
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOne-pot synthesis of liquid photocrosslinkable poly(L-lactide) with terminal triacrylate
Chung-Fu Yu a,
Syang-Peng Rwei*ab and
Yao-Chi Shuc
a,
Syang-Peng Rwei*ab and
Yao-Chi Shuc
aInstitute of Organic and Polymeric Materials, Research, National Taipei University of Technology, 1, Sec. 3, Zhongxiao E. Rd., Taipei 10608, Taiwan, Republic of China. E-mail: f10714@ntut.edu.tw
bResearch and Development Center for Smart Textile Technology, Taiwan
cGraduate School of Fabric Technology Management, Lee-Ming Institute of Technology, Taiwan
First published on 16th January 2023
Abstract
We synthesized a poly(L-lactide)–pentaerythritol triacrylate (PETA) polymer modified with acrylic trifunctional groups using a one-pot method based on ring-opening polymerization of L-lactide and PETA. We calculated the molecular weight and structure of PLA–PETA using gel permeation chromatography (GPC) and nuclear magnetic resonance (NMR) (1H, 13C, heteronuclear multiple bond correlation [HMBC]) spectroscopy. Photocrosslinking PLA–PETA using the Omnirad 1173 photoinitiator yielded a transparent sample with 91% crosslinkage. The crosslinked sample was analyzed using differential scanning calorimetry (DSC), thermogravimetric analysis (TGA), and thermomechanical analysis (TMA) to determine its thermal properties and thermal expansion coefficient. In vitro cell toxicity tests showed an average cell viability >90%, indicating that the PLA–PETA polymer had good biocompatibility with cells after photocrosslinking.
1. Introduction
Polylactic acid (PLA) is a widely used aliphatic polyester material with excellent mechanical properties, high biocompatibility, and good biodegradability. Carothers first synthesized low-molecular-weight PLA in 1932. In 1954, the DuPont company obtained the patent for producing high-molecular-weight polymeric PLA. Since PLA can be produced using renewable resources, it is considered to be environmentally friendly. It can be mixed into various resins using numerous techniques to produce materials that can be used in industrial packaging, agriculture, and medical treatments.1–3PLA has the widest biomedical application among all synthetic aliphatic polyesters, and the US Food and Drug Administration (FDA) approved it for contact with human cells in 1970. In 1974, the DuPont company introduced PLA sutures. Since then, much research and development has been conducted on using methods such as chemical modification, copolymerization, and blending to produce forms of PLA that can be used in biomedical applications, such as bone fixation, stent screw implanting, tissue engineering, and drug delivery.3–8
PLA application is usually limited by its shortcomings of brittleness, low toughness, slow crystallization, poor heat resistance, insufficient mechanical strength, and poor shape stability at high temperatures. Scientific efforts have been made to improve the crystallinity, thermal stability, and mechanical properties of PLA using methods such as chemical modification, copolymerization, or hybrid processing.9–14 A commonly used chemical method for polymer modification involves incorporating appropriate end groups. End groups are introduced into the PLA backbone through polymerization or the post-polymerization end group modification of compounds that already have the desired functionality while retaining the properties of the PLA matrix. Although molecules can be polymerized with desired end groups or introduce various chain-end molecules in polymerization followed by post-polymerization functionalization, the reactions may not be complete. In addition, the end-group functionalization of PLA generally requires multiple reaction steps.15–22
Crosslinking allows PLA chains to chemically bond into a network structure, thereby improving the toughness and thermal properties of the polymer and changing its solubility and gas permeability. Unfortunately, crosslinking reduces degradability; however, the degradation process of PLA can be regulated by controlling process conditions and parameters. The crosslinking methods of PLA consist of chemical, ionizing radiation, and ultraviolet (UV)/visible light crosslinking. The chemical crosslinking of PLA usually requires using crosslinkers or initiators, such as polyisocyanates, polyfunctional anhydrides, and peroxides (for generation of free radicals), to trigger the formation of crosslinked structures.23 Gamma rays or electron beams have been used for crosslinking and grafting polymers, while the structure and length of the PLA chain and radiation dose affect the crosslinking density. Polymer crosslinking by radiation does not require functionalization, considering free radicals can be generated directly in the polymer chains. However, PLA crosslinked by radiation in the absence of a crosslinker will undergo molecular-chain breaking.24,25
Photocrosslinking has biomedical applications as it can be performed under low-viscosity, solvent-free, and low-temperature conditions. The crosslinking of PLA involves double-bond functionalization at chain ends to form linear, branched, or star copolymers, followed by UV irradiation to initiate radical polymerization.16,18,21 Three-dimensional chemical bonding between PLA chains can be formed via photocrosslinkable groups, which enhance the hydrolysis resistance, thermal stability, and mechanical properties of PLA.25–31
Pentaerythritol triacrylate (PETA), a monomer containing side hydroxyl and three acrylic functional groups, has high reactivity, high crosslinking density, high hardness, strong water resistance, and strong chemical resistance. It is mainly used in free radical polymerization and serves as a reactive diluent of polyfunctional acrylate widely used in UV curing. Currently, photocrosslinked PLA with a network structure can be formed in 2–3 steps. Here, we designed a modified-PLA liquid polymer with three acrylic functional groups, where L-lactide (LA) and pentaerythritol triacrylate (PETA) were used as the reaction monomers. To obtain PLA–PETA polymers with low molecular weight, we controlled the molar ratio of the reaction monomer (LA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :PETA) and performed ring-opening polymerization (ROP) using a one-pot method. This study aimed to develop a photocrosslinkable and biodegradable material based on low-molecular-weight PLA and evaluate its chemical and thermal properties and biocompatibility.28,31
:PETA) and performed ring-opening polymerization (ROP) using a one-pot method. This study aimed to develop a photocrosslinkable and biodegradable material based on low-molecular-weight PLA and evaluate its chemical and thermal properties and biocompatibility.28,31
2. Experimental
2.1 Materials
L-Lactide 99.5% (Green Square Materials Inc), pentaerythritol tri and tetraacrylate (Photomer 4335), tin(II) 2-ethylhexanoate 96% (Sn(Oct)2,Alfa Aesar®), 4-methoxyphenol (SHORA), 2-hydroxy-2-methyl-1-phenylpropanone (I.G.M Omnirad 1173), PBS Tablets (MP Biomedicals), tetrahydrofuran (TEDIA), ethyl acetate (TEDIA), acetone (TEDIA), high-purity grade silica gel, pore size 60 Å, 70–230 mesh (AACASH Exports).2.2 Synthesis of PLA–PETA
In this study, we used pentaerythritol tri- and tetraacrylate (Photomer 4335), which contain three and four acrylic functional groups, respectively, to prevent impurities interfering with the polymerization reaction. A glass cylinder was filled with silica gel and mobile-phase 70% ethyl acetate/30% N-hexane, to which we added the Photomer 4335, allowing us to separate out the PETA.The monomer L-lactide and PETA in molar ratios of 3:1 (0.15:0.05), 5:1 (0.25:0.05), 7:1 (0.35:0.05) were respectively placed in 250 mL three-neck flasks and dried under vacuum for 1 h. To the flasks filled with dry nitrogen gas were added Sn(Oct)2 (0.05 wt% of lactide) and 200 ppm of 4-methoxyphenol, and the mixtures were heated at 130 °C in an oil bath for 12 h with stirring (Scheme 1).
 | ||
| Scheme 1 Synthesis of PLA–PETA. | ||
At the end of the reaction, the reactors were cooled to room temperature. The resulting polymers were dissolved in acetone and precipitated in an excess of deionized water while being agitated. The precipitated materials were washed with deionized water and dried under reduced pressure. These polymers were denoted as PLA–PETA, where PLA–PETA is the number-average molecular weight determined by 1H nuclear magnetic resonance (NMR) spectroscopy and gel permeation chromatography (GPC, Fig. 1, see Section 2.4 below and Table 1).
 | ||
| Fig. 1 GPC diagrams of PLA–PETA copolymers, mobile phase: THF, 1 mL min−1, 35 °C. | ||
| Polymer | LA:PETAa |
MnMNRb (g mol−1) | MwGPCc (g mol−1) | MnGPCc (g mol−1) | Mw/MnGPCc | State of matter |
|---|---|---|---|---|---|---|
| a LA:PETA = mol:mol.b Mn = ((HArea8/HArea9) × 72) +298 + 72.c Molecular weights determined by GPC calibrated with PS standards. |
||||||
| PLA–PETA-3:1 |
0.15:0.05 |
519 | 985 | 684 | 1.44 | Liquid |
| PLA–PETA-5:1 |
0.25:0.05 |
801 | 1907 | 1544 | 1.23 | Liquid |
| PLA–PETA-7:1 |
0.35:0.05 |
1371 | 3644 | 2731 | 1.33 | Solid |
We found that the polymer with a 7:1 molar ratio was a solid, so it did not meet the requirements of being able to set in a liquid state. Further, the LA chain segment had a 3:1 molar ratio and its chain length was too short. We therefore set the photopolymerization conditions for PLA–PETA assuming a molar ratio of 5:1 and analyzed the properties of the PLA–PETA based on this molar ratio.
2.3 Photopolymerization conditions21,32
The photocrosslinking process was carried out in a UV cabinet (Philips, HILIPS HPA 400/30S, intensity 10–13 mW cm−2) using 0.5 wt% Omnirad 1173 as a photoinitiator. The following conditions were used: time of irradiation – maximum 5 min, air atmosphere and room temperature. We obtained the transparent PLA–PETA polymer liquid. This PLA–PETA was subjected to photocrosslinking reaction to become transparent solid polymer indicated as PLA–PETA–PO (Fig. 2). | ||
| Fig. 2 Photocrosslinking of PLA–PETA–PO. | ||
2.4 Polymer characterization
| Gel fraction (%) = (WB/WA) × 100% |

3. Results
3.1 1H NMR spectroscopy characterization
Fig. 3 shows the NMR spectrum of PLA–PETA. In the 1H NMR spectrum, the –CH absorption peaks of PLA can be assigned as δH4 = 5.10–5.17 ppm [1H, CH], the methyl-CH3 absorption peaks of PLA can be assigned as δH8, δH9 = 1.45–1.56 ppm [3H, CH3], the absorption peaks of the CH3 at one end of the PLA–PETA polymer can be assigned as δH5 = 4.32–4.34 ppm [1H, –CH], the CH2 absorption peak of PETA can be assigned as δH6, δH7 = 4.22–4.27 ppm [2H, CH2], and the vinyl hydrogen (–CH = CH2) absorption peak of PETA can be assigned as δH1, δH2, δH3 = 5.84–6.42 ppm [3H, CH = CH2]. The molecular weight of lactide monomer (C6H8O4) is 144. After the ring-opening polymerization reaction of lactide monomers, a polymer with a repeat unit of (COCHCH3O)n is formed. The molecular weight of this repeat unit is calculated to be 72. We calculated the number-average molecular weight (Mn) based on that of the repeated unit CH3 (δH8) in the PLA chain segment and the peak integrated area of the CH3 (δH9) at one end of the PLA–PETA polymer (Table 1). The Mn of the PLA–PETA copolymer measured via gel permeation chromatography (GPC) was higher than that calculated via 1H NMR spectroscopy because the hydrodynamic volume of standard polystyrene was lower than that of the aliphatic polyester with the same molar mass, so the measurement data differed.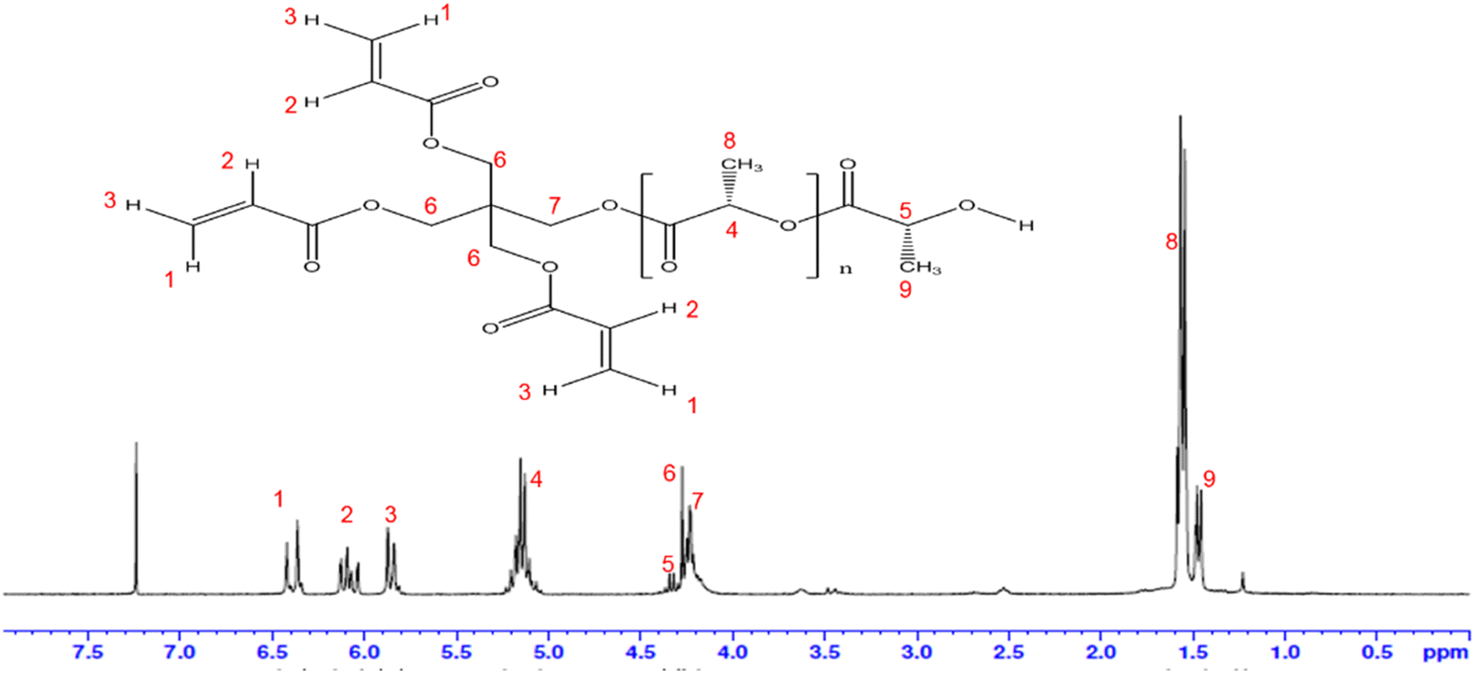 | ||
| Fig. 3 1H NMR (300 MHz, CDCl3, 290 K) spectrum of PLA–PETA. | ||
The integral area of δH8Area: CH3.
The integral area of δH9Area: CH3.
Mn = ((δH8Area/δH9Area) × 72) + 298 + 72 (molecular weight of PETA: 298).
3.2 Spectral characterization of 1H–13C HMBC
A liquid-cooled superconducting NMR system (Bruker Avance III HD-600 MHz) was used to confirm the carbon–hydrogen linkages in the synthesis structure of PLA–PETA via 1H–13C heteronuclear multiple bond correlation (HMBC) spectroscopy. In the 1H–13C HMBC spectrum (Fig. 4), the carbon base unit, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (δCa = 169 ppm), of the PLA chain segment has a distal correlation with the CH2 (δH7 = 4.22–4.24 ppm) of the PETA segment. This suggests that when PETA is mixed with the catalyst Sn(Oct)2, the lactide uses the hydroxyl group of PETA as the initiator for ROP, forming a short-chain polymer containing three acrylic functional groups.34
O (δCa = 169 ppm), of the PLA chain segment has a distal correlation with the CH2 (δH7 = 4.22–4.24 ppm) of the PETA segment. This suggests that when PETA is mixed with the catalyst Sn(Oct)2, the lactide uses the hydroxyl group of PETA as the initiator for ROP, forming a short-chain polymer containing three acrylic functional groups.34
 | ||
| Fig. 4 1H–13C HMBC NMR spectrum of PLA–PETA. NMR correlations are due to the covalent bonding between PLA and PETA. | ||
3.3 Gel formation of PLA–PETA
According to the infrared spectra shown in Fig. 5, the peak at 1635 cm−1 is characteristic of the CC double bond in PLA–PETA. There were two obvious wavelet peaks before the crosslinking was done, and these disappeared afterward. This suggests that when the active radicals react with the monomer double bonds, the molecular chains grow and become highly crosslinked, forming a cross-network polymer. Due to the crosslinking and vitrifying relationship of the polymers, some free radicals and unreacted double bonds were restricted from participating in the crosslinking polymerization reaction, resulting in failure to reach 100% conversion. The gel content test showed that the gel content was 91% after crosslinking.32,34
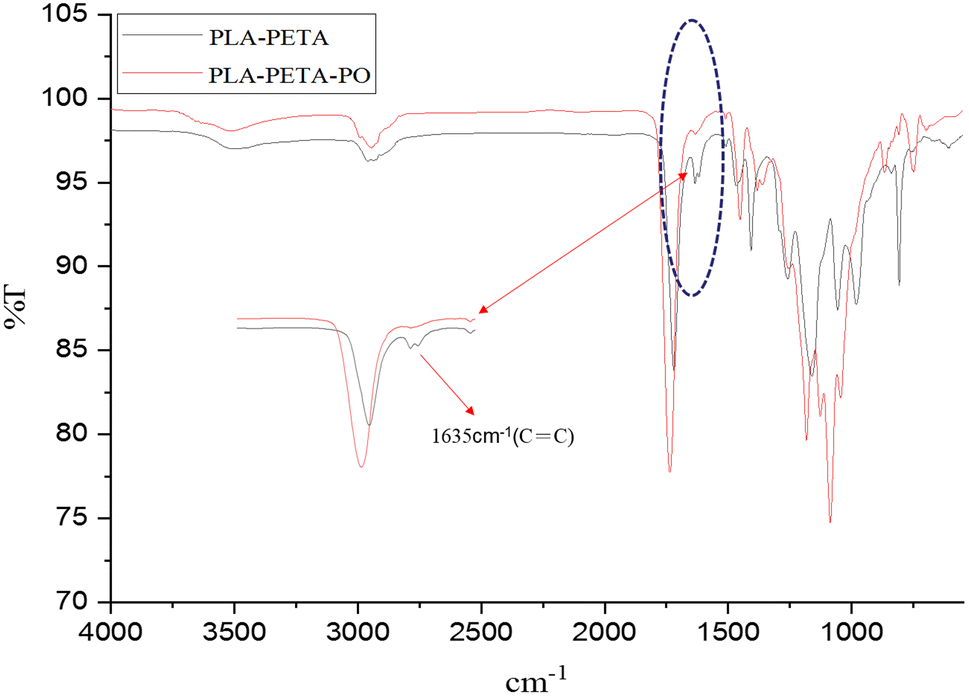 | ||
| Fig. 5 FTIR spectra of PLA–PETA and PLA–PETA–PO, which PLA–PETA–PO is the polymer solid after photocrosslinking reaction of PLA–PETA polymer liquid. | ||
3.4 Thermal analysis
Table 2 shows the thermal properties of PLA–PETA, measured via DSC, TGA, and TMA. The polymer chain structure, molecular weight and degree of crystallization affect the glass transition temperature (Tg) and the thermal degradation temperature. Before crosslinking, PLA–PETA is a short-chain polymer, and after photocrosslinking it forms a transparent network structure. The Tg of PLA–PETA after crosslinking is 47.7 °C, 55.5 °C higher than before crosslinking (−7.8 °C; Fig. 6). The crosslinking of PLA–PETA led to a high crosslinking density of the side acrylate in the copolymer, with the resulting high-density crosslinked network structure preventing the PLA chains from readily relaxing and rotating. Therefore, the Tg still reached 47.7 °C when the molecular chains did not form a high molecular weight structure.| Polymer | Tg,DSCa (°C) | Tg,DSCb (°C) | Tg,TMA (°C) | T5%c (°C) | Tmax1d | Tmax2d | CTE (μm m−1 °C−1) |
|---|---|---|---|---|---|---|---|
| a Non-crosslinked PLA–PETA.b Crosslinked PLA–PETA–PO.c Decomposition temperature of polymers at a weight loss of 5%.d The maximum decomposition temperature of the polymer. | |||||||
| PLA–PETA–PO | −7.8 | 47.7 | 45.8 | 268 | 297 | 452 | 153.6 |
 | ||
| Fig. 6 DSC thermograms of (A) non-crosslinked PLA–PETA and (B) crosslinked PLA–PETA–PO. | ||
It can be seen from Fig. 7 that PLA has only a single-stage thermal degradation curve, with a 5% weight loss temperature (T5%) of 280.78 °C, and its highest thermal decomposition temperature is 299.66 °C. PLA–PETA–PO has two thermal degradation stages: that of the PLA chain segment and that of the PETA chain segment. Specifically, the first stage is the decomposition of the oligomer ester bond in the PLA from 268.53 °C to 298.67 °C. The second stage is the decomposition of the PETA chain segment, starting from the turning point at 400 °C and ending at the maximum thermal decomposition temperature, 452.28 °C. In our experiment, at 500 °C all materials had almost entirely decomposed, leaving only ash.
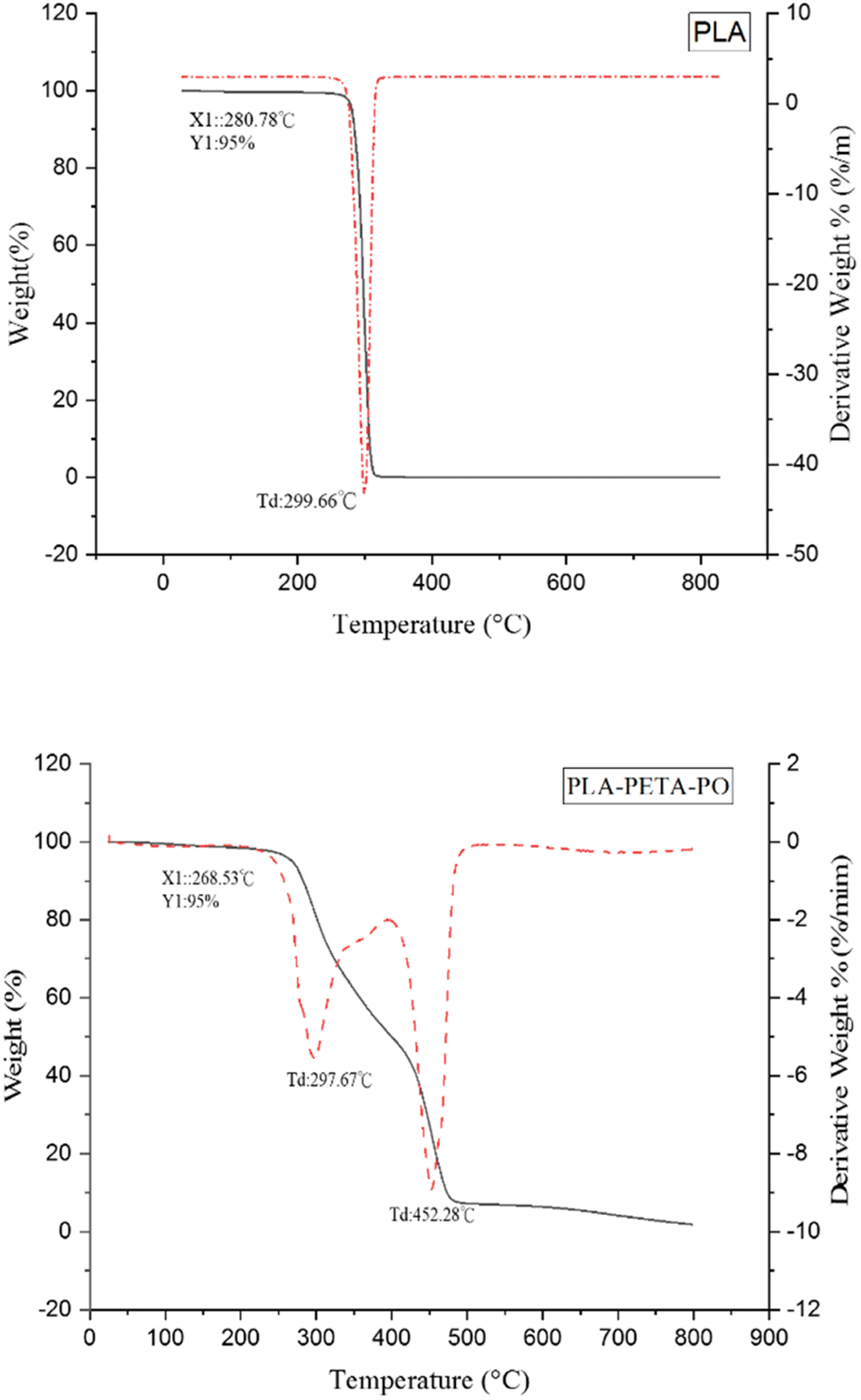 | ||
| Fig. 7 TGA thermograms of PLA and PLA–PETA–PO. | ||
Fig. 8 shows the coefficient of thermal expansion (CTE) and the Tg of the TMA test samples. The CTE indicates the degree of expansion and contraction of materials under temperature changes. According to the MatWeb materials properties datasheet for the PLA biopolymer, the CTE of PLA is 101–120 (μm m−1 °C−1).37 In this study, the CTE of PLA–PETA was 153.6 (μm m−1 °C−1) as the high crosslinking density of the side acrylate in PLA–PETA made the movement of molecular chains difficult. However, the PLA chains that did not react could not form a crosslinked structure so were soft and easier to move. Moreover, the free volume in the network structure was higher than that in the unmodified PLA. Therefore, the CTE increased by 52.6–33.6 (μm m−1 °C−1). The Tg point on the line tangent to the turning point of the thermal expansion curve was 45.8 °C, which was relatively close to that measured via DSC.
 | ||
| Fig. 8 TMA thermograms of PLA–PETA–PO. | ||
3.5 Hydrolytic degradation and cytotoxicity testing
The hydrolysis of PLA is an autocatalytic reaction. In the presence of water, the ester bond segment of PLA is hydrolytically broken to generate oligomers. The carboxyl terminus of the oligomers can catalyze the cleavage of the ester bond to release H+ ions. The ester bond is then broken into a carboxylic acid and alcohol via the chemical hydrolysis of H+ ions.38 According to Fig. 9, the PLA–PETA–PO sample lost 26–28% of its weight after 120 days of hydrolysis, indicating that the PLA chain segment in the crosslinked product was hydrolyzed by water infiltration and diffusion, which then produced water-soluble oligomer products. After 120 days, the rate of weight loss decreased, leaving only the long broken chain segment of the PLA and the PLA chain segment that was coiled within the crosslinked polymer molecular cluster. After 280 days of continuous degradation, the weight loss was 45% (Fig. 10). | ||
| Fig. 9 Mass loss of crosslinked PLA–PETA–PO with degradation time. | ||
 | ||
| Fig. 10 Physical changes in PLA–PETA–PO resulting from hydrolytic degradation after 280 days. | ||
The decomposition products of the polymers and the residues of unreactive groups and crosslinking agents all exhibited good biocompatibility, i.e., they exhibited no toxicity toward cells and had almost no impact on the phenotypic characteristics of cells.39 According to the ISO 10993-5 specification in vitro cytotoxicity testing, the average cell viability of cells treated with PLA–PETA after crosslinking was >90% (Fig. 11), which shows that the polymer exhibits good biocompatibility and can be used in the field of biomedical materials.
 | ||
| Fig. 11 Bar graph of PLA–PETA–PO cytotoxicity testing, where statistical significance was analyzed using Tukey's test. | ||
4. Conclusions
Photocrosslinking by UV or visible light has been extensively explored in the biomedical field.24,25,40 In this study, we modified PLA functional groups with L-lactide and PETA via one-pot ring-opening polymerization. NMR and HMBC spectroscopy confirmed that photocrosslinked, low-molecular-weight PLA–PETA polymers with a network structure are obtainable via one-step synthesis.After the UV photocrosslinking of PLA–PETA, the degree of gelation rose to 91%; however, the thermal cracking temperature did not increase because the PLA chains did not form a high crosslinking density network in the acrylate chains to increase the heat resistance. After crosslinking, the Tg increased by 56.9 °C owing to an increase in crosslinking density, and the thermal expansion coefficient increased by 52.6–33.6 (μm m−1 °C−1).
A degradation experiment was performed at a pH of 7.4 in PBS buffer, with the results showing that it takes 280 days to degrade 45% of the photocrosslinked PLA–PETA. Given that the crosslinking of PLA–PETA molecular chains affected the degradability of the material, it is necessary to further investigate these influencing factors in the future. However, cytotoxicity tests proved that the photocrosslinked polymer showed good biocompatibility.
Conflicts of interest
The authors declare that there is no conflict of interest regarding the publication of this article.References
- K. Madhavan Nampoothiri, N. R. Nair and R. P. John, Bioresour. Technol., 2010, 101, 8493–8501 CrossRef CAS PubMed.
- K. Stefaniak and A. Masek, Materials, 2021, 14 Search PubMed.
- M. S. Singhvi, S. S. Zinjarde and D. V. Gokhale, J. Appl. Microbiol., 2019, 127, 1612–1626 CrossRef CAS PubMed.
- B. Tyler, D. Gullotti, A. Mangraviti, T. Utsuki and H. Brem, Adv. Drug Delivery Rev., 2016, 107, 163–175 CrossRef CAS PubMed.
- A. J. Lasprilla, G. A. Martinez, B. H. Lunelli, A. L. Jardini and R. M. Filho, Biotechnol. Adv., 2012, 30, 321–328 CrossRef CAS PubMed.
- M. S. Lopes, A. L. Jardini and R. M. Filho, Procedia Eng., 2012, 42, 1402–1413 CrossRef.
- E. Capuana, F. Lopresti, M. Ceraulo and V. La Carrubba, Polymers, 2022, 14 Search PubMed.
- I. Manavitehrani, A. Fathi, H. Badr, S. Daly, A. Negahi Shirazi and F. Dehghani, Polymers, 2016, 8(1), 20 CrossRef PubMed.
- S. Farah, D. G. Anderson and R. Langer, Adv. Drug Delivery Rev., 2016, 107, 367–392 CrossRef CAS PubMed.
- M. Nofar, D. Sacligil, P. J. Carreau, M. R. Kamal and M. C. Heuzey, Int. J. Biol. Macromol., 2019, 125, 307–360 CrossRef CAS PubMed.
- F.-L. Jin, R.-R. Hu and S.-J. Park, Composites, Part B, 2019, 164, 287–296 CrossRef CAS.
- X. Zhao, H. Hu, X. Wang, X. Yu, W. Zhou and S. Peng, RSC Adv., 2020, 10, 13316–13368 RSC.
- S. Saeidlou, M. A. Huneault, H. Li and C. B. Park, Prog. Polym. Sci., 2012, 37, 1657–1677 CrossRef CAS.
- O. Mysiukiewicz, M. Barczewski, K. Skorczewska and D. Matykiewicz, Polymers, 2020, 12, 1333 CrossRef CAS PubMed.
- B. Kost, M. Basko, M. Bednarek, M. Socka, B. Kopka, G. Łapienis, T. Biela, P. Kubisa and M. Brzeziński, Prog. Polym. Sci., 2022, 130 Search PubMed.
- X. Zhao, J. Li, J. Liu, W. Zhou and S. Peng, Int. J. Biol. Macromol., 2021, 193, 874–892 CrossRef CAS PubMed.
- K. J. Jem and B. Tan, Adv. Ind. Eng. Polym. Res., 2020, 3, 60–70 Search PubMed.
- S. Corneillie and M. Smet, Polym. Chem., 2015, 6, 850–867 RSC.
- A. Michalski, M. Brzezinski, G. Lapienis and T. Biela, Prog. Polym. Sci., 2019, 89, 159–212 CrossRef CAS.
- L. Phong, E. S. C. Han, S. Xiong, J. Pan and S. C. J. Loo, Polym. Degrad. Stab., 2010, 95, 771–777 CrossRef CAS.
- S. Moeinzadeh, S. N. Khorasani, J. Ma, X. He and E. Jabbari, Polymer, 2011, 52, 3887–3896 CrossRef CAS PubMed.
- T. Maharana, S. Pattanaik, A. Routaray, N. Nath and A. K. Sutar, React. Funct. Polym., 2015, 93, 47–67 CrossRef CAS.
- C. Mangeon, E. Renard, F. Thevenieau and V. Langlois, Mater. Sci. Eng., C, 2017, 80, 760–770 CrossRef CAS PubMed.
- R. Parhi, Adv. Pharm. Bull., 2017, 7, 515–530 CrossRef CAS PubMed.
- M. Bednarek, K. Borska and P. Kubisa, Molecules, 2020, 25(21), 4919 CrossRef CAS PubMed.
- R. D. O'Rorke, O. Pokholenko, F. Gao, T. Cheng, A. Shah, V. Mogal and T. W. Steele, Biomacromolecules, 2017, 18, 674–682 CrossRef PubMed.
- J. Song, J. Xu, S. Pispas and G. Zhang, RSC Adv., 2015, 5, 38243–38247 RSC.
- I. V. Averianov, V. A. Korzhikov-Vlakh, Y. E. Moskalenko, V. E. Smirnova and T. B. Tennikova, Mendeleev Commun., 2017, 27, 574–576 CrossRef CAS.
- M. N. Karim, S. Afroj, M. Rigout, S. G. Yeates and C. Carr, J. Mater. Sci., 2015, 50, 4576–4585 CrossRef CAS.
- J. M. C. Santos, D. S. Marques, P. Alves, T. R. Correia, I. J. Correia, C. M. S. G. Baptista and P. Ferreira, React. Funct. Polym., 2015, 94, 43–54 CrossRef CAS.
- J. Wang, J. Li, X. Wang, Q. Cheng, Y. Weng and J. Ren, React. Funct. Polym., 2020, 155, 104695 CrossRef CAS.
- H. Kaczmarek and I. Vukovic-Kwiatkowska, eXPRESS Polym. Lett., 2012, 6, 78–94 CrossRef CAS.
- J. Wang, L. Zheng, C. Li, W. Zhu, D. Zhang, G. Guan and Y. Xiao, J. Appl. Polym. Sci., 2014, 131 Search PubMed.
- A. J. Ro, S. J. Huang and R. A. Weiss, Polymer, 2008, 49, 422–431 CrossRef CAS.
- K. Dawidziuk, H. Simmons, M. Kontopoulou and J. S. Parent, Polymer, 2018, 158, 254–261 CrossRef CAS.
- H. Simmons and M. Kontopoulou, Polym. Degrad. Stab., 2018, 158, 228–237 CrossRef CAS.
- Overview of materials for Polylactic Acid (PLA) Biopolymer, accessed September 2022, https://www.matweb.com/search/DataSheet.aspx?MatGUID=ab96a4c0655c4018a8785ac4031b9278).
- S. Teixeira, K. M. Eblagon, F. Miranda, M. F. R. Pereira and J. L. Figueiredo, C, 2021, 7(2), 42 CAS.
- S. T. Sikhosana, T. P. Gumede, N. J. Malebo and A. O. Ogundeji, eXPRESS Polym. Lett., 2021, 15, 568–580 CrossRef CAS.
- M. Santos, T. Cernadas, P. Martins, S. P. Miguel, I. J. Correia, P. Alves and P. Ferreira, React. Funct. Polym., 2021, 158 Search PubMed.
| This journal is © The Royal Society of Chemistry 2023 |