
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Anion exchange on hydrous zirconium oxide materials: application for selective iodate removal†
Valtteri Suorsa *a,
Miho Otakia,
Topi Suominena,
Juhani Virkanenb,
Hanna Reijolab,
René Bescd and
Risto Koivulaa
*a,
Miho Otakia,
Topi Suominena,
Juhani Virkanenb,
Hanna Reijolab,
René Bescd and
Risto Koivulaa
aRadiochemistry Unit, Department of Chemistry, University of Helsinki, A.I. Virtasen aukio 1, 00014 Helsinki, Finland. E-mail: valtteri.suorsa@helsinki.fi
bDepartment of Geosciences and Geography, University of Helsinki, Gustaf Hällströmin katu 2, 00014 Helsinki, Finland
cDepartment of Physics, University of Helsinki, PO Box 64, FI-00014 Helsinki, Finland
dHelsinki Institute of Physics, PO Box 64, FI-00014 Helsinki, Finland
First published on 4th January 2023
Abstract
The radioactive 129I is a top-priority radionuclide due to its the long half-life (1.57 × 107 years) and high mobility. Because of the planned and accidental releases to the environment, specific separation technologies are required to limit the potential radiation dose to human beings. Zirconium oxides are known for their adsorption capability and selectivity to oxyanions and here the applicability to selective IO3− removal has been investigated regarding the uptake mechanism, regeneration and competition caused by other anions, like environmentally relevant SO42−. Granular aggregates of hydrous zirconium oxides with and without Sb doping showed high potential for the selective IO3− removal in the presence of competing anions, like the forementioned SO42− (apparent capacity between 0.1–0.4 meq g−1 depending on SO42− concentration). The main uptake mechanism was found to be outer-sphere complexation (ion-exchange) to the protonated hydroxyl groups of hydrous zirconium oxides, but also minor mechanisms were identified including inner-sphere complexation and reduction to I−. The materials were observed to be easily and successively regenerated using dilute acid. Hydrous zirconium oxides showed high potential for IO3− removal from waste solutions regarding technical (high selectivity and apparent capacity) and ecological/economic (feasible regeneration) aspects.
1. Introduction
Iodine is a vital element for human beings and other mammals for the proper functioning of the thyroid gland. Although the most abundant isotope of iodine is stable 127I, also radioactive isotopes are formed during uranium and plutonium fission. Like stable iodine, these radioactive isotopes also concentrate in the thyroid gland if inhaled or digested causing an elevated risk of thyroid cancer. The most relevant risks of the radioactive iodine isotopes can be divided into two categories: the long-term risk of 129I with an extremely long half-life (15.7 million years) and acute risk mainly from 131I with a short half-life (8 days). The former is important from an environmental perspective because of its long half-life and high mobility. Therefore, its risk must be assessed in the case of groundwater contamination and nuclear waste disposal. For example, 129I is regarded as one of the top priority radionuclides in the biosphere safety assessment of the final disposal of spent nuclear fuel in Finland.1 131I is only important in the case of fresh fallout during, for instance, nuclear accidents like at the Chernobyl or Fukushima Daiichi nuclear power plants. The effects of acute iodine uptake and internal dose can be minimized by saturating the thyroid in advance with non-radioactive iodine for which iodine pills are intended.Iodine has complex chemistry in the environment with its three main oxidation states −1, 0, +5. Iodate (IO3−) is the main species with its +5 oxidation state found in oxidizing environments, whereas iodide (I−) prevails with its −1 oxidation state in reducing conditions. For example, in anoxic waters, such as the Baltic sea, iodine exists mainly as I−,2 but in oxidizing waters such as oceans,3 IO3− is the dominant species. Molecular iodine (I2), i.e. 0 oxidation state, is the major species only at low pH.4 In addition, iodine readily reacts with organic molecules forming a wide range of different organo-iodine compounds.5,6
Iodine removal is strongly dependent on its speciation and the immobilization of different iodine species has been comprehensively reviewed in literature.4 Silver-based materials have proved to be feasible for I− decontamination due to the formation of AgI with extremely low solubility (Ksp = 8.5 × 10−17).7–11 The solubility of AgIO3 is however much higher (Ksp = 3.2 × 10−8) which makes silver-based materials inefficient in the removal of IO3−. In fact, the different affinity of I−/IO3− to Ag+ can be utilized in analytical separations of these iodine species.12 Other removal techniques for I− include for example ion exchange resins,13 activated carbons,14 organoclays14–16 and hydrotalcites.17 For the removal of IO3−, a wide range of different adsorbent materials have been studied including zero valent iron,18 hydroxyapatites19,20 and hydrotalcites.17,21,22 Despite the intensive research, no selective, highly performing state-of-art iodate removal technique has yet been established.
ZrO2 is an amphoteric metal oxide known for its cation and anion exchange capabilities depending on the solution pH.23–25 ZrO2 is a widely used material that can be doped with different cations like Y,26,27 Ce28 or Sb29–31 in order to enhance its mechanical, electronic or chemical properties such as the zeta potential of the material. ZrO2 have several crystal structures from which the monoclinic, tetragonal, and cubic phases can be formed in ambient pressures. The monoclinic structure is the most stable in low temperatures, but cubic and tetragonal structures can be stabilized either by the doping or by limiting the crystallite size, i.e. forming nanocrystalline materials. The properties of the crystal structures differ significantly regarding their physical and chemical properties like toughness or the number of anion and oxygen vacancies.32 Sb is known to stabilize the tetragonal form of ZrO2,29 which has been shown to be the active phase for the adsorption of different anions.33–35
Our previous work31 demonstrated the effectiveness of hydrous ZrO2 materials for selective iodate removal. However, the findings could not holistically reveal the mechanism behind the adsorption of IO3− or other anions. Within this study, we have extensively studied the basic ion exchange properties of pure ZrO2 and antimony doped Zr(Sb)O2 with different anions and the basic adsorption experiments have been complemented with supportive XAS (X-ray Absorption Spectroscopy) measurements. The focus has been on the mechanism of IO3− adsorption but also the competition of SO42−, due to its relevance to environmental decontamination and strong affinity to adsorbent materials, has been investigated extensively. Different conditions like concentration and type of competing anions and the reversibility of the adsorption have been investigated to understand the mechanisms of IO3− uptake on ZrO2 materials. The reversibility has not only significance in studying the mechanism of uptake but also regarding the regeneration of the materials, which is an important ecological and economic practical aspect regarding actual application of adsorbents in decontamination processes.
2. Materials and methods
2.1. Chemicals
The reagents used within the study were of analytical grade (Alfa Aesar, Sigma-Aldirch, Riedel de Häen) and were used as received. Deionized water (Type 1: 18.2 MΩ cm or Type 2: 15.0 MΩ cm at 25 °C, Milli-Q ® Merck Millipore or) was used for the solutions in experiments. A radioactive Na125I (PerkinElmer) tracer was used as IO3− probe after oxidation with NaOCl (final concentration ∼2 × 10−4 M). The speciation of 125I was confirmed with the method using silver-impregnated activated carbon (Silcarbon Aktivkohle GmbH, Germany).12 In brief, 10 mL of solution containing 125IO3− was equilibrated for 24 h with 20 mg of the silver -impregnated activated carbon and the solid and the solution were separated. The radioactivity of the solution was measured and compared with the original solution. After successful oxidation, the radioactivity of the original and the separated solution were equal as IO3− adsorption to the material is insignificant whereas I− adsorption is extremely efficient. If the oxidation was not complete, more NaOCl was added, and the analysis step was repeated before use.2.2. Synthesis of materials
The materials were synthesized and characterized as described earlier in literature.31 In brief, two different zirconium oxides were synthesized by the precipitation method. First, Sb doped Zr(Sb)O2 was synthesized by dissolving 45 g of ZrCl4 (Riedel de Häen) and 2 g of SbCl3 (Sigma-Aldrich) to 2 L of 3 M HCl under vigorous stirring using a mechanical stirrer. 1.2 L of 6 M NH3 was added slowly to the solution until pH reached 7.8 and white gel was precipitated. ZrO2 was synthesized similarly but here 100 g of zirconium basic carbonate (Alfa Aesar) was dissolved in 1 L of 6 M HNO3. Precipitates were let to stand in their mother solution overnight and clear supernatants were discarded. The precipitates were washed with deionized water until the conductivity of the supernatant was less than 4.0 mS cm−1. After the wash, the materials were dried in an oven at 70 °C for three days. The dried materials were ground and sieved to particle size 74–149 μm. The syntheses yielded large particles of amorphous zirconia.312.3. Aqueous sample analysis
2.4. Batch ion exchange experiments
In a typical batch experiment, 20 ± 1 mg of the ground and sieved material was weighed into a 20 mL polyethylene scintillation vial to which 10 mL of appropriate solution was pipetted. In some experiments larger masses and volumes were used but the batch factor, i.e. the solution to solid ratio, was kept the same at approximately 500 mL g−1. If needed, pH was adjusted with appropriate volumes of NaOH or HNO3. The samples were equilibrated in a rotary mixer for 24 h and the solids were separated from the liquid phase by a combination of centrifuging (2100 G, 10 min) and filtering with a 0.2 μm filter (PVDF LC. Arcodisc, Gellman Sciences). The equilibration time was chosen for practical reasons and to ensure that equilibrium was attained. Earlier studies31 have shown that IO3− the adsorption on ZrO2 and Zr(Sb)O2 is in equilibrium before 1 hour and pH already after 5 minutes of contact. Finally, 5 mL of filtered solution was measured with a gamma counter as described in previous section (2.3.1 Iodine analyses) and the equilibrium pH measurement was conducted from the remaining filtered solution with an Orion™ 9103BNWP combination pH electrode (Thermo Scientific™). The uncertainty of pH measurements was estimated to be 0.1 units. All the experiments were conducted in normal laboratory air.2.5. Column experiments
In a typical column experiment 0.2–0.5 g of the sieved (74–149 μm) material was weighed to a beaker and rinsed with deionized water. The water was decanted and discarded and remaining white solid was mixed with a small volume (∼5 mL) of fresh deionized water and transferred with a disposable pipette into a low-pressure borosilicate glass column with a diameter of 0.5 or 0.7 cm and a porous polymer bed support at the bottom (Econo-Column®. Bio-Rad Laboratories, Inc.). The feed solution was pumped through the column from the inlet with a flow velocity of about 20 bed volumes (BV) per hour and sample fractions were collected from the outlet. In columns with a smaller mass, a higher flow velocity of 40 BV per hour was used. From the collected fractions, pH and probe (mainly IO3− but also NO3−, Cl− and SO42− in some experiments) concentrations were measured with corresponding methods described earlier in Section 2.3. The feed solution pH and concentration of the elements of interest were monitored throughout the experiments.2.6. Solid sample characterization
3. Results and discussion
For clarity and readability, the results are divided into two parts: (A) basic anion exchange studies regarding ZrO2 and Zr(Sb)O2 interactions with anions in general and (B) the selective adsorption of IO3− to the materials regarding the adsorption mechanism and the effect of competing anions.3.1. Basic anion exchange mechanisms on ZrO2 materials
The basic ion exchange phenomenon on ZrO2 and Zr(Sb)O2 was studied with the series of batch and column experiments, where anion exchange equilibrium of ZrO2 and Zr(Sb)O2 with OH−, Cl−, NO3−, SO42− and IO3− was investigated.| Solution | Eq. pH DI water | Eq. pH 5 mM | Eq. pH 50 mM |
|---|---|---|---|
| NaCl | 3.3 | 3.4 | 3.7 |
| NaNO3 | 3.3 | 3.4 | 3.7 |
| Na2SO4 | 3.3 | 5.4 | 5.9 |
| KIO3 | 3.3 | 5.2 | 6.4 |
Fundamentally, the pH drop can be due either an increase in the concentration of H3O+ or the decrease of the concentration of OH− in the solution. Because pH behaviour was different between the anions (and not the cations) the most probable reason for the pH drop is anion exchange between the OH− of the solution and the exchangeable anions in the materials originated from the synthesis. Evidently, the solutions containing SO42− and IO3− ions prevented this drop of pH, probably because they are exchanged instead of OH−, and in higher concentrations the equilibrium pH even rose which indicates they are exchanged to OH− of the material thus indicating higher selectivity. The higher 50 mM concentration of Cl− and NO3− resulted only in a slightly higher equilibrium pH (3.7) compared to the 5 mM concentration (pH 3.4) and this is likely because with higher concentration more Cl−/NO3− is adsorbed instead of OH− but the selectivity is much lower compared with SO42− and IO3−.
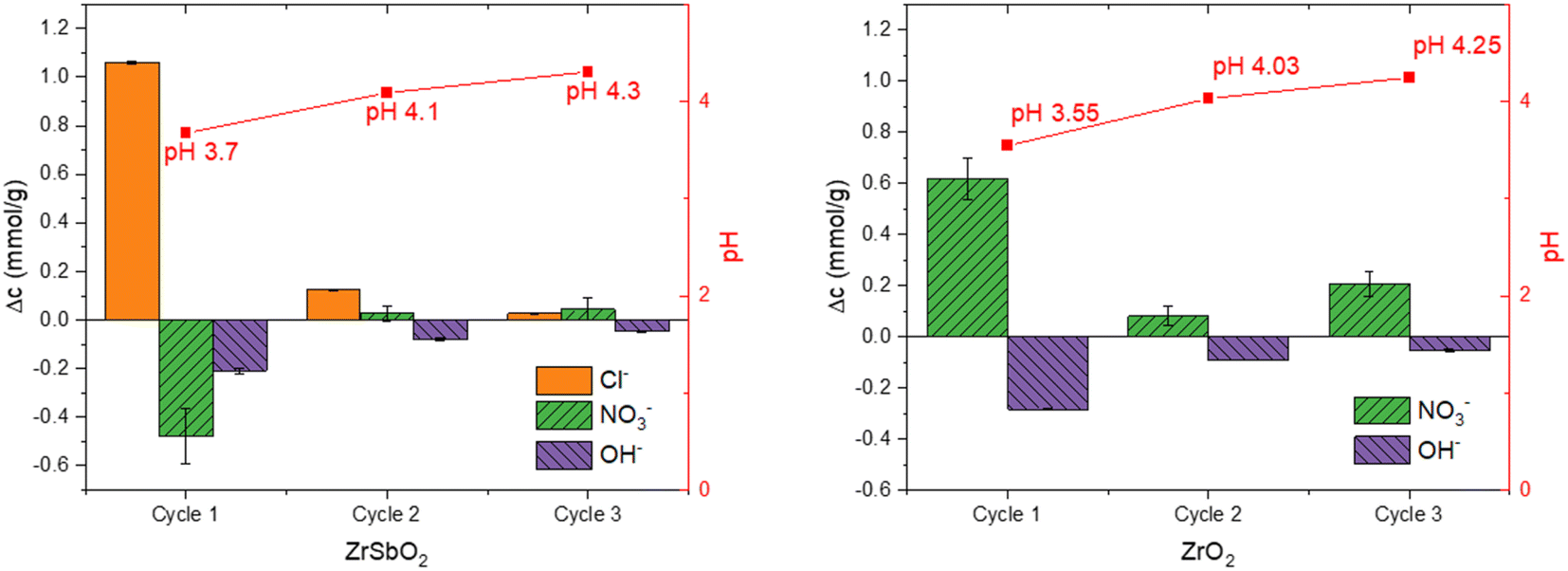 | ||
| Fig. 1 Anions adsorbed and desorbed during successive equilibrations of Zr(Sb)O2 and ZrO2 with 10 mM NaNO3 (pH 5.6). The negative values represent adsorption to the material and the positive values represent desorption to the solution phase. The standard deviations of pH measurements are now showing in figure as they were extremely small related to the measured value. | ||
The ion exchange reaction is described by eqn (1) for Cl− exchange (similarly with NO3− in the case of ZrO2):
| 2H2O + Zr – OH2+Cl− ⇌ H3O+ + Cl− + Zr – OH2+OH− | (1) |
In the exchange reaction, a neutral water molecule is exchanged to hydrochloric (or nitric) acid, which causes the decrease of pH.
The ion exchange between NO3− and Cl− is then described by eqn (2):
| Na+ + NO3− + Zr – OH2+Cl− ⇌ Na+ + Cl− + Zr – OH2+NO3− | (2) |
After the successive batch equilibrations with NaNO3, both materials still showed high affinity to IO3− in trace concentration in the same 10 mM NaNO3 solution: Kd's of IO3− were 240![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 ± 3000 mL g−1 for ZrO2 (sorption-%: 99.58 ± 0.01) and 150000 ± 30000 mL g−1 for Zr(Sb)O2 (sorption-%: 99.30 ± 0.16).
000 ± 3000 mL g−1 for ZrO2 (sorption-%: 99.58 ± 0.01) and 150000 ± 30000 mL g−1 for Zr(Sb)O2 (sorption-%: 99.30 ± 0.16).
 | ||
| Fig. 2 pH of eluate in ZrO2 with 1 mM KIO3 (red squares), Zr(Sb)O2 with 1 mM KIO3 (black squares) and ZrO2 with deionized water (blue squares). The pH of the feed was 5.6 and is shown as dashed horizontal line. The uncertainty of individual pH measurement was estimated to be 0.1 units. | ||
The total OH− uptake calculated from Δ[OH−] + Δ[H3O+] would equal to 0.83 ± 0.04 mmol g−1, which is well in line with the maximum uptake determined in the batch experiments (0.91 ± 0.17 mmol g−1) for other anions. These values agree also well with the apparent capacities published for hydrous zirconium oxides in literature.23,37,38 The decrease of pH is different in the presence of IO3− as the latter seems to inhibit the pH lowering capability of zirconia materials. This indicates that OH− and IO3− compete for the same sites on the material surface.
During the loading of materials with of 1 mM Na2SO4, the release of synthesis derived anions from the materials were studied (Fig. 3). In total 0.49 ± 0.03 meq g−1 SO42− was exchanged to Cl− (0.73 ± 0.02 mmol g−1) and OH−in the case of Zr(Sb)O2. During the first 300 BV's, pH steadily increased and stabilized to pH 5.6 after an instant drop to pH 3 at the beginning of the experiment. The ratio of adsorbed (SO42−) and desorbed (Cl− and OH−) equaled about 1.1. In total, 0.33 ± 0.01 meq g−1 of SO42− was exchanged to NO3− (0.53 ± 0.02 mmol g−1) and OH− in the case of ZrO2 which equals adsorption/desorption ratio of 1.25. Compared with Zr(Sb)O2, pH stabilized to about 6.5 that is slightly higher than the feed solution pH.
 | ||
| Fig. 3 The effluent pH (blue symbols and right y-axis) and concentrations of SO42− (black symbols) and Cl− (red symbols) for Zr(Sb)O2 (closed symbols) and SO42− and NO3− for ZrO2 (open symbols). The uncertainties are not shown in the graph due to the clarity and were 5% (NO3−), 3% (Cl−) and 4.5% (SO42−) of the reported concentration for individual samples. | ||
3.2. Application on selective IO3− removal from waste solutions
The applicability of ZrO2 and Zr(Sb)O2 materials for selective IO3− removal was studied with a series of column experiments with competing ions at different concentrations. In the first set of experiments, the competition of Cl−, NO3− and SO42− with IO3− were studied in respect to the total uptake and breakthrough (BT) profile of IO3−. In the second set, the same was done with SO42− with varying concentrations in the range of 0.1–10 mM. In the third set, the desorption of IO3− from the loaded materials were studied with sequential elution with different solutions (100 mM NaNO3, Na2SO4 and NaOH). Finally, the regeneration of the materials was studied with different solutions (NaCl, NaOH and HCl) and then tested for four successive regeneration cycles. | ||
| Fig. 4 Effect of SO42− and NO3− anions (10 mM) to uptake of IO3− (c = 1 mM) on ZrO2 (open symbols) and Zr(Sb)O2 (filled symbols) and corresponding pH curves. The uncertainties are not shown in the graph due to the clarity but were below 2.5% for the determined apparent capacities. | ||
Due to the strong interfering effect of SO42− and its relevance in decontamination environments, the uptake and competition between SO42− and IO3− was further probed with the additional series of column experiments.
The IO3− BT curves for Zr(Sb)O2 (Fig. 5) and ZrO2 (SI) show a logical trend with a rising SO42−/IO3− ratio: the BT reaches 100% earlier with the higher ratios. Regarding IO3− uptake, a significant decrease in was observed when raising the SO42− concentration from 0.1 (e.g., Zr(Sb)O2: 0.33 meq g−1 ± 1.10%) to 10 mM (0.11 meq g−1 ± 1.96%) (Fig. 6) and the effect was somewhat more profound with ZrO2 material. Although SO42− strongly competes with IO3−, even at SO42−/IO3− ratio of 100 the uptake of IO3− is still about 0.1 meq g−1 which is about 20–30% of the maximum uptake in SO42− solution.
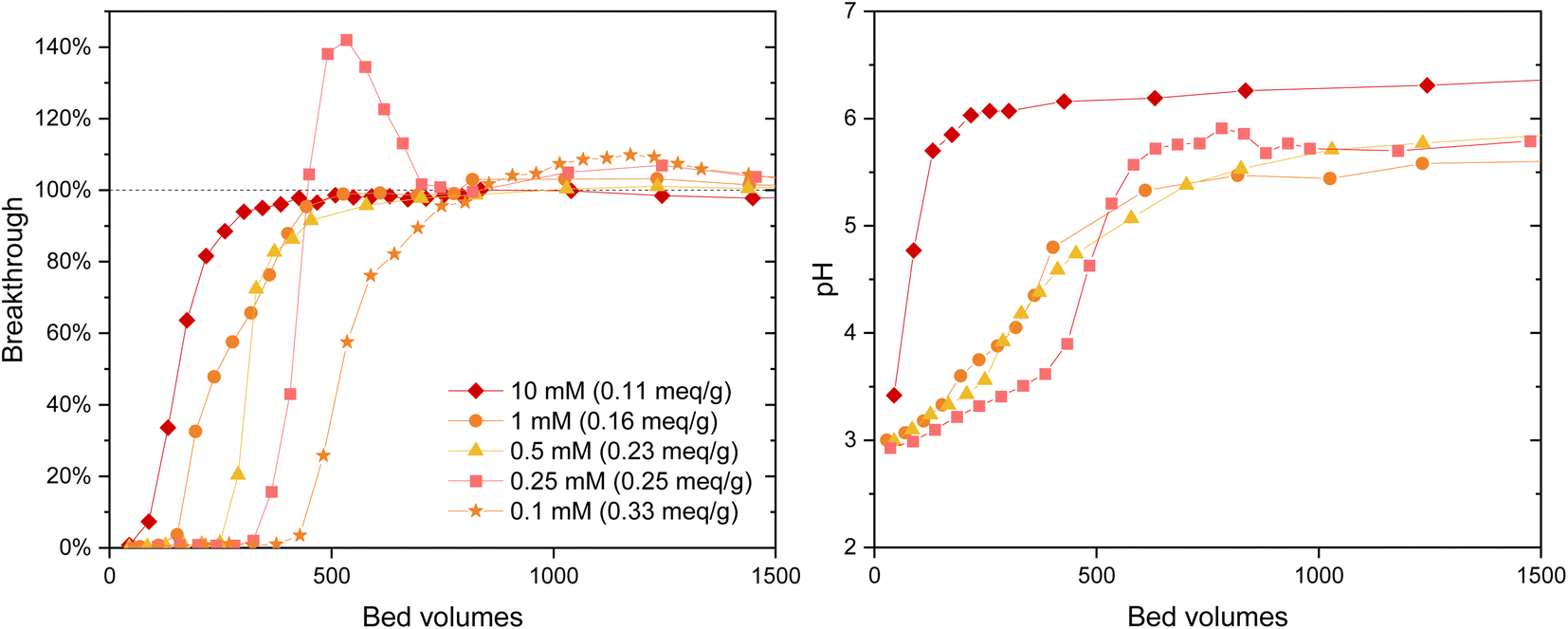 | ||
| Fig. 5 IO3− (c = 1 mM) breakthrough and pH curves for Zr(Sb)O2 columns in different SO42− concentrations. The equilibrium uptake is represented in parentheses of each legend. The uncertainties are not shown in the graph due to the clarity but were below 4.0% for the determined apparent capacities. | ||
 | ||
| Fig. 6 Effect of SO42− concentrations on IO3− (c = 1 mM) uptake on ZrO2 and Zr(Sb)O2. Note the logarithmic scale on x-axis. | ||
In 0.25 mM SO42− solution the IO3− BT reaches above 100% and then stabilizes to 100% (Fig. 5). This experiment was repeated twice with nearly identical results (see ESI†). This is because in the early phase the adsorption sites were not in equilibrium with the solution composition, i.e., more IO3− was adsorbed compared with SO42− because of the deficiency of the latter. Later, when more SO42− was introduced to the column, some adsorbed IO3− was released to the solution by SO42− and the equilibrium was reached. In the more dilute 0.1 mM solution similar behavior was also observed but to a milder extent: the BT reaches over 100% but not as considerably as with 0.25 mM SO42− solution. After the loading, IO3− was desorbed from all columns using 0.1 M NaOH resulting in rather consistent complete desorption of the adsorbed IO3−.
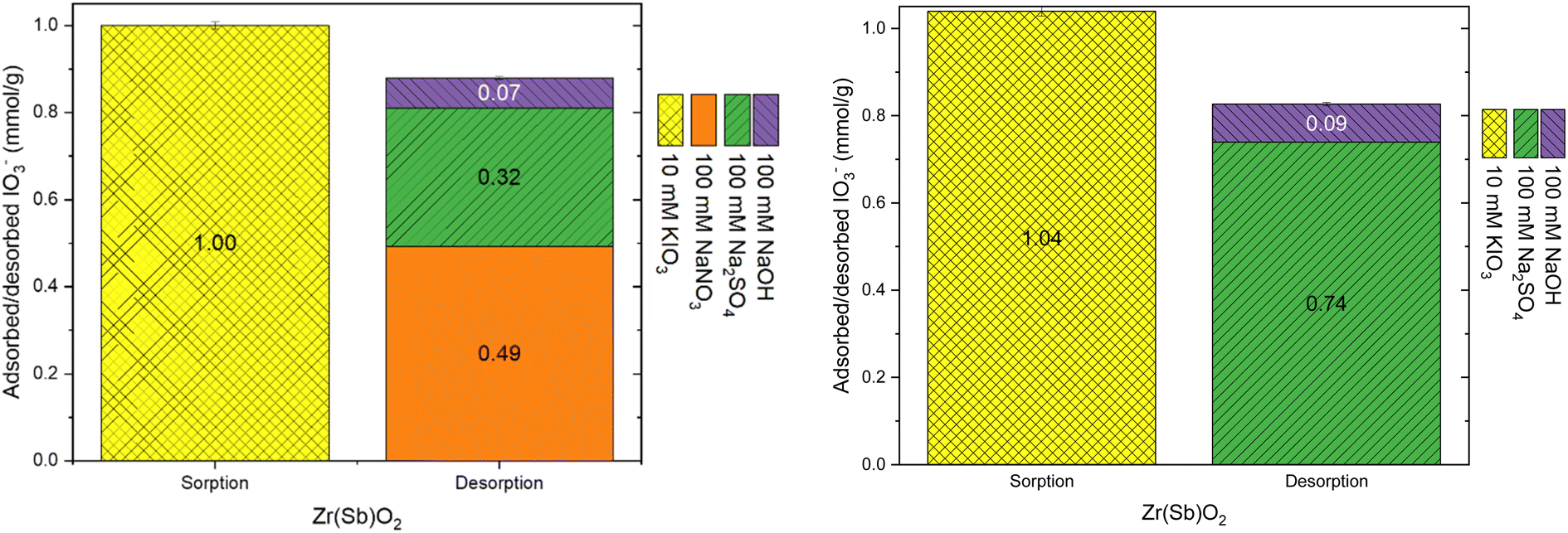 | ||
| Fig. 7 Uptake with 10 mM KIO3 solution and desorption with different solutions on Zr(Sb)O2. | ||
 | ||
| Fig. 8 Uptake with 10 mM KIO3 solution and desorption with different solutions on ZrO2. | ||
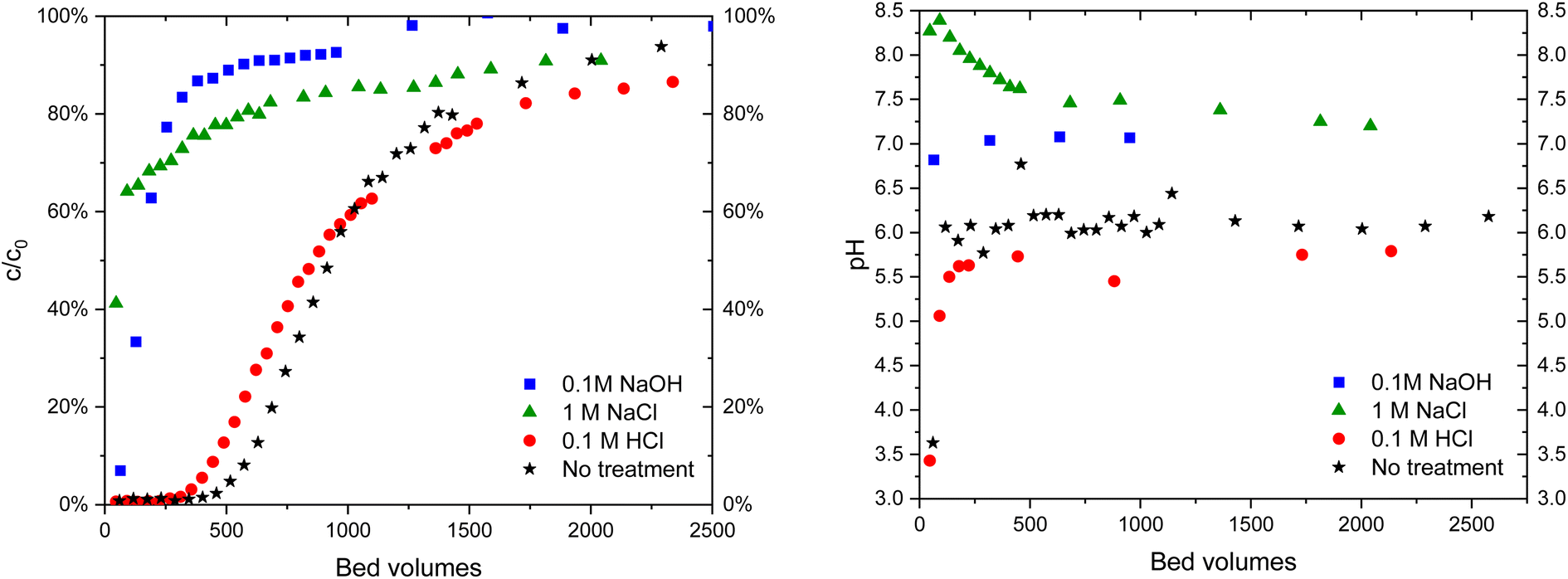 | ||
| Fig. 9 IO3− (c = 10 μM) breakthrough in 10 mM Na2SO4 solution (left) in Zr(Sb)O2 columns of untreated material and after treatments with different solutions and the corresponding pH curves (right). | ||
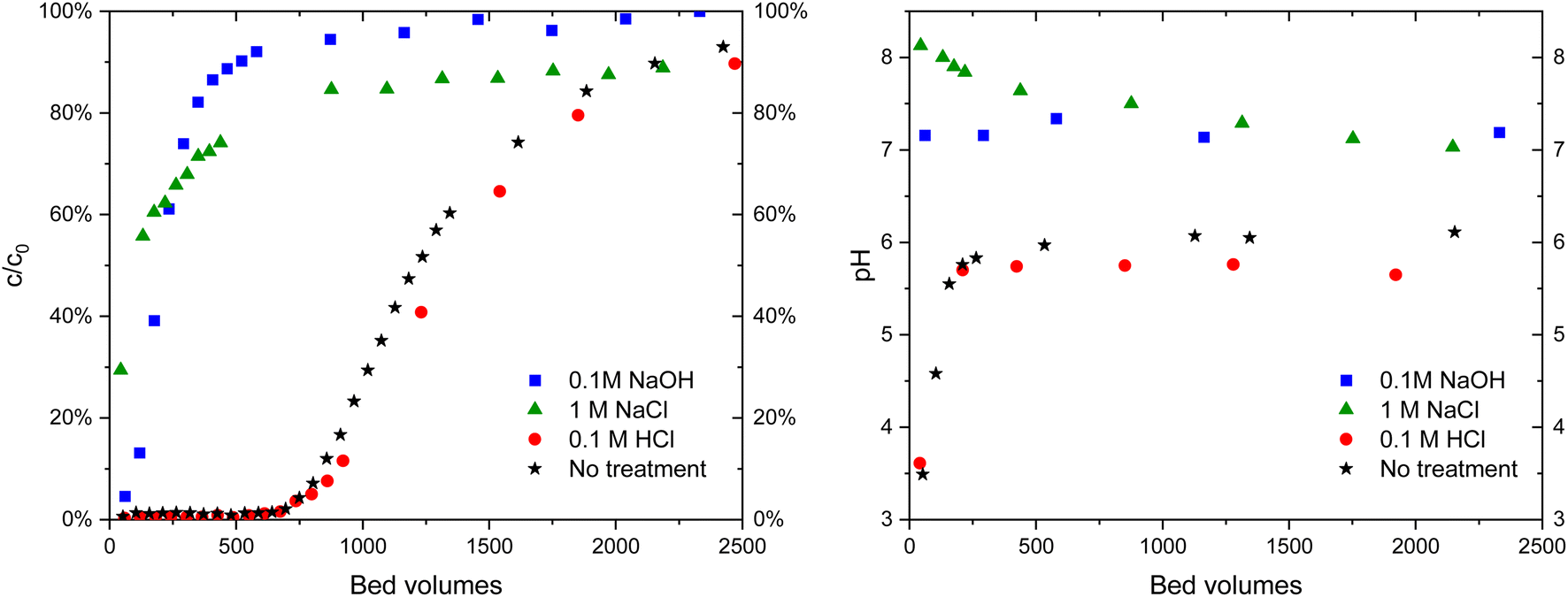 | ||
| Fig. 10 IO3− (c = 10 μM) breakthrough in 10 mM Na2SO4 solution (left) in ZrO2 columns of untreated material and after treatments with different solutions and corresponding pH curves (right). | ||
The regeneration behavior is explained by ion exchange: after the treatment with NaOH the material is in OH− form:
| Na+ + OH− + Zr – OH2+IO3− ⇌ Na+ + IO3− + Zr – OH2+OH− | (3) |
Without any further treatment, IO3− is not able to exchange with OH− in the material to the same extent as with the fresh material. The treatment with 0.1 M HCl returns the material to Cl− form:
| H3O+ + Cl− + Zr – OH2+OH− ⇌ 2H2O + Zr – OH2+Cl− | (4) |
The same applies most probably to 1 M NaCl solution as well, but the conversion is not complete at neutral pH.
The practical regeneration of Zr(Sb)O2 material was tested with four IO3− uptake/eluent cycles using 0.1 M NaOH as an eluent and 0.1 M HCl for the regeneration of the material between the cycles (Fig. 11). The regeneration efficiency remained high for all the cycles and the IO3− uptake was approximately 0.10 ± 0.01 meq g−1 (the uptake and elution curves in ESI†). The successive cycles showed some variation in the eluted IO3− fraction and the largest deviation was associated to the first cycle where non-treated material was used. This resulted in the lower IO3− uptake and the lower elution percentage compared with HCl regenerated material at cycles 2 to 4, due to unexchangeable (inner-sphere complexation) IO3− uptake.
 | ||
| Fig. 11 A total uptake of Zr(Sb)O2 after the several successive load (10 mM Na2SO4, 1 mM KIO3 pH 6), desorption (0.1 M NaOH) and regeneration (0.1 m HCl) cycles. | ||
3.3. Solid sample characterization
| ABET m2 g−1 | Vtotal cm3 g−1 | Dpore nm | |
|---|---|---|---|
| Zr(Sb)O2 | 137 ± 2 | 0.07 | 2.5 |
| ZrO2 | 96 ± 3 | 0.03 | 3.0 |
 | ||
| Fig. 12 Left: I K-edge XANES spectrum of Zr(Sb)O2 (solid line) and ZrO2 (dashed line) loaded with IO3− in deionized (DI) water and different sulfate concentrations along spectra of the KIO3, KI and I2 references. The vertical dashed line shows the position of iodate main absorption peak, as known as the first derivate maximum of the KIO3 reference. Right: The corresponding first derivative relatively to energy spectra. All spectra were vertically shifted for clarity. | ||
The Sb K-edge XANES spectra were measured to see if Sb oxidation state changes in the material during the uptake process (Fig. 13). Sb remained as Sb(III) after the Zr(Sb)O2 synthesis, but after the contact with IO3− solution it partly oxidizes to Sb(V) as seen on the derivative spectra (Fig. 13) which is showing a bimodal white-line. However, the possible oxidation by dissolved oxygen needs to be considered. Since reduction of I was observed in I K-edge XANES, it seems highly probable that Sb is the reason for this. In total context, this must be a secondary adsorption mechanism as 1 gram of 5 molar-% Zr(Sb)O2 could reduce theoretically only 0.04 mmol of IO3− to I− and the fore-mentioned competing oxidation by dissolved oxygen most likely even lowers this value.
 | ||
| Fig. 13 Sb K-edge XANES spectra of Zr(Sb)O2 samples (as prepared and 3.6 mmol g−1 IO3− loaded) compared with Sb2O3 and KSb(OH)6 references. The dashed vertical lines show the absorption peak location of Sb(III) and Sb(V). | ||
 | ||
| Fig. 14 I K-edge EXAFS in R-space (upper graph) and k-space (lower graph) spectra of ZrO2 (solid red lines) and Zr(Sb)O2 (dashed blue lines) after IO3− loading. FT window of 3 to 13.3 Å−1 was used. | ||
In Zr K-edge EXAFS spectra the signal at about 1.5 Å is assigned to the 1st shell oxygen and the second peak at 3.0 Å to 2nd shell Zr atoms (ESI†). These values correspond well to the values found in literature.40,41 Similar fitting as with I EXAFS was done for Zr. However, in all the samples no significant differences were found between the pristine and loaded ZrO2/Zr(Sb)O2 regarding the local coordination environment of Zr. Sb K-edge EXAFS spectra exhibited peaks at about 1.5 Å but the first shell fits of Zr and Sb (ESI†) did not reveal any significant differences, except that σ2 was higher for pure Zr(Sb)O2 compared with the material after IO3− loading.
 | ||
| Fig. 15 TGA curves of HCl and NaOH treated Zr(Sb)O2. | ||
3.4. Consideration on IO3− adsorption mechanism and competition of other anions
The adsorption of IO3− is strongly associated with the pH in the solution and indications on the sorption process can be drawn from the pH changes during the uptake. Firstly, the surface charge of the adsorbent materials changes according to the solution pH, which strongly affects their affinity to anions in the solution. The anion exchange on zirconium oxides have been extensively studied earlier24,25,37,43 and in principle, the anion exchange site on zirconium oxide can be represented as:| ≡Zr – OH + H+ ⇌ ≡Zr – OH2+ | (5) |
In addition, the possible ligand exchange is described as:
|
≡Zr – OH + X− ⇌ ≡Zr − X + OH−
| (6) |
Secondly, the uptake of IO3− (or SO42−) itself affects the solution pH. Zirconium oxides synthesized within this study tend to lower pH even in deionized solutions, e.g., in the column experiments pH was lowered from 5.6 to 2.5 at the beginning of the experiments. The mechanism of this pH change is related to the synthesis derived anions (Cl− and NO3− depending on the synthesis conditions), which remain in the structure due to the incomplete exchange with OH− during the synthesis, i.e. the materials are initially partly in the OH− and partly in the Cl−-form.23 In addition, the form of the material can be changed by regeneration with dilute acid like HCl that changes the material back to the Cl−-form. The exchange of the adsorbed anions with OH− leads to the pH drop in the solution, like shown in eqn (1) earlier. In the presence of IO3− (or SO42−), the competing reaction takes place preventing the drop of pH:
| Zr – OH2+Cl− + IO3− ⇌ Zr – OH2+IO3− + Cl− | (7) |
This explains why pH (i.e., the concentration of OH−) and SO42− concentration are critical parameters regarding the IO3− adsorption on zirconium oxides.31
This study demonstrates that both adsorption mechanisms (eqn (5) and (6)) contribute to the adsorption of IO3−, although the outer-sphere complexation (eqn (7)) is evidently the main mechanism of uptake as the IO3− sorption was observed to be highly reversible and efficient and fast desorption was achieved with relatively dilute NaOH (100 mM). Also, the sorption capability was efficiently regenerated using dilute acid like HCl which changes the material back to the Cl−-form. In addition, certain fractions of adsorbed IO3− were eluted efficiently by solutions containing NO3− and SO42−. However, NO3− eluted less IO3− compared with SO42−, and even combined they were not able to elute all adsorbed IO3− indicating that there are significant selectivity differences between the anions due to e.g., structural properties of the exchange sites. In the high excess of SO42− (10:1 in concentration) the apparent capacity of IO3− remained reasonably high (about 0.10 meq g−1) indicating a certain selectivity to the latter. The inner-sphere complexation (eqn (6)) of IO3− can be regarded as a minor uptake mechanism and it was only observed in the relatively high concentration (10 mM) of IO3− and without any competing anions. This IO3− fraction remained bound to the materials even after successive elutions with NO3−, SO42− or NaOH.
4. Conclusion
Hydrous zirconium oxide materials ZrO2 and its antimony-doped Zr(Sb)O2 counterpart exhibited excellent IO3− adsorption properties regarding apparent capacity (>0.6 meq g−1) and especially selectivity in high excess of competing anions, such as environmentally relevant SO42−. The selectivity differences of zirconium oxides to different anions were observed, as NO3−, SO42− and OH− seem to compete with IO3− for different available adsorption sites (competition decreasing in order OH− > SO42− > NO3−). The materials exhibited the highest IO3− removal when changed to Cl− form with dilute HCl (about 5 times higher apparent capacity compared with the OH−-form). The materials also showed constant uptake performance during three load-regeneration cycles when regenerated with dilute acid (0.1 M HCl) which demonstrates the potential feasibility of the material for practical applications regarding sustainability and financial perspectives.Based on the easy regeneration in dilute conditions and fast uptake, the main mechanism of uptake was concluded to be the ion-exchange between IO3− and anions e.g., NO3−, Cl− and OH− forming the outer-sphere complex with the materials. In the XAS data no external neighboring atoms were observed in the Zr or I K-edge EXAFS. This supports the conclusion regarding the outer-sphere complexation, although certain precautions should be taken as the reason for this could also be the relatively low concentrations of exchangeable anions (e.g., in 4% of total mass Cl− -form) or the amorphous structure of the materials resulting in the homogeneous distribution of the local coordination environments of the adsorption sites. Ligand exchange (inner-sphere complexation) between IO3− and surface OH− was observed to take place as a minor secondary adsorption mechanism in conditions without competing anions. IO3− is known to form both inner- and outer-sphere complexes with oxides and the dominating mechanism depends on the ionic strength and pH of the solution.44 It seems that at least in the case of zirconium oxides, the type of competing anion can affect the proportions of the available sites as well due to electivity differences. In the presence of Sb doping, also a redox reaction between Sb and I was discovered and confirmed by the XANES data, but the mechanism only contributes slightly (theoretical capacity 0.04 mmol g−1) to the overall IO3− (maximum apparent capacity about 1 mmol g−1) uptake. It can, however, become significant in concentrated matrices.
Further studies would be required for the identification of the different ion exchange sites and what is the fundamental chemical or physical explanation for the selectivity of zirconium oxides to IO3− and SO42−. This knowledge could be utilized for the manipulation of the material structure during the synthesis to furthermore improve the IO3− selectivity.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We thank Timo Hatanpää for his help with the TGA experiments and Bree Revill for her help with the regeneration column experiments. We acknowledge DESY (Hamburg, Germany), a member of the Helmholtz Association HGF, for the provision of experimental facilities. Parts of this research were carried out at PETRA III P64, and we would like to thank the beamline scientists Wolfgang Caliebe and Akhil Tayal for their efforts during the XAS measurements. Beamtime was allocated for proposal I-20200428 EC. SEM imaging and XRD measurements were done in ALD Center Finland research infrastructure. The financial support from Doctoral Program in Chemistry and Molecular Sciences (CHEMS) is gratefully acknowledged. Open access funded by Helsinki University Library.References
- T. Hjerpe, A. T. K. Ikonen, and R. Broed, Biosphere assessment report 2009, 2010 Search PubMed.
- X. Hou, H. Dahlgaard and S. Nielsen, Chemical speciation analysis of 129I in seawater and a preliminary investigation to use it as a tracer for geochemical cycle study of stable iodine, Mar. Chem., 2001, 74(2–3), 145–155, DOI:10.1016/S0304-4203(01)00010-X.
- X. Hou, V. Hansen, A. Aldahan, G. Possnert, O. C. Lind and G. Lujaniene, A review on speciation of iodine-129 in the environmental and biological samples, Anal. Chim. Acta, 2009, 632(2), 181–196, DOI:10.1016/j.aca.2008.11.013.
- R. C. Moore, C. I. Pearce, J. W. Morad, S. Chatterjee, T. G. Levitskaia, R. M. Asmussen, A. R. Lawter, J. J. Neeway, N. P. Qafoku, M. J. Rigali, S. A. Saslow, J. E. Szecsody, P. K. Thallapally, G. Wang and V. L. Freedman, Iodine immobilization by materials through sorption and redox-driven processes: A literature review, Sci. Total Environ., 2020, 716, 132820, DOI:10.1016/j.scitotenv.2019.06.166.
- C. Xu, S. Zhang, Y.-F. Ho, E. J. Miller, K. A. Roberts, H.-P. Li, K. A. Schwehr, S. Otosaka, D. I. Kaplan, R. Brinkmeyer, C. M. Yeager and P. H. Santschi, Is soil natural organic matter a sink or source for mobile radioiodine (129I) at the Savannah River Site?, Geochim. Cosmochim. Acta, 2011, 75(19), 5716–5735, DOI:10.1016/j.gca.2011.07.011.
- D. I. Kaplan, et al., Radioiodine biogeochemistry and prevalence in groundwater, Crit. Rev. Environ. Sci. Technol., 2014, 44(20), 2287–2335, DOI:10.1080/10643389.2013.828273.
- R. M. Asmussen, J. J. Neeway, A. R. Lawter, A. Wilson and N. P. Qafoku, Silver-based getters for 129I removal from low-activity waste, Radiochim. Acta, 2016, 104(12), 905–913, DOI:10.1515/ract-2016-2598.
- D. I. Kaplan, et al., Iodine speciation in a silver-amended cementitious system, Environ. Int., 2019, 126, 576–584, DOI:10.1016/j.envint.2019.02.070.
- J. S. Hoskins, T. Karanfil and S. M. Serkiz, Removal and sequestration of iodide using silver-impregnated activated carbon, Environ. Sci. Technol., 2002, 36(4), 784–789, DOI:10.1021/es010972m.
- P. C. Ho and K. A. Kraus, Adsorption on inorganic materials—VIII: Adsorption of iodide on AgCl-filled carbon, J. Inorg. Nucl. Chem., 1981, 43(3), 583–587, DOI:10.1016/0022-1902(81)80507-6.
- T. Karanfil, E. C. Moro and S. M. Serkiz, Development and testing of a silver chloride-impregnated activated carbon for aqueous removal and sequestration of iodide, Environ. Technol., 2005, 26(11), 1255–1262, DOI:10.1080/09593332608618595.
- V. Suorsa, M. Otaki, W. Zhang, J. Virkanen and R. Koivula, A simple method for quantifying iodate and iodide fractions in solution using Ag -impregnated activated carbon, J. Radioanal. Nucl. Chem., 2020, 324(1), 135–142, DOI:10.1007/s10967-020-07061-4.
- K. E. Parker, E. C. Golovich and D. M. Wellman, Iodine adsorption on ion-exchange resins and activated carbons: batch testing, Pacific Northwest National Laboratory, PNNL-23730, U.S. DOE, United States, 2014, DOI:10.2172/1163822.
- D. Li, D. I. Kaplan, A. S. Knox, K. P. Crapse and D. P. Diprete, Aqueous 99Tc, 129I and 137Cs removal from contaminated groundwater and sediments using highly effective low-cost sorbents, J. Environ. Radioact., 2014, 136, 56–63, DOI:10.1016/j.jenvrad.2014.05.010.
- J. Bors, S. Dultz and B. Riebe, Organophilic bentonites as adsorbents for radionuclides: I. Adsorption of ionic fission products, Appl. Clay Sci., 2000, 16(1–2), 1–13, DOI:10.1016/S0169-1317(99)00041-1.
- D. Li, D. I. Kaplan, A. Sams, B. A. Powell and A. S. Knox, Removal capacity and chemical speciation of groundwater iodide (I−) and iodate (IO3−) sequestered by organoclays and granular activated carbon, J. Environ. Radioact., 2018, 192, 505–512, DOI:10.1016/j.jenvrad.2018.08.008.
- T. G. Levitskaia, et al., RedOx-controlled sorption of iodine anions by hydrotalcite composites, RSC Adv., 2016, 6(79), 76042–76055, 10.1039/C6RA13092E.
- A. R. Lawter, W. L. Garcia, R. K. Kukkadapu, O. Qafoku, M. E. Bowden, S. A. Saslow and N. P. Qafoku, Technetium and iodine aqueous species immobilization and transformations in the presence of strong reductants and calcite-forming solutions: Remedial action implications, Sci. Total Environ., 2018, 636, 588–595, DOI:10.1016/j.scitotenv.2018.04.240.
- A. Coulon, D. Laurencin, A. Grandjean, C. Cau Dit Coumes, S. Rossignol, and L. Campayo, Immobilization of iodine into a hydroxyapatite structure prepared by cementation, The Royal Society of Chemistry, 2014, p. 20923, 10.1039/C4TA03236E.
- L. Campayo, A. Grandjean, A. Coulon, R. Delorme, D. Vantelon and D. Laurencin, Incorporation of iodates into hydroxyapatites: a new approach for the confinement of radioactive iodine, J. Mater. Chem., 2011, 21(44), 17609–17611, 10.1039/C1JM14157K.
- L. Liang and L. Li, Adsorption behavior of calcined layered double hydroxides towards removal of iodide contaminants, J. Radioanal. Nucl. Chem., 2007, 273(1), 221–226 CrossRef.
- T. Toraishi, S. Nagasaki and S. Tanaka, Adsorption behavior of IO3− by CO32−- and NO3−- hydrotalcite, Appl. Clay Sci., 2002, 22(1), 17–23, DOI:10.1016/S0169-1317(02)00108-4.
- C. Amphlett, L. A. McDonald and M. Redman, Synthetic inorganic ion-exchange materials—II: hydrous zirconium oxide and other oxides, J. Inorg. Nucl. Chem., 1958, 6(3), 236–245, DOI:10.1016/0022-1902(58)80153-0.
- M. P. Rigney, E. F. Funkenbusch and P. W. Carr, Physical and chemical characterization of microporous zirconia, J. Chromatogr. A, 1990, 499, 291–304, DOI:10.1016/S0021-9673(00)96980-2.
- D. Tichit, D. El Alami and F. Figueras, Preparation and anion exchange properties of zirconia, Appl. Catal., A, 1996, 145(1), 195–210, DOI:10.1016/0926-860X(96)00171-8.
- D. Huang, K. R. Venkatachari and G. C. Stangle, Influence of yttria content on the preparation of nanocrystalline yttria-doped zirconia, J. Mater. Res., 1995, 10(3), 762–773, DOI:10.1557/JMR.1995.0762.
- V. Stubican, R. C. Hink and S. P. Ray, Phase equilibria and ordering in the system ZrO2-Y2O3, J. Am. Ceram. Soc., 1978, 61(1–2), 17–21, DOI:10.1111/j.1151-2916.1978.tb09220.x.
- P. Duwez and F. Odell, Phase relationships in the system zirconia—ceria, J. Am. Ceram. Soc., 1950, 33(9), 274–283, DOI:10.1111/j.1151-2916.1950.tb12798.x.
- A. Gulino, R. G. Egdell and I. Fragala, Low-temperature stabilisation of tetragonal zirconia by antimony, J. Mater. Chem., 1996, 6(11), 1805–1809, 10.1039/JM9960601805.
- S. Lönnrot, J. Paajanen, V. Suorsa, W. Zhang, M. Ritala and R. Koivula, Sb-doped zirconium dioxide submicron fibers for separation of pertechnetate (TcO4–) from aqueous solutions, Sep. Sci. Technol., 2021, 56(14), 2338–2350, DOI:10.1080/01496395.2020.1826967.
- V. Suorsa, M. Otaki, J. Virkanen and R. Koivula, Pure and Sb-doped ZrO2 for removal of IO3− from radioactive waste solutions, Int. J. Environ. Sci. Technol., 2021, 19(6), 5155–5166, DOI:10.1007/s13762-021-03487-9.
- O. A. Graeve, Zirconia, in Ceramic and Glass Materials,ed. J. F. Shackelford and R. H. Doremus, Springer, New York, USA, 2008, pp. 169–197 Search PubMed.
- J. Luo, X. Luo, J. Crittenden, J. Qu, Y. Bai, Y. Peng and J. Li, Removal of Antimonite (Sb(III)) and Antimonate (Sb(V)) from Aqueous Solution Using Carbon Nanofibers That Are Decorated with Zirconium Oxide (ZrO2), Environ. Sci. Technol., 2015, 49(18), 11115–11124, DOI:10.1021/acs.est.5b02903.
- J. Paajanen, S. Lönnrot, M. Heikkilä, K. Meinander, M. Kemell, T. Hatanpää, K. Ainassaari, M. Ritala and R. Koivula, Novel electroblowing synthesis of submicron zirconium dioxide fibers: effect of fiber structure on antimony (V) adsorption, Nanoscale Adv., 2019, 1(11), 4373–4383, 10.1039/C9NA00414A.
- Y. Gao, K. Chen, X. Tan, X. Wang, A. Alsaedi, T. Hayat and C. Chen, Interaction Mechanism of Re(VII) with Zirconium Dioxide Nanoparticles Archored onto Reduced Graphene Oxides, ACS Sustainable Chem. Eng., 2017, 5(3), 2163–2171, DOI:10.1021/acssuschemeng.6b02317.
- B. Ravel and M. Newville, ATHENA, ARTEMIS, HEPHAESTUS: data analysis for X-ray absorption spectroscopy using IFEFFIT, J. Synchrotron Radiat., 2005, 12(4), 537–541, DOI:10.1107/S0909049505012719.
- N. J. Singh and S. N. Tandon, Hydrous zirconium oxide as an anion-exchanger, Talanta, 1977, 24(7), 459–461, DOI:10.1016/0039-9140(77)80129-X.
- A. Clearfield, Crystalline hydrous zirconia, Inorg. Chem., 1964, 3(1), 146–148 CrossRef CAS.
- D. Laurencin, D. Vantelon, V. Briois, C. Gervais, A. Coulon, A. Grandjean and L. Campayo, Investigation of the local environment of iodate in hydroxyapatite by combination of X-ray absorption spectroscopy and DFT modeling, RSC Adv., 2014, 4(28), 14700–14707, 10.1039/C3RA47691J.
- V. V. Kanazhevskii, V. P. Shmachkova, N. S. Kotsarenko, V. N. Kolomiichuk and D. I. Kochubei, Changes in the zirconium local surrounding on ligand substitution in solutions, J. Struct. Chem., 2006, 47(5), 860–868, DOI:10.1007/s10947-006-0401-x.
- S. Foraita, J. L. Fulton, Z. A. Chase, A. Vjunov, P. Xu, E. Barath, D. M. Camaioni, C. Zhao and J. A. Lercher, Impact of the oxygen defects and the hydrogen concentration on the surface of tetragonal and monoclinic ZrO2 on the reduction rates of stearic acid on Ni/ZrO2, Chemistry, 2015, 21(6), 2423–2434, DOI:10.1002/chem.201405312.
- B. Bachiller-Baeza, I. Rodriguez-Ramos and A. Guerrero-Ruiz, Interaction of Carbon Dioxide with the Surface of Zirconia Polymorphs, Langmuir, 1998, 14(13), 3556–3564, DOI:10.1021/la970856q.
- R. Chitrakar, S. Tezuka, A. Sonoda, K. Sakane, K. Ooi and T. Hirotsu, Selective adsorption of phosphate from seawater and wastewater by amorphous zirconium hydroxide, J. Colloid Interface Sci., 2006, 297(2), 426–433, DOI:10.1016/j.jcis.2005.11.011.
- T. Nagata, K. Fukushi and Y. Takahashi, Prediction of iodide adsorption on oxides by surface complexation modeling with spectroscopic confirmation, J. Colloid Interface Sci., 2009, 332(2), 309–316, DOI:10.1016/j.jcis.2008.12.037.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ra06489h |
| This journal is © The Royal Society of Chemistry 2023 |