
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExploring fluoride effects in sterically enhanced cobalt ethylene polymerisation catalysts; a combined experimental and DFT study†
Zilong Li a,
Yanping Ma*ab,
Tian Liua,
Qiuyue Zhanga,
Gregory A. Solan*b,
Tongling Lianga and
Wen-Hua Sun*ac
a,
Yanping Ma*ab,
Tian Liua,
Qiuyue Zhanga,
Gregory A. Solan*b,
Tongling Lianga and
Wen-Hua Sun*ac
aKey Laboratory of Engineering Plastics and Beijing National Laboratory for Molecular Sciences, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, China. E-mail: myanping@iccas.ac.cn; whsun@iccas.ac.cn
bDepartment of Chemistry, University of Leicester, University Road, Leicester LE1 7RH, UK. E-mail: gas8@leicester.ac.uk
cState Key Laboratory for Oxo Synthesis and Selective Oxidation, Lanzhou Institute of Chemical Physics, Chinese Academy of Sciences, Lanzhou 730000, China
First published on 19th December 2022
Abstract
The fluoro-substituted 2,6-bis(arylimino)pyridine dichlorocobalt complexes, [2-{CMeN(2,6-(Ph2CH)2-3,4-F2C6H)}-6-(CMeNAr)C5H3N]CoCl2 (Ar = 2,6-Me2C6H3 Co1, 2,6-Et2C6H3 Co2, 2,6-iPr2C6H3 Co3, 2,4,6-Me3C6H2 Co4, 2,6-Et-4-MeC6H2 Co5), were synthesized in good yield from the corresponding unsymmetrical N,N,N′-ligands, L1–L5. Besides characterization of Co1–Co5 by FT-IR spectroscopy, 19F NMR spectroscopy and elemental analysis, the molecular structures of Co2 and Co5 were also determined highlighting the unsymmetrical nature of the terdentate ligand and the pseudo-square pyramidal geometry about the metal center. When either MAO or MMAO were employed as activators, Co1–Co5 were able to achieve a wide range of catalytic activities for ethylene polymerisation. Co5/MAO exhibited the highest activity of the study at 60 °C (7.6 × 106 g PE mol−1 (Co) h−1) which decreased to 3.3 × 106 g PE mol−1 (Co) h−1 at 80 °C. In addition, it was found that the polymerisation activity increased as the steric hindrance imparted by the ortho groups was enhanced (for MMAO: Co3 > Co5 > Co2 > Co1 > Co4), a finding that was supported by DFT calculations. Furthermore, it was shown that particularly high molecular weight polyethylene could be generated (up to 483.8 kg mol−1) when using Co5/MMAO at 30 °C, while narrow dispersities (Mw/Mn range: 1.8–4.7) and high linearity (Tm > 131.4 °C) were a feature of all polymers produced. By comparison of Co3 with its non-fluorinated analogue using experimental data and DFT calculations, the substitution of fluorides at the meta- and para-positions was demonstrated to boost catalytic activity and improve thermal stability.
Introduction
Bis(imino)pyridine-cobalt and -iron complexes have been thoroughly investigated since the late 1990s, due in large part to their ability to promote the polymerisation/oligomerisation of ethylene with high efficiency.1–3 Against this backdrop, a wide variety of structural variations have been developed that can influence the performance of these late transition metal catalysts and the resulting polymer properties.4–13 With particular regard to the bis(arylimino)pyridine ligand framework, a good proportion of the developments have focused on systematic changes to the N-aryl substituents, with the result that a plethora of different substitution patterns for the N-aryl groups now exist.14–17 Elsewhere, key advances have seen the fusion of cycloalkyl groups to the central pyridine unit, variation of groups on the imine-carbon, and the integration of this N,N,N-ligand into a binucleating framework.18–21In terms of the N-aryl substitution pattern, the introduction of sterically hindered groups or electron donating/withdrawing groups to the ortho-positions of the N-aryl group have proved pivotal in controlling catalytic activity, thermal stability and for the polyethylene generated, molecular weight and various microstructural properties. Likewise, the nature of the substituent on the para-position can also be influential on the catalyst performance and polymer properties. For example, Wu's group22 explored the use of bis(arylimino)pyridine-iron catalysts appended with N-2-R1-4-R2-6-sec-phenethylphenyl groups and found that the nature of both the R1 and R2 substituents to be critical to the thermostability of the catalyst and the molecular weight of the polymer.
Over the years, our group has synthesized an assortment of bis(imino)pyridine-iron and -cobalt complexes appended with sterically hindered benzhydryl (CHPh2) groups to the ortho- and/or para-positions of the N-aryl groups (e.g. A–G, Chart 1).5,14,15,23–29 For example, for symmetrical A, bearing benzhydryl groups at the 2- and 6-positions, both the iron and cobalt species were inactive for ethylene polymerisation. It was proposed that the huge steric hindrance imparted on both sides of the metal hinders the coordination of ethylene, which makes the polymer chain difficult to propagate. On the other hand, unsymmetrical C containing benzhydryl groups at the 2- and 4-positions of just one N-aryl group, is highly active but the molecular weight of polyethylene is lowered.27,28 For its 2,6-substituted comparator D, high thermal stability and high ethylene polymerisation activity (22.4 × 106 g PE mol−1 (Fe) h−1) are displayed and with the added benefit of no oligomeric fractions.24 When the para-methyl substituent in D is substituted with a chloride (E), the thermal stability is slightly lower, but the molecular weight of the polyethylene increases.5
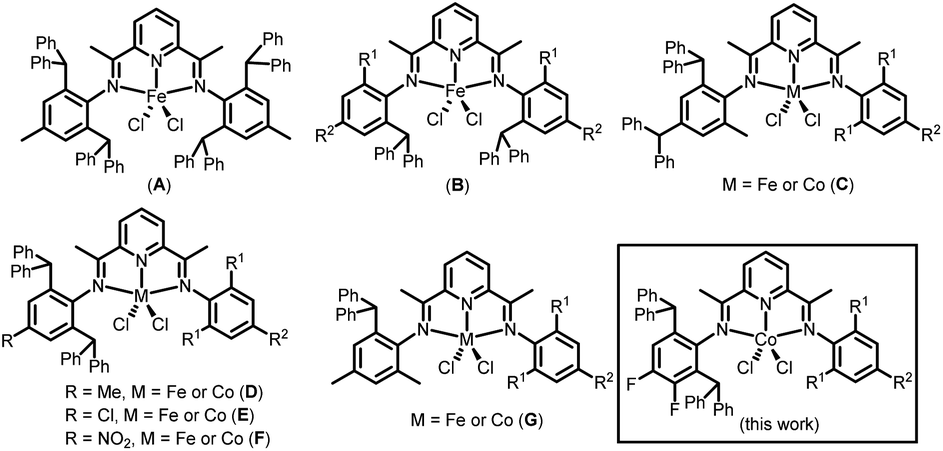 | ||
| Chart 1 Developments in benzhydryl-containing bis(imino)pyridine-iron and cobalt complexes; R1 or R2 = H or alkyl. | ||
In this article we set out to develop the 2,6-dibenzhydryl-substituted class of unsymmetrical bis(imino)pyridine-cobalt(II) complex shown for D–F (Chart 1). In particular, we were interested in exploring the effect of introducing electron withdrawing fluoride groups at the para- and meta-positions groups of the CHPh2-substituted N-aryl group (Chart 1) on catalyst activity, thermal stability and polymer properties. To allow some fine-tuning, the R1/R2 groups on the second N-aryl group are systematically varied (viz. Me/H, Et/H, i-Pr/H, Me/Me, Et/Me). A comprehensive ethylene polymerisation study is then undertaken that probes the influence of changes in the type/amount of co-catalyst, temperature, pressure and run time. In addition, the synthetic details and characterization data for all ligands and cobalt complexes are disclosed. Finally, DFT calculations are performed to probe the experimental findings and in particular how the introduction of fluoride substituents and sterically hindered ortho groups impact on the catalysis.
Results and discussion
Synthesis and characterization of L1–L5 and Co1–Co5
The unsymmetrical bis(imino)pyridines, 2-{CMeN(2,6-((C6H5)2CH)2-3,4-F2C6H)}-6-(CMeNAr)C5H3N (Ar = 2,6-Me2C6H3 L1, 2,6-Et2C6H3 L2, 2,6-iPr2C6H3 L3, 2,4,6-Me3C6H2 L4, 2,6-Et-4-MeC6H2 L5), were prepared via a Schiff base condensation reaction of the corresponding 1-(6(1-(arylimino)ethyl)pyridin-2-yl)ethan-1-one (aryl = 2,6-dimethylphenyl S1, 2,6-diethylphenyl S2, 2,6-diisopropylphenyl S3, 2,4,6-trimethylphenyl S4, 2,6-diethyl-4-methylphenyl S5) with 2,6-dibenzhydryl-3,4-difluoroaniline in toluene at reflux using p-toluenesulfonic acid as the catalyst (Scheme 1).7,30 All five bis(imino)pyridines have been characterized by IR and multinuclear NMR spectroscopy (1H, 13C, 19F) and elemental analysis.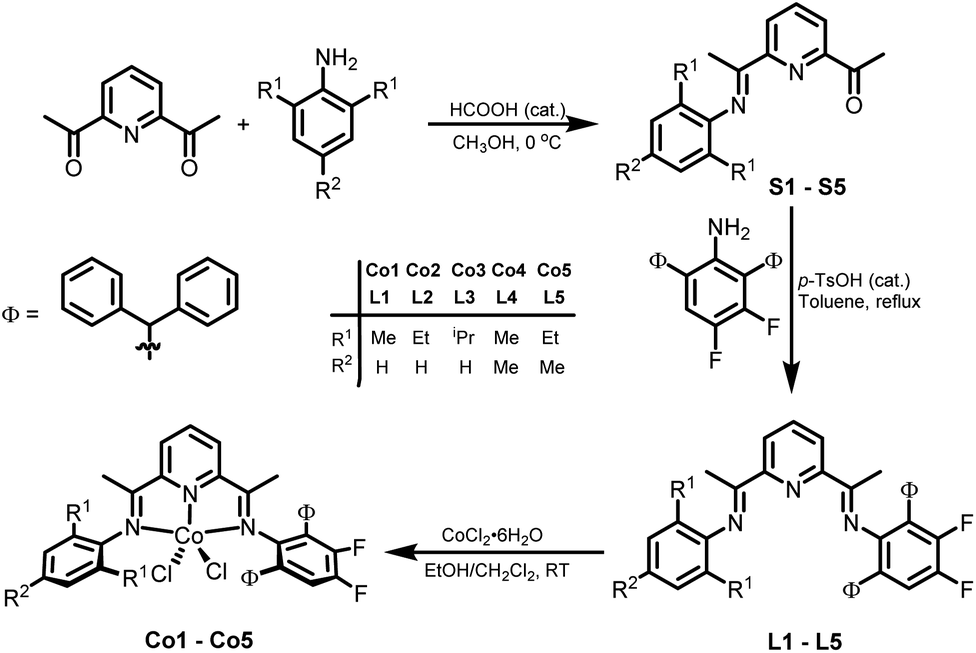 | ||
| Scheme 1 Preparative routes to L1–L5 and their cobalt complexes Co1–Co5. | ||
Subsequently, the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 reaction of cobalt(II) chloride hexahydrate with L1–L5 in a mixture of solvents composed of dichloromethane and ethanol at room temperature gave on work-up, [2-{CMeN(2,6-(Ph2CH)2-3,4-F2C6H)}-6-(CMeNAr)C5H3N]CoCl2 (Ar = 2,6-Me2C6H3 Co1, 2,6-Et2C6H3 Co2, 2,6-iPr2C6H3 Co3, 2,4,6-Me3C6H2 Co4, 2,6-Et-4-MeC6H2 Co5), as green powders in good yields (Scheme 1). All cobalt complexes were characterized by elemental analysis, 19F NMR and FT-IR spectroscopy, while Co2 and Co5 were additionally the subject of single crystal X-ray diffraction studies.
:1 reaction of cobalt(II) chloride hexahydrate with L1–L5 in a mixture of solvents composed of dichloromethane and ethanol at room temperature gave on work-up, [2-{CMeN(2,6-(Ph2CH)2-3,4-F2C6H)}-6-(CMeNAr)C5H3N]CoCl2 (Ar = 2,6-Me2C6H3 Co1, 2,6-Et2C6H3 Co2, 2,6-iPr2C6H3 Co3, 2,4,6-Me3C6H2 Co4, 2,6-Et-4-MeC6H2 Co5), as green powders in good yields (Scheme 1). All cobalt complexes were characterized by elemental analysis, 19F NMR and FT-IR spectroscopy, while Co2 and Co5 were additionally the subject of single crystal X-ray diffraction studies.
In the FT-IR spectra of L1–L5, the vC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretching vibrations appeared in the range 1630–1638 cm−1, while in Co1–Co5 these bands were shifted to lower wavenumber/intensity (1584–1587 cm−1) as is characteristic of metal coordination.26,31 Both ligands and complexes gave distinct signals for the para- and meta-fluoride substituents in their 19F NMR spectra that increased in separation on coordination (e.g. for L1 δ −135.2 and −144.3 ppm vs. δ −94.5 and −158.3 ppm for Co1).
N stretching vibrations appeared in the range 1630–1638 cm−1, while in Co1–Co5 these bands were shifted to lower wavenumber/intensity (1584–1587 cm−1) as is characteristic of metal coordination.26,31 Both ligands and complexes gave distinct signals for the para- and meta-fluoride substituents in their 19F NMR spectra that increased in separation on coordination (e.g. for L1 δ −135.2 and −144.3 ppm vs. δ −94.5 and −158.3 ppm for Co1).
Single crystals of Co2 and Co5 were grown as described in the Experimental Section. Perspective views of Co2 and Co5 are depicted in Fig. 1 and 2; selected bond lengths and angles are listed in Table 1. The structures of Co2 and Co5 are similar differing only in the substitution pattern of one of the N-aryl groups (viz. 2,6-diethylphenyl Co2, 2,6-diethyl-4-methylphenyl Co5) and will be described together. Each structure contains a cobalt center coordinated by two chloride ligands and three nitrogen donors from the chelating N,N,N′-ligand to form a distorted square pyramidal geometry. The square base of the pyramid is filled by N1, N2, N3, and Cl1 with Cl2 occupying the apical position. Above the basal plane sits the cobalt atom at a distance of 0.611 Å in Co2 and 0.556 Å for Co5, which is similar to that seen in a number of structurally related analogs.25,26 Of the three cobalt–nitrogen distances, the central Co–Npyridine bond length [2.077(4) Å for Co2 and 2.072(3) Å for Co5] is the shortest, reflecting the constrictions of the N,N,N′-pincer ligand and the stronger donor properties of the central pyridine unit. The Co–Nimine distances although similar in length show some modest variation with Co1–N1 marginally longer than Co1–N4 [2.183(4) vs. 2.158(4) Å (Co2); 2.220(3) vs. 2.180(3) Å (Co5)]. These differences are in line with the increased steric attributes of the N-2,6-bis(diphenylmethyl)-3,4-difluorophenyl group, while the poorer donor properties of this imine nitrogen due to the electron withdrawing fluoride substituents may also play a contributing factor. As in common for this class of complex, the planes of the N-aryl groups are inclined towards perpendicular with respect to the adjacent pyridine unit, as shown by the dihedral angles (75.3°, 88.1°, Co2; 74.1°, 88.1°, Co5).25,26 There are no noteworthy intermolecular contacts.
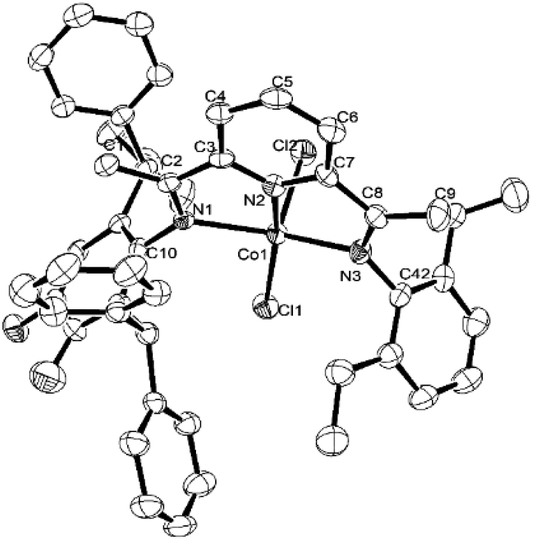 | ||
| Fig. 1 ORTEP representation of Co2. The thermal ellipsoids were set at the 30% probability level, while the hydrogen atoms have been removed for clarity. | ||
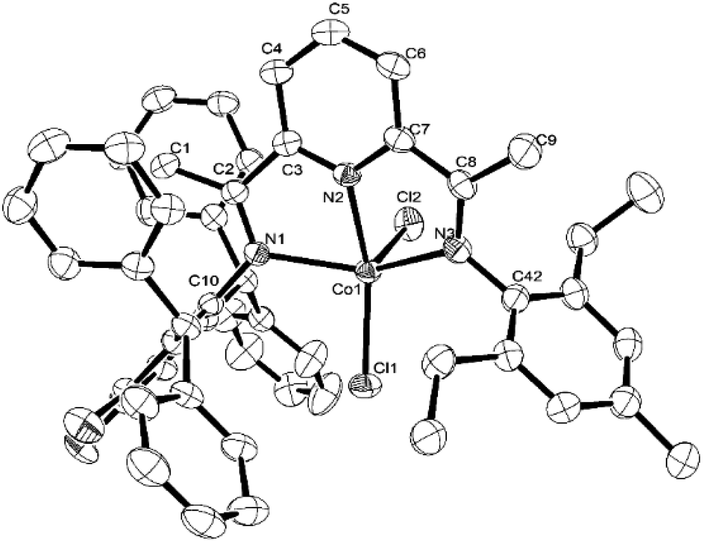 | ||
| Fig. 2 ORTEP representation of Co5. The thermal ellipsoids were set at the 30% probability level, while the hydrogen atoms have been omitted for clarity. | ||
| Co2 | Co5 | |
|---|---|---|
| Bond length (Å) | ||
| Co1–Cl1 | 2.2554(14) | 2.2565(10) |
| Co1–Cl2 | 2.3017(14) | 2.3014(10) |
| Co1–N1 | 2.183(4) | 2.220(3) |
| Co1–N2 | 2.077(4) | 2.072(3) |
| Co1–N3 | 2.158(4) | 2.180(3) |
| Bond angles (deg) | ||
| N2–Co1–N1 | 73.21(15) | 72.58(11) |
| N2–Co1–N3 | 73.72(16) | 74.38(11) |
| N3–Co1–N1 | 139.31(16) | 140.88(11) |
| N1–Co1–Cl1 | 97.72(11) | 97.08(8) |
| N1–Co1–Cl2 | 105.10(11) | 102.94(7) |
| N2–Co1–Cl1 | 159.25(12) | 155.86(8) |
| N2–Co1–Cl2 | 91.96(12) | 92.48(8) |
| N3–Co1–Cl1 | 105.05(13) | 104.77(9) |
| N3–Co1–Cl2 | 99.04(12) | 98.82(8) |
| Cl1–Co1–Cl2 | 108.57(6) | 111.30(4) |
Ethylene polymerisation studies
After a perusal of the literature, it was apparent that nearly all bis(imino)pyridine-cobalt complexes reach optimal catalytic performance for ethylene polymerisation when activated with either modified methylaluminoxane (MMAO) or methylaluminoxane (MAO). Hence, both of these aluminium co-catalysts were used in this work, while Co5 was chosen as the test precatalyst to permit an optimization of the reaction parameters. Systematic changes in the reaction temperature, Al:Co molar ratio, run time, and ethylene pressure are independently undertaken for both Co5/MMAO and Co5/MAO systems before extending the corresponding set of optimized conditions to the remaining precatalysts, Co1–Co4. Typically, the polymerisation runs were performed in toluene at 10 atm ethylene pressure over 30 min run time; the full set of data are collected in Tables 2 and 3. In all cases, differential scanning calorimetry (DSC) and gel permeation chromatography (GPC) are used to measure various polymer properties (e.g. Mw, Mw/Mn and Tm), while high-temperature 1H/13C NMR spectroscopy is employed to explore the microstructural properties of selected polymeric samples.
| Entry | Precat. | Al:Co |
T (°C) | t (min) | Activityb | Tmc (°C) | Mwd | Mw/Mnd |
|---|---|---|---|---|---|---|---|---|
| a Conditions: cobalt precatalyst (2.0 μmol), toluene (100 mL), 10 atm ethylene.b Activity: 106 g PE per mol Co per h.c Measured using differential scanning calorimeter (DSC).d Mw in kg per mol. Mw and Mw/Mn measured by gel permeation chromatography (GPC).e 5 atm. | ||||||||
| 1 | Co5 | 2000 | 20 | 30 | 2.8 | 135.6 | 355.7 | 2.3 |
| 2 | Co5 | 2000 | 30 | 30 | 4.2 | 135.3 | 483.8 | 1.8 |
| 3 | Co5 | 2000 | 40 | 30 | 2.9 | 135.6 | 271.2 | 2.1 |
| 4 | Co5 | 2000 | 50 | 30 | 1.6 | 130.3 | 182.6 | 2.3 |
| 5 | Co5 | 2000 | 60 | 30 | 0.5 | 134.0 | 145.0 | 1.9 |
| 6 | Co5 | 1500 | 30 | 30 | 3.9 | 135.9 | 288.2 | 2.1 |
| 7 | Co5 | 1750 | 30 | 30 | 4.1 | 135.3 | 481.5 | 1.8 |
| 8 | Co5 | 2250 | 30 | 30 | 4.4 | 135.4 | 412.2 | 1.8 |
| 9 | Co5 | 2500 | 30 | 30 | 4.0 | 135.5 | 383.3 | 2.3 |
| 10 | Co5 | 2750 | 30 | 30 | 3.2 | 135.3 | 271.4 | 1.9 |
| 11 | Co5 | 2250 | 30 | 5 | 15 | 136.5 | 422.6 | 1.9 |
| 12 | Co5 | 2250 | 30 | 15 | 6.1 | 135.2 | 480.5 | 1.8 |
| 13 | Co5 | 2250 | 30 | 45 | 3.0 | 134.7 | 511.9 | 2.2 |
| 14 | Co5 | 2250 | 30 | 60 | 2.4 | 135.4 | 573.2 | 2.4 |
| 15e | Co5 | 2250 | 30 | 30 | 2.7 | 136.2 | 327.5 | 3.1 |
| 16 | Co1 | 2250 | 30 | 30 | 3.3 | 135.7 | 274.8 | 2.1 |
| 17 | Co2 | 2250 | 30 | 30 | 3.7 | 135.2 | 418.3 | 2.3 |
| 18 | Co3 | 2250 | 30 | 30 | 4.6 | 135.1 | 332.2 | 2.3 |
| 19 | Co4 | 2250 | 30 | 30 | 3.0 | 135.9 | 253.4 | 2.8 |
| Entry | Precat. | Al:Co |
T (°C) | t (min) | Activityb | Tmc (°C) | Mwd | Mw/Mnd |
|---|---|---|---|---|---|---|---|---|
| a Conditions: cobalt precatalyst (2.0 μmol), toluene (100 mL), 10 atm ethylene.b Activity: 106 g PE per mol Co per h.c Measured using differential scanning calorimeter (DSC).d Mw in kg per mol. Mw and Mw/Mn measured by gel permeation chromatography (GPC).e 5 atm. | ||||||||
| 1 | Co5 | 1750 | 40 | 30 | 3.3 | 134.8 | 132.6 | 2.5 |
| 2 | Co5 | 1750 | 50 | 30 | 2.2 | 132.4 | 41.2 | 4.7 |
| 3 | Co5 | 1750 | 60 | 30 | 7.6 | 132.2 | 45.8 | 3.9 |
| 4 | Co5 | 1750 | 70 | 30 | 5.6 | 133.3 | 48.2 | 2.8 |
| 5 | Co5 | 1750 | 80 | 30 | 3.3 | 133.1 | 41.5 | 1.8 |
| 6 | Co5 | 1250 | 60 | 30 | 3.5 | 131.4 | 23.2 | 3.3 |
| 7 | Co5 | 1500 | 60 | 30 | 5.4 | 133.2 | 33.1 | 2.2 |
| 8 | Co5 | 2000 | 60 | 30 | 2.3 | 132.8 | 48.3 | 3.5 |
| 9 | Co5 | 2250 | 60 | 30 | 2.3 | 131.9 | 39.9 | 3.5 |
| 10 | Co5 | 1750 | 60 | 5 | 10.8 | 135.0 | 38.9 | 1.9 |
| 11 | Co5 | 1750 | 60 | 15 | 13.2 | 135.7 | 42.3 | 2.1 |
| 12 | Co5 | 1750 | 60 | 45 | 6.3 | 134.6 | 50.3 | 2.1 |
| 13 | Co5 | 1750 | 60 | 60 | 5.3 | 133.3 | 62.2 | 2.3 |
| 14e | Co5 | 1750 | 60 | 30 | 2.7 | 132.0 | 29.4 | 2.0 |
| 15 | Co1 | 1750 | 60 | 30 | 1.8 | 133.3 | 51.5 | 3.8 |
| 16 | Co2 | 1750 | 60 | 30 | 1.9 | 132.0 | 30.5 | 2.2 |
| 17 | Co3 | 1750 | 60 | 30 | 6.0 | 135.5 | 112.5 | 2.1 |
| 18 | Co4 | 1750 | 60 | 30 | 2.3 | 131.4 | 45.5 | 3.8 |
:Co molar ratio fixed at 2000:1 (entries 1–5, Table 2). The highest activity of 4.2 × 106 g PE mol−1 (Co) h−1 was observed at 30 °C which then declined as the temperature was further increased reaching a minimum of 0.5 × 106 g PE mol−1 (Co) h−1 at 60 °C. It would appear that catalyst deactivation accounts for the loss of activity at higher operating temperatures. The highest molecular weight polyethylene (Mw = 483.8 kg mol−1) was obtained at 30 °C and then steadily fell as the temperature was raised reaching 145.0 kg mol−1 at 60 °C (Fig. 3). This finding would suggest that chain transfer becomes more important as the temperature is increased.31–33 In terms of the polymer dispersities obtained across the temperature range, these remained narrow and unimodal in all cases (Mw/Mn range: 1.8–2.3), indicative of single-site active species.
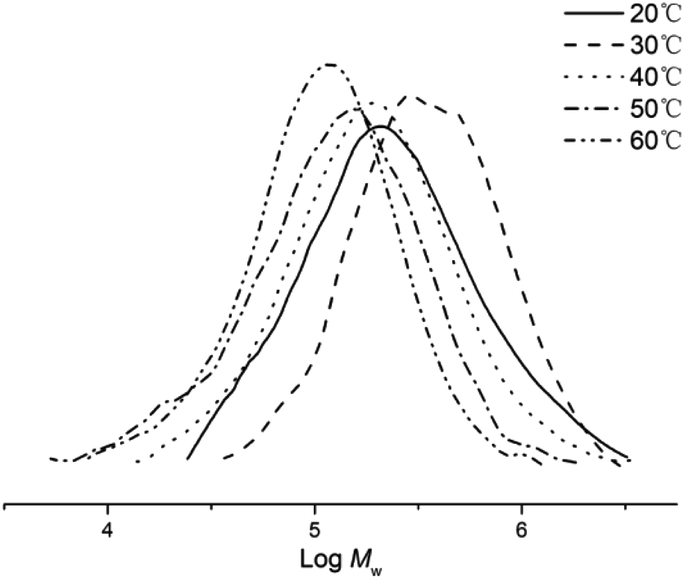 | ||
| Fig. 3 GPC traces of the polyethylene obtained using Co5/MMAO at different temperatures (entries 1–5, Table 2). | ||
Subsequently, the influence of the Al:Co molar ratio on catalyst performance and polymer properties was investigated using Co5/MMAO (entries 2, 6–10, Table 2). On varying the ratio from 1500:1 to 2750:1, it was found that the peak polymerisation activity (4.4 × 106 g PE mol−1 (Co) h−1) was attained using 2250 equivalents of the co-catalyst. Nonetheless, the range in activities (3.9–4.4 × 106 g PE mol−1 (Co) h−1) was narrow which would suggest the amount of co-catalyst employed had limited effect on polymerisation activity. On the other hand, the molecular weight of the polyethylene was more influenced by the Al:Co molar ratio. By increasing the ratio from 1500:1 to 2000:1, the molecular weight increased reaching a maximum value of 483.8 kg mol−1, whereas at ratios in excess of 2000:1, it decreased achieving a value of 271.4 kg mol−1 at 2750:1 (Fig. 4). This latter observation would suggest that chain transfer from the cobalt catalyst to the alkyl aluminium species starts to become operational with larger amounts of co-catalyst.17,34,35
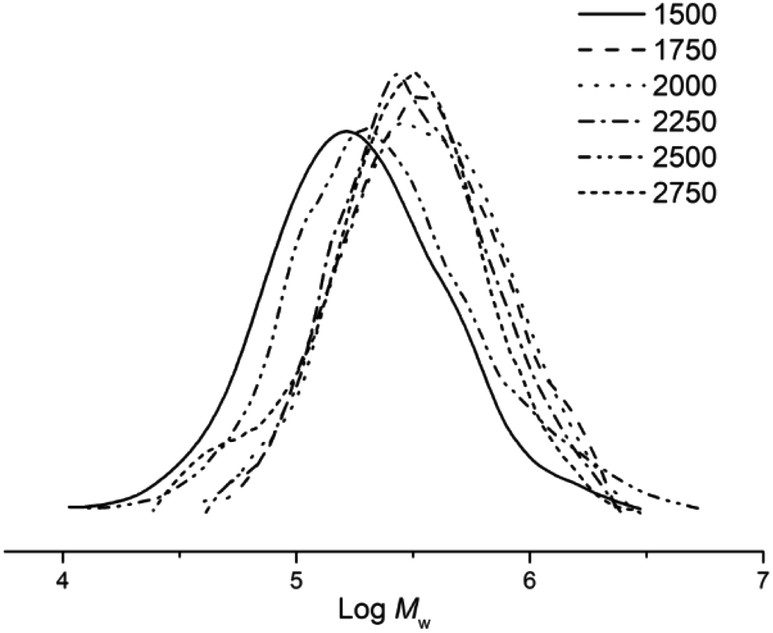 | ||
| Fig. 4 GPC traces of the polyethylene obtained using Co5/MMAO at different Al:Co molar ratios (entries 2, 7–11, Table 2). | ||
Next, we examined the activity/time profile of Co5/MMAO, by performing the polymerisation runs over pre-determined times of 5, 15, 30, 45, and 60 min (entries 8 and 11–14, Table 2); the run temperature and the molar ratio of Al:Co were kept at 30 °C and 2250:1, respectively. After 5 min (entry 11, Table 2), the highest activity of 15 × 106 g PE mol−1 (Co) h−1 was reached, which then subsided reaching a value of 2.4 × 106 g PE mol−1 (Co) h−1 after 60 min. Indeed, such high activity early in the polymerisation is commonplace in cobalt ethylene polymerisation catalysis with gradual catalyst deactivation considered to occur over longer run times.7,36
When the ethylene pressure was reduced from 10 atm to 5 atm (entries 8 and 15 in Table 2), the catalytic activity declined significantly, while the polymer dispersities broadened. Nevertheless, the polyethylene exhibited similar molecular weights over the two different pressures.
On the basis of the most effective set of polymerisation conditions established for Co5/MMAO, the performance of the remaining catalysts Co1–Co4 was then studied (entries 8, 16–19, Table 2). Overall, the catalytic activities for all five complexes fell between 3.0 × 106 g PE mol−1 (Co) h−1 and 4.6 × 106 g PE mol−1 (Co) h−1, with their relative levels decreasing in the order: Co3 > Co5 > Co2 > Co1 > Co4 (Fig. 5). Conversely, the impact of the para-R2 group (R2 = H: Co1–Co3 vs. R2 = Me: Co4, Co5) is less obvious. In addition, all catalysts produced high molecular weight polyethylene (253.4–418.3 kg mol−1) with narrow dispersities (Mw/Mn range: 1.8–2.8).
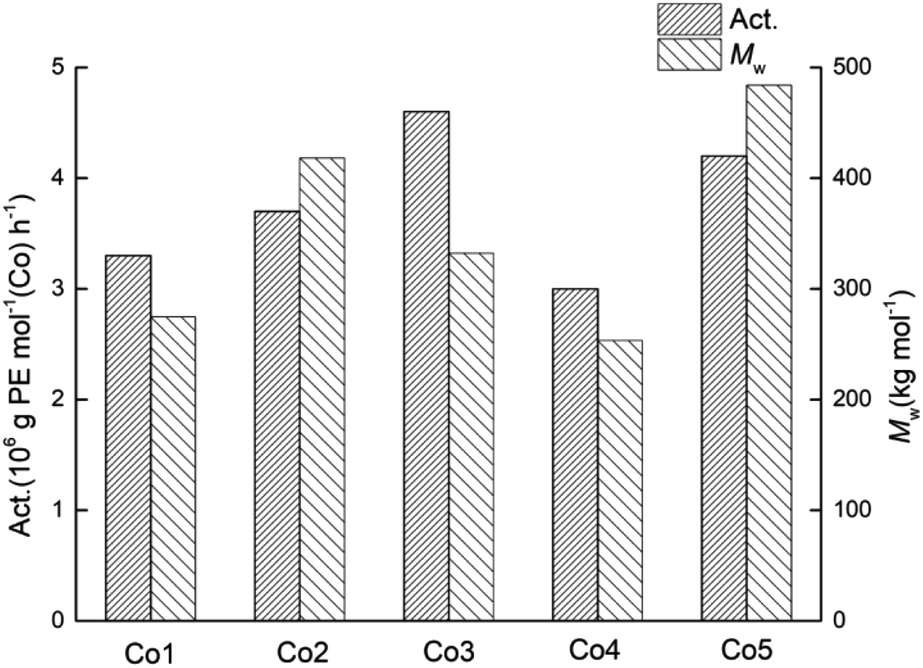 | ||
| Fig. 5 Variations in catalytic activity and molecular weight of the polyethylene obtained using Co1–Co5 (entries 8, 16–19, Table 2); MMAO used as the co-catalyst in each case. | ||
Initially we looked at the effect of the run temperature on the performance of Co5/MAO. With the Al:Co molar ratio set at 1750:1, the highest activity of 7.6 × 106 g PE mol−1 (Co) h−1 was reached at 60 °C (entry 3, Table 3). Notably, this catalytic activity far exceeds the maximum level seen for Co5/MMAO (4.2 × 106 g PE mol−1 (Co) h−1) at 30 °C. Clearly, Co5/MAO displays better thermal stability which would point towards the beneficial effects of MAO as a co-catalyst and its impact on the active species. On the other hand, the molecular weight of the polyethylene dropped to 45.8 kg mol−1, which was almost one-tenth of that seen with MMAO. The reason behind this decrease in Mw most likely stems from temperature induced chain transfer.37 However, when the operating temperature was increased to 80 °C, the activity decreased by more than a half, while the molecular weight of the polymer also decreased (entry 5, Table 3).
With the reaction temperature retained at 60 °C, the Al:Co molar ratio of Co5/MAO was incrementally adjusted between 1250:1 and 2250:1 (entries 3 and 6–9, Table 3). Examination of the data revealed the optimal activity was established with the ratio at 1750:1 which coincides with the value selected during the temperature screening step. When the molar ratio was increased above 1750:1, the catalytic activity converged to only 30% of the optimal activity (entries 8, 9, Table 3). As for the molecular weight of the polymers, these reached a peak of 48.3 kg mol−1 at a ratio of 2000:1 and then decreased at higher ratios (Fig. 6), a finding that can be attributed to termination pathway involving chain transfer of the polymer to the co-catalyst.38,39
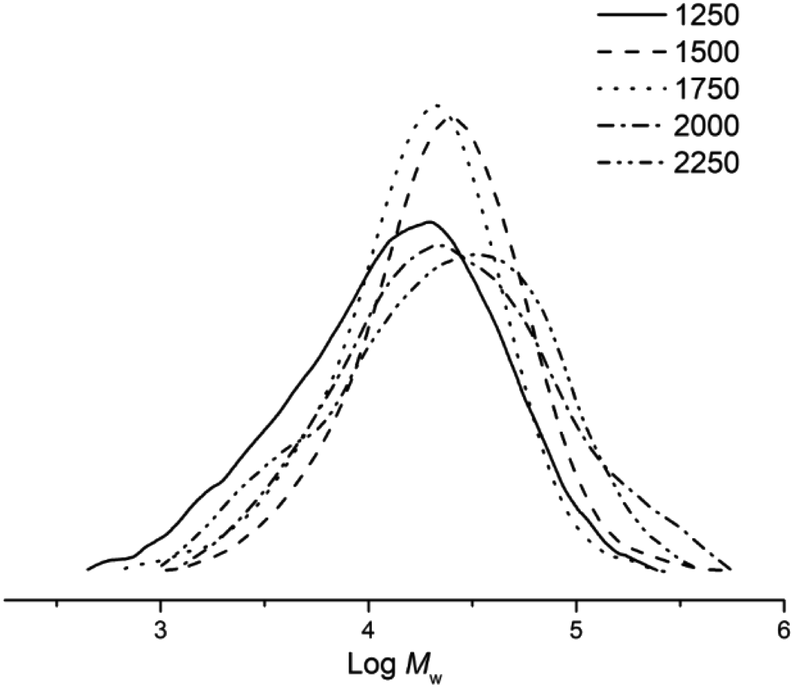 | ||
| Fig. 6 GPC traces of the polyethylene obtained using Co5/MAO at different Al:Co molar ratios (entries 3 and 6–9, Table 3). | ||
Next the effect of run time on the activity of Co5/MAO was investigated with the temperature kept at 60 °C and the molar ratio at 1750:1. Dissimilar to the Co5/MMAO study, the activity reached a peak after 15 min (13.2 × 106 g of PE mol−1 (Co) h−1) and then slowly declined attaining a value of 5.3 × 106 g of PE mol−1 (Co) h−1 after 1 h (entries 3, 10–13, Table 3). It is evident that Co5/MAO displays not only higher activity than Co5/MMAO, but also maintains a more uniform activity/time profile and requires a longer induction period to achieve optimal performance.
With the most effective set of conditions identified for Co5/MAO (viz. Al:Co molar ratio of 1750:1, run temperature of 60 °C, run time of 30 min), the four other cobalt catalysts, Co1–Co4, were similarly evaluated; the results are displayed alongside Co5 in Table 3 (entries 3, 15–18). Inspection of the data revealed that these cobalt catalysts exhibited a wider range in activities spanning from 1.8 to 7.6 × 106 g of PE mol−1 (Co) h−1 when compared to that found with MMAO (3.0 to 4.6 × 106 g PE mol−1 (Co) h−1). In terms of their relative performance, their catalytic activities decreased in the order: Co5 > Co3 > Co4 > Co2 ∼ Co1 (Fig. 7). This order differs to some degree from that found with MMAO, which plausibly reflects the stability of the active species formed using MMAO over MAO under the higher temperature conditions. Nevertheless, the correlation between steric hindrance and activity remains largely the same with bulkier ortho-substituents leading to higher activity. Furthermore, the dispersities of the polymers remained narrow for all five catalysts (Mw/Mn range: 2.1–3.9), highlighting the well-controlled nature of these polymerisations.
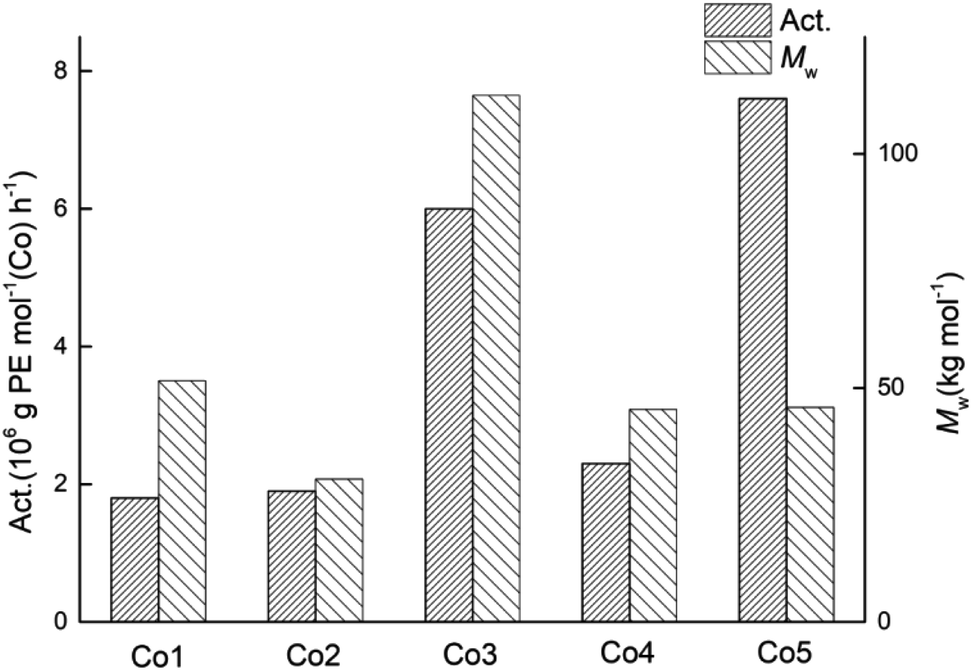 | ||
| Fig. 7 Variations in catalytic activity and molecular weight of the polyethylene displayed using Co1–Co5 (entries 3, 15–18, Table 3); MAO used as the co-catalyst in each case. | ||
Influence of the para- and meta-fluorides
To appreciate the influence posed by the introduction of fluorides to the para- and meta-positions of the N-aryl group in this work, the activity and molecular weight of Co3 were compared with those of structurally related cobalt-containing precatalysts D,23 E24 and F25 (Fig. 8); MAO was employed as co-catalyst in each case. At first glance, it is clear that the catalytic systems bearing electron withdrawing para-groups (Cl (E), NO2 (F) and F (Co3)) displayed higher polymerisation activity than with para-methyl D. With particular regard to Co3, this proved the second most productive system (6.0 × 106 g of PE mol−1 (Co) h−1) behind Cl-containing E, highlighting how variations in electronegativity of the halide can impact on the overall effectiveness of the catalyst. On the other hand, Co3 produced polyethylene with the lowest molecular weight of this series which relates the relative ease of chain termination.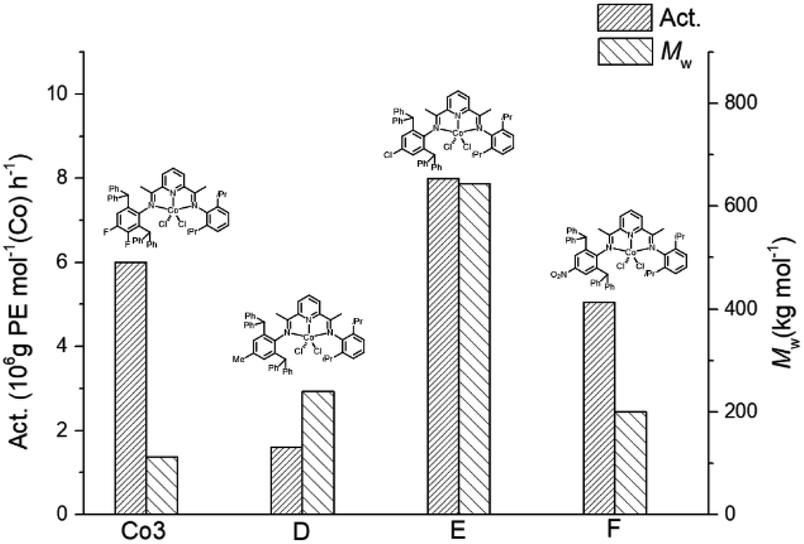 | ||
| Fig. 8 Comparison of catalytic activity and polymer molecular weight obtained using Co3 and that for cobalt-containing D, E, F; MAO used as a co-catalysts in each case under comparable polymerisation conditions. | ||
Microstructural properties of the polyethylene
As given in Tables 2 and 3, the melting points displayed by the polyethylenes fell between 130.3 °C and 136.5 °C, which is characteristic of highly linear polyethylene.16,17,30 In order to confirm this and obtain more information about their microstructural properties (e.g. polymer end groups and branching), representative samples obtained using Co5/MAO and Co5/MMAO have been analyzed by 1H and 13C NMR spectroscopy (recorded in tetrachloroethane-d2 at 100 °C).For the polymer sample produced using Co5/MAO (Mw = 45.8 kg mol−1; entry 3, Table 3), the 13C NMR spectrum revealed as the only visible resonance for the polymer, a high-intensity singlet at δ 30.00 ppm assignable to the –(CH2)n– repeat unit in line with the high linearity of the material (Fig. 9). By contrast, the 1H NMR spectrum gave information on end group composition with downfield resonances typical of an alkenyl chain group end (RCH = CH2). This observation indicates that β-H elimination or β-H transfer provides a key chain termination pathway.37 It is uncertain why the 13C NMR spectrum did not reveal vinylic carbons but may relate to the strength of the sample and solubility.
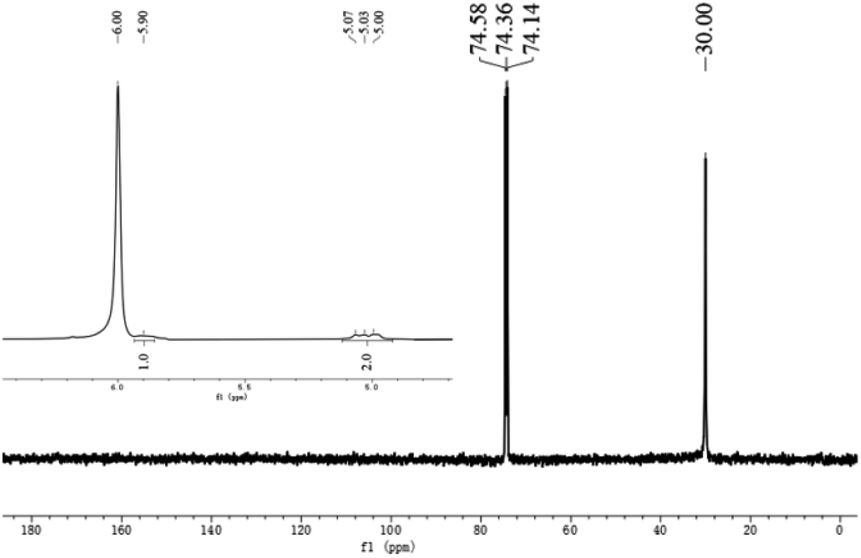 | ||
| Fig. 9 13C NMR spectrum of the polyethylene obtained using Co5/MAO along with an inset showing the vinylic region of its 1H NMR spectrum (entry 3, Table 3); both spectra were recorded in tetrachloroethane-d2 at 100 °C. | ||
Additionally, the polymer sample with higher molecular weight prepared using Co5/MMAO (Mw = 412.2 kg mol−1; entry 8, Table 2) was characterized by 13C NMR spectroscopy (Fig. S22†). Once again the only detectable signal was for the –(CH2)n– repeat unit at δ 29.60 ppm. However, in this case the 1H NMR spectrum showed no evidence for vinylic protons which is likely due to the higher molecular weight of this sample.
DFT calculations
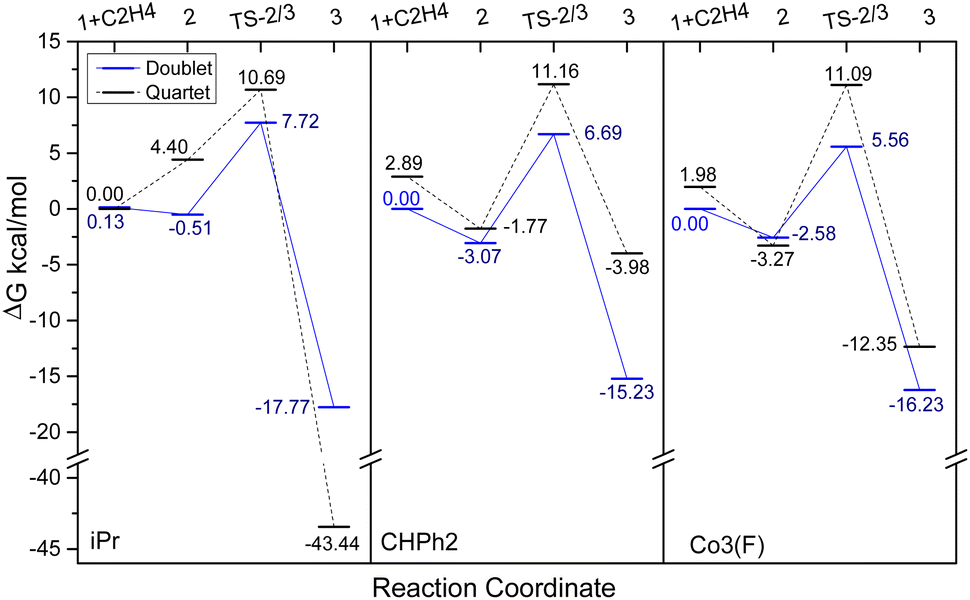
To study the influence of the para- and meta-fluoride substituents in Co3 along with the steric hindrance exerted by the ortho groups on the catalytic activity, the ethylene insertion pathway was computed by DFT calculations using the M06/6-31G** method. In previous works, active catalysts can be generated by treating Co(I) and Co(II) species with strong Lewis acid activators like MAO, but the oxidation state of the active species is still unclear.40–42 In this study we are concerned with making a comparison of the transition state that contain a coordinated ethylene and alkyl group.As a cationic cobalt-alkyl species can display higher catalytic activity than its neutral counterpart,43 we selected cationic cobalt-alkyl species 1 in Scheme 2 (viz. 1-iPr, 1-CHPh2 and 1-Co3) as the starting structure to react with the ethylene. The chain propagated structure 3 was produced via coordinated intermediate 2 and transition state TS2/3. The Gibbs energy profiles of the reaction pathways (Scheme 2) for the three cobalt species are displayed in Fig. 10 (left, middle and right); both the doublet and quartet states were calculated. On inspection of the figure, is apparent that the doublet state possesses a lower energy than the quartet state in most cases. Exceptionally, the chain propagated intermediate 3-iPr is particularly stable in its quartet state (ΔG −43.44 kcal mol−1), as has been found in our previous calculations.43 On the other hand, a similar finding is not observed where the system contains the more sterically demanding CHPh2 groups in the ortho positions. It would seem plausible that the catalysts incorporating CHPh2 groups (1-CHPh2 and 1-Co3) induce a less stable intermediate that reacts further to generate polymers with higher molecular weight.
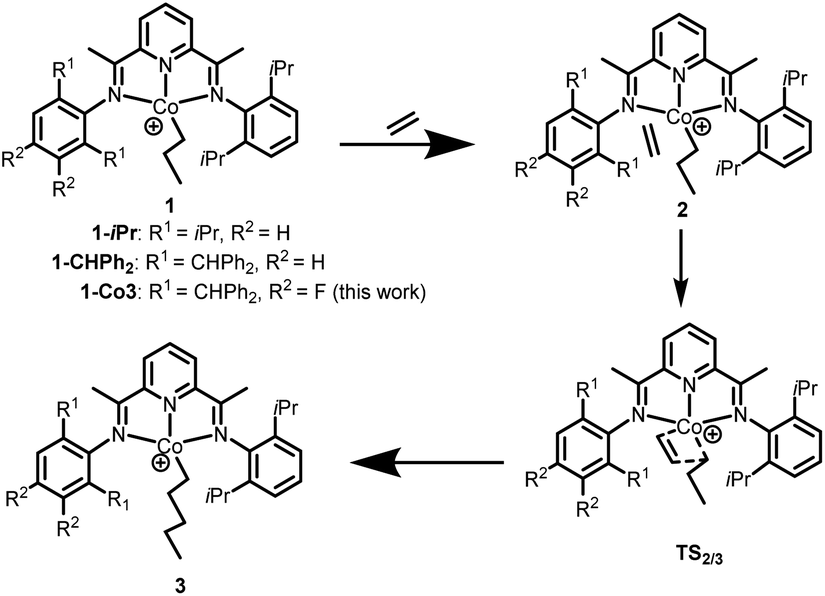 | ||
| Scheme 2 Intermediates and transition states involved in the proposed reaction pathway: 1 + C2H4 → 2 → TS2/3 → 3. | ||
 | ||
| Fig. 10 The Gibbs energy profiles for the ethylene insertion pathway 1 + C2H4 → 2 → TS2/3 → 3, using 1-iPr (left) 1-CHPh2 (middle) and 1-Co3 (right) as shown Scheme 2. Units in kcal mol−1. | ||
In terms of the energy barriers for the ethylene insertion step (i.e. via TS2/3), these were found to decrease in the order: iPr (7.72 kcal mol−1) > CHPh2 (6.69 kcal mol−1) > Co3 (5.56 kcal mol−1). This would suggest that the increase in the steric hindrance at the ortho-positions makes the barrier to ethylene insertion more facile, leading to higher polymerisation activity. Furthermore, the relatively lower barrier in Co3 over CHPh2 would suggest the electron withdrawing fluorides in the meta and para positions are contributing cooperatively with the bulky CHPh2 groups. Moreover, these computational results support the experimental observations, where the catalytic activities for these three systems follow the order: Co3 (6.0 × 106 g of PE mol−1 (Co) h−1) > CHPh2 (1.8 × 106 g of PE mol−1 (Co) h−1) > iPr (4.6 × 105 g of PE mol−1 (Co) h−1).1
Conclusions
In summary, the bis(arylimino)pyridine dichlorocobalt complexes, Co1–Co5, comprising one N-2,6-bis(diphenylmethyl)-3,4-difluorophenyl group and one sterically variable N-aryl group have been successfully synthesized. In addition, all five complexes and their ligand precursors have been fully characterized including in the cases of Co2 and Co5 by single crystal X-ray diffraction. On activation with either MAO or MMAO, all complexes showed good catalytic activity for ethylene polymerisation forming highly linear polyethylenes with narrow dispersities (Mw/Mn range: 1.8–4.7); vinyl-end groups were identified for lower molecular weight samples. Notably, it was found that the polymerisation activity was enhanced with an increase of the steric hindrance at the ortho positions which was further supported by DFT calculations. Moreover, the highest activity (7.6 × 106 g PE mol−1 (Co) h−1) was achieved at 60 °C for Co5/MAO, highlighting the appreciable thermal stability of this catalyst. On the other hand, the highest molecular weight polymer (483.8 kg mol−1) was achieved using Co5/MMAO at 30 °C. On the basis of experimental data and DFT calculations for Co3 and its non-fluorinated analogue, the substitution of fluorides at the meta- and para-positions was shown to enhance catalytic activity and raise thermal stability.Experimental
General considerations
All experimental operations that make use of compounds which are sensitive to air and/or moisture were performed under a nitrogen atmosphere using standard Schlenk techniques. The toluene used for the ethylene polymerisation evaluation was heated to reflux over sodium for at least 10 h before distillation was undertaken under a nitrogen atmosphere. Methylaluminoxane (MAO, 1.38 M in toluene) and modified methylaluminoxane (MMAO, 1.93 M in heptane) were provided by Albemarle Corp. High purity ethylene was provided by Beijing Yanshan Petrochemical Company and used as received. Other reagents were purchased from Aldrich, Acros or local suppliers. The 1H, 13C NMR and 19F NMR spectroscopic measurements for L1–L5 as well as the 19F NMR spectra for cobalt complexes Co1–Co5 were performed on a Bruker DMX 400 and 500 MHz (19F NMR) instrument at room temperature using TMS as an internal standard for 1H/13C NMR and CF3COOH as an external standard. All chemical shifts and coupling constants are given in ppm and in Hz, respectively. Elemental analyses (C, H and N) were performed on a Flash EA 1112 microanalyzer. FT-IR spectra were recorded using a PerkinElmer System 2000 FT-IR spectrometer. The molecular weights and molecular weight distributions (Mw/Mn) of the polyethylenes were measured using a PL-GPC220 instrument at 160 °C with 1,2,4-trichlorobenzene as the solvent. Data collection and processing were performed using Cirrus GPC Software (Beijing, China) and Multi Detector Software (Beijing, China). The calibrants employed for constructing conventional calibration (Polystyrene Calibration KitS-M-10) were provided by PL Company (Beijing, China). The true average molecular weights of polyethylene were achieved by inputting the Mark-Houwink constants of polyethylene; K (0.727) and α (40.6) were provided by PL Company (Beijing, China). The samples were dissolved at a concentration of 1.0 to 2.5 mg mL−1 depending on the molecular weights. The DSC traces and melting points of the polyethylene were obtained from the second scanning run on a PerkinElmer TA-Q2000 DSC analyzer under a nitrogen atmosphere. During the procedure, a sample of about 4.0–6.0 mg was heated to 160 °C at a heating rate of 20 °C min−1, followed by 5 min at 150 °C to remove the thermal history and then cooled at a rate of 20 °C min−1 to −20 °C. High temperature 1H and 13C NMR spectra of the polyethylenes were recorded on a Bruker DMX 500 MHz instrument at 100 °C in deuterated 1,1,2,2-tetrachloroethane with TMS as an internal standard. All computational work was carried out by density functional theory (DFT) using the Gaussian 09 software.44 A hybrid meta-GGA level density functional M06 (ref. 45) in conjunction with 6-31G** basis set was adopted for all atoms. Solvent effect was considered for the fully optimized structures by using the integral equation formalism polarizable continuum model (IEFPCM)46 with the radii and cavity-dispersion-solvent structure terms given in Truhlar and co-workers' SMD solvation model47 for the solvent effect correction of toluene: ε = 2.3741. An ultrafine integration grid (99590) was used for numerical integrations. Thermal corrections were calculated within the harmonic potential approximation on optimized structures under T = 298.15 K and 1 atm pressure. The keto-imine compounds, 1-(6(1-(arylimino)ethyl)pyridin-2-yl)ethan-1-one (aryl = 2,6-dimethylphenyl S1, 2,6-diethylphenyl S2, 2,6-diisopropylphenyl S3, 2,4,6-trimethylphenyl S4, 2,6-diethyl-4-methylphenyl S5), were prepared according to literature routes via the interaction of 2,6-diacetylpyridine with the corresponding aniline in methanol with formic acid as promoter at 0 °C.48
(b) Ar = 2,6-Et2C6H3 L2. Using a similar procedure as described for L1 but with S2 as the ketone, L2 was isolated as a green powder (0.54 g, 37%). FT-IR (cm−1): 3028(w), 2963(m), 2935(w), 2869(w), 1970(w), 1631(m), 1595(m), 1494(m), 1475(m), 1447(m), 1420(m), 1366(m), 1321(w), 1292(m), 1261(s), 1193(m), 1080(s), 1017(s), 925(m), 865(m), 797(vs), 767(s), 743(m), 696(vs). 1H-NMR (400 MHz; CDCl3, TMS): δ 8.44–8.42 (d, J = 7.6 Hz 1H, Py-H), 8.23–8.21 (d, J = 7.6 Hz, 1H, Py), 7.91–7.87 (t, J = 7.8 Hz, 1H, Py), 7.30–6.73 (m, 24H, Ph), 5.40 (s, 1H, –CHPh2), 5.31 (s, 1H, –CHPh2), 2.48–2.30 (m, 4H, –CH2CH3), 2.11 (s, 3H, CH3), 1.18–1.13 (m, 6H, CH3), 1.11 (s, 3H, CH3). 13C NMR (100 MHz; CDCl3; TMS): δ 169.92, 164.80, 153.08, 152.42, 145.63, 140.96, 140.58, 139.54, 138.30, 134.76, 129.07, 129.03, 127.61, 127.21, 126.42, 126.39, 126.22, 125.94, 124.47, 124.41, 124.07, 123.87, 121.28, 120.25, 120.08, 119.96, 119.85, 114.67, 49.79, 47.10, 22.49, 15.07, 14.64, 11.66. 19F NMR (470 MHz, CDCl3): δ −135.6, −144.1. Anal. calcd. for C51H45F2N3 (737.94): C, 83.01; H, 6.15; N, 5.69. Found: C, 82.92; H, 5.95; N, 5.62%.
(c) Ar = 2,6-iPr2C6H3 L3. Using a similar procedure as described for L1 but with S3 as the ketone, L3 was isolated as a green powder (0.84 g, 22%). FT-IR (cm−1): 3060(w), 3027(w), 2960(m), 2923(w), 2866(w), 1947(w), 1637(m), 1605 w), 1575(w), 1494(m), 1473(s), 1449(s), 1420(m), 1365(m), 1324(m), 1244(m), 1194(m), 1104(s), 1078(s), 1003(m), 967(m), 930(m), 907(w), 861(w), 818(m), 792(m), 765(s), 695(vs). 1H NMR (400 MHz; CDCl3, TMS): δ 8.43–8.41 (d, J = 7.6 Hz 1H, Py-H), 8.22–8.20 (d, J = 7.6 Hz, 1H, Py), 7.91–7.87 (t, J = 7.8 Hz, 1H, Py), 7.31–6.76 (m, 24H, Ph), 5.34 (s, 1H, –CHPh2), 5.31 (s, 1H, –CHPh2), 2.79–2.12 (m, 2H, –CH2(CH3)2), 2.12 (s, 3H, CH3), 1.19–1.16 (m, 12H, CH3), 1.12 (s, 3H, CH3). 13C NMR (100 MHz, CDCl3, TMS): δ 169.99, 164.91, 153.12, 152.48, 144.35, 140.99, 140.61, 139.59, 138.34, 134.81, 133.73, 133.69, 127.66, 127.25, 126.46, 126.42, 126.26, 125.98, 124.51, 124.45, 124.12, 121.58, 120.98, 120.33, 120.10, 120.00, 49.79, 49.84, 47.14, 26.26, 21.20, 20.87, 20.85, 15.12, 15.04. 19F NMR (470 MHz, CDCl3): δ −135.6, −144.1. Anal. calcd. for C53H49F2N3 (765.99): C, 83.11; H, 6.45; N, 5.49. Found: C, 82.94; H, 6.27; N, 5.36%.
(d) Ar = 2,4,6-Me3C6H2 L4. Using a similar procedure as described for L1 but with S4 as the ketone, L4 was isolated as a green powder (0.26 g, 7%). FT-IR (cm−1): 3030(w), 2959(w), 2913(w), 2880(vw), 2162(w), 1973(w), 1639(s), 1602(m),1 566(m), 1472(s), 1447(m), 1417(m), 1363(m), 1322(m), 1261(m), 1214(m), 1145(m), 1111(s), 1077(s), 1005(s), 931(m), 905(m), 854(m), 817(s), 794(s), 744(m), 696(vs). 1H NMR (CDCl3, TMS): δ 8.44–8.42 (d, J = 7.6 Hz, 1H, Py-H), 8.21–8.19 (d, J = 7.6 Hz, 1H, Py), 7.89–7.85 (t, J = 7.8 Hz, 1H, Py), 7.30–6.73 (m, 23H, Ph), 5.39 (s, 1H, –CHPh2), 5.30 (s, 1H, –CHPh2), 2.30 (s, 3H, CH3), 2.09 (s, 3H, CH3), 2.02–2.01 (d, J = 4.4 Hz, 6H, CH3), 1.10 (s, 3H, CH3). 13C NMR (100 MHz, CDCl3, TMS): δ 169.99, 165.31, 153.28, 152.45, 144.15, 141.05, 140.66, 139.61, 138.37, 134.78, 130.26, 127.68, 127.28, 126.57, 126.51, 126.47, 126.30, 126.01, 124.54, 124.49, 124.15, 123.23, 123.18, 120.35, 120.13, 114.93, 114.75, 49.87, 47.17, 18.72, 15.86, 15.14, 14.30. 19F NMR (470 MHz, CDCl3): δ −135.6, −144.1. Anal. calcd. for C50H43F2N3 (723.91): C, 82.96; H, 5.99; N, 5.80. Found: C, 82.82; H, 5.87; N, 5.74%.
(e) Ar = 2,6-Et2-4-MeC6H2 L5. Using a similar procedure as described for L1 but with S5 as the ketone, L5 was isolated as a green powder (0.45 g, 30%). FT-IR (cm−1): 3029(w), 2963(m), 2932(w), 2870(w), 1953(w), 1638(s), 1602(m), 1566(m), 1494(m), 1475(s), 1450(s), 1420(m), 1363(s), 1322(m), 1296(m), 1260(m), 1243(m), 1221(m), 1296(m), 1260(m), 1243(m), 1221(m), 1208(m), 1146(m), 1118(s), 1076(s), 1007(m), 931(m), 863(m), 802(s), 764(m), 744(m), 697(vs). 1H NMR (CDCl3, TMS): δ 8.43–8.41 (d, J = 7.2 Hz 1H, Py-H), 8.21–8.19 (d, J = 7.6 Hz, 1H, Py), 7.89–7.86 (t, J = 7.8 Hz, 1H, Py), 7.30–6.75 (m, 23H, Ph), 5.39 (s, 1H, –CHPh2), 5.30 (s, 1H, –CHPh2), 2.39–2.31 (m, 3H, CH3), 2.36 (s, 4H, –CH2CH3), 2.10 (s, 3H, CH3), 1.25–1.12 (m, 6H, –CH2CH3), 1.11 (s, 3H, CH3). 13C NMR (100 MHz, CDCl3, TMS): δ 165.03, 153.25, 152.41, 141.00, 140.62, 139.58, 138.34, 134.75, 130.44, 128.97, 127.65, 127.24, 126.46, 126.42, 126.25, 125.97, 124.63, 124.45, 124.10, 120.27, 120.04, 49.82, 22.52, 18.95, 15.11, 14.62, 11.80. 19F NMR (470 MHz, CDCl3): δ −135.6, −144.1. Anal. calcd. for C52H47F2N3 (751.97): C, 83.06; H, 6.30; N, 5.59%. Found: C, 83.14; H, 6.15; N, 5.66%.
(b) Ar = 2,6-Et2C6H3 Co2. The synthesis of Co2 was carried out using a procedure and molar ratios similar to that described for Co1, but with L2 used in place of L1. Following work-up, Co2 was isolated as a green powder (0.095 g, 94%). FT-IR (KBr, cm−1): 3063(w), 2972(w), 2937(w), 2878(w), 2161(m), 1974(w), 1617(w), 1587(m), 1496(m), 1476(s), 1449(m), 1424(m), 1365(m), 1324(m), 1266(m), 1207(m), 1078(m), 1007(m), 940(m), 862(m), 812(m), 770(s), 739(m), 703(vs). 19F NMR (470 MHz, CDCl3): δ −93.1, −158.0. Anal. calcd. for C51H45F2N3Cl2Co (867.77): C, 70.59; H, 5.23; N, 4.84. Found: C, C, 70.34; H, 5.45; N, 4.74%.
(c) Ar = 2,6-iPr2C6H3 Co3. The synthesis of Co3 was carried out using a procedure and molar ratios similar to that described for Co1, but with L3 used in place of L1. Following work-up, Co3 was isolated as a green powder (0.051 g, 94%). FT-IR (KBr, cm−1): 3057(w), 2961(m), 2920(w), 2866(w), 2161(w), 1971(w), 1584(m), 1495(m), 1473(s), 1450(s), 1371(m), 1323(m), 1269(m), 1208(m), 1103(m), 1078(m), 1028(m), 1003(m), 938(m), 863(w), 806(m), 769(s), 699(vs). 19F NMR (470 MHz, CDCl3): δ −92.5, −157.6. Anal. calcd. for C53H49F2N3Cl2Co (895.83): C, 71.06; H, 5.51; N, 4.69. Found: C, 70.96; H, 5.57; N, 4.64%.
(d) Ar = 2,4,6-Me3C6H2 Co4. The synthesis of Co4 was carried out using a procedure and molar ratios similar to that described for Co1, but with L4 used in place of L1. Following work-up, Co4 was isolated as a green powder (0.038 g, 50%). FT-IR (KBr, cm−1): 3061(w), 3034(w), 2951(w), 2923(w), 2916(w), 2840(w), 2160(w), 1974(w), 1617(w), 1588(m), 1496(m), 1477(s), 1449(m), 1425(m), 1365(m), 1325(m), 1265(m), 1221(m), 1196(m), 1079(m), 1009(m), 939(w), 858(m), 816(m), 771(m), 736(m), 703(vs). 19F NMR (470 MHz, CDCl3): δ −94.7, −158.0. Anal. calcd. for C50H43F2N3Cl2Co (853.75): C, 70.34; H, 5.08; N, 4.92. Found: C, 70.12; H, 5.23; N, 4.74%.
(e) Ar = 2,6-Et2-4-MeC6H2 Co5. The synthesis of Co5 was carried out using a procedure and molar ratios similar to that described for Co1, but with L5 used in place of L1. Following work-up, Co5 was isolated as a green powder (0.13 g, 94%). FT-IR (KBr, cm−1): 3032(w), 2972(w), 2925(w), 2878(w), 2162(w), 1974(w), 1619(w), 1586(m), 1496(m), 1477(s), 1449(m), 1423(m), 1363(m), 1325(m), 1263(s), 1219(m), 1078(m), 1031(m), 1010(m), 939(m), 862(m), 814(m), 7701(s), 735 (s), 702(vs). 19F NMR (470 MHz, CDCl3): δ −93.3, −157.9. Anal. calcd. for C52H47F2N3Cl2Co (881.80): C, 70.83; H, 5.37; N, 4.77. Found: C, 70.74; H, 5.59; N, 4.55%.
X-ray crystallographic studies
Single crystals of Co2 and Co5 for X-ray diffraction studies were grown by diffusing diethyl ether onto dichloromethane solutions of the corresponding complex at ambient temperature. By employing a XtaLAB Synergy R HyPix diffractometer equipped with graphite monochromated Cu-Kα radiation (λ = 1.54184 Å) at 173(2) K, cell parameters were obtained by global refinement of the positions of all collected reflections. Intensities were corrected for Lorentz and polarization effects and empirical absorption. Structure solution was performed by direct methods and refined by full-matrix least squares on F2. All non-hydrogen atoms were refined anisotropically and all hydrogen atoms were placed in calculated positions. Using Olex2,49 the structure was solved by the SHELXT50 structure solution program employing Intrinsic Phasing by and refined with the SHELXL51 refinement package using the Least Squares minimization. Details of the X-ray structure determinations and refinements are provided in Table 4.| Co2 | Co5 | |
|---|---|---|
| Crystal colour | Brown | Brown |
| Empirical formula | C52H47Cl4CoF2N3 | C53H49Cl4CoF2N3 |
| Formula weight | 952.65 | 966.68 |
| Temperature/K | 170.00(11) | 169.97(11) |
| Crystal system | Monoclinic | Monoclinic |
| Space group | Cc | Cc |
| a/Å | 23.8272(3) | 24.2166(3) |
| b/Å | 14.5493(2) | 14.6821(2) |
| c/Å | 16.0514(2) | 16.4823(2) |
| α/° | 90 | 90 |
| β/° | 125.0950(10) | 127.0490(10) |
| γ/° | 90 | 90 |
| Volume/Å3 | 4552.89(11) | 4677.22(11) |
| Z | 4 | 4 |
| ρcalc g cm−3 | 1.39 | 1.373 |
| μ/mm−1 | 5.498 | 5.36 |
| F(000) | 1972 | 2004 |
| Crystal size/mm3 | 0.15 × 0.1 × 0.05 | 0.15 × 0.12 × 0.08 |
| 2Θ range for data collection/° | 7.582 to 150.576 | 7.562 to 150.714 |
| Index ranges | −29 ≤ h ≤ 29, −18 ≤ k ≤ 14 | −30 ≤ h ≤ 30, −17 ≤ k ≤ 18 |
| −19 ≤ l ≤ 19 | −19 ≤ l ≤ 20 | |
| Reflections collected | 16783 |
16345 |
| Independent reflections | 7537 [Rint = 0.0351, Rsigma = 0.0390] | 5983 [Rint = 0.0268, Rsigma = 0.0267] |
| Data/restraints/parameters | 7537/2/563 | 5983/2/573 |
| Goodness-of-fit on F2 | 1.041 | 1.027 |
| Final R indexes [I >= 2σ(I)] | R1 = 0.0509, wR2 = 0.1403 | R1 = 0.0379, wR2 = 0.1039 |
| Final R indexes [all data] | R1 = 0.0523, wR2 = 0.1416 | R1 = 0.0389, wR2 = 0.1046 |
| Largest diff. peak/hole/e Å−3 | 2.66/−0.61 | 1.38/−0.49 |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 21871275). G.A.S. thanks the Chinese Academy of Sciences for a President's International Fellowship for Visiting Scientists. Y. M. thanks the Chinese Academy of Sciences for the support of her visiting scholarship to the UK.Notes and references
- G. J. P. Britovsek, V. C. Gibson, B. S. Kimberley, P. J. Maddox, S. J. McTavish, G. A. Solan, A. J. P. White and D. J. Williams, Chem. Commun., 1998, 7, 849 RSC.
- B. L. Small, M. Brookhart and A. M. A. Bennett, J. Am. Chem. Soc., 1998, 120, 4049 CrossRef CAS.
- G. J. P. Britovsek, M. Bruce, V. C. Gibson, B. S. Kimberley, P. J. Maddox, S. Mastroianni, S. J. McTavish, C. Redshaw, G. A. Solan, S. Strömberg, A. J. P. White and D. J. Williams, J. Am. Chem. Soc., 1999, 121, 8728 CrossRef CAS.
- V. C. Gibson, C. Redshaw and G. A. Solan, Chem. Rev., 2007, 107, 1745 CrossRef CAS PubMed.
- X. Cao, F. He, W. Zhao, Z. Cai, X. Hao, T. Shiono, C. Redshaw and W.-H. Sun, Polymer, 2012, 53, 1870 CrossRef CAS.
- M. Han, Q. Zhang, I. I. Oleynik, H. Suo, I. V. Oleynik, G. A. Solan, Y. Ma, T. Liang and W.-H. Sun, Catalysts, 2020, 10, 1002 CrossRef CAS.
- Q. Zhang, Z. Li, M. Han, J. Xiang, G. A. Solan, Y. Ma, l. tongling and W.-H. Sun, Catal. Sci. Technol., 2020, 11, 656 RSC.
- F. Huang, Q. Xing, T. Liang, Z. Flisak, B. Ye, X. Hu, W. Yang and W.-H. Sun, Dalton Trans., 2014, 43, 16818 RSC.
- Z. Wang, R. Zhang, W. Zhang, G. A. Solan, Q. Liu, T. Liang and W.-H. Sun, Catal. Sci. Technol., 2019, 9, 1933 RSC.
- W. Zhang, S. Wang, S. Du, C.-Y. Guo, X. Hao and W.-H. Sun, Macromol. Chem. Phys., 2014, 215, 1797 CrossRef CAS.
- C. Bariashir, Z. Wang, S. Du, G. A. Solan, C. Huang, T. Liang and W.-H. Sun, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 3980 CrossRef CAS.
- S. Ahmed, W. Yang, Z. Ma and W.-H. Sun, J. Phys. Chem. A, 2018, 122, 9637 CrossRef CAS PubMed.
- V. K. Appukuttan, Y. Liu, B. C. Son, C.-S. Ha, H. Suh and I. Kim, Organometallics, 2011, 30, 2285 CrossRef CAS.
- Q. Mahmood, J. Guo, W. Zhang, Y. Ma, T. Liang and W.-H. Sun, Organometallics, 2018, 37, 957 CrossRef CAS.
- Q. Mahmood, E. Yue, J. Guo, W. Zhang, Y. Ma, X. Hao and W.-H. Sun, Polymer, 2018, 159, 124 CrossRef CAS.
- F. Huang, W. Zhang, E. Yue, T. Liang, X. Hu and W.-H. Sun, Dalton Trans., 2016, 45, 657 RSC.
- H. Suo, I. I. Oleynik, C. Bariashir, I. V. Oleynik, Z. Wang, G. A. Solan, Y. Ma, T. Liang and W.-H. Sun, Polymer, 2018, 149, 45 CrossRef CAS.
- Y. Huang, R. Zhang, T. Liang, X. Hu, G. A. Solan and W.-H. Sun, Organometallics, 2019, 38, 1143 CrossRef CAS.
- C. Bariashir, Z. Wang, G. A. Solan, C. Huang, X. Hao and W.-H. Sun, Polymer, 2019, 171, 87 CrossRef CAS.
- Q. Xing, T. Zhao, Y. Qiao, L. Wang, C. Redshaw and W.-H. Sun, RSC Adv., 2013, 3, 26184 RSC.
- Q. Chen, W. Zhang, G. A. Solan, R. Zhang, L. Guo, X. Hao and W.-H. Sun, Organometallics, 2018, 37, 4002 CrossRef CAS.
- L. Guo, H. Gao, L. Zhang, F. Zhu and Q. Wu, Organometallics, 2010, 29, 2118 CrossRef CAS.
- J. Yu, W. Huang, L. Wang, C. Redshaw and W.-H. Sun, Dalton Trans., 2011, 40, 10209 RSC.
- J. Yu, H. Liu, W. Zhang, X. Hao and W. H. Sun, Chem. Commun., 2011, 47, 3257 RSC.
- F. He, W. Zhao, X.-P. Cao, T. Liang, C. Redshaw and W.-H. Sun, J. Organomet. Chem., 2012, 713, 209 CrossRef CAS.
- Q. Mahmood, Y. Ma, X. Hao and W.-H. Sun, Appl. Organomet. Chem., 2019, 33, e4857 CrossRef.
- W. Zhao, J. Yu, S. Song, W. Yang, H. Liu, X. Hao, C. Redshaw and W.-H. Sun, Polymer, 2012, 53, 130 CrossRef CAS.
- J. Lai, W. Zhao, W. Yang, C. Redshaw, T. Liang, Y. Liu and W.-H. Sun, Polym. Chem., 2012, 3, 787 RSC.
- C. Bariashir, R. Zhang, A. Vignesh, Y. Ma, T. Liang and W.-H. Sun, ACS Omega, 2021, 6, 4448 CrossRef CAS.
- R. Zhang, Y. Ma, M. Han, G. A. Solan, Y. Pi, Y. Sun and W.-H. Sun, Appl. Organomet. Chem., 2019, 33, e5157 Search PubMed.
- M. Han, Q. Zhang, I. I. Oleinik, H. Suo, G. A. Solan, I. V. Oleinik, Y. Ma, T. Liang and W.-H. Sun, Dalton Trans., 2020, 49, 4774 RSC.
- M. Han, I. I. Oleynik, Y. Ma, I. V. Oleynik, G. A. Solan, T. Liang and W.-H. Sun, Appl. Organomet. Chem., 2021, 35, e6429 CAS.
- R. Zhang, Y. Huang, G. A. Solan, W. Zhang, X. Hu, X. Hao and W.-H. Sun, Dalton Trans., 2019, 48, 8175 RSC.
- J. Guo, W. Zhang, I. I. Oleynik, G. A. Solan, I. V. Oleynik, T. Liang and W.-H. Sun, Dalton Trans., 2020, 49, 136 RSC.
- Q. Zhang, R. Zhang, M. Han, W. Yang, T. Liang and W.-H. Sun, Dalton Trans., 2020, 49, 7384 RSC.
- T. Liu, Y. Ma, G. A. Solan, T. Liang and W.-H. Sun, Appl. Organometal. Chem., 2021, 35, e6259 CAS.
- R. Zhang, Y. Huang, Y. Ma, G. A. Solan, X. Hu, T. Liang and W.-H. Sun, Polymer, 2021, 222, 123684 CrossRef CAS.
- H. Suo, I. I. Oleynik, C. Bariashir, I. V. Oleynik, Z. Wang, G. A. Solan, Y. Ma, T. Liang and W.-H. Sun, Polymer, 2018, 149, 45 CrossRef CAS.
- L. Guo, M. Zada, W. Zhang, A. Vignesh, D. Zhu, Y. Ma, T. Liang and W.-H. Sun, Dalton Trans., 2019, 48, 5604 RSC.
- P. Margl, L. Deng and T. Ziegler, Organometallics, 1999, 18, 5701 CrossRef CAS.
- K. P. Tellmann, M. J. Humphries, H. S. Rzepa and V. C. Gibson, Organometallics, 2004, 23, 5503 CrossRef CAS.
- I. E. Soshnikov, N. V. Semikolenova, A. N. Bushmelev, K. P. Bryliakov, O. Y. Lyakin, C. Redshaw, V. A. Zakharov and E. P. Talsi, Organometallics, 2009, 28, 6003 CrossRef CAS.
- Z. Li, Y. Ma and W.-H. Sun, Catalysts, 2020, 10, 1396 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, and D. J. Fox, Gaussian 09, Revision E.01, Gaussian, Inc., Wallingford, CT, 2013 Search PubMed.
- Y. Zhao and D. G. Truhlar, J. Chem. Phys., 2006, 125, 194101 CrossRef PubMed.
- B. Mennucci, Tomasi and R. Cammi, Chem. Rev., 2005, 105, 2999 CrossRef PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378 CrossRef CAS PubMed.
- C. Bianchini, G. Mantovani, A. Meli, F. Migliacci, F. Zanobini, F. Laschi and A. Sommazzi, Eur. J. Inorg. Chem., 2003, 8, 1620 CrossRef.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Figures and additional characterization data for ligands, complexes, and resultant polymers; coordinates of all optimized structures by DFT calculations. CCDC 2201737 and 2201738. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2ra05806e |
| This journal is © The Royal Society of Chemistry 2023 |