
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInsight into TLR4 receptor inhibitory activity via QSAR for the treatment of Mycoplasma pneumonia disease†
Zemin Zhua,
Ziaur Rahmana,
Muhammad Aamira,
Syed Zahid Ali Shah b,
Sattar Hamidc,
Akhunzada Bilawald,
Sihong Lie and
Muhammad Ishfaq*a
b,
Sattar Hamidc,
Akhunzada Bilawald,
Sihong Lie and
Muhammad Ishfaq*a
aCollege of Computer Science, Huanggang Normal University, Huanggang 438000, China. E-mail: muhammad@hgnu.edu.cn; Tel: +86 15972855212
bDepartment of Pathology, Faculty of Veterinary and Animal Sciences, The Islamia University of Bahawalpur-Pakistan, Pakistan
cThe University of Agriculture Peshawar, Khyber Pakhtunkhwa, 25130, Pakistan
dCollege of Food Science, Northeast Agricultural University, Harbin, China
eKey Laboratory of Applied Technology on Green-Eco-Healthy Animal Husbandry of Zhejiang Province, College of Animal Science and Technology, College of Veterinary Medicine, Zhejiang A&F University, Hangzhou 311300, China
First published on 11th January 2023
Abstract
Mycoplasma pneumoniae (MP) is one of the most common pathogenic organisms causing upper and lower respiratory tract infections, lung injury, and even death in young children. Toll-like receptors (TLRs) play an important role in innate immunity by allowing the host to recognize pathogens invading the body. Previous studies demonstrated that TLR4 is a potential therapeutic target for the treatment of MP pneumonia. Therefore, the present study aimed to screen biologically active ingredients that target the TLR4 receptor pathway. We first used molecular docking to screen out the active compounds inhibiting the TLR4 pathway, and then used regression and classification machine learning algorithms to establish a quantitative structure–activity relationship (QSAR) model to predict the biological activity of the screened compounds. A total of 78 molecules were used in QSAR modelling, which were retrieved from the ChEMBL database. The QSAR models had acceptable correlation coefficients of R2 on the training and testing dataset in the range of 0.96 to 0.91 and 0.93 to 0.76, respectively. The multiclass classification models showed accuracy on training and testing data within ranges of 1.0 to 0.70, 0.96 to 0.63, and log loss ranges from 0.27 to 8.63, respectively. In addition, molecular descriptors and fingerprints have been studied as structural elements involved in increased and decreased inhibitory activities. These results provide a quantitative analysis of QSAR and classification models applicable for high-throughput screening, as well as insights into the mechanisms of inhibition of TLR4 antagonists.
1. Introduction
Mycoplasmas are highly pleomorphic microorganisms, contain a very small genome ranging from 0.58–2.20 Mb and cannot flourish without cholesterol.1 As a human pathogen, Mycoplasma pneumoniae (MP) lack a cell wall and has a specialized tip organelle responsible for its cytoadherence.2 MP causes asthma, bronchitis, pneumonia, and pharyngitis in humans. Young children and young adults are most likely to be affected by the MP infection.3 There were outbreaks of MP infections worldwide from 2010 to 2012 in different regions of the World.4,5 The pathophysiological changes that take place because of MP infection are attributed to several factors, including membrane lipoproteins, polysaccharides, and viral nucleases.6 Previous studies demonstrated that pattern recognition receptors (PRRs) on immune cells detect pathogen-associated molecular patterns (PAMPs) on the surface of pathogens when they invade the host.7 The toll-like receptor family (TLRs) is a family of type I transmembrane transport receptors that are primarily expressed on the surface of epithelial cells and immune cells. They play an important role in the early recognition and inflammatory response of the host against pathogens.8 Among the TLRs, TLR4 is the first in the TLR family. It can recognize pathogenic microorganisms, triggering the production of cytokines by cells, chemokines, adhesion factors, and acute phase proteins that regulate inflammatory responses,9 which plays an important role in the occurrence of inflammation, especially the activation of the immune system.10 A previous study had demonstrated that exposure of mouse macrophages to MP lipids activated the TLR4 signaling pathway, resulting in the expression of tumor necrosis factor (TNF)-α and interleukin-1β mediated by autophagy and nuclear factor kappa B (NF-κB).11 Accordingly, these findings suggest that TLR4 is not only involved in the body's ability to resist pathogen invasion, but that it may also play an important role in the development and occurrence of inflammation-mediated injuries. Thus, the inhibition of TLR4 in MP infections needs to be further explored. Meanwhile, anti-inflammatory compounds that target the TLR4 signaling pathway have received much attention, and it has become a research hotspot in this field, which can help in discovering new therapeutic targets.Recently, more and more active compounds in natural products have been found to inhibit the TLR4 pathway. Studies revealed that natural products are rich in molecules that possess the potential to inhibit the TLR4 protein and have attracted the attention of researchers.12–15 Furthermore, TLR4 inhibition mediated by small molecules led to an array of research focusing on the molecular mechanism of action of TLR4 inhibitors.16 However, future studies are needed to confirm these findings. Besides, the emergence of antibiotic resistance represents another challenge in the context of the treatment of MP infections.17 It is therefore crucial to find alternatives to antibiotics to prevent the emergence of resistance against anti-mycoplasma drugs. Hence, active ingredients of natural products could be used to modulate the host immune inflammatory response. The active ingredients of natural products require more experimental data on its toxicology and pharmacology before its use in clinical trials, which is a lengthy process. To screen TLR4 inhibitors in a limited period of time, it is necessary to use fast, accurate, and reliable screening methods based on detailed study of TLR4 inhibition and regulation. In this context, modern machine learning technologies make better use of information obtained from several sources to predict the bioactivities of drugs for several diseases, thus facilitating the discovery of new drugs more efficiently. In recent years, computer-aided drug screening is advancing towards practicality and is emerging as a core technology for innovative drug research. Several drug libraries were screened in a very short time-period, leading to the discovery of many active compounds in traditional Chinese medicines and the successful repurposing of several approved drugs.18,19 Based on previous research, virtual screening paved the way for the future development of improved chemical analogs for use in treating a wide variety of human and animal diseases through medicinal chemistry structure–activity relationships and drug screenings.20 Molecular docking and quantitative structure activity relationship (QSAR) models provide structural information and insight into TLR4 inhibitors that can be used to guide more effective drug development, including screening and rational drug discovery of TLR4 inhibitors. Therefore, the objective of the present study was to identify lead compounds that can inhibit the TLR4 protein for the treatment of MP pneumonia. The regression and classification QSAR models were developed from a set of known chemical TLR4 inhibitors. These QSAR models will be used to predict and classify the bioactive compounds based on their predicted bioactivity (pIC50) values and provide theoretical foundations to enable the development of potent drugs from natural products for the prevention and treatment of MP-pneumonia. The flow chart for the experimental process is shown in Fig. 1.
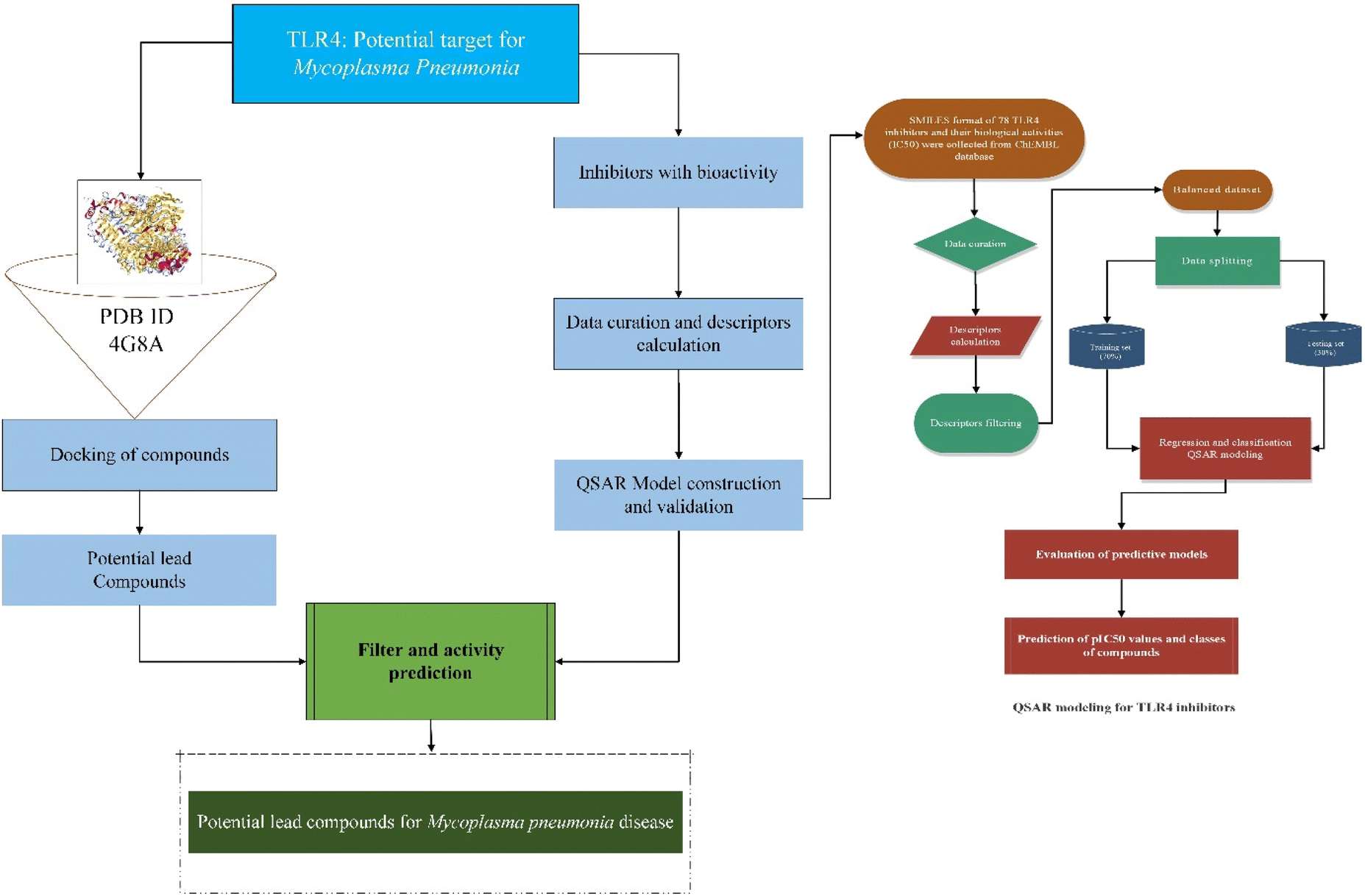 | ||
| Fig. 1 Schematic diagram represents the workflow of experimental process. | ||
2. Materials and methods
2.1. Molecular docking
The set of 50.2 K Discovery Diversity Set (DDS) compounds were used for docking to the crystal structure of human TLR4 (PDB ID: 4G8A), which is taken from the RCSB PDB database. The structure of each ligand as well as the structure of the protein were both preprocessed prior to docking. The docking process was visualized with PyMol (version 11.4) using Schrödinger Maestro (version 11.4). Schrödinger's Protein Preparation Wizard tool was used to prepare the protein structure.21 The water molecules have been deleted, and the hydrogens, resides and side chains have been added. Using the force field (OPLS2005, RMSD is 0.30 Å), the energy optimization was carried out in order to minimize the protein structure.22,23 Maestro (version 11.4) was used to generate the receptor grid, selecting amino acid residues with hydrogen and ionic bonding Glu42/Asp60/Arg87/Glu135/Ser183/Arg234/Arg264/Asn265/Glu266/Arg289 based on the complex formed by the TLR4 with the extracellular adaptor MD-2 (a glycoprotein) and the hydrophobic portion of lipid polysaccharide (LPS), and the grid size was set to 20 Å × 20 Å × 20 Å so that new compounds could be attached to the same binding site and co-crystallized ligands could be excluded.24 In Schrödinger Maestro, the LigPrep module was used to prepare the ligand structure. After the ligands were loaded and optimized, and the 3D structure was obtained.25 For docking, the prepared molecules were imported into Glide. In the generated grid of the target protein, the ligand molecules are docked through geometric and energy matching. In the Glide module, standard precision (SP) was utilized with the high-throughput screening (HTVS) setting, and the top 10% of compounds were selected. Thereafter, extra precision (XP) screenings were performed in the Glide module and the top 10% of small molecules were selected for further screening.26–28 Best docking pose of each ligand was kept for the further analysis.292.2. Dataset
From the ChEMBL database (version 29),30 we compiled a list of 78 TLR4 inhibitors along with their IC50 values. CHEMBL is a searchable database of more than 2 million compounds, molecule bioactivities, and information on bioactive molecules exhibiting drug-like properties. In ESI S1† (Excel sheet), the list of TLR4 inhibitory molecules used in the QSAR model, simplified molecular input line entry system (SMILES) notation, pIC50 and ChEMBL ID as well as Lipinski's descriptors are presented. For a more uniform distribution of the data, the IC50 values were converted to the pIC50 by taking the negative logarithm of the IC50 values by using the equation pIC50 = −log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 10(IC50). Here, pIC50 is the negative logarithm of IC50. As this study is primarily concerned with developing regression and classification models of biological activity. Thus, the IC50 values were divided into three classes (high (<1 μM), moderate (≤3 μM and >1 μM) and low (>3 μM)) for a clear distinction between the potency of these compounds. The geometry is optimized using MOPAC (Molecular Orbital Package) using the AM1 method, the 3D coordinates are preserved, and the energy is minimized in Merck Molecular Force Field (MMFF94) for all ligands before descriptors and molecular fingerprints are calculated. Molecular descriptors were calculated, and the data was curated based on the calculated descriptors. Model building relies on qualitative and/or quantitative chemical information that can be obtained from molecular fingerprints. The PaDel molecular fingerprints and descriptors were calculated using ChemDes web-based platform.31 To clean the initial data collected from the ChEMBL database, data preprocessing has been performed as described previously.32–35 As part of the data curation process, some key steps are performed including (i) structural cleaning and conversion, (ii) removal of duplicates, (iii) removal of mixtures and inorganics, (iv) normalization of specific chemotypes, and (v) manual verification of the data. All other inhibitory targets except for TLR4, as well as the pIC50 values for each, were removed from the data. Molecules with missing values for the pIC50 and SMILES notation, as well as duplicate value entries, were removed. The feature selection process is also known as variable selection or attribute selection. Predictive modeling involves automatic selection of attributes most relevant to the problem. As a first step, all the descriptors and molecular fingerprints were checked manually for any missing values, and all the columns containing zero values were removed from the files using a Python script. The fingerprints were then combined into a single csv (Comma Separated Values) file. A variation selection method was initially applied in order to remove the redundant features from the list of combined features.
10(IC50). Here, pIC50 is the negative logarithm of IC50. As this study is primarily concerned with developing regression and classification models of biological activity. Thus, the IC50 values were divided into three classes (high (<1 μM), moderate (≤3 μM and >1 μM) and low (>3 μM)) for a clear distinction between the potency of these compounds. The geometry is optimized using MOPAC (Molecular Orbital Package) using the AM1 method, the 3D coordinates are preserved, and the energy is minimized in Merck Molecular Force Field (MMFF94) for all ligands before descriptors and molecular fingerprints are calculated. Molecular descriptors were calculated, and the data was curated based on the calculated descriptors. Model building relies on qualitative and/or quantitative chemical information that can be obtained from molecular fingerprints. The PaDel molecular fingerprints and descriptors were calculated using ChemDes web-based platform.31 To clean the initial data collected from the ChEMBL database, data preprocessing has been performed as described previously.32–35 As part of the data curation process, some key steps are performed including (i) structural cleaning and conversion, (ii) removal of duplicates, (iii) removal of mixtures and inorganics, (iv) normalization of specific chemotypes, and (v) manual verification of the data. All other inhibitory targets except for TLR4, as well as the pIC50 values for each, were removed from the data. Molecules with missing values for the pIC50 and SMILES notation, as well as duplicate value entries, were removed. The feature selection process is also known as variable selection or attribute selection. Predictive modeling involves automatic selection of attributes most relevant to the problem. As a first step, all the descriptors and molecular fingerprints were checked manually for any missing values, and all the columns containing zero values were removed from the files using a Python script. The fingerprints were then combined into a single csv (Comma Separated Values) file. A variation selection method was initially applied in order to remove the redundant features from the list of combined features.
2.3. Constructing regression and classification models
In QSAR modeling, Random Forest (RF) regression and classification algorithms are frequently used since they provide good predictive performance and high model interpretability. The RF algorithm is an ensemble of regression and classification trees.36 It is a robust algorithm with higher performance in the presence of very high-dimensional parameters, spaces, and outliers than other machine learning algorithms.37 In addition, random sampling reduces overfitting, and the RF algorithm recognizes important features that affect the QSAR model, allowing for further evaluation of the model and improved prediction accuracy. The dataset is divided into two main subsets, the training subset (70%) and the test subset (30%). The training subset of the data is used to train the model, whereas the test subset is used to cross-validate the model. First, the model was evaluated using 5-fold cross-validation, providing a more general model;38 then, the model's hyperparameters were tuned using the Optune framework (Optune-A hyperparameter optimization framework).39 For the training, testing, and cross-validation sets of compounds, the performance of regression models was assessed using root mean squared error (RMSE), mean absolute error (MAE), and squared correlation coefficient (R2). The predictability of the model was also assessed by calculating the residual error between the predicted and experimental pIC50 values. The performance of the classification model was evaluated in terms of classification accuracy, RMSE, MAE, and Log loss over the training, testing, and validation sets of compounds. Additionally, the RF model was compared with Decision Tree Regression (DTR), AdaBoost Regression (ABR), Gradient Boosting Regression (GBR) and Extra Trees Regression (ETR) models trained on the same data. RF classification model was also compared with KNeighbors classifier, Support Vector Classifier (SVC), Decision Tree Classifier, AdaBoost Classifier, Gradient Boosting Classifier, Linear Discriminant Analysis and Quadratic Discriminant Analysis, on both training and testing data. The R2, accuracy, RMSE, MAE, Log Loss, and the performance of these models are analyzed and compared. For these compounds, a set of descriptors and fingerprints was selected in the same manner as for the fingerprints that were used to build the QSAR model. Based on these features, pIC50 values were predicted for DDS set of compounds.2.4. Statistical analysis for assessing predictive models
For the training, testing, and cross-validation sets of compounds, the performance of regression models was evaluated using the root mean squared error (RMSE), mean absolute error (MAE), and squared correlation coefficient (R2). The predictive performance of the model was further examined by calculating the residual error between the predicted and experimental pIC50 values. While the performance of the classification model for the training, testing, and validation set of compounds was evaluated in terms of classification accuracy, RMSE, MAE, and log loss.2.5. Prediction of small molecule compounds
Based on 50.2 K DDSs of compounds screened by docking, the top 30 compounds were selected. ChemDes online website was used to compute chemical descriptors and molecular fingerprints for all these compounds using canonical smiles format. A set of fingerprints and descriptors were selected for these compounds, like those on which the QSAR model was based. Using these fingerprints, the pIC50 values and potencies of DDS compounds were predicted.3. Results and discussion
3.1. Molecular docking
The docking technique is a computational method for predicting the orientation of a ligand or protein when bound to an enzyme or receptor protein.40 In the field of drug discovery and design, docking is extremely useful as it provides understanding of the best binding affinity of the potent ligand.41 Here, docking of the DDS compounds to TLR4 receptor protein provides insights and highlights important protein-ligand interactions. The 2D and 3D docking poses of the representative top 2 compounds are represented in Fig. 2. It has been noted that longer bond results in weaker hydrogen bonding and vice versa. Compound 1 (Fig. 2(A)) formed five hydrogen bonds with TLR4 protein, showing that the carboxyl group forms two hydrogen bonds with ASP181 and ASN156 as hydrogen bond donor and acceptor, respectively, with distances of 1.7 Å and 1.8 Å, respectively. Further, the NH of the sulfonamide forms a hydrogen bond with SER183 at a distance of 1.9 Å and the NH2 group forms two hydrogen bonds with ASN265 and GLU266 with distances of 1.7 Å and 2.1 Å, respectively. Compound 2 (Fig. 2(D)) forms 4 hydrogen bonds with the TLR4 protein that is the NH on the bridged ring acts as a hydrogen bond donor to form a hydrogen bond with SER184 with a distance of 2.0 Å and the carbonyl group acts as a hydrogen bond acceptor to form a hydrogen bond with SER184 with a distance of 2.1 Å. Besides, the NH form a hydrogen bond with GLU266 with a distance of 1.9 Å and the NH of indole form a hydrogen bond with GLU266 with a distance of 1.8 Å. Furthermore, the docking scores of the top 30 compounds of DDS set of compounds along with their prediction results are shown in Table 1. It is worthy to mention here that docking results needs further verification by biological testing to further confirm and modify the experimental process for these potent ligands for the treatment of MP pneumonia.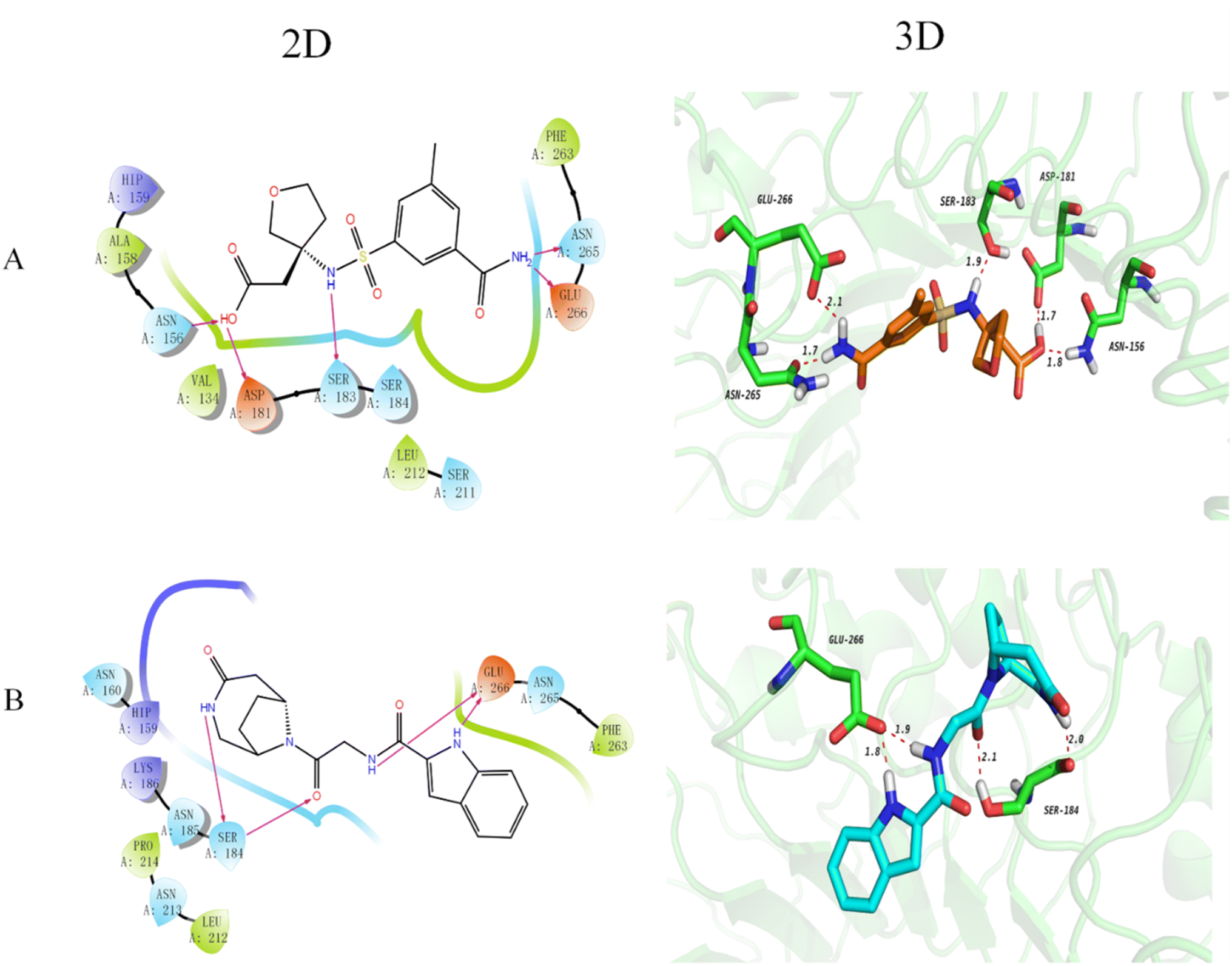 | ||
| Fig. 2 2D and 3D representation of the docked compounds. Panel (A) and (B) represents top 2 top 2 compounds ((R)-2-(3-3-carbamoyl-5-methylphenylsulfonamido) tetrahydrofuran-3-yl)acetic acid and N-(2-oxo-2-((6R)-4-oxo-3,9-diazabicyclo[4.2.1]nonan-9-yl)ethyl)-1H-indole-2-carboxamide, respectively. The hydrogen bonds and their lengths (Å) are displayed in these cartoons. | ||
| S. No. | Drug names | MW | Docking score | RFR model | ETR model | DTR model | ABR model | GBR model |
|---|---|---|---|---|---|---|---|---|
| 1 | (R)-2-(3-(3-Carbamoyl-5-methylphenylsulfonamido)tetrahydrofuran-3-yl)acetic acid | 342.37 | −5.228 | 5.430829277 | 6.314821411 | 6.958607315 | 4.813787229 | 5.136554754 |
| 2 | N-(2-Oxo-2-((6R)-4-oxo-3,9-diazabicyclo[4.2.1]nonan-9-yl)ethyl)-1H-indole-2-carboxamide | 340.38 | −5.127 | 4.815372966 | 6.16129079 | 4.978810701 | 4.644380954 | 4.864479656 |
| 3 | 4-(N-(2-Carbamoylphenyl)sulfamoyl)-3-fluorobenzoic acid | 338.31 | −5.014 | 5.131434015 | 5.942968037 | 4.779891912 | 4.644380954 | 4.848622926 |
| 4 | (R)-3-(5-Oxo-2,5-dihydro-1H-1,2,4-triazol-3-yl)-N-((3-(trifluoromethyl)-1H-1,2,4-triazol-5-yl)methyl)piperidine-1-carboxamide | 360.30 | −4.888 | 5.335803885 | 4.311301872 | 5 | 5.128070842 | 5.154233598 |
| 5 | 1-(((1S,2R)-2-Hydroxy-1,2,3,4-tetrahydronaphthalen-1-yl)carbamoyl)cyclopent-3-enecarboxylic acid | 301.34 | −4.857 | 5.162769848 | 5.498993506 | 5.229147988 | 5.853871964 | 5.136223419 |
| 6 | 3-(3-Amino-5-methylisoxazole-4-sulfonamido)-4-methoxybenzoic acid | 327.31 | −4.841 | 5.42557839 | 5.515008442 | 7.096910013 | 6.198970004 | 4.768100468 |
| 7 | 5-(2-((2-Aminoethyl)amino)thiazol-4-yl)-2-hydroxybenzamide | 278.33 | −4.788 | 5.062946566 | 4.451147788 | 6.795880017 | 4.676057904 | 4.715750754 |
| 8 | (R)-3-((4-Amino-6,7-dimethoxyquinazolin-2-yl)amino)-2-hydroxy-2-methylpropanoic acid | 322.32 | −4.765 | 5.384378737 | 4.723592771 | 5 | 5.551578992 | 5.318842099 |
| 9 | 4-((3-(1-(3-Methylbutanoyl)piperidin-4-yl)ureido)methyl)benzoic acid | 361.44 | −4.729 | 5.352768581 | 4.604814799 | 6.958607315 | 5.128070842 | 4.930009052 |
| 10 | (S)-3-(5-Fluoropyridine-3-sulfonamido)-2-hydroxypropanoic acid | 264.23 | −4.626 | 5.469392305 | 5.131277132 | 6.958607315 | 5.857242359 | 5.598004671 |
| 11 | 3,4-Difluoro-N-(2-((2R,4R)-4-hydroxy-2-(hydroxymethyl)pyrrolidin-1-yl)-2-oxoethyl)benzamide | 314.29 | −4.559 | 5.303713126 | 4.866537918 | 6.958607315 | 5.121293699 | 4.907629008 |
| 12 | (S)-3-(2,3-Dichlorophenylsulfonamido)-2-hydroxypropanoic acid | 314.14 | −4.530 | 5.448314838 | 4.792741186 | 6.958607315 | 5.121293699 | 4.849140284 |
| 13 | 2-(((3R,4R)-3-Methyltetrahydro-2H-pyran-4-yl)amino)-5-sulfamoylbenzoic acid | 314.36 | −4.524 | 5.737739993 | 4.597185429 | 6.795880017 | 4.644380954 | 5.211471563 |
| 14 | (R)-2-(3-Methyl-1H-1,2,4-triazol-5-yl)-N-(4-oxochroman-3-yl)acetamide | 286.29 | −4.508 | 5.198586102 | 4.835972975 | 6.795880017 | 5.121293699 | 4.841100257 |
| 15 | 4-(N-(2-Amino-2-oxoethyl)-N-benzylsulfamoyl)-1H-pyrrole-2-carboxylic acid | 337.35 | −4.507 | 4.93587296 | 5.078963707 | 4.256568635 | 4.644380954 | 5.278315448 |
| 16 | (R)-4-Isopropoxy-3-(1-(tetrahydrofuran-3-yl)-1H-pyrazole-4-sulfonamido)benzoic acid | 395.43 | −4.504 | 5.659485927 | 5.673346573 | 5 | 4.676057904 | 5.283553443 |
| 17 | 3-(N-(5-Cyano-2-(methylamino)phenyl)sulfamoyl)-5-fluorobenzoic acid | 349.34 | −4.489 | 5.2393042 | 6.270021811 | 4.586700236 | 4.676057904 | 5.202654077 |
| 18 | 1-((2-(5-Fluoro-1H-indol-3-yl)ethyl)carbamoyl)azetidine-3-carboxylic acid | 305.30 | −4.463 | 5.138107357 | 5.712863539 | 6.958607315 | 5.121293699 | 4.859899139 |
| 19 | 4-(3-Ethylphenylsulfonamido)-3-hydroxybenzoic acid | 321.35 | −4.456 | 5.326559803 | 5.193816275 | 5 | 5.266000713 | 5.426620204 |
| 20 | (R)-3-(3-(5,6-Dimethyl-4-oxo-1,4-dihydrothieno[2,3-d]pyrimidin-2-yl)propanamido)-2-hydroxy-2-methylpropanoic acid | 353.39 | −4.450 | 5.372020827 | 5.783184637 | 5 | 5.121293699 | 5.442711298 |
| 21 | (2R,3S,4R)-1-(Tert-butoxycarbonyl)-3,4-dihydroxypyrrolidine-2-carboxylic acid | 247.25 | −4.443 | 5.469071944 | 5.613893366 | 5 | 5.121293699 | 4.953810874 |
| 22 | 2,4-Difluoro-N-(2-((2R,4R)-4-hydroxy-2-(hydroxymethyl)pyrrolidin-1-yl)-2-oxoethyl)benzamide | 314.29 | −4.411 | 5.278958496 | 4.61805273 | 6.958607315 | 4.813787229 | 4.931341817 |
| 23 | (3R,5R)-1-(6-(((3-Cyclopropyl-1H-pyrazol-5-yl)methyl)amino)pyrimidin-4-yl)-5-((dimethylamino)methyl)pyrrolidin-3-ol | 357.45 | −4.399 | 5.284046701 | 4.564151855 | 6.602059991 | 5.121293699 | 5.125098228 |
| 24 | (R)-2-(1-Oxo-1,2-dihydroisoquinoline-3-carboxamido)-3-phenylpropanoic acid | 336.34 | −4.386 | 5.163010804 | 5.302824295 | 5.853871964 | 5.595633098 | 5.484515313 |
| 25 | (R)-4-(N-(2-Oxo-2-((tetrahydro-2H-pyran-3-yl)amino)ethyl)sulfamoyl)benzoic acid | 342.37 | −4.380 | 5.820985738 | 5.221450444 | 6.958607315 | 5.035830554 | 5.35223672 |
| 26 | (R)-2-(6,7-Dihydro-5H-pyrrolo[1,2-a]imidazole-3-sulfonamido)-2-(3-methoxyphenyl)acetic acid | 351.38 | −4.365 | 5.275202795 | 5.291271987 | 5 | 4.676057904 | 5.012575341 |
| 27 | (R)-N-(1-Amino-3-methoxy-1-oxopropan-2-yl)-7-methyl-1H-indole-2-carboxamide | 275.30 | −4.342 | 5.130831094 | 5.946522112 | 6.795880017 | 5.121293699 | 4.992894312 |
| 28 | N-((2R,3R)-4-Hydroxy-3-(methylthio)butan-2-yl)-2-oxo-2,3-dihydrobenzo[d]oxazole-6-sulfonamide | 332.40 | −4.339 | 5.36250487 | 5.963766686 | 6.958607315 | 4.813787229 | 4.866545583 |
| 29 | 1-(2-Morpholino-2-oxoethyl)-3-(pyridin-3-yl)urea | 264.28 | −4.278 | 5.323176653 | 4.938421827 | 6.795880017 | 5.121293699 | 4.845185655 |
| 30 | (3R,4R)-1-((3-Carbamoylphenethyl)carbamoyl)-4-methylpiperidine-3-carboxylic acid | 333.38 | −4.260 | 5.316126193 | 4.52083702 | 6.476253533 | 5.121293699 | 4.632121561 |
3.2. Exploring the chemical space of TLR4 inhibitory compounds
The QSAR paradigm is based on the link between the molecular structure of compounds and their respective biological activities. Molecular descriptors play a critical role in providing analytical descriptions of the physicochemical properties of molecules. In order to adequately account for these structural features, it is of utmost importance to select appropriate descriptors for a QSAR analysis. Todeschini et al.42 have compiled a handbook providing comprehensive coverage of molecular descriptors. This study selected a subset of molecular descriptors and fingerprints representative of the general characteristics of molecules (i.e., flexibility, molecular size, solubility, polarity, electronic properties, and charge, as well as chemical reactivity) that are mostly correlated with bioactivities. Using such descriptors, we can explore the chemical space of inhibitors as mentioned earlier.43 Lipinski's rule-of-five (Ro5) descriptors are used to examine the general chemical space of the investigated data set. According to previous studies have suggested that chemical space analysis provides a mechanistic explanation of the correlation between MW, ALogP, number of hydrogen bond donors and number of hydrogen bond acceptors of a compound and its degree of potency and pIC50 values.44 In order to visualize the relative distribution of the bioactivity classes and Ro5, scatter and box plots were created, as shown in Fig. 3(A–G). The results showed that 60% of compounds have molecular weight of less than 500 Da (Fig. 3(G)). Whereas the ALogP for the majority of compounds varies between 2 and 7. Molecular lipophilicity can be measured by ALogP where a high ALogP value indicates high lipophilicity whereas a low value suggests low lipophilicity. The ALogP is a computational estimator of the logarithm of the partition coefficient between water and octanol, which has been indispensable in determining molecular hydrophobicity. The boxplots indicate distribution and frequency of high-, moderate- and Low-class compounds over the Ro5 (Fig. 3 (A–D)). The Ro5 index may be used to distinguish compounds based on their pharmacological effects based on their molecular properties, namely the octanol–water partition coefficient (logP < 5), the molecular weight (<500), the number of hydrogen bond donors (>5), and the number of hydrogen bond acceptors (<10). The Ro5 were found to be of limited use in contributing to our understanding of the targets–ligands relationship (i.e., their affinity towards the target) as they were based solely on general ligand properties. Oprea et al. demonstrated that the Ro5 criteria are not effective in discriminating between drugs and non-drugs based on the availability of more than 90% of the chemical reagents listed in the Available Chemical Directory that meets Ro5 criteria.45 The Ro5 criteria, however, do not eliminate the possibility that they may be used to narrow the pharmacokinetic space for therapeutically relevant compounds. In addition, Benet et al. have demonstrated that a QSAR model developed using the Ro5 criteria can effectively predict drug disposition characteristics for drugs that meet or do not meet the Ro5.46
 | ||
| Fig. 3 Panels (A–F) show exploratory TLR4 inhibitors data analysis and panel (G) shows chemical space analysis. The scatter plot showed the diversity of ALogP versus MW of TLR4 inhibitory compounds. | ||
3.3. Regression QSAR models of TLR4 inhibitors
The development of QSAR models was conducted using curated data sets containing 78 structurally diverse compounds spanning several scaffolds to predict TLR4 inhibitory activity. The molecular features of compounds are described using several 1D and 2D descriptors and fingerprint types. Several rounds of data splitting were used to test the generalization and intrapolation abilities of QSAR model. This model was designed using a data split ratio of 70/30, in which 70% of the data set was used as the internal set, while 30% of the data set was used as the external set. In the first subset of 70% of the data, the model was internally validated by using it both as the training set as well as the cross-validation set, and its performance was evaluated according to R2, RMSE, and MAE. This second set of data containing 30% of the bioactivity data was used for external validation, and the performance of the models was based on R2, RMSE, and MAE. Table 2 summarizes the results of the models constructed using the RF algorithm. The descriptors and fingerprints including feature importance score on which the final RF model was trained and tested is provided in ESI S2.† The feature importance score was calculated from the RF algorithm based on its built-in function for determining feature importance. The descriptors and fingerprints which have a score greater than 0.00011 were included in the training of the model. While the descriptors and fingerprints whose score is below 0.00011 were discarded because these descriptors and fingerprints do not affect the model performance significantly based on our pre-experiments. The predictive power of the model was assessed according to the thresholds suggested by previous studies,47,48 in which acceptable models possess R2 (train) > 0.6 and R2 (test) > 0.5. Meanwhile, it is necessary to develop a generalized QSAR model that is capable of automatically identifying informative features from a large pool of chemical descriptors that will result in a better comprehension of the mechanism of chemical compounds. The model has to be simple, user-friendly, and consistent with acceptable prediction results. Thus, the RF algorithm was utilized since it is easily able to calculate the relative importance of molecular fingerprints used during model construction. Further, the RF method was employed in order to select informative molecular descriptors because of its built-in function for measuring the importance of descriptors. Additionally, an insight into the applicability domain of the TLR4 QSAR model is established in the current study for the prediction of pIC50 values for DDS set of compounds. In the present study, it was taken into account the robustness of the model, goodness-of-fit and predictability were used to determine how well the model was performing. In this study, the model was further evaluated against several other algorithms and developed QSAR models for the prediction of potent compounds. The performance summary of the RF regression model and its comparison with other models (extra-trees regression (ETR), decision tree regression (DTR), adaboost regression (ABR), gradient boosting regression (GBR)) is shown in Table 2. While the difference (residual error) between the predicted and experimental bioactivity is represented in Fig. 4. From these results, it has been suggested that these models performed better on training, testing and validation data. The comparison results showed that the proposed models are quite promising and hold potential for the prediction of TLR4 inhibitory compounds. To the best of our knowledge, this is the first study that proposed QSAR models for TLR4 receptor as a target for MP pneumonia. In a previous study, novel antagonists of Toll-Like Receptor 4 (TLR4) were identified using structure and ligand-based virtual screening. They noted that three in silico hits exhibited promising anti-TLR4 activities with micromolar IC50 values, and significantly reduced the production of TNF-α.49 Incredibly, several TLR4 antagonists are already being investigated as potential anti-sepsis drugs. The most advanced of these compounds, TAK-242 and eritoran, have shown promising results in preclinical studies. These two strategies failed to meet their primary objective which was to reduce the mortality rate in patients with sepsis.50,51 However, a critical need remains for the identification of new TLR4 antagonists that can serve as novel therapeutics.| Models | R2 (train) | RMSE (train) | MAE (train) | R2 (test) | RMSE (test) | MAE (test) | R2 (CV) | RMSE (CV) | MAE (CV) |
|---|---|---|---|---|---|---|---|---|---|
| RFR | 0.91 | 0.36 | 0.25 | 0.89 | 0.39 | 0.3 | 0.71 | 0.65 | 0.68 |
| ETR | 0.96 | 0.23 | 0.06 | 0.76 | 0.53 | 0.41 | 0.76 | 0.56 | 0.41 |
| DTR | 0.96 | 0.22 | 0.04 | 0.82 | 0.53 | 0.42 | 0.63 | 0.69 | 0.51 |
| ABR | 0.91 | 0.36 | 0.26 | 0.89 | 0.39 | 0.29 | 0.74 | 0.59 | 0.42 |
| GBR | 0.96 | 0.23 | 0.06 | 0.93 | 0.29 | 0.21 | 0.79 | 0.51 | 0.36 |
 | ||
| Fig. 4 Plot of experimental versus predicted pIC50 values of TLR4 inhibition for QSAR regression models generated by RF (A), ETR (B), DTR (C), ABR (D) and GBR (E) for the testing set (24 compounds). The X-axis shows compounds. The Y-axis on the left represent the pIC50 values and the Y-axis on the right shows residual error. | ||
3.4. Classification QSAR models of TLR4 inhibitors
The models were constructed in a similar manner to those for regression, with the exception that quantitative values were replaced by semi-quantitative labels (high, moderate and low) for bioactivity. We present a simple and general-purpose RF method for predicting the low, medium, and high-class DDS set of compounds. The analysis of the training, testing, and cross validation performance of the RF classification model is presented in Table 3. The performance of machine learning models was evaluated by evaluation metrics such as sensitivity and specificity including the classification accuracy, confusion matrixes, true positive rates for a given model and false positive rates as mentioned previously.52 Confusion matrixes are very popular in solving classification problems. Fig. 5(A and B) depicts the confusion matrix related to the test and 5-fold cross validation. An important tool for evaluating a machine learning model's performance is the receiver operating characteristic curve (ROC curve). ROC curves demonstrate the correlation between true positive rates (TPR) for a given model and false positive rates (FPR). The Fig. 5(C) shows the multiclass ROC curves for the RF classifier. On a class-by-class basis, the classification report presents a summary of the principal classification metrics. Table 4 provides a classification report that includes precision, recall, and F1-scores on both test data and cross validation for each of the three classes. A deeper understanding of the classifier behavior over global accuracy may be gained by assessing the behavior of a specific class within a multiclass problem. All the evaluating parameters (classification report, performance summary, ROC curve, and confusion matrix) confirmed that the developed RF classification model performed better on training, testing, and validation data. For further verification, it is necessary to compare the proposed method with other existing methods. The developed RF model was evaluated against a variety of different classifiers including KNeighbors, support vectors, decision trees, adaboost, gradient boosting, linear discriminant analysis, and quadratic discriminant analysis. The performance accuracy and log loss of each classifier are illustrated in Fig. 5(D) and (E) respectively. The performance summary of all these classifiers is presented in Table 5. The results clearly indicate that the RF classifier outperforms all other data-based classifiers both on the test and on the trained data. It is evident from the comparison results that the proposed RF classifier, as presented in this study, is quite promising. It has the potential to be a useful tool for discriminating low, moderate, and high potency compounds. Furthermore, the proposed RF model can be viewed as complementary to the existing method in the same area.| Accuracy (train) | RMSE (train) | MAE (train) | Accuracy (test) | RMSE (test) | MAE (test) | Accuracy (CV) | RMSE (CV) | MAE (CV) | Log loss |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 0 | 0 | 0.96 | 0.2 | 0.04 | 0.82 | 0.46 | 0.19 | 0.27 |
 | ||
| Fig. 5 (A and B) Depicts the confusion matrix related to the test and 5-fold cross validation. (C) Shows the multiclass ROC curves for the RF classifier. The performance accuracy and log loss of each classifier are illustrated in (D) and (E) respectively. | ||
| Precision | Recall | F1-score | Support | |
|---|---|---|---|---|
| Classification report of RF model on test data | ||||
| 0 | 0.888889 | 1 | 0.941176 | 8 |
| 1 | 1 | 0.933333 | 0.965517 | 15 |
| 2 | 1 | 1 | 1 | 1 |
| Accuracy | 0.958333 | 0.958333 | 0.958333 | 0.958333 |
| Macro avg. | 0.962963 | 0.977778 | 0.968898 | 24 |
| Weighted avg. | 0.962963 | 0.958333 | 0.95884 | 24 |
|
||||
| Classification report of RF model on cross validation data | ||||
| 0 | 0.736842 | 0.777778 | 0.756757 | 18 |
| 1 | 0.844828 | 0.924528 | 0.882883 | 53 |
| 2 | 1 | 0.142857 | 0.25 | 7 |
| Accuracy | 0.820513 | 0.820513 | 0.820513 | 0.820513 |
| Macro avg. | 0.860557 | 0.615054 | 0.62988 | 78 |
| Weighted avg. | 0.833834 | 0.820513 | 0.79698 | 78 |
| Models | Accuracy (train) | RMSE (train) | MAE (train) | Accuracy (test) | RMSE (test) | MAE (test) | Log loss |
|---|---|---|---|---|---|---|---|
| RF model | 1 | 0 | 0 | 0.96 | 0.2 | 0.04 | 0.27 |
| KNeighbors classifier | 0.76 | 0.64 | 0.3 | 0.75 | 0.5 | 0.25 | 5.95 |
| SVC | 0.7 | 0.54 | 0.29 | 0.63 | 0.61 | 0.37 | 0.87 |
| Decision-tree classifier | 1 | 0 | 0.35 | 0.88 | 0.35 | 0.13 | 4.31 |
| AdaBoost classifier | 0.98 | 0.14 | 0.02 | 0.92 | 0.46 | 0.13 | 0.54 |
| Gradient boosting classifier | 1 | 87.5 | 0 | 0.88 | 0.35 | 0.13 | 0.97 |
| Linear discriminant analysis | 0.98 | 0.27 | 0.04 | 0.71 | 0.54 | 0.29 | 5.79 |
| Quadratic discriminant analysis | 1 | 0 | 0 | 0.75 | 0.5 | 0.25 | 8.63 |
3.5. Predictions of DDS set of compounds
To predict the bioactivity of compounds, we applied the final selected QSAR models as prediction models. The top 30 compounds selected from DDS set on the basis of docking scores and their predicted pIC50 bioactivities derived from machine learning models are summarized in Table 1, respectively. RF model has been compared with several other models, in order to reduce redundancy in the predicted results. A comparison of the results generated by the proposed models suggests that these models have promising results and can be used to predict compounds that inhibit TLR4. This is the very first study that comes to our knowledge that proposes QSAR study to be used as a target in MP pneumonia based on a TLR4 receptor. Additionally, these results can be also used as references to find the structures of compounds that are potentially TLR4 modulating as well as saving time and money compared to the traditional biological screening procedures. In addition, previous studies have explored the importance of these QSAR models for in silico prediction of novel compounds that can be used to predict the activity of a target or receptor when this is a computationally intensive process that requires learning from bioactivity data.53–564. Conclusion
The pathological process of MP pneumonia has been identified as being associated with TLR4 and has been described as one of the most difficult and threatening diseases of the human population. TLR4 can be successfully inhibited therapeutically for the prevention of MP. A widespread use of traditional antibiotics in clinical settings has led to an increase in drug resistance and other adverse effects. Thus, natural products may have the potential to be used to modulate the host immune inflammatory response in the prevention of TLR4-driven diseases. Hence, regulating the host immune inflammatory response by means of natural products might represent a new strategy for preventing MP infection. As a result, the project can serve as a theoretical base for the treatment of TLR4-related illnesses by regulating the immune response of the host species. Hence, the present research aims to screen medicinal compounds to find new candidate drugs to treat TLR4-mediated inflammatory diseases. Recently, machine learning is reshaping research in various fields.57–60 Furthermore, molecular docking analysis of the inhibitors and key residues in the binding pocket of the TLR4 protein also suggested the presence of several hydrogen bonds and hydrophobic interactions between inhibitors and key residues. Nevertheless, further research will be performed to test the screened and selected candidates, and in-depth molecular research will be conducted in order to understand the mechanism of action, to provide new ideas for new anti-inflammation medications targeting TLR4. This study explores the relationship between structure and activity for a library of 78 compounds from the ChEMBL database that are known TLR4 inhibitors. Using SMILES-based descriptors, we propose a simple method for constructing both regression and classification QSAR models. For the numerical description of the compounds, several 1D and 2D chemical descriptors and molecular fingerprints were employed. By analyzing the calculated molecular descriptors, the chemical space of TLR4 inhibitors was explored thus providing important insights into their molecular characteristics. Presented models have the advantage of being simple and easy to use and interpret. Key features that are correlated with pIC50 values, as determined by the RF algorithm. In the future, such information can be used to guide the design of novel molecular structures that act as TLR4 inhibitors. These classification and regression QSAR models have demonstrated robust statistical results and are interpretable, which strongly supports their use for rapid screening of TLR4 inhibitors.Author contributions
All authors contributed to the study conception and design. Data collection and formal analysis were performed by Muhammad Ishfaq, Ziaur Rahman, Akhunzada Bilawal and Sattar Hamid. Zemin Zhu, Sihong Li and Muhammad Ishfaq supervised and provided the resources and funding. Muhammad Aamir performed visualization and data curation. Syed Zahid Ali Shah helps in critical revision of the manuscript. The first draft of the manuscript was written by Muhammad Ishfaq and all authors commented on the manuscript. All authors read and approved the final version of the manuscript.Conflicts of interest
The authors report no conflicts of interest in this work.Acknowledgements
This project is supported by the Huanggang Normal University Project Number (2042022005).References
- (a) R. Chaudhry, A. K. Varshney and P. Malhotra, Adhesion proteins of Mycoplasma pneumoniae, Front. Biosci., 2007, 12, 690–699, DOI:10.2741/2093; (b) T. A. Tsai, C. K. Tsai, K. C. Kuo and H. R. Yu, Rational stepwise approach for Mycoplasma pneumoniae pneumonia in children, J Microbiol Immunol Infect, 2021, 54(4), 557–565, DOI:10.1016/j.jmii.2020.10.002.
- B. Abdulhadi and J. Kiel, Mycoplasma Pneumonia, in StatPearls [Internet], StatPearls Publishing, Treasure Island (FL), 2022 Jan 24 Search PubMed.
- P. M. Meyer Sauteur and C. Berger, Proadrenomedullin in Mycoplasma pneumoniae Community-Acquired Pneumonia in Children, Clin. Infect. Dis., 2021, 73(7), e1769–e1771, DOI:10.1093/cid/ciaa1888.
- S. Pereyre, A. Charron, C. Hidalgo-Grass, A. Touati, A. E. Moses, R. Nir-Paz and C. Bebear, The spread of Mycoplasma pneumoniaeis polyclonal in both an endemic setting in France and in an epidemic setting in Israel, PLoS One, 2012, 7, e38585 CrossRef CAS PubMed.
- N. Miyashita, Atypical pneumonia: Pathophysiology, diagnosis, and treatment, Respir. Invest., 2022, 60(1), 56–67, DOI:10.1016/j.resinv.2021.09.009.
- D. Li and M. Wu, Pattern recognition receptors in health and diseases, Signal Transduction Targeted Ther., 2021, 6(1), 291 CrossRef CAS PubMed.
- T. Kawai and S. Akira, The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors, Nat. Immunol., 2010, 11, 373–384 CrossRef CAS PubMed.
- A. Oblak and R. Jerala, The molecular mechanism of species-specific recognition of lipopolysaccharides by the MD-2/TLR4 receptor complex, Mol. Immunol., 2015, 63(2), 134–142 CrossRef CAS PubMed.
- E. M. Pålsson-McDermott and L. A. O'Neill, Signal transduction by the lipopolysaccharide receptor, Toll-like receptor-4, Immunology, 2004, 113(2), 153–162 CrossRef PubMed.
- H. Luo, J. He, L. Qin, Y. Chen, L. Chen, R. Li, Y. Zeng, C. Zhu, X. You and Y. Wu, Mycoplasma pneumoniae lipids license TLR-4 for activation of NLRP3 inflammasome and autophagy to evoke a proinflammatory response, Clin. Exp. Immunol., 2021, 203(1), 66–79, DOI:10.1111/cei.13510.
- M. Hu, Y. Zhang and X. Li, et al., TLR4-Associated IRF-7 and NFκB Signaling Act as a Molecular Link Between Androgen and Metformin Activities and Cytokine Synthesis in the PCOS Endometrium, J. Clin. Endocrinol. Metab., 2021, 106(4), 1022–1040 CrossRef PubMed.
- S. Qu, M. Liu and C. Cao, et al., Chinese Medicine Formula Kai-Xin-San Ameliorates Neuronal Inflammation of CUMS-Induced Depression-like Mice and Reduces the Expressions of Inflammatory Factors via Inhibiting TLR4/IKK/NF-κB Pathways on BV2 Cells, Front. Pharmacol., 2021, 12, 626949 CrossRef CAS PubMed.
- S. Shao, R. Jia and L. Zhao, et al., Xiao-Chai-Hu-Tang ameliorates tumor growth in cancer comorbid depressive symptoms via modulating gut microbiota-mediated TLR4/MyD88/NF-κB signaling pathway, Phytomedicine, 2021, 88, 153606 CrossRef CAS PubMed.
- B. R. Selfridge, X. Wang and Y. Zhang, et al., Structure-Activity Relationships of (+)-Naltrexone-Inspired Toll-like Receptor 4 (TLR4) Antagonists, J. Med. Chem., 2015, 58(12), 5038–5052 CrossRef CAS PubMed.
- C. Y. Chen, TCM Database@Taiwan: the world's largest traditional Chinese medicine database for drug screening in silico, PLoS ONE, 2011, 6(1), e15939 CrossRef CAS PubMed.
- B. Cao, J. X. Qu and Y. D. Yin, et al., Overview of antimicrobial options for Mycoplasma pneumoniae pneumonia: focus on macrolide resistance, Clin. Respir. J., 2017, 11(4), 419–429 CrossRef CAS PubMed.
- S. Jiang, Q. Cui and B. Ni, et al., Databases for facilitating mechanistic investigations of traditional Chinese medicines against COVID-19, Pharmacol. Res., 2020, 159, 104989 CrossRef CAS PubMed.
- H. D. Pan, X. J. Yao and W. Y. Wang, et al., Network pharmacological approach for elucidating the mechanisms of traditional Chinese medicine in treating COVID-19 patients, Pharmacol. Res., 2020, 159, 105043 CrossRef CAS PubMed.
- String, (accessed on 28 December 2021), Available online: https://string-db.org/ Search PubMed.
- G. Madhavi Sastry and M. Adzhigirey, Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments, J. Comput.-Aided Mol. Des., 2013, 27, 221–234, DOI:10.1007/s10822-013-9644-8.
- M. H. M. Olsson, C. R. SØndergaard and M. Rostkowski, PROPKA3: Consistent treatment of internal and surface residues in empirical p K a predictions, J. Chem. Theory Comput., 2011, 7, 525–537, DOI:10.1021/ct100578z.
- M. Rostkowski, M. H. Olsson and C. R. Søndergaard, Graphical analysis of pH-dependent properties of proteins predicted using PROPKA, BMC Struct Biol, 2011, 11, 6, DOI:10.1186/1472-6807-11-6.
- V. Sharma, P. C. Sharma and V. Kumar, In silico molecular docking analysis of natural pyridoacridines as anticancer agents, Adv. Chem., 2016, 1–9, DOI:10.1155/2016/5409387.
- G. M. Sastry, M. Adzhigirey, T. Day, R. Annabhimoju and W. Sherman, Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments, J. Comput. Aided Mol. Des., 2013, 27, 221–234, DOI:10.1007/s10822-013-9644-8.
- J. Singh, M. Kumar, R. Mansuri, G. C. Sahoo and A. J. Deep, Inhibitor designing, virtual screening, and docking studies for methyltransferase: A potential target against dengue virus, Pharm. Bioallied. Sci., 2016, 8, 188–194, DOI:10.4103/0975-7406.171682.
- R. A. Friesner, R. B. Murph, M. P. Repasky, L. L. Frye, J. R. Greenwood, T. A. Halgren, P. C. Sanschagrin and D. T. Mainz, Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein–ligand complexes, J. Med. Chem., 2006, 49, 6177–6196, DOI:10.1021/jm051256o.
- E. B. Lenselink, J. Louvel, A. F. Forti, J. P. D. van Veldhoven, H. de Vries, T. Mulder-Krieger, F. M. McRobb, A. Negri, J. Goose, R. Abel, H. W. T. van Vlijmen, L. Wang, E. Harder, W. Sherman, A. P. IJzerman and T. Beuming, Predicting binding affinities for GPCR ligands using free-energy perturbation, ACS Omega, 2016, 1, 293–304, DOI:10.1021/acsomega.6b00086.
- Ç. K. Atay, T. Tilki and B. Dede, Design and synthesis of novel ribofuranose nucleoside analogues as antiproliferative agents: a molecular docking and DFT study, J. Mol. Liq., 2018, 269, 315–326, DOI:10.1016/j.molliq.2018.08.009.
- M. Davies, M. Nowotka, G. Papadatos, N. Dedman, A. Gaulton, F. Atkinson, L. Bellis and J. P. Overington, ChEMBL web services: streamlining access to drug discovery data and utilities, Nucleic Acids Res., 2015, 43, W612–W620, DOI:10.1093/nar/gkv352.
- J. Dong, D. S. Cao, H. Y. Miao, S. Liu, B. C. Deng, Y. H. Yun, N. N. Wang, A. P. Lu, W. B. Zeng and A. F. Chen, ChemDes: an integrated web-based platform for molecular descriptor and fingerprint computation, J. Cheminf., 2015, 7, 60, DOI:10.1186/s13321-015-0109-z.
- D. Fourches, E. Muratov and A. Tropsha, Trust, but verify: on the importance of chemical structure curation in cheminformatics and QSAR modeling research, J. Chem. Inf. Model., 2010, 50, 1189–1204, DOI:10.1021/ci100176x.
- C. Steinbeck, Y. Han, S. Kuhn, O. Horlacher, E. Luttmann and E. Willighagen, The Chemistry Development Kit (CDK): an open-source Java library for Chemo- and Bioinformatics, J. Chem. Inf. Comput. Sci., 2003, 43, 493–500, DOI:10.1021/ci025584y.
- L. H. Hall and L. B. Kier, Electrotopological state indices for atom types: A novel combination of electronic, topological, and valence state information, J. Chem. Inf. Comput. Sci., 1995, 35, 1039–1045, DOI:10.1021/ci00028a014.
- J. L. Durant, B. A. Leland, D. R. Henry and J. G. Nourse, Reoptimization of MDL keys for use in drug discovery, J. Chem. Inf. Comput. Sci., 2002, 42, 1273–1280, DOI:10.1021/ci010132r.
- A. A. Malik, S. C. Ojha, N. Schaduangrat and C. Nantasenamat, ABCpred: a webserver for the discovery of acetyl- and butyryl-cholinesterase inhibitors, Mol. Diversity, 2021, 26, 467–487, DOI:10.1007/s11030-021-10292-6.
- A. Sarica, A. Cerasa and A. Quattrone, Random Forest Algorithm for the Classification of Neuroimaging Data in Alzheimer's Disease: A Systematic Review, Front. Aging Neurosci., 2017, 9, 329, DOI:10.3389/fnagi.2017.00329.
- R. Caruana and A. Niculescu-Mizil, An empirical comparison of supervised learning algorithms, in 23rd International Conference on Machine Learning: ACM Press, Pittsburgh, PA, 2006, pp. 161–168 Search PubMed.
- S. Yousefinejad, F. Honarasa and H. Montaseri, Linear solvent structure-polymer solubility and solvation energy relationships to study conductive polymer/carbon nanotube composite solutions, RSC Adv., 2015, 5(53), 42266–42275, 10.1039/C5RA05930E.
- P. Srinivas and R. Katarya, hyOPTXg: OPTUNA hyper-parameter optimization framework for predicting cardiovascular disease using XGBoost, Biomedical Signal Processing and Control, 2022, 73, 103456, DOI:10.1016/j.bspc.2021.103456.
- M. Zheng, X. Liu, Y. Xu, H. Li, C. Luo and H. Jiang, Computational methods for drug design and discovery: focus on China, Trends Pharmacol. Sci., 2013, 34, 549–559, DOI:10.1016/j.tips.2013.08.004.
- Y. C. Chen, Beware of docking, Trends Pharmacol. Sci., 2015, 36, 78–95, DOI:10.1016/j.tips.2014.12.001.
- R. Todeschini and V. Consonni, Molecular Descriptors for Chemoinformatics, Wiley-VCH Verlag, Weinheim, Germany, 2009 Search PubMed.
- C. Nantasenamat, H. Li, P. Mandi, A. Worachartcheewan, T. Monnor, C. Isarankura-Na-Ayudhya and V. Prachayasittikul, Exploring the chemical space of aromatase inhibitors, Mol. Diversity, 2013, 17(4), 661–677, DOI:10.1007/s11030-013-9462-x.
- W. Shoombuatong, V. Prachayasittikul, N. Anuwongcharoen, N. Songtawee, T. Monnor, S. Prachayasittikul, V. Prachayasittikul and C. Nantasenamat, Navigating the chemical space of dipeptidyl peptidase-4 inhibitors, Drug Des., Dev. Ther., 2015, 9, 4515–4549, DOI:10.2147/DDDT.S86529.
- T. I. Oprea, Property distribution of drug-related chemical databases, J. Comput.-Aided Mol. Des., 2000, 14(3), 251–264, DOI:10.1023/a:1008130001697.
- L. Z. Benet, C. M. Hosey, O. Ursu and T. I. Oprea, BDDCS, the Rule of 5 and drugability, Adv. Drug Delivery Rev., 2016, 101, 89–98, DOI:10.1016/j.addr.2016.05.007.
- A. Golbraikh and A. Tropsha, Beware of q2, J. Mol. Graphics Modell., 2002, 20(4), 269–276, DOI:10.1016/s1093-3263(01)00123-1.
- A. Tropsha and A. Golbraikh, Predictive QSAR modeling workflow, model applicability domains, and virtual screening, Curr. Pharm. Des., 2007, 13(34), 3494–3504, DOI:10.2174/138161207782794257.
- U. Švajger, B. Brus, S. Turk, M. Sova, V. Hodnik, G. Anderluh and S. Gobec, Novel Toll-Like Receptor 4 (TLR4) Antagonists Identified by Structure- and Ligand-Based Virtual Screening, Eur. J. Med. Chem., 2013, 70, 393–399, DOI:10.1016/j.ejmech.2013.10.019.
- A. Barochia, S. Solomon, X. Cui, C. Natanson and P. Q. Eichacker, Eritoran tetrasodium (E5564) Treatment for Sepsis: Review of Preclinical and Clinical Studies, Expert Opin. Drug Metab. Toxicol., 2011, 7, 479–494 CrossRef CAS PubMed.
- T. W. Rice, A. P. Wheeler, G. R. Bernard, J.-L. Vincent, D. C. Angus and N. Aikawa, et al., A randomized, double-blind, placebo-controlled trial of TAK-242 for the treatment of severe sepsis, Crit. Care Med., 2010, 38, 1685–1694 CrossRef CAS PubMed.
- S. Yousefinejad, L. Aalizadeh and F. Honarasa, Application of ATR-FTIR spectroscopy and chemometrics for the discrimination of furnace oil, gas oil and mazut oil, Anal. Methods, 2016, 8(23), 4640–4647, 10.1039/C6AY00051G.
- C. Nantasenamat, C. Isarankura-NaAyudhya and T. Naenna, et al, A practical overview of quantitative structure activity relationship, EXCLI J., 2009, 874–888, DOI:10.17877/DE290R-690.
- C. Nantasenamat, C. Isarankura-NaAyudhya and V. Prachayasittikul, Advances in computational methods to predict the biological activity of compounds, Expert Opin. Drug Discovery, 2010, 5(7), 633–654, DOI:10.1517/17460441.2010.492827.
- S. Yousefinejad and B. Hemmateenejad, Chemometrics tools in QSAR/QSPR studies: A historical perspective, Chemom. Intell. Lab. Syst., 2015, 149, 177–204, DOI:10.1016/j.chemolab.2015.06.016.
- M. Bahadori, B. Hemmateenejad and S. Yousefinejad, Quantitative sequence-activity modeling of ACE peptide originated from milk using ACC-QTMS amino acid indices, Amino acids, 2019, 51(8), 1209–1220, DOI:10.1007/s00726-019-02761-y.
- A. Mahmood, A. Irfan and J. L. Wang, Machine learning and molecular dynamics simulation-assisted evolutionary design and discovery pipeline to screen efficient small molecule acceptors for PTB7-Th-based organic solar cells with over 15% efficiency, J. Mater. Chem. A, 2022, 10, 4170–4180, 10.1039/D1TA09762H.
- A. Mahmood, A. Irfan and J. L. Wang, Developing efficient small molecule acceptors with sp2-hybridized nitrogen at different positions by density functional theory calculations, molecular dynamics simulations and machine learning, Chem. Eur J., 2021, 28(2), e202103712, DOI:10.1002/chem.202103712.
- A. Mahmood and J. L. Wang, A time and resource efficient machine learning assisted design of non-fullerene small molecule acceptors for P3HT-based organic solar cells and green solvent selection, J. Mater. Chem. A, 2021, 9, 15684–15695, 10.1039/D1TA04742F.
- A. Mahmood and J. L. Wang, Machine learning for high performance organic solar cells: current scenario and future prospects, Energy Environ. Sci., 2021, 14, 90–105, 10.1039/D0EE02838J.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ra06178c |
| This journal is © The Royal Society of Chemistry 2023 |