
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrochemical stripping voltammetrical sensor based on polypyrrole exfoliated polyetheramine–montmorillonite nanocomposite for nanomolar detection of nifuroxazide†
Mona Elfiky *,
Mohamed Ghoneim,
Hanaa El-Desoky,
Amera Hassanein and
Nehal Salahuddin
*,
Mohamed Ghoneim,
Hanaa El-Desoky,
Amera Hassanein and
Nehal Salahuddin
Department of Chemistry, Faculty of Science, Tanta University, 31527, Egypt. E-mail: Elfiky_mona@science.tanta.edu.eg; Fax: +20403350804; Tel: +201004155414
First published on 9th February 2023
Abstract
A series of polypyrrole/polyetheramine–montmorillonite nanocomposites have been fabricated by the intercalation of different types of polyoxyalkylene amine hydrochloride (Jeffamines: D400, D2000, T5000, and T403) into montmorillonite layers via the cation-exchange process followed by in situ polymerization of pyrrole. The physicochemical characteristics of as-prepared nanocomposites were investigated using Fourier transform infrared (FT-IR), X-ray diffraction (XRD), scanning electron microscopy (SEM), transmission electron microscopy (TEM), Brunauer–Emmett–Teller (BET), and electrochemical impedance spectroscopy (EIS) instruments. The change of the types of Jeffamine causes a change in the geometrical structure, and surface area of nanocomposites. Noteworthy, the resulting polypyrrole/D2000–montmorillonite ([PDM-50]) nanocomposite exhibited a cauliflower-like shape with a specific surface area (116.2 m2 g−1) with the highest conductivity. Furthermore, the modified stripping voltammetric carbon paste sensor was fabricated based on 1.0% [PDM-50] nanocomposites to detect the drug nifuroxazide (NF). The sensor achieved detection limits (LD) of 0.24, and 0.9 nM of NF in the medication, and human urine fluid, respectively. This sensor showed appropriate repeatability, reproducibility, stability, and selectivity for NF sensing in different fluids accompanied by other interferents.
1. Introduction
4-Hydroxy-N′-[(5-nitrofuran-2-yl)methylene]benzohydrazide (nifuroxazide (NF))1 is an oral nitrofuran antibiotic, with a wide range of bactericidal activity against Gram-positive and Gram-negative entero-pathogenic organisms (Scheme S1†). NF is extensively used as an intestinal antiseptic in the treatment of susceptible gastrointestinal infections.2 But NF is not absorbed from the gastrointestinal tract and, consequently rapidly removed from the body without metabolization. Due to the therapeutic significance of the NF, various analytical systems have been applied to evaluate its validity in commercial and biological fluids.2–25 It is noteworthy that the two spectrofluorimetric methods3,4 allow NF to be detected in either a single form or in a binary mixture with drotaverine. However, the drawback of the former3 is that the method can not be applied for the determination of NF in presence of drotaverine, since the alkaline conditions required would lead to degradation of drotaverine4 and thus the method requires intensive sample pretreatment.4 Furthermore, most reported methods2,5–12,15 were successfully applied for analysis of NF in its dosage forms with no attempt for its assay in biological fluids. Only one HPLC method15 was applied to measure NF concentrations (down to 7.27 nM), which were extracted from buffered plasma with chloroform. However, the required high-strength ionic buffered mobile phases are hazardous for column efficiency and need prolonged time for column saturation and washing as well as insufficient sensitivity and selectivity.In contrast, the electrochemical sensors have higher sensitivity and proper selectivity in the field of analytical applications without the need for complex sample preparation with a short analysis time.26–30 The recent reports describe the detection of NF in commercial products (capsules and tablets) via differential pulse polarographic (DPP), biological fluids (urine and human serum) via adsorptive stripping voltammetry (AdSV) upon mercury, and carbon paste sensors.27,30 Also, the electro-oxidation of NF was investigated via differential pulse voltammetry (DPV) upon bare, and Sephadex-modified carbon paste sensors with LOD of 20 nM; this direct detection of NF in urine is not efficient upon unmodified and modified sensors due to the large oxidation peak of blank urine. Recently, J. D. Mozo et al.29 has been successfully achieved LOD 10 ng mL−1 in a bulk form via linear-sweep adsorptive stripping voltammetry (LS-AdSV) upon a screen-printed carbon nanofiber modulated sensor, and revealed an average recovery of 100.7 ± 2.5% in commercial tablets.29 Therefore, the modification of sensor has a great importance in electrochemistry as they enhance the sensitivity of specific analytes.
Montmorillonite (MMT) is natural clay, which is one of the very promising modifiers, which belongs to the smectite group of clays with a layer lattice. MMT has a high chemical, mechanical stability, a well-layered structure, and strong adsorptive properties attributed to the expandability of the internal layers. Moreover, the SW-AdSV method has been well characterized as an extremely sensitive source for electroanalytical applications of ultra-trace determinations of a variety of organic or inorganic substances.31–33 However, to date no square-wave adsorptive anodic stripping voltammetric (SW-AdASV) method is available in the literature for the determination of NF using a modified CPE with Na-MMT clay.
Also, slices of MMT clay can interact with different guest species including metal oxides to form hybrids,31 or/and conducting polymers to form nanocomposites34 with different morphologies for different sorts of applications. Notably, MMT clay was successfully used as a modified material to improve the adsorptive behaviour of the carbon paste sensor (CPS) for the electrochemical sensing of some inorganic materials.31,35,36 However, the usage of clays in electrochemical applications is limited due to their poor electrical conductivity.37 Furthermore, the organophilic MMT (OMMT) could be fabricated via the ion-exchange process of organic compounds (e.g., surfactants and multifunctional organic amines chains), resulting in stable OMMT with powerful adsorption, and considerable dispersibility in organic dispersion medium.38–40 Various reports have been made to modify MMT with different types of poly(oxypropylene)amines;41–44 the resulting material showed potential use in various applications, such as a curing agent for epoxy,45,46 and drug-delivery vehicles.47
On the other hand, scientific innovations have recently gained extensive attention to the usage of polypyrrole (Ppy) in electrochemical sensing applications48,49 due to its excellent conductivity up to 1000 S cm−1, and environmental stability.32,48 Recently, many research efforts have been made to prepare montmorillonite/polypyrrole (MMT/Ppy) nanocomposites.50–54 But, to achieve a better degree of miscibility with the polymer matrix, it is essential to adjust them with organic cations to improve their hydrophobicity. In addition, some functional organic cations can react with the polymer matrix.55,56 N. Salahuddin et al.,57 have fabricated nanocomposites with higher resistivity, and hydrophobicity in comparison with pristine polyurethane; the syncretization was carried out via in situ polymerization of toluene diisocyanate and butanediol with different content of OMMT. As well, Doris Pospiech et al.58 studied the dispersion property of MMT/poly(oxyalkylene)s in polypropylene. Recently, Dos Santos et al.59 has used polyester-based thermoplastic polyurethane (TPU) as matrix and Ppy or MMT/Ppy composite in the presence of surfactant to fabricate polymer composites via melt mixing technique.
Herein, a series of polypyrrole/polyetheramine–montmorillonite [Ppy/organoclay] nanocomposites were fabricated by intercalation of various molecular masses of polyoxyalkylene amine hydrochloride (Jeffamine D400, D2000, T403, and T5000) (Scheme S2†) into Na-MMT via ion-exchange treatment followed by the in situ polymerization of Py with different organoclay contents. Afterward, sensitive electrochemical sensors based on CP blended with the prepared nanocomposites were fabricated and examined to detect NF using square-wave adsorptive anodic stripping voltammetry (SW-AdAS) in commercial, and human urine fluid.
2. Experimental
2.1. Materials, equipments, electrochemical liquids, and fabrication of unmodified and modified CPSs
All details are mentioned in the ESI† section.2.2. Preparation of polyetheramine–montmorillonite
The polyetheramine–montmorillonite (organoclay) was synthesized via cation-exchange process between the Na+ in the MMT matrix and –NH3+ groups in Jeffamine. 5 g of MMT was swelled in 300 mL distilled water (DW) with steady stirring for 3 h at 60 °C and for 24 h at 25 °C. 6.2 g of acidified Jeffamine D2000 with an aqueous solution of HCl was inserted into the swelled clay solution with steady stirring for 24 h. The precipitate was thoroughly rinsed with DW until no Cl− ions were detected. The precipitate of (D2000-MMT) was dried at 60 °C to give 8.191 g. The same procedure was carried out to prepare other types of organoclay, as listed in Table 1.| Code | Jeffamine | MMT wt (g) | Yield wt (g) | |
|---|---|---|---|---|
| Type | Wt (g) | |||
| D2000-MMT | D2000 | 6.2 | 5 | 8.2 |
| D400-MMT | D400 | 1.4 | 5 | 5.6 |
| T5000-MMT | T5000 | 11.6 | 5 | 7.9 |
| T403-MMT | T403 | 1.0 | 5 | 5.6 |
2.3. Preparation of Ppy and Ppy/polyetheramine–montmorillonite nanocomposites
(0.98 g, 0.14 M) of Py was added into 50 mL HCl (0.1 M). Then, 0.29 M of FeCl3 (7.8 g dissolved in 50 mL HCl, 0.1 M) was inserted into Py solution dropwise with steady stirring for 24 h. The molar ratio between Py and FeCl3 was (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2). The precipitate was thoroughly rinsed with DW, and dried at 70 °C for 24 h in a vacuum oven. 0.1 g of D2000-MMT was stirred in 5 mL DMF for 24 h, and 3 h at 60 °C. Then, 0.29 M of FeCl3 (7.8 g dissolved in 50 mL HCl, 0.1 M) was inserted into the organoclay suspension with constant stirring for 24 h. The Py (0.98 g, 0.14 M) dissolved in 50 mL HCl (0.2 M), and was added into the organoclay solution dropwise with constant stirring for 24 h at 30 °C. Finally, the nanocomposite precipitate was thoroughly rinsed with DW and dried at 60 °C in the oven for 48 h to afford 0.7424 g of [PDM-10]. The same procedure was carried out to prepare different types of nanocomposites with different contents of organoclay, as listed in (Table S1†).
:2). The precipitate was thoroughly rinsed with DW, and dried at 70 °C for 24 h in a vacuum oven. 0.1 g of D2000-MMT was stirred in 5 mL DMF for 24 h, and 3 h at 60 °C. Then, 0.29 M of FeCl3 (7.8 g dissolved in 50 mL HCl, 0.1 M) was inserted into the organoclay suspension with constant stirring for 24 h. The Py (0.98 g, 0.14 M) dissolved in 50 mL HCl (0.2 M), and was added into the organoclay solution dropwise with constant stirring for 24 h at 30 °C. Finally, the nanocomposite precipitate was thoroughly rinsed with DW and dried at 60 °C in the oven for 48 h to afford 0.7424 g of [PDM-10]. The same procedure was carried out to prepare different types of nanocomposites with different contents of organoclay, as listed in (Table S1†).
3. Results and discussion
3.1. FTIR and XRD
FTIR spectra of Na-MMT, organoclay, Ppy, and Ppy/organoclay nanocomposites with different content of organoclay were illustrated in (Fig. S1(a–d)†). The spectrum of Na-MMT displayed absorption bands at 3633, and 1050 cm−1, which could be attributed to the stretching vibration of Al–OH, and Si–O–Si, respectively.60 While bands at 1635, 921, and 523 cm−1 are ascribed to bending of Al–OH, Al–Al–O, and Si–O–Al respectively. The FT-IR spectrum of D2000-MMT displayed a shift in the band at 1658, and 1485 cm−1, which could be corresponded to the asymmetrical and symmetrical bending of N–H in NH3+; affirming the electrostatic attraction between silicate sheets and polyoxypropylene.61 Furthermore, the spectrum of Ppy exhibited bands at 1543, 1456, 1168, and 1046 cm−1, which could be assigned to the stretching C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, stretching of C–N, C–C in-plane deformation, and bending of N–H in Py rings, respectively.62 On the other hand, the spectrum of [PDM] displayed the band of the C–C stretching of Ppy at 1543 cm−1. Notably, the N–H stretching band shifted from 3422 to 3441 cm−1 for [PDM-50] confirming the interaction between sheets of MMT and Ppy matrix.63,64 The same results were observed for other nanocomposites.
C, stretching of C–N, C–C in-plane deformation, and bending of N–H in Py rings, respectively.62 On the other hand, the spectrum of [PDM] displayed the band of the C–C stretching of Ppy at 1543 cm−1. Notably, the N–H stretching band shifted from 3422 to 3441 cm−1 for [PDM-50] confirming the interaction between sheets of MMT and Ppy matrix.63,64 The same results were observed for other nanocomposites.
XRD patterns of Na-MMT, Ppy, organoclay, and Ppy/organoclay nanocomposites were shown in (Fig. 1(a–d)) at various organoclay contents. In the XRD pattern of Ppy, the broad band at 2θ = 18.3° corresponded to the amorphous crosslinked chains of the polymer matrix.48 The crystalline band corresponds to d-spacing between layers (d001) in the pristine clay sample 9.6 Å was shifted after preparation of organoclay, owing to the intercalation of polyoxyalkylene between the sheets of MMT.65 The diffraction patterns displayed in (Fig. 1) indicate a regular lattice spacing of 20.1 Å for D2000, 15.2 Å for D400, 14.0 Å for T5000, and 14.5 Å for T403 and with an expansion of the clay galleries by 10.8, 4.9, 4.89, and 5.2 Å respectively. Furthermore, Ppy/organoclay nanocomposites with 10, 20, and 50% of organoclay show no distinguishable band in the XRD pattern, confirming the exfoliation of the organoclay during the in situ polymerization of Py. While the polymerization of Py within D2000-MMT, D400-MMT, and T403-MMT 50 a broad d001 was observed, which could be related to the partially exfoliated structure.65 It should emphasize that Ppy is more adequate to intercalate between clay sheets modified by T5000 even at high concentrations. The disappearance of the basal reflection indicates the formation of an exfoliated nanostructure.
 | ||
| Fig. 1 X-ray diffraction pattern of Na-MMT, Ppy, organoclay, and Ppy/organoclay nanocomposites at different content of organoclay. | ||
3.2. The preliminary electrochemical sensing test and SEM micrographs of the surface of sensors
AdAS-SW voltammograms of 30.0 nM NF in pH 5 (B–R buffer) were evaluated upon [B], 7.0% [Na-MMT], and 1.0% of various nanocomposites MCPSs at Eacc = 0.6 V vs., Ag/AgCl/KCls for 10 s, as shown in (Fig. S2(a)†). The maximal peak was obtained upon the 1.0% [PDM-50] MCPS with about 7- and 2.7-time increase compared with [B], and 7.0% [Na-MMT] CPSs, respectively. Whereas, the organoclay was obtained via a simple ion-exchange process of hydrophilic MMT with polyoxyalkylene amine, leading to opening up the lamellae of MMT and causing profound changes in the sorption and/or intercalation of organic molecules including drug.66,67 The adsorption of the drug increases with increasing the hydrophobicity of nanocomposites. Whereas longer organo-chains (e.g., Jeffamine D2000, and T5000) have more active sites available for adsorption and are more hydrophobic than shorter chains.In general, the change in the sensitivity of sensors could arise from the significant difference in the geometrical structure of the surface of sensors accompanied by improvement in the conductivity. As demonstrated in (Fig. 2), SEM micrographs of the surface of 1.0% [PT1M-50] (Fig. 2(a)), and [PTM-50] (Fig. 2(b)) MCPSs appeared as smooth domains assembled in small necklace-like structure. As well, 1.0% [PT1M-50] MCPS has compact particles relative to [PTM-50] MCPS, which shows more voids to improve the adsorption of NF upon its surface. Furthermore, the morphological structure of 1.0% [PDM-50] MCPS (Fig. 2(c)) shows irregular granular structures with long coral reefs-like shapes as well as more voids and vacuoles, providing the largest surface area of CPS and more improvement in the adsorption, and sensitive detection of NF compared with the other MCPSs.
 | ||
| Fig. 2 SEM micrographs of sensor surface with 1.0% of [PT1M-50] (a), [PTM-50] (b), [PD1M-50] (c), and [PDM-50] (d). | ||
3.3. The conductivity and morphological properties of nanocomposites
The high conductivity of nanocomposites refers to the relatively slow rate of Ppy polymerization in the presence of organoclay, which may promote the higher-ordered morphology of Ppy with limited defects. Therefore, the preliminary investigation of the electrical conductivity of all prepared materials was carried out as a function of Ppy content via a 4-probe system of a direct current circuit (DC circuit), as shown in (Fig. S3†). Noteworthy, the conductivity of nanocomposites was increased with increasing Ppy content up to 50%, and nanocomposites with Ppy content up to 50% were higher than pure Ppy.The morphological aspects of Ppy, MMT, organoclay, and Ppy/organoclay nanocomposites were also investigated via SEM, as shown in (Fig. 3). The morphology of Ppy, and Na-MMT (Fig. 3(a and b)) resembled a tree-like and flake-like structure, respectively. Moreover, micrographs of the surface of Ppy/organoclay nanocomposites exhibited a cauliflower-like structure with a homogenous dispersion of the mineral domains in the Ppy matrix without the presence of any inorganic aggregations (Fig. 3(c–f)) with round shape domains in the range of 200–600 nm. Notably, micrographs of nanocomposites with 50% of organoclay exhibited more dense structure compared with that of 10%. This proves that the increase in the organoclay% affects the compactness of the materials.
 | ||
| Fig. 3 SEM micrographs of Ppy (a), Na-MMT (b), [PTM-10] (c), [PTM-50] (d), [PDM-10] (e), and [PDM-50] (f) nanocomposites. | ||
To give direct evidence of the nanometer-scale exfoliation of organoclay in the Ppy matrix, TEM of some nanocomposites was investigated, as shown in (Fig. 4). In [PDM], and [PTM] nanocomposites, some clay stacks appeared with thickness in the range of 15–100 nm. Since the functional amine groups of polyoxypropylene can form hydrogen bonds with the silicate hydroxylated edge groups, confirming the strong interaction between silicate layers and polymer matrix. Some intercalated organoclay stacks present in the TEM of [PDM-50] (Fig. 4(a)), and [PTM-50] (Fig. 4(b)) micrographs are dispersed uniformly in the matrix. Nanocomposite micrographs revealed a necklace-like shape of organoclay. Meanwhile, the BET analysis (Fig. S4†) indicated that the [PDM-50], and [PTM-50] have a proper specific surface area (SA) of 183.0, and 165.3 m2 g−1, respectively compared with Ppy (28.6 m2 g−1) and MMT (215.6 m2 g−1), which confirms the improvements in the geometrical structure of proposed nanocomposites.
 | ||
| Fig. 4 TEM micrographs of [PDM-50] (a), and [PTM-50] (b). | ||
3.4. Composition, adsorption, affinity, and electrochemical impedance measurements of the constructed sensors
AdAS-SW voltammograms of 30.0 nM NF in pH 5 (B–R buffer) were evaluated upon different compositions (0.5–3.0%) of [PDM-50] MCPS, as plotted in (Fig. S2(b)†). The maximal peak current was obtained at 1.0% [PDM-50] MCPS, owing to its geometrical structure, as displayed in the SEM micrograph (Fig. 5(b)). | ||
| Fig. 5 SEM micrograph of 0.5 (a), 1.0 (b), 2.0 (c), and 3.0% (d) of [PDM-50] MCPSs. | ||
The fabrication of ultrasensitive MCPS does not only depends on the geometrical structure but also mainly on the degree of resistance (Rct) of the sensor. As demonstrated in (Fig. S5†), EIS measurements were evaluated to be 257, and 184 Ω for [B] CPS, and 1.0% [PDM-50] MCPS, respectively.
Also, the absorption affinity of 10.0 nM NF (pH 5) upon the surface of the [B], and 1.0% [PDM-50] MCPSs were tested, as shown in (Fig. S6†). CVs were recorded at Eacc = 0.6 V for 0.0 s [Cycle (I)], subsequently for 10 s (Cycles 1st; (II) and 2nd; (III)). In the preceding measurement, a small ox. peak was observed upon the [B] CPS, as shown in (Fig. S6(a)†). While a well-defined peak was revealed upon the 1.0% [PDM-50] MCPS (Cycles 1st; (II) and 2nd; (III)) (Fig. S5(b)†), which signifies the remarkable enhancements of adsorption property towards NF even at tacc = 0.0 s [Cycle (I)]. Besides, the noteworthy decay of the peak was observed in the [2nd cycle; (II)], which denotes the desorption of NF from the surface of the sensor. Therefore, the 1.0% [PDM-50] MCPS has the strongest adsorptive character towards NF with about a 12-time increase in peak current compared with [B] CPS. According to the previous results, 1.0% [PDM-50] MCPS exhibited the highest sensitivity, and adsorption properties for subsequent electroanalytical measurements.
3.5. Validation studies
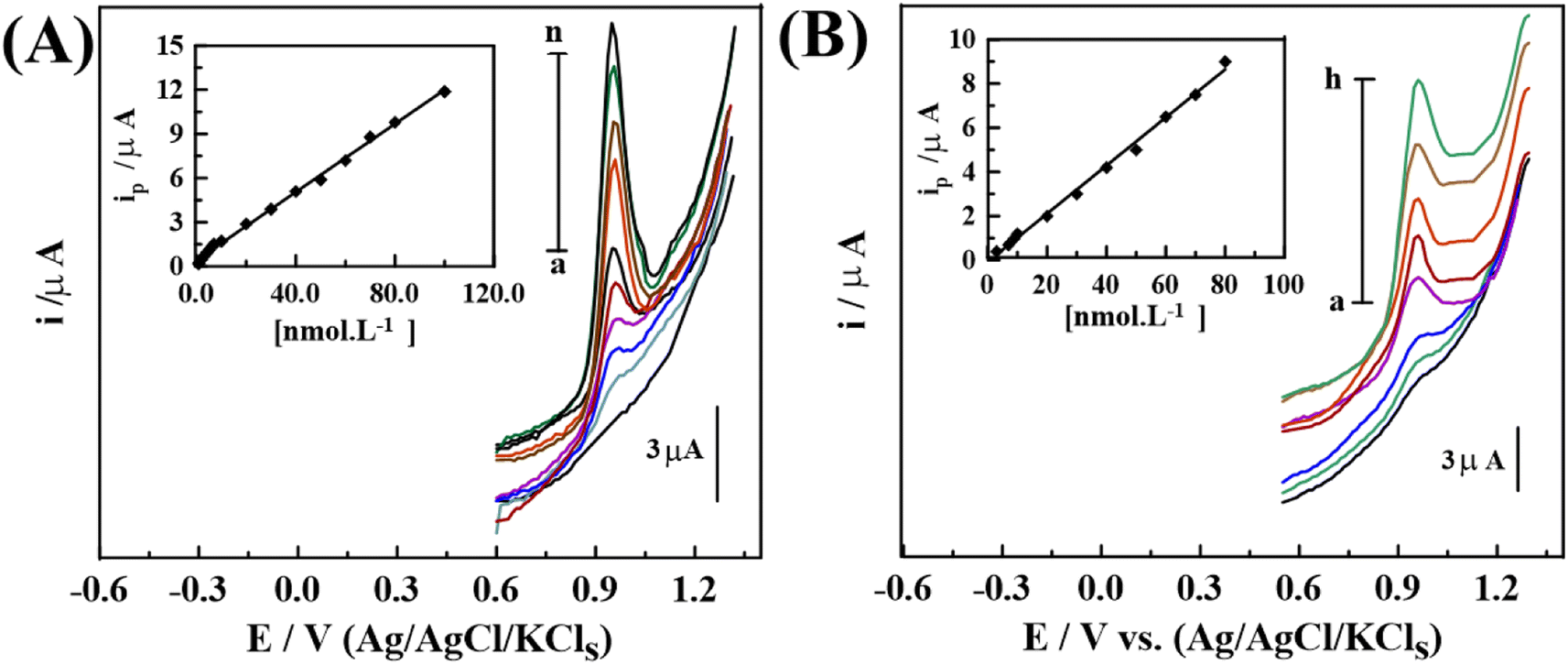 | ||
| Fig. 6 SW-AdAS voltammograms of various concentrations of NF in pH 5 upon 1.0% [PDM-50] MCPS in (A) bulk: (a) background, (b) 2.0, (c) 6.0, (d) 8.0, (e) 20.0, (f) 30.0, (g) 60.0, (h) 70.0, (i) 80.0 and (j) 100.0 nM of NF and, (B) spiked human urine: (a) background, (b) 7.0, (c) 9.0, (d) 10.0, (e) 30.0, (f) 50.0, (g) 60.0, and (h) 80.0 nM of NF (Eacc = 0.6 V for 10 s, f = 120 Hz, ΔEs = 12 mV, and a = 30 mV). | ||
| Sensor | Technique | LOD (nM)/LR (nM) | Type of fluid | Ref. |
|---|---|---|---|---|
| a Carbon paste electrode (CPE), differential pulse voltammetry (DPV), linear sweep adsorptive stripping voltammetry (LS-AdSV), carbon nanofiber modified screen-printed electrode (CN-SPCE), adsorptive stripping voltammetry (AdSV), hanging mercury drop electrode (HMDE). | ||||
| Hg and CPEs | AdSV | 36.3 | Urine | 30 |
| Sephadex-modified CPE | DPV | 8.0/15.0–2000 | Bulk | 26 |
| 50.0–400.0 | Urine | |||
| HMDE | HMDE | 5.0/100–2000 | Bulk | 27 |
| 181.7–908.4 | Serum | |||
| CNF-SPCEs | LS-AdSV | 36.3/1800–10900 |
Bulk | 29 |
| 1.0% [PDM-50] MCPS | SW-AdASV | 0.24/0.8–100 | Bulk | This work |
| 0.90/3.0–80 | Human urine | |||
In addition, recovery (R%), precision (RSD%), and accuracy (RE%)68 of the applied SW-AdASV method were estimated by measuring 5 replicates at 3 concentrations levels of bulk NF upon each sensor over 1 day (intra-day), and for 3 days (inter-day), respectively, as shown in (Table 3). There are no significant differences between the taken, and found concentrations of NF. As well as, satisfactory mean (R ± RSD), and RE% were accomplished, confirming the validity of the proposed method for the detection of NF.
| Day | [Taken]/(nM) | Mean [found]/(nM) | Recovery (R%) | Accuracy (RE%) | Precision (RSD%) |
|---|---|---|---|---|---|
| Intra-day | |||||
| 1 | 20.0 | 20.03 | 100.15 | 0.15 | 1.84 |
| 60.0 | 60.67 | 101.12 | 1.12 | 0.60 | |
|
|||||
| Inter-day | |||||
| 1 | 20.0 | 20.03 | 100.15 | 0.15 | 1.97 |
| 60.0 | 60.8 | 101.33 | 1.33 | 0.92 | |
| 2 | 20.0 | 20.006 | 100.03 | 0.03 | 2.12 |
| 60.0 | 60.8 | 101.33 | 1.33 | 0.933 | |
| 3 | 20.0 | 19.773 | 98.87 | −1.13 | 1.05 |
| 60.0 | 60.59 | 100.98 | 0.98 | 0.63 | |
On the other hand, the reusability of the sensor was tested by recording five consecutive measurements, which retained 97% of the performance, as shown in (Fig. S11(a)†). After 5 measurements, the performance significantly decreased, owing to the fullness of the binding cavities on the surface of the sensor. Furthermore, the stability of the sensor was checked by storing the sensor at 4 °C for 1 month accompanied by weekly measurements of the performance. The proposed sensor demonstrated a suitable performance for up to 15 days by retaining its performance up to about 96.3%, as illustrated in (Fig. S11(b)†). After that, the performance decreases gradually up to 90.3% after 30 days. This means that the sensor needs to be stored at a low temperature to avoid damage to the contents of the sensor, and prevent moisture absorption. All these results proved that the sensor has proper selectivity, and sufficient stability for the detection of NF.
3.6. Applications
Furthermore, the RSD% of the sensor was pointed out to be 1.12% (Antinal® capsules), and 1.18% (Drotazide® capsules) for 6.0 nM NF utilizing the standard addition method, as illustrated in (Table 4). This result was statistically compared with a reference spectrofluorimetric method.3 The calculated F-value did not exceed the theoretical one at the 95.0% confidence level (n = 5), and by the t-test value (Student's t-test)69 exhibited non-significant variance in values regarding accuracy and precision. Accordingly, the proposed sensor offered satisfactory recovery in medical drugs even in the presence of drug excipients.
| Method | SW-AdASV method | Reference method |
|---|---|---|
| Antinal® capsules | ||
| Mean Cfound/(nM) ± SD | 5.88 ± 0.066 | |
| Mean% recovery ± RSD | 98.00 ± 1.12 | 98.44 ± 1.06 |
| F-Value | 1.12 | |
| t-Test | 0.64 | |
|
||
| Drotazide® capsules | ||
| Mean Cfound/(nM) ± SD | 5.98 ± 0.071 | |
| Mean% recovery ± RSD | 99.67 ± 1.18 | 100.62 ± 1.12 |
| F-Value | 1.11 | |
| t-Test | 1.30 | |
| Electrode | Linearity range (nM) | ip (μA) = (b ± SD) C (μM) + (a ± SD) | R2 | LOD (nM) | LOQ (nM) |
|---|---|---|---|---|---|
| Volunteer 1 | 3.0–80 | ip = 0.109 ± 0.35 × 10−3 C −0.070 ± 1.90 × 10−2 | 0.998 | 0.9 | 3.0 |
| Volunteer 2 | 3.0–80 | ip = 0.110 ± 0.80 × 10−3 C −0.044 ± 2.36 × 10−2 | 0.999 | 0.9 | 3.0 |
| Volunteer 3 | 3.5–80 | ip = 0.106 ± 1.35 × 10−3 C −0.042 ± 2.75 × 10−2 | 0.997 | 1.05 | 3.5 |
Noteworthy, these results revealed that the optimized method upon 1.0% [PDM-50] MCPS has the suitable reliability of direct detection of NF in spiked human urine. As shown in (Table 2), the attained LOD in spiked human urine using 1.0% [PDM-50] MCPS is about 60, and 83 times lower than that in published works by W. Buchberger et al.30 and A. Radi,26 respectively.
4. Conclusion
In this study, we have first introduced a new kind of nanocomposite that was used as a modifier for CPS. 1.0% [PDM-50] MCPS shows a remarkable enhancement in the peak current compared with that of bare CPS. The proper reproducibility, wide concentrations ranges and low detection limits in bulk form and urine, make this modified sensor very attractive for assay of NF than other sensors reported in the literature. An additional advantage of the proposed method may be related to its high level of selectivity that allows assay of the NF in the nanomolar level of concentration without the intervention of another compound drug or common excipients. In addition, this method ables to detect the drug in human urine samples without any interference from endogenous materials compared with other published voltammetric methods. Furthermore, the optimized method is rapid, accurate, precise, and economic. The 1.0% [PDM-50] MCPS could be recommended for use in quality control of medical drugs.Conflicts of interest
There are no conflicts to declare.References
- G. R. Greenstein, The Merck Index: An Encyclopedia of Chemicals, Drugs, and Biologicals Ref. Rev., 2007, 21, 40 Search PubMed.
- H. M. Maher and T. S. Belal, J. Liq. Chromatogr. Relat. Technol., 2012, 35, 2001–2020 CrossRef CAS.
- T. S. Belal, A simple and sensitive spectrofluorimetric method for analysis of some nitrofuran drugs in pharmaceutical preparations, J. Fluoresc., 2008, 18, 771–780 CrossRef CAS PubMed.
- A. A. El-Zaher and M. A. Mahrouse, A validated spectrofluorimetric method for the determination of nifuroxazide through coumarin formation using experimental design, Chem. Cent. J., 2013, 7, 90 CrossRef PubMed.
- A. Abul Khier, M. M. Elhenawee and M. S. Elmasry, Spectrophotometric method for the determination of some drugs using fast red B salt, J. Chem., 2008, 5, 1087–1097 Search PubMed.
- K. Emara, I. Refaat and O. Abdel mageed, HPLC and spectrophotometric determination of nifuroxazide and its pharmaceutical formulations, Egypt. J. Pharm. Sci., 1994, 35, 313 CAS.
- E. Szuminska and A. Cisak, Polarographic and spectrophotometric determination of nifuroxazide in pharmaceuticals. I. Nifuroxazide determination in capsules, Acta Pol. Pharm., 1988, 45, 551–558 CAS.
- E. Szumińska and A. Cisak, Polarographic and spectrophotometric determination of nifuroxazide in pharmaceuticals. II. Determination of nifuroxazide in suspensions, Acta Pol. Pharm., 1990, 47, 1–2 Search PubMed.
- M. I. Toral, M. Paine, P. Leyton and P. Richter, Determination of attapulgite and nifuroxazide in pharmaceutical formulations by sequential digital derivative spectrophotometry, J. AOAC Int., 2004, 87, 1323–1328 CrossRef CAS PubMed.
- F. H. Metwally, M. Abdelkawy and I. A. Naguib, Determination of nifuroxazide and drotaverine hydrochloride in pharmaceutical preparations by three independent analytical methods, J. AOAC Int., 2006, 89, 78–87 CrossRef CAS.
- F. H. Metwally, Simultaneous determination of nifuroxazide and drotaverine hydrochloride in pharmaceutical preparations by bivariate and multivariate spectral analysis, Spectrochim. Acta, Part A, 2008, 69, 343–349 CrossRef PubMed.
- M. A. Hegazy, W. A. Hassanain and L. E. Abdel-Fattah, Stability Indicating Spectrophotometric and Chemometric Methods for Determination of Nifuroxazide in Presence of Its Alkaline Degradation Products, Pharm. Anal. Acta, 2011, 2, 127 Search PubMed.
- A. W. Sobanska, Rapid determination of nifuroxazide in tablets by near-infrared spectroscopy, Chem. Anal., 2009, 54, 1021–1033 CAS.
- M. M. Ayad, N. F. Youssef, H. E. Abdellatif and S. M. Soliman, A comparative study on various spectrometries with thin layer chromatography for simultaneous analysis of drotaverine and nifuroxazide in capsules, Chem. Pharm. Bull., 2006, 54, 807–813 CrossRef CAS PubMed.
- P. Guinebault, M. Broquaire and R. Braithwaite, Determination of nifuroxazide in biological fluid by automated high-performance liquid chromatography with large-volume injection, J. Chromatogr. A, 1981, 204, 329–333 CrossRef CAS PubMed.
- N. S. Abdelwahab, N. W. Ali, M. M. Zaki, M. Abdelkawy and M. T. El-Saadi, Quantitative Determination of Synthesized Genotoxic Impurities in Nifuroxazide Capsules by Validated Chromatographic Methods, J. AOAC Int., 2018, 101, 385–393 CrossRef PubMed.
- J. Squella, I. Lemus and G. Lonza, Electrochemical study of nifuroxazide and its analytical determination in tablets, Bol. Soc. Chil. Quim., 1991, 36, 109–116 CAS.
- A. Radi, S. El-Laban and I. Kenawy, Determination of nifuroxazide in capsules by differential pulse polarography, Anal. Sci., 1998, 14, 607–608 CrossRef CAS.
- N. Y. Sreedhar, K. R. Samatha and P. R. K. Reddy, Electrochemical reduction behaviour and analysis of nifuroxazide in formulations and urine samples, Indian Drugs, 1999, 36, 509–512 CAS.
- G. R. Naidu, Electrochemical sensing of paracetamol and its simultaneous resolution in the presence of dopamine and folic acid at a multi-walled carbon nanotubes/poly (glycine) composite modified electrode, Anal. Methods, 2014, 6, 9459–9468 RSC.
- W. Buchberger, G. Niessner and R. Bakry, Determination of nifuroxazide with polarography and adsorptive stripping voltammetry at mercury and carbon paste electrodes, Fresenius' J. Anal. Chem., 1998, 362, 205–208 CrossRef CAS.
- A. Radi and M. El Ries, Determination of nifuroxazide in human serum by adsorptive stripping voltammetry, Anal. Sci., 1999, 15, 385–388 CrossRef CAS.
- J. Mozo, J. Carbajo, J. Sturm, L. Núñez-Vergara, P. Salgado and J. Squella, Determination of Nifuroxazide by Flow Injection Linear Adsorptive Stripping Voltammetry on a Screen-Printed Carbon Nanofiber Modified Electrode, Electroanalysis, 2012, 24, 676–682 CrossRef CAS.
- A. Radi, Voltammetric study of nifuroxazide at unmodified and Sephadex-modified carbon paste electrodes, Fresenius' J. Anal. Chem., 1999, 364, 590–594 CrossRef CAS.
- J. Squella Serrano, M. Letelier, L. Lindermeyer and L. Núñez Vergara, Redox behaviour of nifuroxazide: generation of the one-electron reduction product, Chem.-Biol. Interact., 1996, 99, 227–238 CrossRef PubMed.
- A. Radi, Voltammetric study of nifuroxazide at unmodified and Sephadex-modified carbon paste electrodes, Fresenius’ J. Anal. Chem., 1999, 364, 590–594 CrossRef CAS.
- A. Radi and M. El Ries, Determination of nifuroxazide in human serum by adsorptive stripping voltammetry, Anal. Sci., 1999, 15, 385–388 CrossRef CAS.
- S. Zayed and Y. J. B. Issa, Cathodic adsorptive stripping voltammetry of drotaverine hydrochloride and its determination in tablets and human urine by differential pulse voltammetry, Bioelectrochemistry, 2009, 75, 9–12 CrossRef CAS PubMed.
- J. Mozo, J. Carbajo, J. Sturm, L. Núñez-Vergara, P. Salgado and J. Squella, Determination of Nifuroxazide by Flow Injection Linear Adsorptive Stripping Voltammetry on a Screen-Printed Carbon Nanofiber Modified Electrode, Electroanalysis, 2012, 24, 676–682 CrossRef CAS.
- W. Buchberger, G. Niessner and R. Bakry, Determination of nifuroxazide with polarography and adsorptive stripping voltammetry at mercury and carbon paste electrodes, Fresenius’ J. Anal. Chem., 1998, 362, 205–208 CrossRef CAS.
- M. Elfiky, N. Salahuddin, A. Matsuda, M. Elfiky, N. Salahuddin and A. Matsuda, Mater. Sci. Eng., C, 2020, 110773 CrossRef CAS PubMed.
- A. Hassanein, N. Salahuddin, A. Matsuda, G. Kawamura and M. Elfiky, Fabrication of biosensor based on chitosan-ZnO/polypyrrole nanocomposite modified carbon paste electrode for electroanalytical application, Mater. Sci. Eng., C, 2017, 80, 494–501 CrossRef CAS PubMed.
- A. H. Naggar, G. A. Saleh, M. A. Omar, A. M. Haredy and S. Derayea, Square wave adsorptive anodic stripping voltammetric determination of antidiabetic drug linagliptin in pharmaceutical formulations and biological fluids using pencil graphite electrode, Anal. Sci., 2020, 36, 1031–1038 CrossRef CAS PubMed.
- L. Pontes, J. De Souza, A. Galembeck and C. De Melo, Gas sensor based on montmorillonite/polypyrrole composites prepared by in situ polymerization in aqueous medium, Sens. Actuators, B, 2013, 177, 1115–1121 CrossRef CAS.
- H. S. El-Desoky, I. M. Ismail and M. M. Ghoneim, Stripping voltammetry method for determination of manganese as complex with oxine at the carbon paste electrode with and without modification with montmorillonite clay, J. Solid State Electrochem., 2013, 17, 3153–3167 CrossRef CAS.
- H. S. El-Desoky and M. M. Ghoneim, Stripping voltammetric determination of silymarin in formulations and human blood utilizing bare and modified carbon paste electrodes, Talanta, 2011, 84, 223–234 CrossRef CAS PubMed.
- G. Kibria and S. Hossain, Electrical resistivity of compacted clay minerals, Environ. Geotech., 2017, 6, 18–25 CrossRef.
- Z. Yang, W. Wang, X. Tai and G. Wang, Preparation of modified montmorillonite with different quaternary ammonium salts and application in Pickering emulsion, New J. Chem., 2019, 43, 11543–11548 RSC.
- L. Zhang, B. Zhang, T. Wu, D. Sun and Y. Li, Adsorption behavior and mechanism of chlorophenols onto organoclays in aqueous solution, Colloids Surf., A, 2015, 484, 118–129 CrossRef CAS.
- J. L. Alves, P. Rosa and A. R. Morales, A comparative study of different routes for the modification of montmorillonite with ammonium and phosphonium salts, Appl. Clay Sci., 2016, 132, 475–484 CrossRef.
- L. Wang, S. Liu, T. Wang, D. Sun, L. Wang, S. Liu, T. Wang and D. Sun, Colloids Surf., A, 2011, 381, 41–47 CrossRef CAS.
- J.-J. Lin, I.-J. Cheng, R. Wang and R.-J. Lee, Tailoring basal spacings of montmorillonite by poly (oxyalkylene) diamine intercalation, Macromolecules, 2001, 34, 8832–8834 CrossRef CAS.
- Y. Wang, Y. Duan, Y. Liu and S. Du, Modification of montmorillonite with poly (oxypropylene) amine hydrochlorides: basal spacing, amount intercalated, and thermal stability, Clays Clay Miner., 2011, 59, 507–517 CrossRef CAS.
- L. Yuzhong, W. Xiaoqun, C. Yichi and D. Shanyi, Co-intercalation of montmorillonite by poly (oxypropylene) amine hydrochlorides with different chain length, Acta Chim. Sin., 2012, 70, 911–916 CrossRef.
- D. Yifeng, W. Xiaoqun, L. Yuzhong and D. Shanyi, Effect of montmorillonite modified by poly (oxypropylene) amines on curing process of epoxy nanocomposites, Acta Chim. Sin., 2012, 70, 1179–1186 CrossRef.
- L. Li, H. Zou, M. Liang and Y. Chen, Study on the effect of poly (oxypropylene) diamine modified organic montmorillonite on curing kinetics of epoxy nanocomposites, Thermochim. Acta, 2014, 597, 93–100 CrossRef CAS.
- N. Salahuddin, E. R. Kenawy and R. Abdeen, Polyoxypropylene–montmorillonite nanocomposites for drug-delivery vehicles: preparation and characterization, J. Appl. Polym. Sci., 2012, 125, E157–E166 CrossRef CAS.
- A. Hassanein, N. Salahuddin, A. Matsuda, T. Hattori and M. Elfiky, Fabrication of electrochemical sensor based on layered double hydroxide/polypyrrole/carbon paste for determination of an alpha-adrenergic blocking agent terazosin, Electroanalysis, 2018, 30, 459–465 CrossRef CAS.
- N. Salahuddin, M. Elfiky and A. Matsuda, Sensors and biosensors nanocomposites based on polymer/inorganic nanostructures, in Handbook of Polymer Nanocomposites for Industrial Applications, Elsevier, 2021, pp. 709–731 Search PubMed.
- K. Boukerma, J.-Y. Piquemal, M. M. Chehimi, M. Mravčáková, M. Omastová and P. Beaunier, Synthesis and interfacial properties of montmorillonite/polypyrrole nanocomposites, Polymer, 2006, 47, 569–576 CrossRef CAS.
- S. D. Ramôa, G. M. Barra, C. Merlini, W. H. Schreiner, S. Livi and B. G. Soares, Production of montmorillonite/polypyrrole nanocomposites through in situ oxidative polymerization of pyrrole: effect of anionic and cationic surfactants on structure and properties, Appl. Clay Sci., 2015, 104, 160–167 CrossRef.
- G. Contri, G. Barra, S. Ramoa, C. Merlini, L. Ecco, F. Souza and A. Spinelli, Epoxy coating based on montmorillonite-polypyrrole: electrical properties and prospective application on corrosion protection of steel, Prog. Org. Coat., 2018, 114, 201–207 CrossRef CAS.
- A. Olad, A. Rashidzadeh and M. Amini, Preparation of polypyrrole nanocomposites with organophilic and hydrophilic montmorillonite and investigation of their corrosion protection on iron, Adv. Polym. Technol., 2013, 32, 21337 CrossRef.
- Y. Han, Synthesis and characterization of montmorillonite/polypyrrole nanocomposite, Polym. Compos., 2009, 30, 66–69 CrossRef CAS.
- J. Cuppoletti, Nanocomposites and Polymers with Analytical Methods, BoD – Books on Demand, 2011 Search PubMed.
- M. Ayad, N. Salahuddin and M. Ali, Polyaniline–organoclay nanocomposites as curing agent for epoxy: preparation and characterization, Polym. Compos., 2009, 30, 467–473 CrossRef CAS.
- N. Salahuddin, S. Abo-El-Enein, A. Selim and O. S. El-Dien, Synthesis and characterization of polyurethane/organo-montmorillonite nanocomposites, Appl. Clay Sci., 2010, 47, 242–248 CrossRef CAS.
- D. Pospiech, B. Kretzschmar, M. Willeke, A. Leuteritz, D. Jehnichen, A. Janke and M. Omastová, Exfoliation behavior of montmorillonite modified by poly (oxyalkylene) s in polypropylene and the properties of the resulting nanocomposites, Polym. Eng. Sci., 2007, 47, 1262–1271 CrossRef CAS.
- A. M. dos Santos, C. Merlini, S. D. S. Ramôa and G. M. Barra, The Role of Inorganic Fillers in Electrostatic Discharge Composites, Polym. Compos., 2020, 41, 2003–2012 CrossRef CAS.
- K. Xu, K. Li, D. Tu, T. Zhong and C. Xie, Reinforcement on the mechanical-, thermal-, and water-resistance properties of the wood flour/chitosan/poly (vinyl chloride) composites by physical and chemical modification, J. Appl. Polym. Sci., 2014, 131, 40757 CrossRef.
- N. Salahuddin, E. R. Kenawy and R. Abdeen, Polyoxypropylene–montmorillonite nanocomposites for drug-delivery vehicles: preparation and characterization, J. Appl. Polym. Sci., 2012, 125, E157–E166 CrossRef CAS.
- A. Kassim, H. E. Mahmud and F. Adzmi, Polypyrrole–montmorillonite clay composites: an organic semiconductor, Mater. Sci. Semicond. Process., 2007, 10, 246–251 CrossRef CAS.
- H. Wu, G. Li and J. Gao, Template synthesis flake polypyrrole and montmorillonite/polypyrrole composites by vapor–liquor phase polymerization, Acta Mater. Compositae Sin., 2009, 26, 86–92 CAS.
- J. Liu and M. Wan, Synthesis, characterization and electrical properties of microtubules of polypyrrole synthesized by a template-free method, J. Mater. Chem., 2001, 11, 404–407 RSC.
- M. Elfiky, R. Kumar and A. Beltagi, Anthropogenic greenhouse CO2 gas sensor based on glassy carbon modified with organoclay/polypyrrole-alginate nanocomposites in brackish water and seawater, J. Electroanal. Chem., 2022, 926, 116926 CrossRef CAS.
- K. R. Srinivasan and H. S. Fogler, Use of inorgano-organo-clays in the removal of priority pollutants from industrial wastewaters: adsorption of benzo (a) pyrene and chlorophenols from aqueous solutions, Clays Clay Miner., 1990, 38, 287–293 CrossRef CAS.
- N. L. Dias Filho, D. R. do Carmo, N. L. Dias Filho and D. R. do Carmo, Talanta, 2006, 68, 919–927 CrossRef CAS PubMed.
- T. Rao, Validation of analytical methods, Calibration and Validation of Analytical Methods – A Sampling of Current Approaches, 2018, pp. 131–141 Search PubMed.
- G. D. Christian, Sequential injection analysis for electrochemical measurements and process analysis, Analyst, 2014, 119, 2309–2314 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ra06160k |
| This journal is © The Royal Society of Chemistry 2023 |