DOI:
10.1039/D2RA06423E
(Paper)
RSC Adv., 2023,
13, 9109-9118
Theoretical study on structural and mechanical properties of Si-containing ternary transition metal nitrides M0.5Si0.5N (M = Ti, Zr, Hf)†
Received
12th October 2022
, Accepted 9th March 2023
First published on 20th March 2023
Abstract
Si-containing transition-metal nitrides Ti0.5Si0.5N, Zr0.5Si0.5N and Hf0.5Si0.5N with conventional rock salt B1 structure exhibit superior hardness, strength and oxidation resistance. However, the potential phases of the ternary systems at various pressures remain unexplored. In this work, we firstly studied the potential structures of Ti0.5Si0.5N, Zr0.5Si0.5N and Hf0.5Si0.5N in pressures of 0–100 GPa. A hexagonal phase with P63/mmc symmetry was uncovered and verified to be quenchable in the ambient conditions. The structural, mechanical and electronic properties were systematically studied and compared with the well-known ordered B1 structure. We surprisingly found that Ti0.5Si0.5N within this hexagonal phase displayed much improved ideal indentation shear strength from about 10 GPa for a B1 structure to 30 GPa. The estimated hardness based on the empirical formula is up to 38 GPa, greatly exceeding that of the B1 structure. By the detailed electronic analysis, the underlying atomic mechanism for the outstanding mechanical properties was also studied.
1. Introduction
Binary transition metal (TM) nitrides such as TiN, ZrN, TaN, VN, etc. are widely used as protective coatings for cutting tools.1 However, these nitrides have poor oxidation resistance in oxygen or air when the temperature is beyond 500 °C.2,3 In cutting applications such as high speed milling, the temperature can be easily improved to exceed 800 °C.4 To address this problem, an effective design strategy to enhance the oxidation resistance and mechanical properties is introducing a third element Al or Si into the TM nitrides to form ternary or quaternary systems like TiAlN, TiSiN, TiAlCrN, TiAlCN, etc.5–10 These aluminum or silicon nitrides have a distinct improvement of oxidation resistance. Furthermore, the introduction of the third element also induces the formation of a nanocomposite structure containing a non-miscible nanocrystalline or amorphous structures and results in high hardness.11
As is known, these binary or ternary TM nitrides adopt the metastable NaCl-type B1 structure at room temperature thanks to the stabilization of the anharmonic effect.12 This B1 structure can't sustain its dynamic stability when the temperature descends to 0 K due to the appearance of unphysical longitudinal acoustic phonon energy.12 Most of these ternary B1 nitrides containing two TM elements often have a disordered occupation of TM atoms in the cation locations. As a result of this disordered occupation, the mechanical anisotropy is further improved on the basis of the ordered metastable B1 structure, which is not favorable in engineering application. Additionally, though the cubic B1 structure has relatively high crystal symmetry, the mechanical anisotropy is strong and this has been verified in our previous work for ternary VAlN and quaternary VWCN systems.13,14 However, these TM nitrides within hexagonal structure often behave less mechanical anisotropy, which can be ascribed to the better distribution of the bonds between TM and nitrogen atoms. Meanwhile, the electronic structure analysis shows the “pseudo-gap” separating the bonding and antibonding states is present in the most B1-structure compounds and the EF often lies in the antibonding region, which improves the formation enthalpy of the systems and induces the metastability. The occupition of d–d antibonding states also weakens the p–d hybridization between TM and light elements. However, this p–d hybridization is indispensable in determining the anti-shear properties and macroscopic hardness for this type of compounds. Thus, the hardness of these B1 structure TM nitrides is mostly in the range of 10–30 GPa much less than the threshold of the superhard standard (40 GPa).15,16 For the Si-containing ternary TM nitrides within B1 structure, Si atoms are introduced for replacing partial TM atoms. This replacement can't prominently tune the EF position due to the structural limitation. Thus, it also can't significantly improve the mechanical properties. Therefore, it is greatly meaningful to search new configuration for these Si-containing nitrides with tuned EF position and enhanced mechanical properties.
In this work, we systematically explored structural, mechanical and electronic properties of the ternary Ti0.5Si0.5N, Zr0.5Si0.5N and Hf0.5Si0.5N systems using the first-principle and structure searching methods implemented in the CALYPSO code17,18 in the pressure of 0–100 GPa. The hexagonal P63/mmc phase was firstly uncovered. To clarify the structural effects on hardness, strengths, mechanical anisotropies and crystal stability of these three systems within P63/mmc and B1 structures, we simulated the uniaxial and biaxial stains to obtain the corresponding stress and analyzed the thermal and dynamic stabilities as well as electronic structures.
2. Calculation methods and details
The structure searching process for Ti0.5Si0.5N, Zr0.5Si0.5N and Hf0.5Si0.5N systems was performed in the pressure of 0–100 GPa, by the method of CALYPSO,17,18 which has been fully confirmed as an reliable and effective structural searching approach.19–25 We adopted 0, 30, 50 and 100 GPa as the searching pressure points. The crystal cell containing 1–4 formula units (f.u.) was used in the searching process. All the calculations were carried out by the first-principle density functional theory (DFT) method implemented in the Vienna ab initio simulation package (VASP).26 The generalized gradient approximation (GGA) was used in calculating elastic and structural properties.27 The exchange correlation potential within the GGA proposed by Perdew, Burke and Ernzerhoff (PBE) with projector augmented wave (PAW) method28,29 was used in all the calculations. The kinetic cutoff energy was set as 600 eV during the optimization process of all the structures. The convergence criteria of the total energy in all the structural relaxations were set as 10−5 eV. The 7 × 7 × 7 and 12 × 12 × 3 k-point meshs in Monkhorst Pack scheme and Gamma centered grid30 were used for ordered pseudo-B1 and P63/mmc sturcutes, respectively. The ternary disordered B1 systems were calculated with 4 × 4 × 4 k-point meshs and 64 atoms supercells using the special quasi-random structure (SQS) method.31,32 The SQS structure was constructed for ternary Ti0.5Si0.5N system using unit cells containing 32 cations and 32 anions within fcc structure. The 2 × 2 × 2 supercell containing 64 atoms was generated. The evaluation of the generated model was performed by the objective function provided by the SQS approach. The simulated SQS model as well as the predicted structures were set in the ESI data.† The phonon spectra were calculated by the density functional perturbation theory (DFPT) approach implemented in the PHONOPY code.33 The elastic constants Cij were calculated by applying a small strain onto the crystal structures with a finite variation. All the elastic moduli were calculated according to the Voigt–Reuss–Hill approximation.34 The 3D and 2D distributions of elastic moduli were investigated by calculating the directional Young's and shear moduli, which were obtained by calculating the Young's and shear moduli along various crystal directions based on the formulas provided by the literature35 using the elastic constants. The Chen empirical formula was adopted in assessing the Vickers hardness.36 The electron localization function (ELF)37 is also calculated to further analyze the bonding nature.
3. Results and discussions
3.1. Structure, property and stability
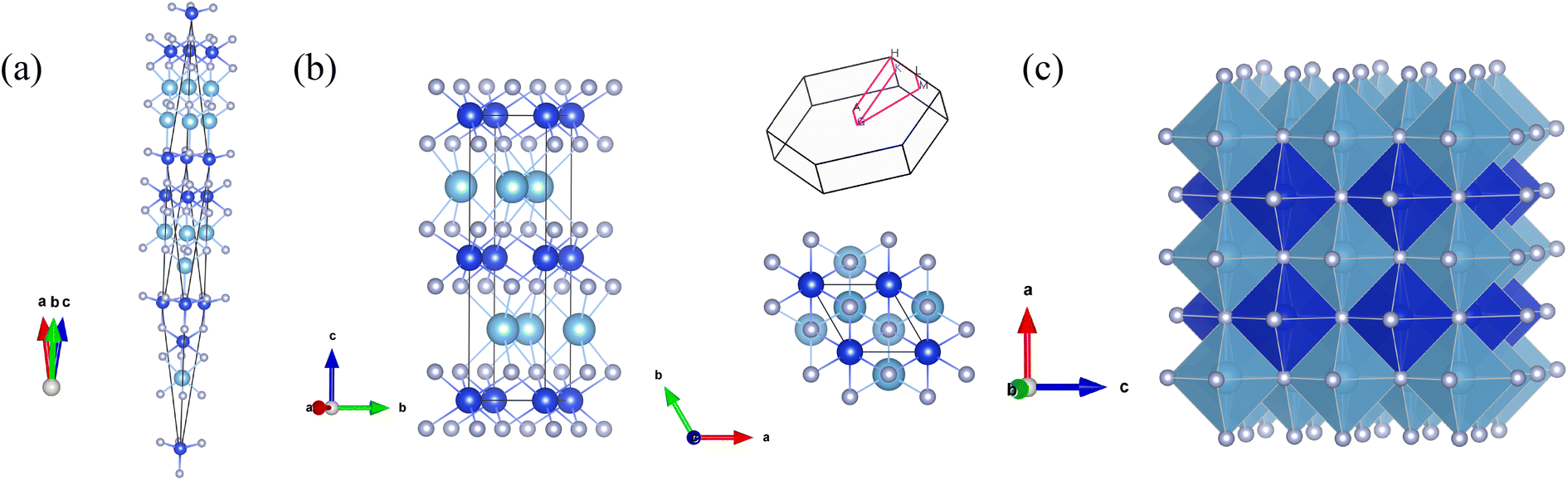
The CALYPSO method was employed to searching potential structures at the pressure points of 0, 30, 50 and 100 GPa, respectively. A trigonal structure with the space group of R3m (No. 160) was confirmed to be the ground-state phase of these three Si-containing ternary systems. However, this phase cannot sustain its thermal stability when the pressure ascends up to 30 GPa. At the pressures of 30, 50 and 100 GPa, the results of the structure searches showed the most favorable structure of these systems tuned to be the hexagonal P63/mmc (No.194) phase, which is displayed in Fig. 1. The TM and Si atoms have an alternate layered distribution. As a comparison, the ordered B1 structure was also exhibited in the figure. It can be seen there are six light element N atoms surrounding the TM and Si atoms, which is consistent with the B1 structure. The difference is the bond angles and lengths of the TM-N and Si–N bonds have some variations. Here, we take Ti0.5Si0.5N as an example. It is clearly seen the bond angle of all the adjacent Ti–N and Si–N bonds is 90° in the B1 structure. After full relaxation of both lattice constants and the positions of intra atoms, the obtained bond angles are 77.6°, 87.2° for N–Ti–N and 94.0°, 86.0° for N–Si–N. The bond lengths of Ti–N and Si–N are 2.227 Å and 1.909 Å, respectively, which are all different from those of ordered B1 structure. The Wyckoff sites of Ti, Si and N are 2d (0.667, 0.333, 0.25), 2a (0, 0, 0) and 4f (0.333, 0.667, 0.100) respectively. As listed in Table 1, these three ternary systems within the same structures have approximately identical lattice constants. The lattice constants of hexagonal Ti0.5Si0.5N are a = b = 2.793 Å and c = 10.236 Å. The evaluated density is 3.007 × 103 kg m−3, larger than the ordered B1 structure (2.726 × 103 kg m−3) and the trigonal structure (2.705 × 103 kg m−3) presented in Table 1. Thus, the average TM–N and Si–N bond lengths are smaller than B1 structure and the trigonal structure. For other two Zr0.5Si0.5N and Hf0.5Si0.5N systems, the structural properties are similar to the Ti0.5Si0.5N system.
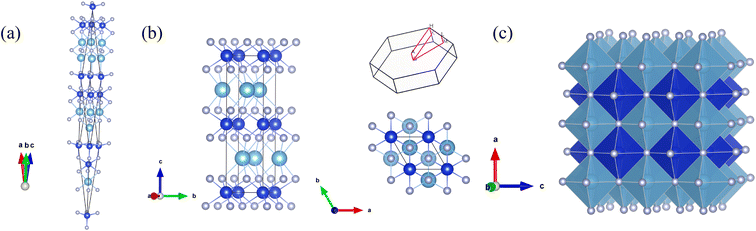 |
| | Fig. 1 Crystal structures of (a) trigonal R3m, (b) side and top views of hexagonal P63/mmc as well as the corresponding first Brillouin zone and (c) ordered B1 structures of Ti0.5Si0.5N. The large cyan, blue and small silvery spheres represent Ti, Si and N atoms respectively. | |
Table 1 Calculated lattice constants, M–N bond lengths (Å), formation enthalpies (eV/f.u.), and densities (kg m−3)
| |
Symmetry |
Source |
a |
c |
M–N |
Si–N |
Hf |
Density |
| Ti0.5Si0.5N |
R3m |
This work |
10.449 |
|
|
|
−2.092 |
2.705 × 103 |
| |
P63/mmc |
This work |
2.793 |
10.236 |
2.227 |
1.909 |
−1.918 |
3.007 × 103 |
| |
B1 |
This work |
4.230 |
|
2.151 |
2.105 |
−1.172 |
2.726 × 103 |
| Zr0.5Si0.5N |
R3m |
This work |
10.820 |
|
|
|
−1.846 |
3.375 × 103 |
| |
P63/mmc |
This work |
2.898 |
10.891 |
2.397 |
1.953 |
−1.666 |
3.720 × 103 |
| |
B1 |
This work |
4.524 |
|
2.243 |
2.281 |
−1.050 |
3.210 × 103 |
| Hf0.5Si0.5N |
R3m |
This work |
10.709 |
|
|
|
−1.866 |
5.494 × 103 |
| |
P63/mmc |
This work |
2.878 |
10.861 |
2.378 |
1.946 |
−1.596 |
6.021 × 103 |
| |
B1 |
This work |
4.521 |
|
2.226 |
2.226 |
−1.104 |
5.237 × 103 |
To verify the thermal stability, we calculated the formation enthalpy using the following formula: 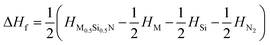 , (M = Ti, Zr, Hf), where HM0.5Si0.5N, HM, HSi, and HN2 are the enthalpies per f.u. of M0.5Si0.5N, pure hcp-Ti/Zr/Hf, diomand-type Si and α-N2. The calculated results are −1.918, −1.666 and −1.596 eV/f.u. for hexagonal Ti0.5Si0.5N, Zr0.5Si0.5N and Hf0.5Si0.5N systems respectively. For trigonal R3m structure, the formation enthalpies are −2.092, −1.846 and −1.866 eV/f.u. respectively. As a contrast, the formation enthalpies of them within ordered B1 structure are −1.172, −1.050 and −1.104 eV/f.u., respectively, indicating the trigonal phases are more thermodynamically stable than the hexagonal and ordered B1 phases in principle at ambient condition. Fig. 2 displays the calculated results of enthalpy differences of these three systems within B1 and R3m structures relative to the hexagonal P63/mmc structure as a function of pressure. It is clearly seen that with the pressure ascending beyond 15 15 and 25 GPa, P63/mmc structure transforms to be the most energetically favourable phase for them, as it has the lowest total enthalpy. Meanwhile, the enthalpy differences have an increasing tendency with pressure, implying the thermodynamic stability of P63/mmc structure can be maintained up to 100 GPa.
, (M = Ti, Zr, Hf), where HM0.5Si0.5N, HM, HSi, and HN2 are the enthalpies per f.u. of M0.5Si0.5N, pure hcp-Ti/Zr/Hf, diomand-type Si and α-N2. The calculated results are −1.918, −1.666 and −1.596 eV/f.u. for hexagonal Ti0.5Si0.5N, Zr0.5Si0.5N and Hf0.5Si0.5N systems respectively. For trigonal R3m structure, the formation enthalpies are −2.092, −1.846 and −1.866 eV/f.u. respectively. As a contrast, the formation enthalpies of them within ordered B1 structure are −1.172, −1.050 and −1.104 eV/f.u., respectively, indicating the trigonal phases are more thermodynamically stable than the hexagonal and ordered B1 phases in principle at ambient condition. Fig. 2 displays the calculated results of enthalpy differences of these three systems within B1 and R3m structures relative to the hexagonal P63/mmc structure as a function of pressure. It is clearly seen that with the pressure ascending beyond 15 15 and 25 GPa, P63/mmc structure transforms to be the most energetically favourable phase for them, as it has the lowest total enthalpy. Meanwhile, the enthalpy differences have an increasing tendency with pressure, implying the thermodynamic stability of P63/mmc structure can be maintained up to 100 GPa.
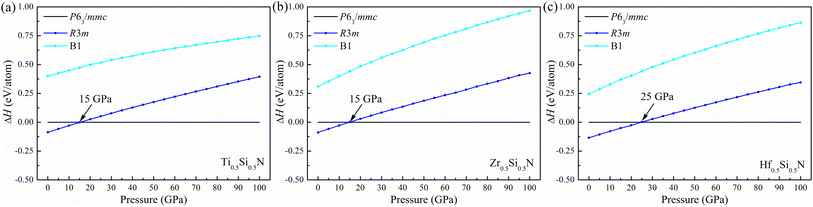 |
| | Fig. 2 Enthalpy difference of (a) Ti0.5Si0.5N, (b) Zr0.5Si0.5N and (c) Hf0.5Si0.5N within P63/mmc structure relative to R3m and B1 structures as a function of pressure. | |
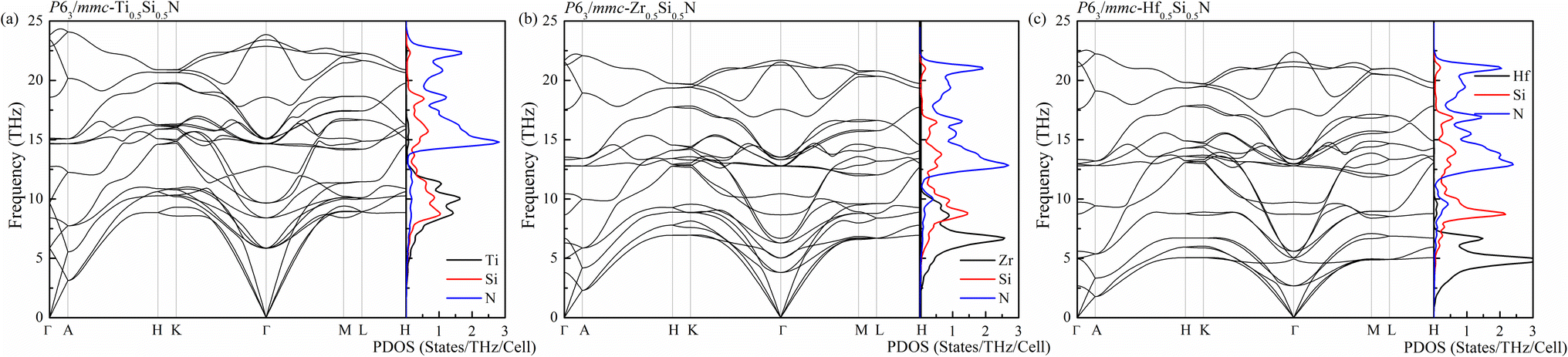
To further check the stability and the possibility of artificial synthesis of these systems with new uncovered hexagonal structure, the study of dynamic properties are necessary. Therefore, using the DFPT method which is implemented in the PHONOPY code, the phonon spectra as well as the corresponding projected density of states (PDOS) are calculated by simulating the 3 × 3 × 1 supercells for hexagonal Ti0.5Si0.5N, Zr0.5Si0.5N and Hf0.5Si0.5N systems. As is plotted in Fig. 3, there are no imaginary modes in the whole Brillouin zone for all of them, confirming their dynamic stability at ambient condition. For the primitive cell of uncovered hexagonal structure contains 8 atoms, the corresponding phonon spectra have 24 modes for each wave vector q. There are 3 acoustic and 21 optical branches in the spectra. It is obviously seen that the frequency distributions of these three systems are different with each other. The highest frequency of the atomic vibrations is up to near 25 THz for Ti0.5Si0.5N, exceeding those of Zr0.5Si0.5N and Hf0.5Si0.5N. Thus, it can be concluded the atomic interactions of Ti0.5Si0.5N are the strongest in them, implying this type of Ti0.5Si0.5N has better mechanical property and stability. The PDOS plots provide the information of frequency distributions of each element contained in the systems. One can see for these three Si-containing systems, the vibrations of Si and TM atoms dominate the lower frequency regions and the N atoms mainly supplies the higher frequency ranges.
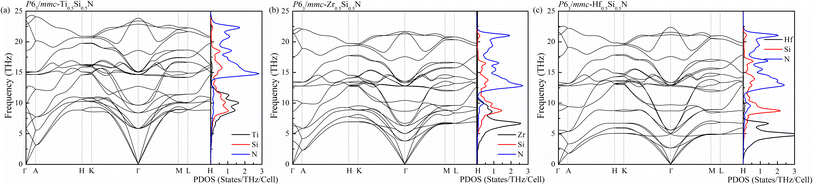 |
| | Fig. 3 Phonon spectra of Ti0.5Si0.5N (a), Zr0.5Si0.5N (b) and Hf0.5Si0.5N (c) within P63/mmc structure at ambient pressure. | |
3.2. Mechanical property
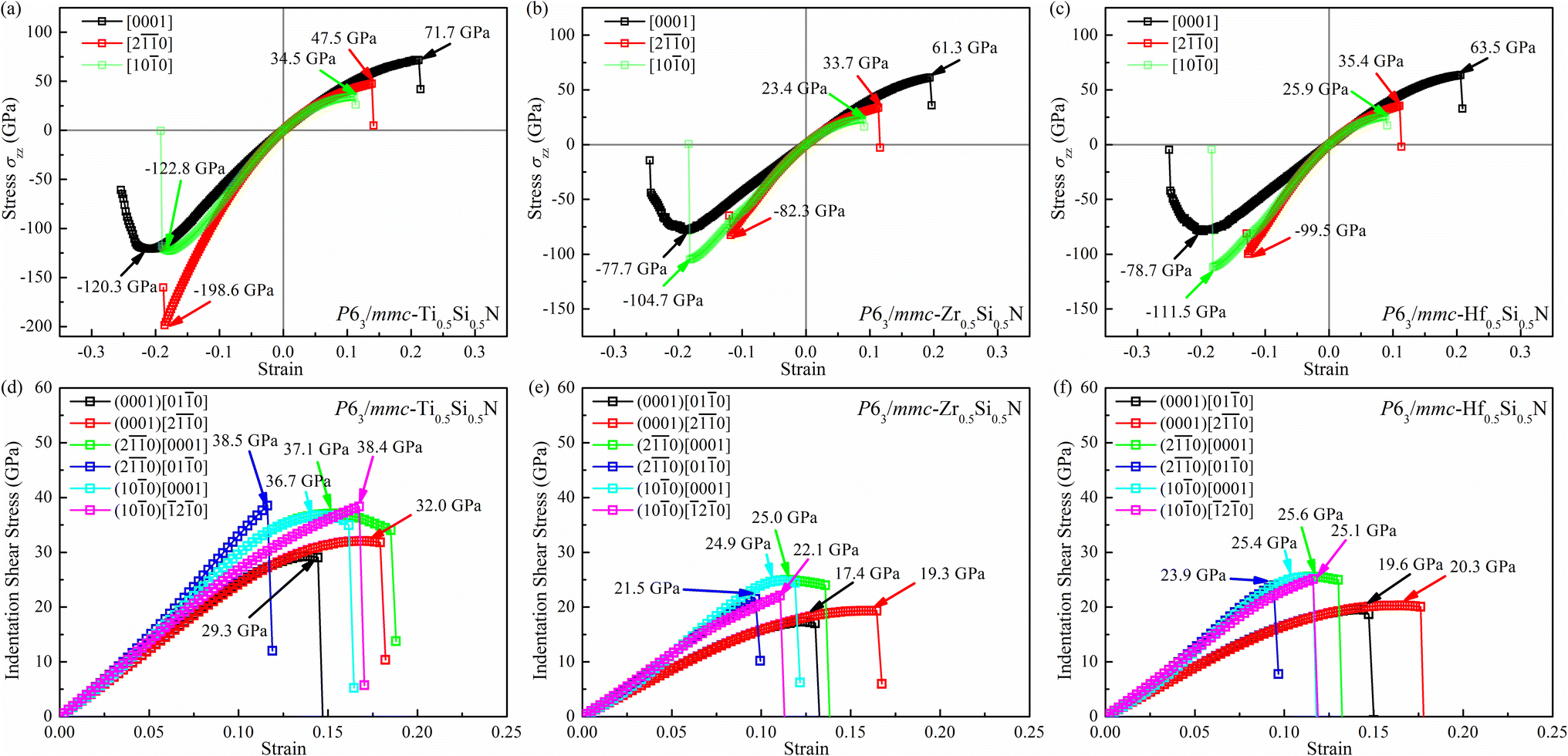
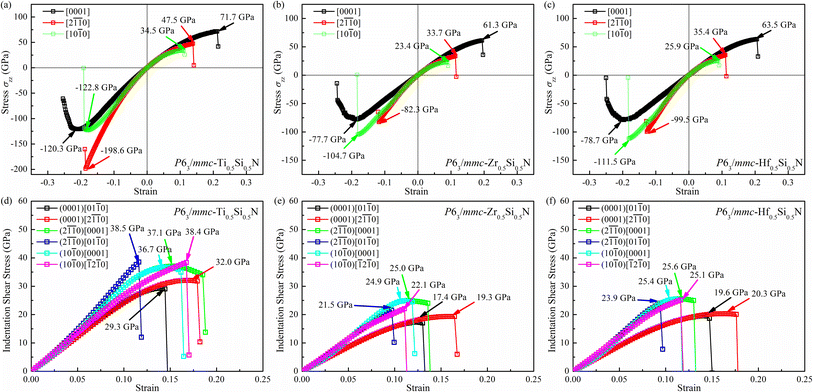
Let's focus our attention on the mechanical properties of these three ternary Si-containing systems in this section. As is known, ideal strength is an important mechanical property in definiting the macro physical natures of the materials such as hardness, toughness, brittleness and ductility etc. By applying tensile/compressive strains along certain directions while relaxing the crystal along the rest four orientations, the tensile/compressive stress–strain relations can be obtained. In stress–strain curves, the corresponding stress value of the yield point is defined as the ideal strength in this direction. Due to the hexagonal crystal symmetry, stress–strain relations in three directions ([0001], 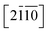 and
and 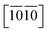 ) are necessary and plotted in Fig. 4a–c. Here, the tensile/compressive stress is denoted as σzz. It can be seen the nonlinear tensile/compressive stress occurs at the strains of about 0.04 and all these three Si-containing systems have the largest/smallest tensile strengths along [0001]/
) are necessary and plotted in Fig. 4a–c. Here, the tensile/compressive stress is denoted as σzz. It can be seen the nonlinear tensile/compressive stress occurs at the strains of about 0.04 and all these three Si-containing systems have the largest/smallest tensile strengths along [0001]/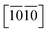 directions, reflecting their strongest and weakest stress response are along [0001] and
directions, reflecting their strongest and weakest stress response are along [0001] and 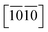 directions as shown in Fig. S1,† respectively. The ideal tensile strengths along [0001]/
directions as shown in Fig. S1,† respectively. The ideal tensile strengths along [0001]/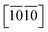 directions are 71.7/34.5 GPa, 61.3/23.4 GPa and 63.5/25.9 GPa on the strains of 0.212/0.111, 0.194/0.089 and 0.206/0.089 for Ti0.5Si0.5N, Zr0.5Si0.5N and Hf0.5Si0.5N systems respectively. The compressive strengths are all beyond 75 GPa for them in all these three directions, confirming they all have ultraincompressibility. The fracture is present near the yield points upon tensile strains and the plastic deformation is absent for all of them, implying they all behave brittleness. Because of the longest tensile curves for Ti0.5Si0.5N, that means it has the largest toughness nature relative to the rest two. By comparing the largest and the smallest strengths (denoted as σmax and σmin), the strength anisotropies can also be assessed by the ratios of σmax and σmin. The calculated tensile σmax/σmin values are 2.1, 2.6 and 2.5 for Ti0.5Si0.5N, Zr0.5Si0.5N and Hf0.5Si0.5N systems respectively, suggesting Ti0.5Si0.5N has the smallest strength anisotropy.
directions are 71.7/34.5 GPa, 61.3/23.4 GPa and 63.5/25.9 GPa on the strains of 0.212/0.111, 0.194/0.089 and 0.206/0.089 for Ti0.5Si0.5N, Zr0.5Si0.5N and Hf0.5Si0.5N systems respectively. The compressive strengths are all beyond 75 GPa for them in all these three directions, confirming they all have ultraincompressibility. The fracture is present near the yield points upon tensile strains and the plastic deformation is absent for all of them, implying they all behave brittleness. Because of the longest tensile curves for Ti0.5Si0.5N, that means it has the largest toughness nature relative to the rest two. By comparing the largest and the smallest strengths (denoted as σmax and σmin), the strength anisotropies can also be assessed by the ratios of σmax and σmin. The calculated tensile σmax/σmin values are 2.1, 2.6 and 2.5 for Ti0.5Si0.5N, Zr0.5Si0.5N and Hf0.5Si0.5N systems respectively, suggesting Ti0.5Si0.5N has the smallest strength anisotropy.
 |
| | Fig. 4 Tensile/compressive (a–c) and indentation shear (d–f) stress–strain curves for Ti0.5Si0.5N, Zr0.5Si0.5N and Hf0.5Si0.5N. | |
By simulating the real experimental measuring process of Vickers hardness, we also evaluated the values of hardness for these three systems. According to the measuring process, a biaxial strain field containing two stress components (shear σzx and compressive σzz) forms in the tested materials. The relation of the two stress components obeys the formula: σzx = σzz tan![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) φ, where φ (=68°) denotes the face-to-centerline angle of the diamond indenter.38 Using this method, the Vickers hardness of them should be well assessed. The calculated results of indentation shear stress–strain relations are plotted in Fig. 4d–f. In accord with the tensile strength, the weakest stress response occurs in the (0001) for all of these three systems, suggesting the (0001) plane can be viewed as their first cleavage plane. To obtain the indentation shear strength along as many crystal directions as possible, three typical planes along six slip directions were adopted to calculate the indentation shear strength. The largest/smallest strengths are 38.5/30.0, 25.0/17.4 and 25.6/19.6 on the strains of 0.116/0.139, 0.113/0.122 and 0.116/0.141 for Ti0.5Si0.5N, Zr0.5Si0.5N and Hf0.5Si0.5N systems respectively. Correspondingly, the indentation shear σmax/σmin values are about 1.28, 1.44 and 1.31 respectively, meaning Ti0.5Si0.5N has the best shear strength anisotropy again. To be noted that this ideal strength calculations don't take the defect and impurity inside the real crystals into consideration. However, it is effective to assess the Vickers hardness for the ideal crystalline materials. Thus, it can be concluded that this hexagonal P63/mmc Ti0.5Si0.5N possesses high Vickers hardness up to 38.5 GPa under real experimental conditions in principle, very close to the threshold of superhard bound (40 GPa). Meanwhile, Ti0.5Si0.5N, Zr0.5Si0.5N and Hf0.5Si0.5N systems all have larger tensile/compressive strengths in various directions relative to their shear strengths, indicating the failure modes of them are mainly induced by the shear loadings in the future engineering applications.
φ, where φ (=68°) denotes the face-to-centerline angle of the diamond indenter.38 Using this method, the Vickers hardness of them should be well assessed. The calculated results of indentation shear stress–strain relations are plotted in Fig. 4d–f. In accord with the tensile strength, the weakest stress response occurs in the (0001) for all of these three systems, suggesting the (0001) plane can be viewed as their first cleavage plane. To obtain the indentation shear strength along as many crystal directions as possible, three typical planes along six slip directions were adopted to calculate the indentation shear strength. The largest/smallest strengths are 38.5/30.0, 25.0/17.4 and 25.6/19.6 on the strains of 0.116/0.139, 0.113/0.122 and 0.116/0.141 for Ti0.5Si0.5N, Zr0.5Si0.5N and Hf0.5Si0.5N systems respectively. Correspondingly, the indentation shear σmax/σmin values are about 1.28, 1.44 and 1.31 respectively, meaning Ti0.5Si0.5N has the best shear strength anisotropy again. To be noted that this ideal strength calculations don't take the defect and impurity inside the real crystals into consideration. However, it is effective to assess the Vickers hardness for the ideal crystalline materials. Thus, it can be concluded that this hexagonal P63/mmc Ti0.5Si0.5N possesses high Vickers hardness up to 38.5 GPa under real experimental conditions in principle, very close to the threshold of superhard bound (40 GPa). Meanwhile, Ti0.5Si0.5N, Zr0.5Si0.5N and Hf0.5Si0.5N systems all have larger tensile/compressive strengths in various directions relative to their shear strengths, indicating the failure modes of them are mainly induced by the shear loadings in the future engineering applications.
In comparison, the tensile strengths of Ti0.5Si0.5N in ordered B1 and disordered SQS-B1 structures are also studied. As is plotted in Fig. S2,† it can be obviously seen both of them exhibit much weaker tensile and compressive strengths relative to those of hexagonal phase. The largest/smallest strengths are 13.6/6.5 GPa and 12.4/6.4 GPa for ordered and disordered structures respectively, which are very close to each other, implying there is little effect on the mechanical strengths for the occupations of the TM and Si atoms. However, most experimental hardness values of this B1–Ti0.5Si0.5N are up to beyond 20 GPa, which is mainly ascribed to the hardening effect of superlattice formed as nanocomposite.
Calculated elastic constants Cij, corresponding elastic moduli and the assessed Vickers hardness are given in Table 2. The hexagonal P63/mmc structure has five independent elastic constants (C11, C33, C44, C12 and C13) and the cubic B1 has three (C11, C12 and C44). However, this ternary systems have three elements with different radii, thus, the ordered B1 structure have different lattice constants of a and b due to the layered and alternated distribution of TM and Si atoms. Thus, the cubic B1 structure degenerates to be the tetragonal phase, correlating to the six independent elastic constants. For the firstly uncovered P63/mmc structure, the mechanical stability was checked using the criteria:34 C44 > 0, C11 > |C12|, (C11 + 2C12)C33 > 2C213. All these three Si-containing systems well satisfy the criteria, suggesting their mechanical stabilities. As is seen in Table 2, the present calculated three elastic constants for binary TiN are excellently agreement with the previous theoretical and experimental results, confirming the high reliability of our calculations. For the hexagonal ternary systems, the firstly calculated elastic constants can be used as good references for future theoretical or experimental study. Notably, with the addition of Si element, there is a decreasing trend of bulk modulus, which is attributed to the reduction of electron concentrations. However, due to the enhanced p–d hybridization and weakened d–d metallic states between TM and N atoms, the shear moduli are maintained or improved at large values, leading to larger G/B values for them. Thus, these three ternary systems all behave brittle nature (G/B > 0.57).39,40 Meanwhile, based on the elastic moduli, the Vickers hardness is calculated using the Chen formula.35 Their hardness values are 38.1, 27.3 and 30.2 GPa for Ti0.5Si0.5N, Zr0.5Si0.5N and Hf0.5Si0.5N respectively. Remarkably, despite the fact that these hardness values are evaluated in the elastic limit of the crystals, they are highly consistent with our results using the indentation shear stress–strain method above. Such high hardness supports they can be viewed as potential superhard materials. Also, it can be seen the hardness of B1 structure of Ti0.5Si0.5N is much smaller, though it was reported to possess the high hardness up to 30 GPa. The reason of the deviation can be mainly ascribed to the superlattice effect in the experimental condition. Additionally, it can be seen the mechanical properties of these three ternary systems such as the G/B values, hardness as well as strengths are not linear while going from Ti0.5Si0.5N to Zr0.5Si0.5N to Hf0.5Si0.5N. The hardness of the hexagonal Hf0.5Si0.5N is slightly higher than Zr0.5Si0.5N while Ti0.5Si0.5N has the largest value. Similar results can be seen in the “indentation shear” strengths. This seeming irregularity may be ascribed to the nonlinear distribution of the atomic radii from Ti to Zr to Hf. Zr has the largest atomic radius resulting in the largest lengths of Zr–N bonds which leads to the weaker bond strength. Thus, Zr0.5Si0.5N has the smallest hardness and lowest strengths.
Table 2 Calculated elastic constants (Cij, GPa), bulk modulus (B, GPa), shear modulus (G, GPa), Young's modulus (E, GPa), Poisson's ratio ν, G/B ratio and the Vickers hardness (Hv, GPa)
| |
Symmetry |
Source |
C11 |
C22 |
C33 |
C44 |
C55 |
C66 |
C12 |
C13 |
C23 |
B |
G |
E |
ν |
G/B |
Hv |
| Ref. 41. Ref. 42. Ref. 43. Ref. 44. |
| Ti0.5Si0.5N |
P63/mmc |
This work |
640 |
|
570 |
223 |
|
251 |
137 |
101 |
|
280 |
240 |
559 |
0.17 |
0.86 |
38.1 |
| |
B1 |
This work |
316 |
399 |
|
156 |
47 |
|
129 |
|
101 |
203 |
89 |
232 |
0.31 |
0.44 |
7.5 |
| |
B1 |
Expa |
|
|
|
|
|
|
|
|
|
|
|
260 |
|
|
25 |
| |
B1 |
Expb |
|
|
|
|
|
|
|
|
|
|
|
334 |
|
|
26 |
| Zr0.5Si0.5N |
P63/mmc |
This work |
528 |
|
469 |
168 |
|
197 |
134 |
107 |
|
246 |
185 |
443 |
0.20 |
0.75 |
27.3 |
| Hf0.5Si0.5N |
P63/mmc |
This work |
568 |
|
477 |
174 |
|
216 |
136 |
96 |
|
251 |
197 |
468 |
0.19 |
0.79 |
30.2 |
| TiN |
B1 |
This work |
598 |
|
|
155 |
|
|
144 |
|
|
295 |
181 |
451 |
0.25 |
0.61 |
20.6 |
| |
B1 |
Theoryc |
590 |
|
|
169 |
|
|
145 |
|
|
294 |
189 |
466 |
0.24 |
0.51 |
22.6 |
| |
B1 |
Expd |
625 |
|
|
163 |
|
|
165 |
|
|
320 |
|
|
|
|
23 |
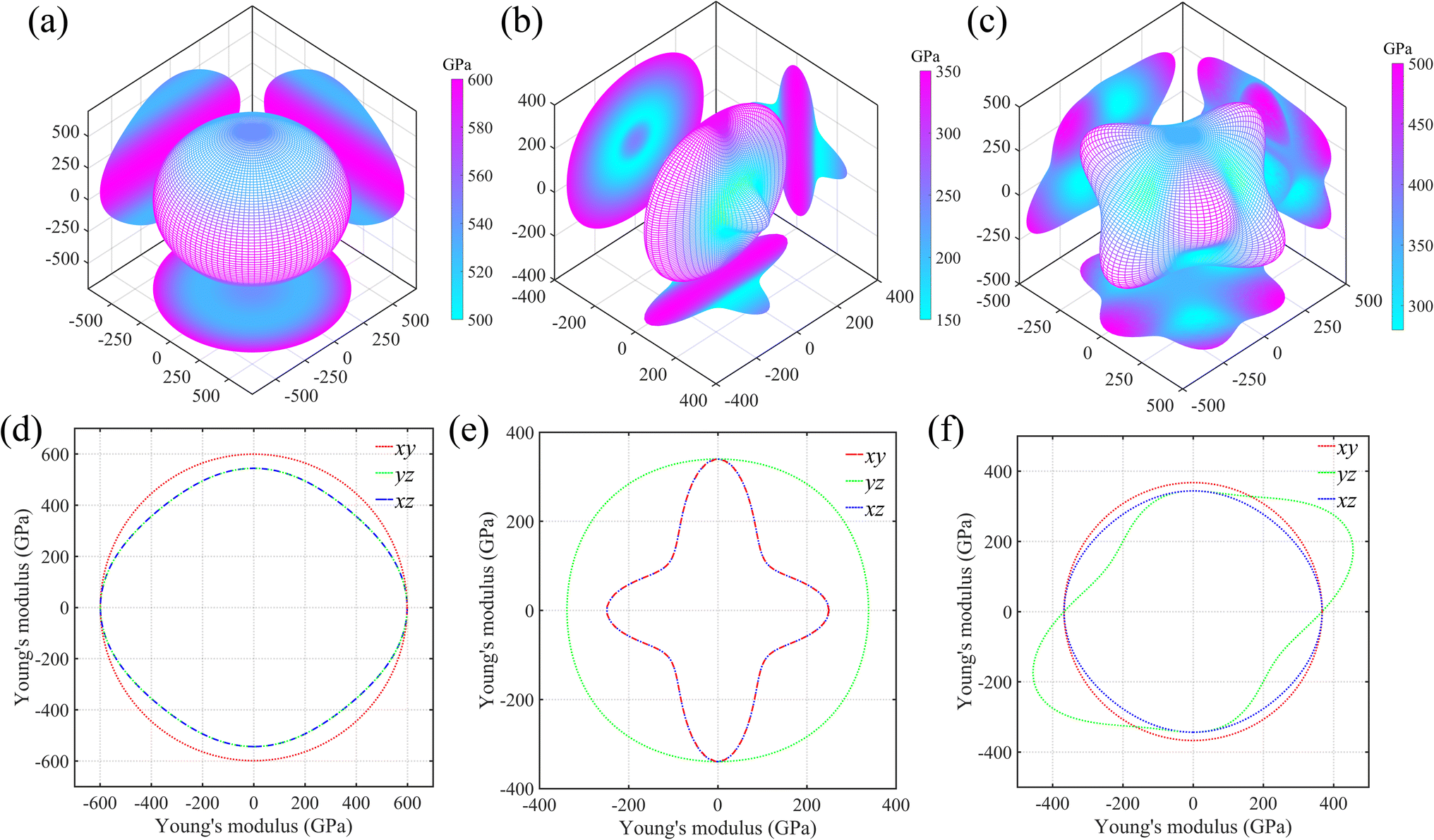
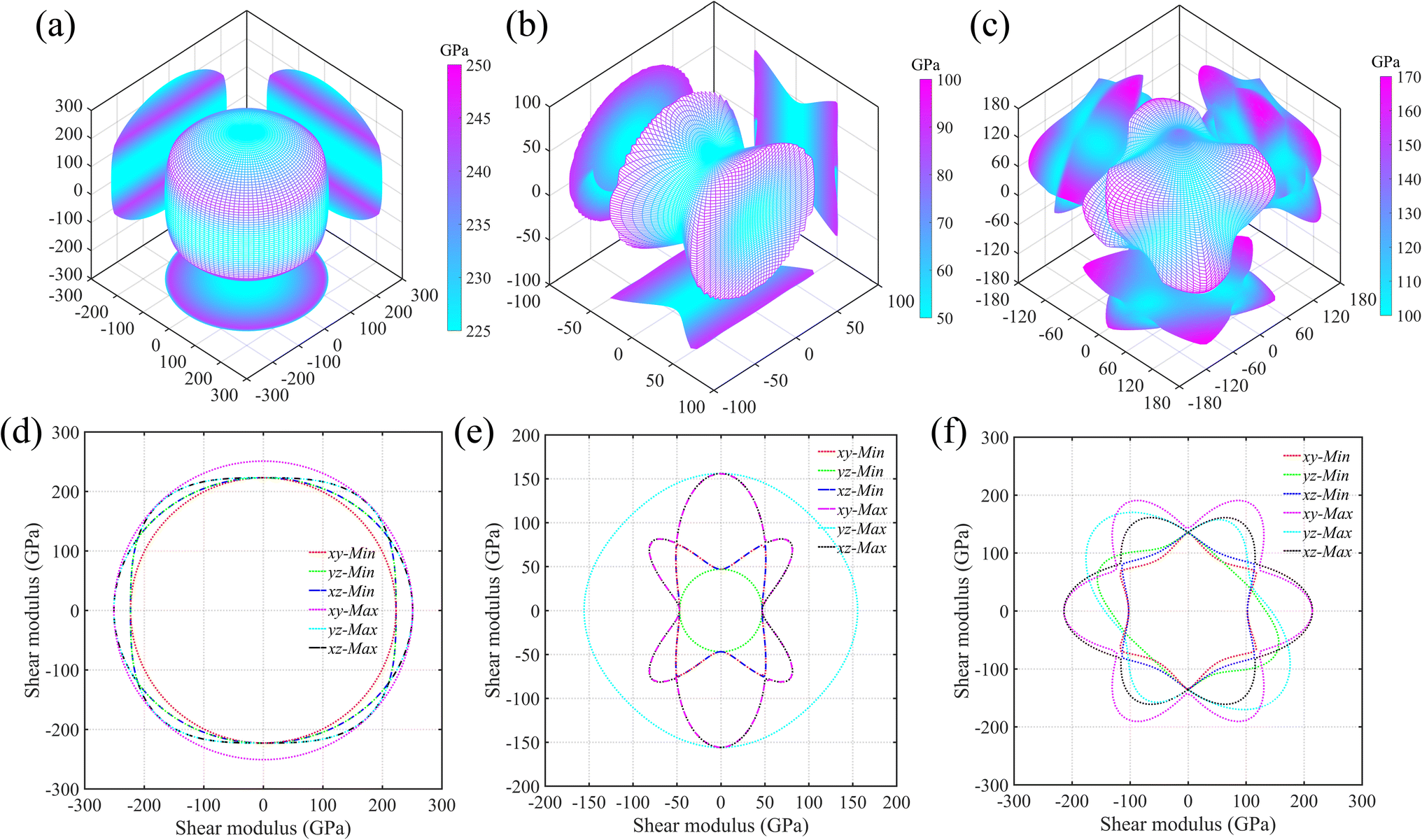
As is known, the elastic properties are achieved by estimating the stress response of the crystals under micro strains. Normally, there is elastic anisotropy depending on the crystal symmetry and the bond distributions in the materials. Here, we adopt Ti0.5Si0.5N as the representative to further compare the elastic anisotropies of hexagonal P63/mmc, B1 and trigonal R3m structures through calculating the Young's (E) and shear (G) moduli in various directions based on the formulas provided by the literature.35 The results are displayed in Fig. 5 (Young's modulus E) and Fig. 6 (shear modulus G) using the three-dimensional (3D) surface representations and two-dimensional (2D) projections on xy, xz and yz crystal planes. Here, we use the distance from the coordinate origin to the 3D surface denote the directional modulus E/G. Thus, if a material behaves isotropic elastic properties, the 3D plots would be a perfect sphere. The deviation from the sphere can reflect the elastic anisotropy. It is obviously deduced the P63/mmc structure possesses the best isotropic elastic properties thanks to the smallest deviation from the perfect sphere. This is in accordance with the calculated tensile/compressive stress–strain curves, in which the curve slopes of those three typical crystal directions near the coordinate origin are close with each other. The largest/smallest Young's moduli (Emax/Emin) for P63/mmc, B1 and R3m structures are 599/528 GPa, 348/152 GPa and 500/277 GPa, respectively. The quantitative elastic anisotropies can be compared by the ratios of Emax and Emin. The Emax/Emin values of them are 1.13, 2.29 and 1.81, meaning the P63/mmc structure has the best elastic isotropy. Also, 3D and 2D directional dependence of the shear modulus (G) are plotted in Fig. 6. Notably, the shear modulus is different from the Young's modulus, because the G value varies with the slipping direction for a given crystal plane. Therefore, each crystal plane has minimum and maximum G values (Gmin and Gmax). Here, the 3D directional dependence of Gmin are plotted in Fig. 6a–c and the 2D projections of Gmin and Gmax on xy, xz and yz planes are plotted in Fig. 6d–f. Obviously, P63/mmc structure exhibits the best shear anisotropy due to the fact that its 3D plot is close to a perfect sphere. The ratios of Gmax/Gmin are 251 GPa/223 GPa ≈ 1.13 (P63/mmc), 156 GPa/47 GPa ≈ 3.32 (B1) and 216 GPa/105 GPa ≈ 2.06 (R3m). It can be seen the ordered B1 structure has large shear anisotropy. For the Zr0.5Si0.5N and Hf0.5Si0.5N systems within these three structures, the results of the strength and elastic anisotropies are similar to the Ti0.5Si0.5N. Hence, they are not repetitively discussed here.
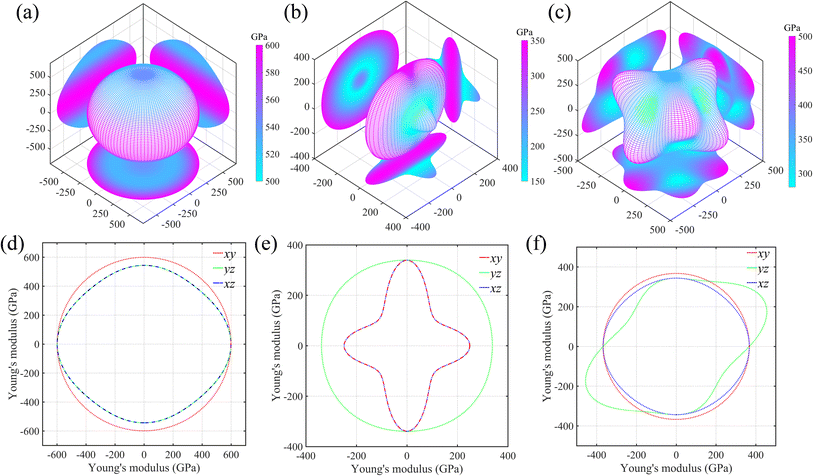 |
| | Fig. 5 Three-dimensional (3D) surface representations of Young's moduli for Ti0.5Si0.5N within P63/mmc (a), B1 (b) and R3m (c) structures and two-dimensional (2D) projections (d–f) of the Young's moduli on the xy, yz and xz planes. | |
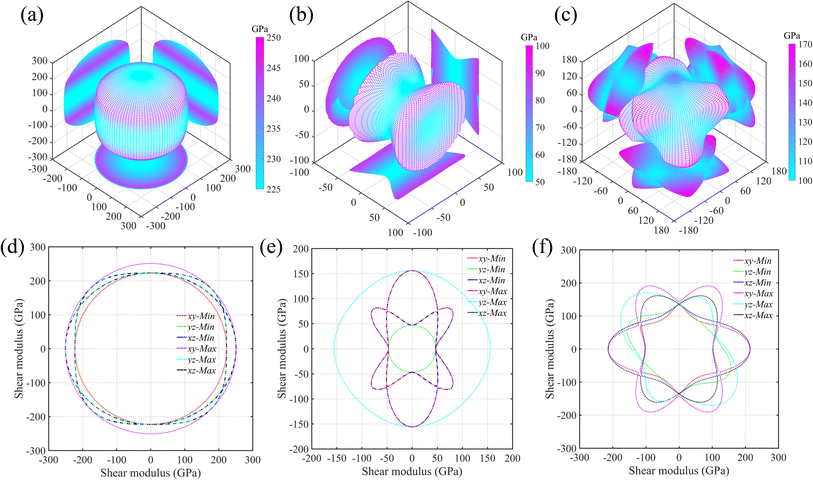 |
| | Fig. 6 Three-dimensional (3D) surface representations of shear moduli for Ti0.5Si0.5N within P63/mmc (a), B1 (b) and R3m (c) structures and two-dimensional (2D) projections (d–f) of the shear moduli on the xy, yz and xz planes. | |
3.3. Electronic properties
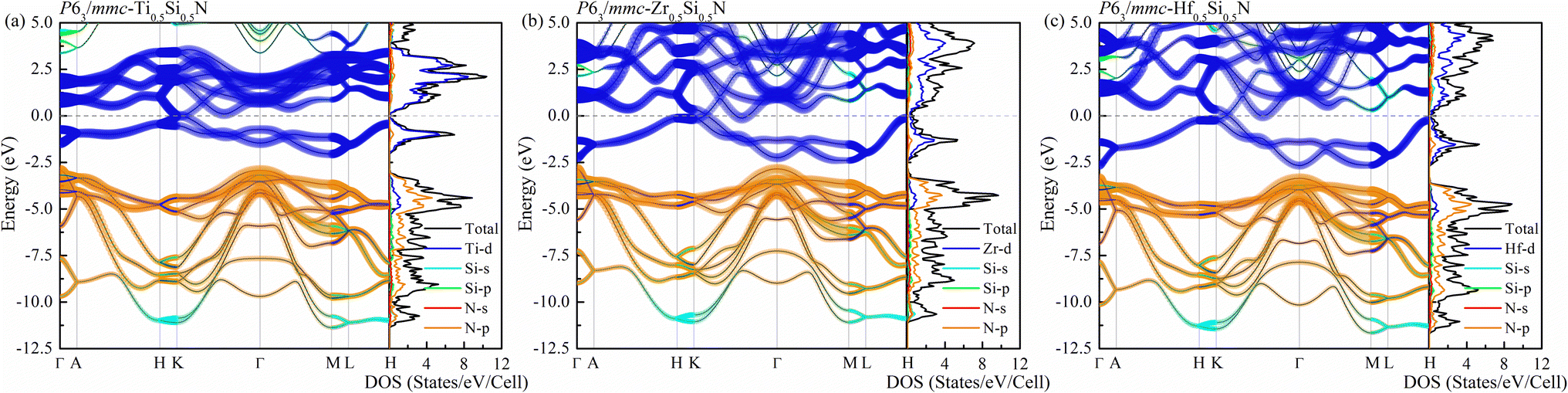
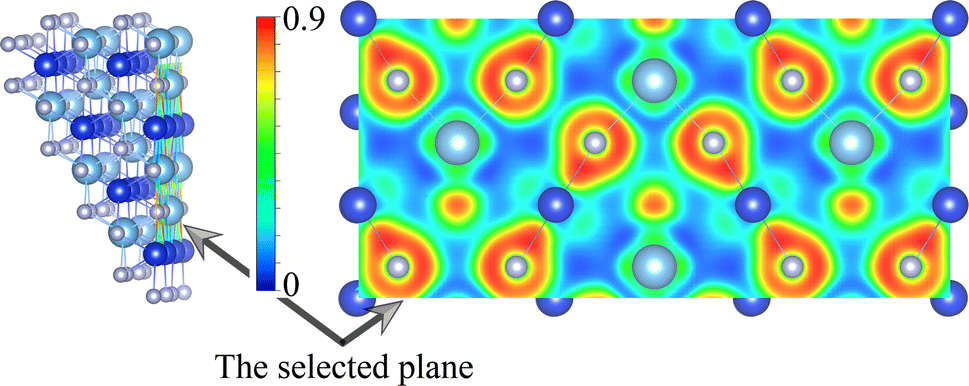
We now turn to discuss the electronic properties of these Si-containing ternary systems. Due to the same valence electrons for Ti, Zr and Hf elements, the electronic structures of Ti0.5Si0.5N, Zr0.5Si0.5N and Hf0.5Si0.5N systems are similar with each other in the same structures. Fig. 7 shows the calculated projected band structures and partial density of states (PDOS) of Ti0.5Si0.5N, Zr0.5Si0.5N and Hf0.5Si0.5N within P63/mmc structure. It can be seen their band structures are analogous with each other. The band gaps are absent for all of them, indicating their metallic nature. In particular, it can be seen the conduction bands are mainly dominated by the M–d orbitals, the valence bands by partial M–d and N–p orbitals. The p–d hybridization can be obviously observed near the EF. Meanwhile, one can see the difference of crystal structure enormously impact the band structures. As shown in Fig. S3(a),† for Ti0.5Si0.5N within B1 phase, the EF lies at the antibonding regions, in contrast, it is modified effectively at the overlap of valence band top and conduction band bottom for P63/mmc structure, leading to the enhanced electronic stability and mechanical property. In comparison, TiN and SiN within hexagonal P63/mmc structure are also simulated to study the difference of the band structure from the ternary system. As seen in Fig. S3(b and c),† it can be seen some conduction bands descend below the EF for TiN, as a contrast, the valence bands spread through the EF for SiN. That means the ternary hexagonal phase possesses the best electronic stability and the maximized covalent p–d hybridization, inducing the enhanced anti-shear property. The PDOS plots show that the pseudo-gaps near the EF separating the bonding and antibonding states can be obviously seen for Ti0.5Si0.5N within P63/mmc and B1 structures. However, thanks to the introduction of Si element, the EF is exactly modulated at the valley of the pseudo-gap for P63/mmc structure. It suggests the bonding states are fully occupied, whereas the antibonding states are not. The p–d hybridization can be seen in the energy range of 0 to −5 eV for Ti0.5Si0.5N. B1 structure exhibits the obvious occupation of d–d states near the EF, resulting in the improved total formation enthalpy and the metallic nature, which is not favorable for the mechanical properties of compounds. Though the Si element induces the modulated electronic structure, it has weaker interactions with N element due to its small contributions to the bonding states for both s-orbitals and p-orbitals in the related energy range. The plot of calculated ELF is presented in Fig. 8. To clearly exhibit the Ti–N and Si–N bonding nature, herein, we selected a crystal plane holding the Ti, Si and N atoms within a common plane. The ELF value is dimensionless and it has a range of 0–1. ELF = 1/ELF = 0 have the meaning of highly localized/lack of electrons. Hence, the bonding nature can be evaluated by discerning the color of the region among the atoms in the plot. It can be seen the electrons are highly localized between Si and N atoms, suggesting the covalent Si–N bonding. Thanks to the highly p–d coupling, the Ti–N bonds also exhibit strong covalent feature, though comprising some of metallic component.
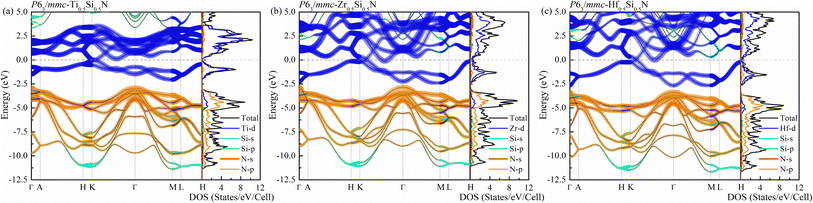 |
| | Fig. 7 Projected band structures and projected density of states (PDOS) for (a) Ti0.5Si0.5N, (b) Zr0.5Si0.5N and (c) Hf0.5Si0.5N within P63/mmc structure. | |
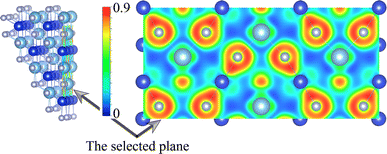 |
| | Fig. 8 Calculated ELF slice of the selected plane of P63/mmc-Ti0.5Si0.5N. | |
4. Conclusions
In summary, the structure diversity of Ti0.5Si0.5N, Zr0.5Si0.5N and Hf0.5Si0.5N systems under the pressure of 0–100 GPa are firstly explored using the developed crystal structure searching method combined with the first-principle calculations. Theoretical explorations on their structural stability, electronic and mechanical properties were systematically performed by first-principle calculations. Our results confirmed that the P63/mmc structure of these three Si-containing ternary systems has surprisingly enhanced ideal strengths and larger hardness up to 38.1 GPa, as well as better mechanical isotropy relative to the ordered B1 structure. Electronic analysis in depth was also carried out to further study the mechanism of the hardening inducement. For this new hexagonal P63/mmc structure, the addition of Si element well tuned the electronic structure to make the EF position optimized and maximized the p–d hybridization and minimized the occupation of antibonding d–d states. Meanwhile, the structural stability was also improved by this electronic adjust. All these findings would be expected to stimulate further experimental and theoretical works on these Si-containing systems and provide effective strategy in designing new type of hard materials with composite combination of beneficial properties.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by State Key Laboratory of Precision Blasting and Hubei Key Laboratory of Blasting Engineering, Jianghan University (No. PBSKL2022D03), the Natural Science Foundation of China (Grant Nos. 51808554), the Natural Science Basic Research plan in Shaanxi Province of China (Grant No. 2020JQ-892).
References
- R. F. Bunshah, Handbook of Hard Coatings, William Andrew Publishing, New York, 2002 Search PubMed.
- J. H. Hsieh, A. L. K. Tan and X. T. Zeng, Oxidation and wear behaviors of Ti-based thin films, Surf. Coat. Technol., 2006, 201, 4094–4098 CrossRef CAS.
- M. Wittmer, J. Noser and H. Melchior, Oxidation kinetics of TiN thin films, J. Appl. Phys., 1981, 52, 6659 CrossRef CAS.
- T. Altan, Process simulation using finite element method-prediction of cutting forces, tool stresses and temperatures in high-speed flat end milling, Int. J. Mach. Tools Manuf., 2000, 40, 713–738 CrossRef.
- B. D. Beake, L. Isern, J. L. Endrino and G. S. Fox-Rabinovich, Micro-impact testing of AlTiN and TiAlCrN coatings, Wear, 2019, 418–419, 102–110 CrossRef CAS.
- G. S. Fox-Rabinovich, A. I. Kovalev, M. H. Aguirre, B. D. Beake, K. Yamamoto, S. C. Veldhuis, J. L. Endrino, D. L. Wainstein and A. Y. Rashkovskiy, Design and performance of AlTiN and TiAlCrN PVD coatings for machining of hard to cut materials, Surf. Coat. Technol., 2009, 204(4), 489–496 CrossRef CAS.
- D. G. Sangiovanni, V. Chirita and L. Hultman, Toughness enhancement in TiAlN-based quarternary alloys, Thin Solid Films, 2012, 520(11), 4080–4088 CrossRef CAS.
- D. Becker, Wear of nanostructured composite tool coatings, Wear, 2013, 304, 88–95 CrossRef CAS.
- G. Greczynski, S. Mráz, M. Hans, D. Primetzhofer, J. Lu, L. Hultman and J. M. Schneider, Unprecedented Al supersaturation in single-phase rock salt structure VAlN films by Al+ subplantation, J. Appl. Phys., 2017, 121(17), 171907 CrossRef.
- I. Endler, M. Höhn, J. Schmidt, S. Scholz, M. Herrmann and M. Knaut, Ternary and quarternary TiSiN and TiSiCN nanocomposite coatings obtained by Chemical Vapor Deposition, Surf. Coat. Technol., 2013, 215, 133–140 CrossRef CAS.
- S. Vepřek and S. Vepr, The search for novel, superhard materials, J. Vac. Sci. Technol., A, 1999, 17, 2401 CrossRef.
- A. B. Mei, O. Hellman, N. Wireklint, C. M. Schlepütz, D. G. Sangiovanni, B. Alling, A. Rockett, L. Hultman, I. Petrov and J. E. Greene, Dynamic and structural stability of cubic vanadium nitride, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 91(5), 054101 CrossRef.
- L. Chen, J. Xu, M. Zhang, T. Rong, Z. Jiang and P. Li, Systematic study on mechanical and electronic properties of ternary VAlN, TiAlN and WAlN systems by first-principles calculations, Ceram. Int., 2021, 47(6), 7511–7520 CrossRef CAS.
- L. Chen and Z. Jiang, First-principles study on a new type of quaternary carbonitride VWCN with outstanding mechanical properties, Int. J. Refract. Met. Hard Mater., 2020, 92, 105319 CrossRef CAS.
- Y. Tian, B. Xu and Z. Zhao, Microscopic theory of hardness and design of novel superhard crystals, Int. J. Refract. Met. Hard Mater., 2012, 33, 93–106 CrossRef CAS.
- X. Guo, L. Li, Z. Liu, D. Yu, J. He, R. Liu, B. Xu, Y. Tian and H. T. Wang, Hardness of covalent compounds Roles of metallic component and d valence electrons, J. Appl. Phys., 2008, 104, 023503 CrossRef.
- Y. Wang, J. Lv, L. Zhu and Y. Ma, Crystal structure prediction via particle-swarm optimization, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82(9), 1–8 Search PubMed.
- Y. Wang, J. Lv, L. Zhu and Y. Ma, CALYPSO: A method for crystal structure prediction, Comput. Phys. Commun., 2012, 183, 2063–2070 CrossRef CAS.
- M. Zhang, H. Yan and Q. Wei, Unexpected ground-state crystal structures and mechanical properties of transition metal pernitrides MN2 (M= Ti, Zr, and Hf), J. Alloys Compd., 2019, 774, 918–925 CrossRef CAS.
- Q. Wei, W. Tong, B. Wei, M. Zhang and X. Peng, Six new silicon phases with direct band gaps, Phys. Chem. Chem., 2019, 21, 19963–19968 RSC.
- L. Chen, J. Xu, M. Zhang, Z. Wen and Z. Jiang, Structural , mechanical and electronic properties study on group 5 transition metals ternary mononitrides from first-principles calculations, J. Alloys Compd., 2020, 813, 152246 CrossRef CAS.
- L. Chen, M. Zhang, J. Chang and Z. Jiang, Theoretical investigation on vanadium dinitrides from first-principles calculations, Ceram. Int., 2018, 45(2), 2457–2465 CrossRef.
- Y. R. Zhao, T. T. Bai, L. N. Jia, W. Xin, Y. F. Hu, X. S. Zheng and S. T. Hou, Probing the Structural and Electronic Properties of Neutral and Anionic Lanthanum-Doped Silicon Clusters, J. Phys. Chem. C, 2019, 123(47), 28561–28568 CrossRef CAS.
- L. Chen, J. Xu, M. Zhang and Y. Zhang, Theoretical study on structural, mechanical and electronic properties of ternary mononitride Ti0.5W0.5N from first-principles calculations, Mater. Chem. Phys., 2020, 242, 122476 CrossRef CAS.
- L. Chen, J. Xu, M. Zhang, G. Zhang, Y. Zhao and Y. Zhang, Theoretical study on novel orthorhombic ternary monocarbides M0.5Re0.5C (M = V, Nb, Ta) from first-principles calculations, Ceram. Int., 2020, 46(15), 24624–24634 CrossRef CAS.
- G. Kresse and J. Furthmüller, Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54(16), 11169–11186 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- P. E. Blöchl, Projector augmented-wave method, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- G. Kresse and D. Joubert, From ultrasoft pseudopotentials to the projector augmented-wave method, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59(3), 1758–1775 CrossRef CAS.
- H. J. Monkhorst and J. D. Pack, Special points for Brillonin-zone integrations, Phys. Rev. B: Solid State, 1976, 13, 5188–5192 CrossRef.
- S. H. Wei, L. G. Ferreira, J. E. Bernard and A. Zunger, Electronic properties of random alloys: Special quasirandom structures, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 42(15), 9622–9649 CrossRef CAS.
- S. H. W. A. Zunger, L. G. Ferreira and J. E. Bernard, Special Quasirandom Structures, Phys. Rev. Lett., 2012, 65(3), 353–356 CrossRef.
- A. Togo and I. Tanaka, First principles phonon calculations in materials science, Scr. Mater., 2015, 108, 1–5 CrossRef CAS.
- K. H. M. Born, Dynamical Theory of Crystal Lattices, Oxford University Press, Inc., London, 1954 Search PubMed.
- T. C. T. Ting, Anisotropic Elasticity Theory and Applications, Oxford University Press, Inc., 1996 Search PubMed.
- X.-q. Chen, H. Niu, D. Li and Y. Li, Intermetallics Modeling hardness of polycrystalline materials and bulk metallic glasses, Intermetallics, 2011, 19(9), 1275–1281 CrossRef CAS.
- A. S. B. Silvi, Classification of chemical bonds based on topological analysis of electron localization functions, Nature, 1994, 371, 683–686 CrossRef.
- C. Zang, H. Sun, J. S. Tse and C. Chen, Indentation strength of ultraincompressible rhenium boride , carbide , and nitride from first-principles calculations, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 86, 014108 CrossRef.
- S. F. Pugh, XCII. Relations between the elastic moduli and the plastic properties of polycrystalline pure metals, Philos. Mag., 1954, 45, 823–843 CAS.
- H. Yan, Z. Wei, M. Zhang and Q. Wei, Exploration of stable stoichiometries, ground-state structures, and mechanical properties of the W–Si system, Ceram. Int., 2020, 46(10), 17034–17043 CrossRef CAS.
- G. Greczynski, B. Bakhit, L. Hultman and M. Odén, High Si content TiSiN films with superior oxidation resistance, Surf. Coat. Technol., 2020, 398, 126087 CrossRef CAS.
- A. Al-rjoub, A. Cavaleiro and F. Fernandes, Influence of Ag alloying on the morphology, structure, mechanical properties, thermal stability and oxidation resistance of multilayered TiSiN/Ti(Ag)N films, Mater. Des., 2020, 192, 108703 CrossRef CAS.
- S. Yu, Q. Zeng, A. R. Oganov, G. Frapper and L. Zhang, Phase stability, chemical bonding and mechanical study, Phys. Chem. Chem. Phys., 2015, 17, 11763–11769 RSC.
- R. Ahuja, O. Eriksson and J. Wills, Structural, elastic, and high-pressure properties of cubic TiC, TiN, and TiO, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 53(6), 3072–3079 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence abc,
Quanmin Xie*ab,
Yongsheng Jia*ab and
Yingkang Yaoab
abc,
Quanmin Xie*ab,
Yongsheng Jia*ab and
Yingkang Yaoab
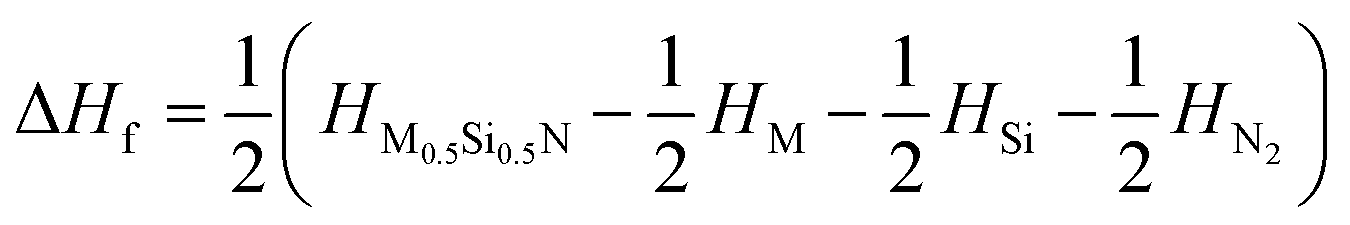 , (M = Ti, Zr, Hf), where HM0.5Si0.5N, HM, HSi, and HN2 are the enthalpies per f.u. of M0.5Si0.5N, pure hcp-Ti/Zr/Hf, diomand-type Si and α-N2. The calculated results are −1.918, −1.666 and −1.596 eV/f.u. for hexagonal Ti0.5Si0.5N, Zr0.5Si0.5N and Hf0.5Si0.5N systems respectively. For trigonal R3m structure, the formation enthalpies are −2.092, −1.846 and −1.866 eV/f.u. respectively. As a contrast, the formation enthalpies of them within ordered B1 structure are −1.172, −1.050 and −1.104 eV/f.u., respectively, indicating the trigonal phases are more thermodynamically stable than the hexagonal and ordered B1 phases in principle at ambient condition. Fig. 2 displays the calculated results of enthalpy differences of these three systems within B1 and R3m structures relative to the hexagonal P63/mmc structure as a function of pressure. It is clearly seen that with the pressure ascending beyond 15 15 and 25 GPa, P63/mmc structure transforms to be the most energetically favourable phase for them, as it has the lowest total enthalpy. Meanwhile, the enthalpy differences have an increasing tendency with pressure, implying the thermodynamic stability of P63/mmc structure can be maintained up to 100 GPa.
, (M = Ti, Zr, Hf), where HM0.5Si0.5N, HM, HSi, and HN2 are the enthalpies per f.u. of M0.5Si0.5N, pure hcp-Ti/Zr/Hf, diomand-type Si and α-N2. The calculated results are −1.918, −1.666 and −1.596 eV/f.u. for hexagonal Ti0.5Si0.5N, Zr0.5Si0.5N and Hf0.5Si0.5N systems respectively. For trigonal R3m structure, the formation enthalpies are −2.092, −1.846 and −1.866 eV/f.u. respectively. As a contrast, the formation enthalpies of them within ordered B1 structure are −1.172, −1.050 and −1.104 eV/f.u., respectively, indicating the trigonal phases are more thermodynamically stable than the hexagonal and ordered B1 phases in principle at ambient condition. Fig. 2 displays the calculated results of enthalpy differences of these three systems within B1 and R3m structures relative to the hexagonal P63/mmc structure as a function of pressure. It is clearly seen that with the pressure ascending beyond 15 15 and 25 GPa, P63/mmc structure transforms to be the most energetically favourable phase for them, as it has the lowest total enthalpy. Meanwhile, the enthalpy differences have an increasing tendency with pressure, implying the thermodynamic stability of P63/mmc structure can be maintained up to 100 GPa.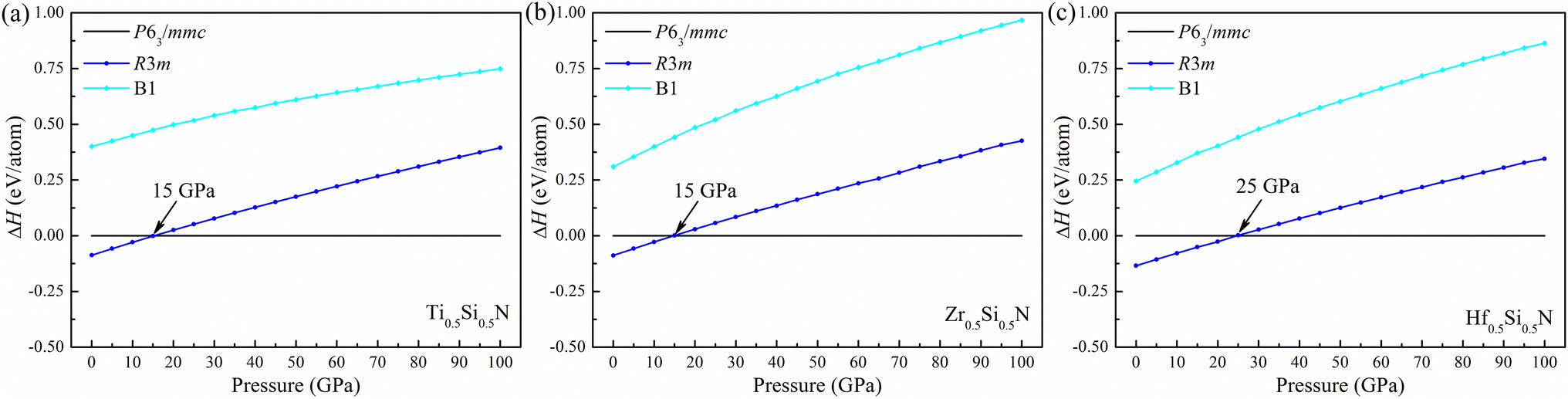
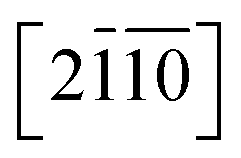 and
and 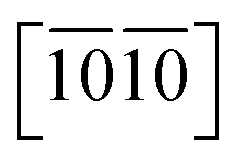 ) are necessary and plotted in Fig. 4a–c. Here, the tensile/compressive stress is denoted as σzz. It can be seen the nonlinear tensile/compressive stress occurs at the strains of about 0.04 and all these three Si-containing systems have the largest/smallest tensile strengths along [0001]/
) are necessary and plotted in Fig. 4a–c. Here, the tensile/compressive stress is denoted as σzz. It can be seen the nonlinear tensile/compressive stress occurs at the strains of about 0.04 and all these three Si-containing systems have the largest/smallest tensile strengths along [0001]/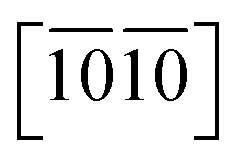 directions, reflecting their strongest and weakest stress response are along [0001] and
directions, reflecting their strongest and weakest stress response are along [0001] and 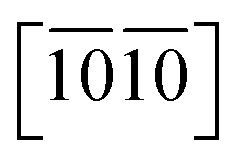 directions as shown in Fig. S1,† respectively. The ideal tensile strengths along [0001]/
directions as shown in Fig. S1,† respectively. The ideal tensile strengths along [0001]/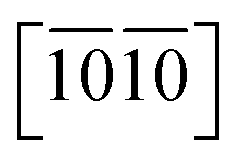 directions are 71.7/34.5 GPa, 61.3/23.4 GPa and 63.5/25.9 GPa on the strains of 0.212/0.111, 0.194/0.089 and 0.206/0.089 for Ti0.5Si0.5N, Zr0.5Si0.5N and Hf0.5Si0.5N systems respectively. The compressive strengths are all beyond 75 GPa for them in all these three directions, confirming they all have ultraincompressibility. The fracture is present near the yield points upon tensile strains and the plastic deformation is absent for all of them, implying they all behave brittleness. Because of the longest tensile curves for Ti0.5Si0.5N, that means it has the largest toughness nature relative to the rest two. By comparing the largest and the smallest strengths (denoted as σmax and σmin), the strength anisotropies can also be assessed by the ratios of σmax and σmin. The calculated tensile σmax/σmin values are 2.1, 2.6 and 2.5 for Ti0.5Si0.5N, Zr0.5Si0.5N and Hf0.5Si0.5N systems respectively, suggesting Ti0.5Si0.5N has the smallest strength anisotropy.
directions are 71.7/34.5 GPa, 61.3/23.4 GPa and 63.5/25.9 GPa on the strains of 0.212/0.111, 0.194/0.089 and 0.206/0.089 for Ti0.5Si0.5N, Zr0.5Si0.5N and Hf0.5Si0.5N systems respectively. The compressive strengths are all beyond 75 GPa for them in all these three directions, confirming they all have ultraincompressibility. The fracture is present near the yield points upon tensile strains and the plastic deformation is absent for all of them, implying they all behave brittleness. Because of the longest tensile curves for Ti0.5Si0.5N, that means it has the largest toughness nature relative to the rest two. By comparing the largest and the smallest strengths (denoted as σmax and σmin), the strength anisotropies can also be assessed by the ratios of σmax and σmin. The calculated tensile σmax/σmin values are 2.1, 2.6 and 2.5 for Ti0.5Si0.5N, Zr0.5Si0.5N and Hf0.5Si0.5N systems respectively, suggesting Ti0.5Si0.5N has the smallest strength anisotropy.