or 1 μM tracked closely with the molecules' activity against HSV-1 (Fig. 2A vs. Fig. 2B). For example, 3 of the 6 potent inhibitors of HSV-1 at 1 μM were capable of inhibiting HSV-2 replication at the same concentration. Only 1 molecule tested did not inhibit HSV-1 replication at 1 μM but could inhibit HSV-2 at that concentration (9, αHT-834, Fig. 2B). Thus, amide-appended αHTs are also capable of potently suppressing replication of the closely related HSV-2. In addition, synthetic αHTs can inhibit drug-resistant HSV mutants.6d To ensure that amide-appended αHTs also inhibit drug-resistant virus, 3 of the 6 potently inhibitory molecules (19, αHT-539; 40, αHT-1017; and 20, αHT-807) were tested against a mutant of HSV-1 resistant to acyclovir (ACV) (Fig. 2C). Whereas ACV inhibited only the wild-type virus, the three amide-appended αHTs strongly inhibited both wild-type and ACV-resistant viruses. This result suggests amide-appended αHTs could eventually be therapeutically useful in patients with nucleoside analogue-resistant infections.
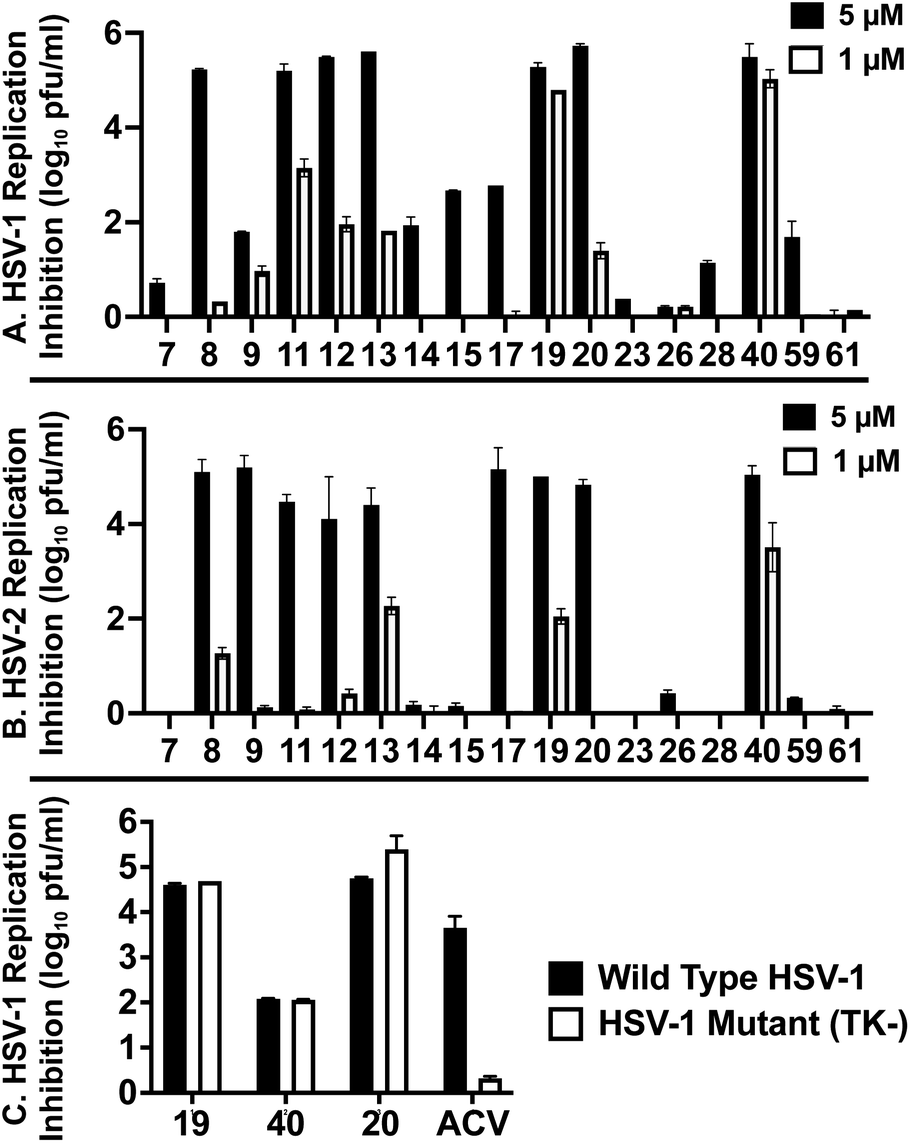 | ||
| Fig. 2 HSV replication inhibition at constant concentration. HSV-1 (A) vs. HSV-2 (B) replication inhibition of select αHTs. (C) Inhibition of αHTs and acyclovir (ACV) against wild-type HSV-1 and an acyclovir-resistant mutant (TK−). Data shown are the average and standard deviation of duplicate wells from a representative experiment. | ||
The 6 most potent molecules were also evaluated more quantitatively to determine EC50 and CC50 values (Table 2A). The two most powerful inhibitors, αHT-798 and αHT-539 (13 and 19, entries 3 and 4), were isomeric, and both possessed para substitution patterns. The only difference was the placement of the methylene linker on one side of the aryl ring or the other. αHT-799 (12), another isomer, was 6-fold less potent (entry 2). Thus, placements of substituents on the ring appear to be important for the higher activity. This trend for para preference was also observed with ether-containing molecules αHT-807 (20) and αHT-836 (11), although these were closer in activity to one another (entries 5 and 1). Finally, αHT-1017 (40) was the only piperazine-containing molecule to be among the most potent molecules (entry 6), and also showed potency on par with αHT-798 (13) and αHT-539 (19). Some modest cytotoxicity was observed against the host cell line, Vero, after 48 h for some of the molecules. However, even in these instances, selectivity indexes were all in excess of 100.
| Entry | Amide (#) name | HSV-1 EC50 (μM) ± SD | Vero CC50 (μM) ± SD | Selectivity CC50/EC50 | Lipophilicity (clogP) |
|---|---|---|---|---|---|
| (A) Top molecules to emerge from repurposing studies on 57 amide αHTs | |||||
| 1 | 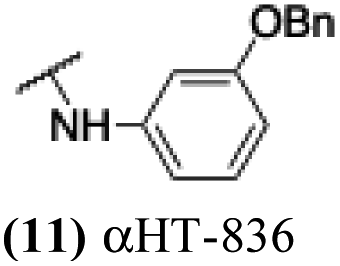 |
0.824 ± 0.264 | 29.6 ± 5.2 | 36 | −1.28 |
| 2 | 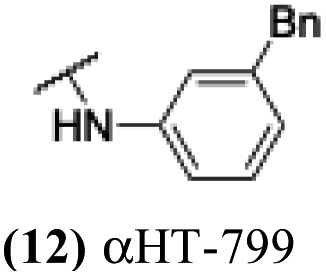 |
1.274 ± 0.227 | >100 | >83 | −0.9 |
| 3 | 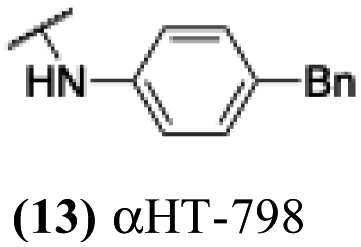 |
0.217 ± 0.048 | 31.6 ± 6.1 | 140 | −0.9 |
| 4 | 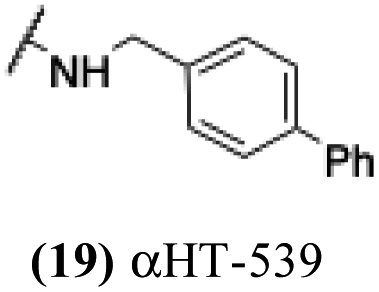 |
0.251 ± 0.059 | 90.2 ± 7.4 | 360 | −0.9 |
| 5 | 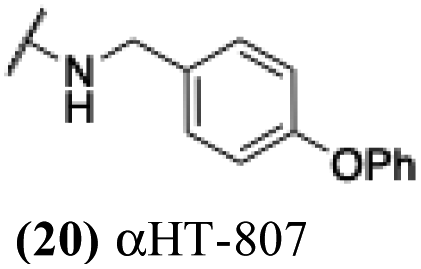 |
0.462 ± 0.106 | >100 | >213 | −0.69 |
| 6 | 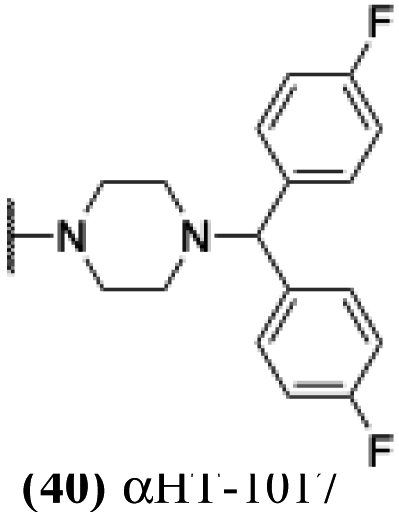 |
0.326 ± 0.024 | 49.5 ± 7.8 | 152 | −0.59 |
|
|||||
| (B) Newly created amide αHTs with increased lipophilicity | |||||
| 7 | 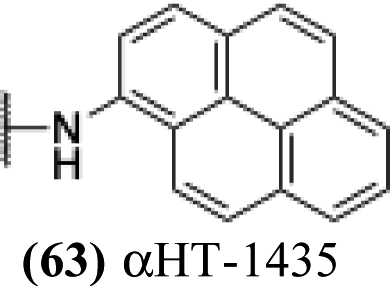 |
0.244 ± 0.097 | 74.2 ± 8.7 | 304 | −0.157 |
| 8 | 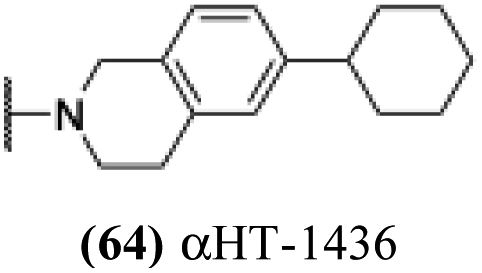 |
0.072 ± 0.021 | 75.7 ± 5.3 | 1050 | 0.425 |
| 9 | 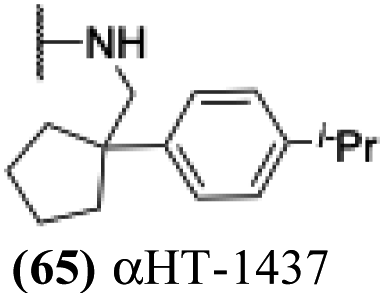 |
0.306 ± 0.016 | 92.5 ± 7.5 | 302 | 0.322 |
| 10 | 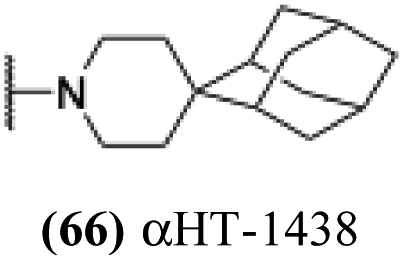 |
0.296 ± 0.054 | 23.7 ± 3.8 | 80 | 0.184 |
Finally, with confirmation that amide-appended αHTs could serve as potent anti-HSV agents, and close correlation between this activity and lipophilicity, we sought to make a targeted library of 4 αHTs with enhanced lipophilicity (Table 2B). In addition, 3 of the molecules were designed to have limited rotational freedom (entries 7, 8 and 10). Consistent with our expectation that these would be potent antivirals against HSV-1, all 4 molecules inhibited viral replication > 1.0log10 at a 1 μM concentration (data not shown). The most potent of the molecules tested was αHT-1436 (64, entry 8), with an EC50 measurement of 72 nM, in line with the most potent anti-HSV αHT we have described to date (5, αHT-360, Fig. 1). Cytotoxicity after 48 h remained low, providing a selectivity index for αHT-1436 (64) of over 1000. αHT-1436 (64) was the most lipophilic molecule tested, but was also highly rigid, with only a single rotatable bond on its amide appendage.
Structure–function studies on amide-appended αHTs provide additional data that demonstrates a close correlation between αHT lipophilicity and their potency against HSV. It remains unclear why this is the case, although when considering highly lipophilic anti-HSV agents, one must note 1-docosanol, an active ingredient in the over-the-counter cold sore medication Abreva. It is believed that the activity of 1-docosanol may be related to its disruption of viral fusion with the cell membrane,32 though we have previously demonstrated no impact of αHTs on viral infectivity.14 A close correlation also exists between lipophilicity and anti-HSV activity of simple alkyl gallate derivatives, which have a 3-oxygen motif analogous to the αHTs.33 Interest in the anti-HSV activity of gallates stems from the potent inhibitory activity of the epigallocatechin-3-O-gallate and its structural derivatives, which have multiple mechanisms of action including fusion inhibition, and they are not known to inhibit the activity of either pUL12 or pUL15.34 There is little structure–function analysis for pUL12 or pUL15 inhibition outside of the tropolones. The pUL12 inhibitors emodin35 and 1,2 benzisothiazolin-3-one-type36 are the only non-αHT molecules known to inhibit either enzyme, and no studies have been published to date detailing synthetic chemistry-driven, targeted structure–function studies on these molecules. As a result, the potent anti-HSV activity of amide-appended αHTs – with cell-based antiviral activity on par or exceeding the clinically used anti-HSV drug acyclovir (ECHSV-150 = 170 nM, ECHSV-250 = 1.4 μM)26 – coupled with our rapid strategy for synthesizing them, should be highly valuable in novel anti-HSV therapeutic development.
Conclusion
Several amide-appended αHTs have been identified with activity profiles on par with the most potent anti-HSV-1 tropolones described to date. Given the ease of their synthesis, this class warrants further investigation for HSV-1 and -2 antiviral development. Furthermore, given the broad range of biological activity and therapeutic potential of the αHT chemotype, amide-appended αHTs could be highly useful for a broader range of drug-development pursuits. For example, recent studies have shown that certain αHTs possess antiviral activity against Rift Valley fever virus, and amide-appended αHTs 1017 (40) 867 (47) and 1039 (56) were some of the more potent compounds.37Materials and methods
General information
All starting materials and reagents were purchased from commercially available sources and used without further purification, with the exception of THF, which was purified on a solvent purification system prior to the reaction. 1H NMR shifts are measured using the solvent residual peak as the internal standard (CDCl3 δ 7.26, THF-d8 δ 3.58), and reported as follows: chemical shift, multiplicity (s = singlet, br s = broad singlet, d = doublet, t = triplet, p = pentet, dd = doublet of doublets, hept = heptet, q = quartet, m = multiplet), coupling constant (Hz), and integration. Microwave reactions were performed via the Biotage Initiator 2.5. Purification via column chromatography was performed on the Biotage Isolera Prime, with Biotage SNAP 12 g C18 cartridges, in a solvent system of acetonitrile (MeCN) and water (H2O), with each containing 0.05% trifluoroacetic acid (TFA). Column gradients are measured in terms of column volumes (CV). Other abbreviations used: THF = tetrahydrofuran; DMF = dimethylformamide; DMSO = dimethyl sulfoxide; HATU = 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate.Amide-appended αHT synthesis
All αHTs described in Table 1 were obtained as described previously.27,28 αHT-1435–1438 (63–66) were synthesized in an analogous fashion and a description of their synthesis follows:Cells and viruses
Vero cells were originally obtained from D. Knipe and were maintained in Dulbecco's modified Eagle's medium supplemented to contain 3% bovine growth serum, 3% newborn calf serum, and 100 IU per mL penicillin/0.1 mg per mL streptomycin. HSV-1 and HSV-2 strains were de-identified, minimally passaged clinical isolates obtained from the Saint Louis University Hospital Lab.HSV replication inhibition assay
Vero cells were seeded into 24-well plates and used when monolayers became confluent. Compounds were diluted in PBS supplemented to contain 2% newborn calf serum and 2 mM L-glutamine and added to cell monolayers in duplicate. HSV-2 was diluted in supplemented PBS medium and added so that the final concentrations of compound were 50 μM or 5 μM per well and of virus were 2 × 104 PFU per well. The cells were incubated at 37 °C for 1 h, the virus-containing inoculum was removed, the wells were washed once in PBS, and compound (50 μM or 5 μM) in supplemented DMEM was added. Cells were incubated at 37 °C for an additional 24 h, and the plates were then inspected by phase-contrast microscopy for toxicity. Cells in wells showing no obvious toxicity compared with DMSO-control wells were frozen at −80 °C for later determination of virus titers by standard plaque assay on Vero cells. Cell monolayers were fixed with methanol, stained with Giemsa, and plaques were visualized and counted under low power magnification. The 50% effective concentration (EC50) was determined as described above except that serial dilutions of the compounds were employed in 96-well plates. Values were calculated with GraphPad Prism using the three-parameter log(inhibitor)-versus-response algorithm with the bottom value set to zero. Representative dose–response curves can be found on Page s4 of the ESI.†Cytotoxicity assay
Vero cells were seeded in 96-well plates and incubated in DMEM as indicated above. The compounds were serially diluted in medium containing 1% DMSO and added to the cells 24 h after plating, with each concentration tested in duplicate. Twenty-four or 48 h after compound addition, 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) was added, the cultures were incubated for 90 min, and the absorbance was read at 490 nm. 50% cytotoxic concentration (CC50) values were calculated with GraphPad Prism using the four-parameter variable slope algorithm with the bottom value set to zero. Representative dose–response curves can be found on Page s5 of the ESI.†Author contributions
Conceptualization, L. A. M. and R. P. M.; chemical synthesis, A. J. B., D. V. S., D. M. K., R. P. M.; biological studies, A. G. C., A. J. Y., H. E. W., L. A. M.; writing, review and editing, L. A. M. and R. P. M. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
LAM and RPM are inventors on patents describing the HSV antiviral activity of αHTs.Acknowledgements
This research was funded by the National Institutes of Health, grant number SC1-GM111158 (R. P. M.) and the Pershing Trust (L. A. M.). The authors would like to thank David Knipe (Harvard) for providing Vero cells, and John Tavis and his lab (SLU) for help with the organization and maintenance of the α-HT library used herein.Notes and references
- R. J. Whitley, Herpes Simplex Viruses, in Fields Virology, ed. D. Knipe and P. Howley, Lippincott Williams & Wilkins, Philadelphia, PA, US, 4th edn, 2001, vol. 2, pp. 2461–2509 Search PubMed.
- C. James, M. Harfouche, N. J. Welton, K. M. Turner, L. J. Abu-Raddad, S. L. Gottlieb and K. J. Looker, Bull. World Health Organ., 2020, 98, 315–329 CrossRef PubMed.
- K. J. Looker, A. S. Magaret, K. M. Turner, P. Vickerman, S. L. Gottlieb and L. M. Newman, PLoS One, 2015, 10, e114989 CrossRef PubMed.
- (a) S. G. Pinninti and D. W. Kimberlin, Am. J. Perinatol., 2013, 30, 113–119 CrossRef PubMed; (b) N. Dabestani, D. Katz, J. Dombrowski, A. Magaret, A. Wald and C. Johnston, Sex. Transm. Dis., 2019, 46, 795–800 CrossRef PubMed.
- C. Johnston, M. Saracino, S. Kuntz, A. Magaret, S. Selke, M. L. Huang, J. T. Schiffer, D. M. Koelle, L. Corey and A. Wald, Lancet, 2012, 379, 641–647 CrossRef CAS PubMed.
- (a) S. H. James and M. N. Prichard, Curr. Opin. Virol., 2014, 8, 54–61 CrossRef CAS PubMed; (b) J. Piret and G. Boivin, Rev. Med. Virol., 2014, 24, 186–218 CrossRef CAS PubMed; (c) J. Piret and B. Guy, Antimicrob. Agents Chemother., 2011, 55, 459–472 CrossRef CAS PubMed; (d) M. J. Lee, T. H. Bacon and J. J. Leary, Clin. Infect. Dis., 2004, 39, S248–S257 CrossRef PubMed.
- (a) R. A. V. Hodge and H. J. Field, Adv. Pharmacol., 2013, 67, 1–38 CAS; (b) S. K. Mamidyala and S. V. Firestine, Expert Opin. Ther. Pat., 2006, 16, 1463–1480 CrossRef CAS; (c) K. Szczubiatka, K. Pyrc and M. Nowakowska, RSC Adv., 2016, 6, 1058–1075 RSC.
- (a) C. S. Crumpacker and P. A. Schaffer, Nat. Med., 2002, 8, 327–328 CrossRef CAS PubMed; (b) G. Kleymann, R. Fischer, U. A. K. Betz, M. Hendrix, W. Bender, U. Schneider, G. Handke, P. Eckenberg, G. Hewlett, V. Pevzner, J. Baumeister, O. Weber, K. Henninger, J. Keldenich, A. Jensen, J. Kolb, U. Bach, A. Popp, J. Mäben, I. Frappa, D. Haebich, O. Lockhoff and H. Rübsamen-Waigmann, Nat. Med., 2002, 8, 392–398 CrossRef CAS PubMed; (c) J. J. Crute, C. A. Grycon, K. D. Hargrave, B. Simoneau, A.-M. Faucher, G. Bolger, P. Kibler, M. Liuzzi and M. G. Cordingley, Nat. Med., 2002, 8, 386–391 CrossRef CAS PubMed.
- A. Manikowski, A. Verri, A. Lossani, B. M. Gebhardt, J. Gambino, F. Focher, S. Spadari and G. E. Wright, J. Med. Chem., 2005, 48, 3919–3929 CrossRef CAS PubMed.
- T. E. Antoine, P. J. Park and D. Shukla, Rev. Med. Virol., 2013, 23, 194–208 CrossRef CAS PubMed.
- (a) Z. Jiang, Q. You and X. Zhang, Eur. J. Med. Chem., 2019, 165, 172–197 CrossRef CAS PubMed; (b) S. M. A. Cohen, Acc. Chem. Res., 2017, 50, 2007–2016 CrossRef CAS PubMed.
- (a) R. A. Bentley, Nat. Prod. Rep., 2008, 25, 118–138 RSC; (b) N. Liu, W. Song and W. Tang, Tetrahedron, 2014, 70, 9281–9305 CrossRef CAS PubMed; (c) H. Guo, D. Roman and C. Beemelmanns, Nat. Prod. Rep., 2019, 36, 1137–1155 RSC.
- (a) S. N. Ononye, M. D. VanHeyst, E. Z. Oblak, W. Zhou, M. Ammar, A. C. Anderson and D. L. Wright, ACS Med. Chem. Lett., 2013, 4, 757–761 CrossRef CAS PubMed; (b) J. L. Fullagar, A. L. Garner, A. Struss, J. A. Day, D. P. Martin, J. Yu, X. Cai, K. D. Janda and S. M. Cohen, Chem. Commun., 2013, 49, 3197–3199 RSC; (c) A. S. Grillo, A. M. SantaMaria, M. D. Kafina, A. G. Cioffi, N. C. Huston, M. Han, Y. A. Seo, Y. Y. Yien, C. Nardone, A. V. Menon, J. Fan, D. C. Svoboda, J. B. Anderson, J. D. Hong, B. G. Nicolau, K. Subedi, A. A. Gewirth, M. Wessling-Resnick, J. Kim, B. H. Paw and M. D. Burke, Science, 2017, 356, 608–616 CrossRef CAS PubMed.
- J. E. Tavis, H. Wang, A. E. Tollefson, B. Ying, M. Korom, X. Cheng, F. Cao, K. L. Davis, W. S. W. Wold and L. A. Morrison, Antimicrob. Agents Chemother., 2014, 58, 7451–7461 CrossRef PubMed.
- (a) E. A. Semenova, A. A. Johnson, C. Marchand, D. A. Davis, R. Yarchoan and Y. Pommier, Mol. Pharmacol., 2006, 69, 1454–1460 CrossRef CAS PubMed; (b) S. F. Martin, B. C. Follows, P. J. Hergenrother and C. L. Franklin, J. Org. Chem., 2000, 65, 4509–4514 CrossRef CAS PubMed; (c) S. R. Piettre, A. Ganzhorn, J. Hoflack, K. Islam and J.-M. Hornsperger, J. Am. Chem. Soc., 1997, 119, 3201–3204 CrossRef CAS; (d) N. E. Allen, W. E. Alborn, J. N. Hobbs Jr and J. H. A. Kirst, Antimicrob. Agents Chemother., 1982, 22, 824–831 CrossRef CAS PubMed; (e) For a review, see: C. M. Meck, M. P. D'Erasmo, D. R. Hirsch and R. P. Murelli, Med. Chem. Commun., 2014, 5, 842–852 RSC.
- T. Masaoka, H. Zhao, D. R. Hirsch, M. P. D'Erasmo, C. M. Meck, B. Varnado, A. Gupta, M. J. Meyers, J. D. Baines, J. A. Beutler, R. P. Murelli, L. Tang and S. F. J. Le Grice, Biochemistry, 2016, 55, 809–819 CrossRef CAS PubMed.
- L. M. Grady, R. Szczepaniak, R. P. Murelli, T. Masaoka, S. F. J. Le Grice, D. L. Wright and S. K. Weller, J. Virol., 2017, 91, e01380 CrossRef CAS PubMed.
- (a) P. M. Beard, N. S. Taus and J. D. Baines, J. Virol., 2002, 76, 4785–4791 CrossRef CAS PubMed; (b) D. Yu and S. Weller, J. Virol., 1998, 72, 7428–7439 CrossRef CAS PubMed.
- (a) F. Foolad, S. L. Aitken and R. F. Chemaly, Expert Rev. Clin. Pharmacol., 2018, 10, 931–941 CrossRef PubMed; (b) L. Yang, Q. Yang, M. Wang, R. Jia, S. Chen, D. Zhu, M. Liu, Y. Wu, X. Zhao, S. Zhang, Y. Liu, Y. Yu, L. Zhang, X. Chen and A. Cheng, Viruses, 2019, 11, 219 CrossRef CAS PubMed.
- J. N. Goldstein and S. K. Weller, Virology, 1998, 244, 442–457 CrossRef CAS PubMed.
- (a) J. Zinser, S. Henkel and B. Fohlisch, Eur. J. Org. Chem., 2004, 1344–1346 CrossRef; (b) M. G. Banwell and M. P. Collis, J. Chem. Soc., Chem. Commun., 1991, 1343–1345 RSC.
- G. Sennari, R. Saito, T. Hirose, M. Iwatsuki, A. Ishiyama, R. Hokari, K. Otoguro, S. Omura and T. Sunazuka, Sci. Rep., 2017, 7, 7259 CrossRef PubMed.
- (a) J. Didierjean, C. Isel, F. Querre, J.-F. Mouscadet, A.-M. Aubertin, J.-Y. Valnot, S. R. Piettre and R. Marquet, Antimicrob. Agents Chemother., 2005, 49, 4884–4894 CrossRef CAS PubMed; (b) S. R. Piettre, C. Andre, M.-C. Chanal, J.-B. Ducep, B. Lesur, F. Piriou, P. Raboisson, J.-M. Rondeau and C. Schelcher, J. Med. Chem., 1997, 40, 4208–4221 CrossRef CAS PubMed.
- S. Chung, D. M. Himmel, J.-K. Jiang, K. Wojtak, J. D. Bauman, J. W. Rausch, J. A. Wilson, J. A. Beutler, C. J. Thomas, E. Arnold and S. F. J. Le Grice, J. Med. Chem., 2011, 54, 4462–4473 CrossRef CAS PubMed.
- C. Meck, N. Mohd and R. P. Murelli, Org. Lett., 2012, 14, 5988–5991 CrossRef CAS PubMed.
- P. J. Ireland, J. E. Tavis, M. P. D’Erasmo, D. R. Hirsch, R. P. Murelli, M. M. Cadiz, B. S. Patel, A. K. Gupta, T. C. Edwards, M. Korom, E. A. Moran and L. A. Morrison, Antimicrob. Agents Chemother., 2016, 60, 2140–2149 CrossRef CAS PubMed.
- A. J. Berkowitz, A. D. Franson, A. G. Casals, K. A. Donald, A. J. Yu, A. K. Garimallaprabhakaran, L. A. Morrison and R. P. Murelli, Med. Chem. Commun., 2019, 10, 1173–1176 RSC.
- A. J. Berkowitz, R. G. Abdelmessih and R. P. Murelli, Tetrahedron Lett., 2018, 59, 3026–3028 CrossRef CAS PubMed.
- Q. Li, E. Lomonosoca, M. Donlin, F. Cao, A. O'Dea, B. Milleson, A. J. Berkowitz, J.-C. Baucom, J. P. Stasiak, D. V. Schiavone, R. G. Abdelmessih, A. Lyubimova, A. J. Fraboni, L. P. Bejcek, J. A. Villa, E. Gallicchio, R. P. Murelli and J. E. Tavis, Antiviral Res., 2020, 177, 104777 CrossRef CAS PubMed.
- R. Mannhold, G. I. Poda, C. Ostermann and I. V. Tetko, J. Pharm. Sci., 2009, 98, 861–893 CrossRef CAS PubMed.
- (a) J. P. Stasiak, A. Grigoryan and R. P. Murelli, Tetrahedron Lett., 2019, 60, 1643–1645 CrossRef CAS PubMed; (b) N. Yui, Sci. Rep. Tohoku Univ. First Ser., 1956, 40, 114–120 CAS.
- D. H. Katz, J. F. Marcelletti, M. H. Khalil, L. E. Pope and L. R. Katz, Proc. Natl. Acad. Sci. U. S. A., 1991, 88, 10825–10829 CrossRef CAS PubMed.
- M. Uozaki, H. Yamasaki, Y. Katsuyama, M. Higuchi, Y. Higuti and A. H. Koyama, Antiviral Res., 2007, 73, 85–91 CrossRef CAS PubMed.
- K. Kaihatsu, M. Yamabe and Y. Ebara, Molecules, 2018, 23, 2475 CrossRef PubMed.
- C.-Y. Hsiang and T.-Y. Ho, Br. J. Pharmacol., 2008, 155, 227–235 CrossRef CAS PubMed.
- J. C. Bronstein and P. C. A. Weber, Anal. Biochem., 2001, 293, 239–245 CrossRef CAS PubMed.
- E. Geerling, V. Murphy, M. C. Mai, E. T. Stone, A. G. Casals, M. Hassert, A. T. O'Dea, F. Cao, M. J. Donlin, M. Elagawany, B. Elgendy, V. Pardali, E. Giannakopoulou, G. Zoidis, D. V. Schavone, N. B. Agyemang, R. P. Murelli, J. E. Tavis, A. K. Pinto and J. D. Brien, PLoS One, 2022, 17, e0274266 CrossRef CAS PubMed.
- Complex splitting arises from the molecule's atropdiastereotopicity. This is due to hindered rotation about the Ar–C(O) bond combined with the presence of inequivalent E and Z amide rotamers.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ra06749h |
| ‡ Authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |