
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Highly sensitive pork meat detection using copper(II) tetraaza complex by electrochemical biosensor†
Noraisyah Abdul Kadir Jilani ,
Emma Izzati Zakariah,
Eda Yuhana Ariffin,
Suhaila Sapari,
Devika Nokarajoo,
Bohari Yamin and
Siti Aishah Hasbullah*
,
Emma Izzati Zakariah,
Eda Yuhana Ariffin,
Suhaila Sapari,
Devika Nokarajoo,
Bohari Yamin and
Siti Aishah Hasbullah*
Department of Chemical Sciences. Faculty Science and Technology, Universiti Kebangsaan Malaysia, 43600 Bangi, Selangor Darul Ehsan, Malaysia. E-mail: aishah80@ukm.edu.my
First published on 11th January 2023
Abstract
Three copper(II) tetraaza complexes [Cu(II)LBr]Br (1a), [Cu(II)L(CIO4)](CIO4) (2a) and [Cu(II)L](CIO4)2 (2b), where L = 5,5,7,12,12,14-hexamethyl-1,4,8,11-tetraazacyclotetradeca-7,14-diene were prepared and confirmed by FTIR, 1HNMR and 13CNMR. The binding interaction of complex (1a, 2a, 2b) with calf thymus DNA (CT-DNA) was investigated using UV-vis absorption, luminescence titrations, viscosity measurements and molecular docking. The findings suggested that complex 1a, 2a and 2b bind to DNA by electrostatic interaction, and the strengths of the interaction were arranged according to 2b > 1a > 2a. The differences in binding strengths were certainly caused by the complexes' dissimilar charges and counter anions. Complex 2b, with the biggest binding strength towards the DNA, was further applied in developing the porcine sensor. The developed sensor exhibits a broad linear dynamic range, low detection limit, good selectivity, and reproducibility. Analysis of real samples showed that the biosensor had excellent selectivity towards the pork meat compared to chicken and beef meat.
1. Introduction
Adulteration of pork or its derivatives in food, particularly in meatballs and sausage, continuously occurs due to its cheaper availability than other meats.1,2 As some religious laws do not permit consuming any food containing pork, this matter should not be taken lightly. In addition, carelessly consuming this contaminated food sometimes induces allergic effects in certain individuals. It harms people, especially those with diabetes and cardiovascular disease, since it contains high cholesterol and saturated fats.3 Besides, excessive intake of pork products also can lead to an increased likelihood of developing other dangerous diseases such as arthritis, osteoporosis, Alzheimer's, asthma, and infertility. Therefore, recognizing pork in foods is essential for meat authentication and satisfying market demand for protection against falsely branded foods.4In this regard, a reliable approach is required for food ingredients analysis in order to identify the existence of any pork derivatives in food products. Because of this, DNA biosensors that provide simple, specific, fast, on-site analysis and easier verification is one of the suitable techniques to solve the detection of pork in food or materials. Although pork DNA electrochemical biosensors have been reported, the detection is not really sensitive.5
In order to improve the sensitivity, the tetraaza complexes having four amine groups were investigated. Interestingly, copper(II) tetraaza complexes and tetraaza ligands have excellent anticancer activities.6 This ability is due to the presence of amine functional groups capable of becoming a host molecule that provides hydrogen bonding and can bind to biopolymers such as nucleic acid and proteins.6 The binding mechanism between complexes and DNA is essential for developing biosensors.
Thus, DNA interaction studies have been investigated extensively. Basically, transition metal complexes such as copper(II) complex can typically bind to DNA through intercalation, groove binding, electrostatic and other forms of interactions.7,8 As cofactor agents, this complex can generate redox signals before and after the hybridization event.9 This characteristic allows the complex to be used as an electroactive compound to track the DNA hybridization.10 Previous research investigated the DNA interaction with copper(II) macrocyclic polyamines bearing acridine moieties. The complex bind to DNA through intercalation mode due to the synthesized complex's planar moieties, which can cause it to stack between DNA base pairs.11 A similar mode of binding has been reported by Arthi et al., where the tetraaza macrocycles with benzoyl pendant-arms with the planar environment interacted with DNA through intercalation mode.12 X. Li et al. have described a development of electrochemical detection of DNA hybridization based on copper(II) complex via intercalative interaction [Cu(phen)2]2+.13 Meanwhile, G. Li et al. have reported the potential of a copper(II) complex [Cu(dafone)2]2+ bind with double-stranded salmon sperm DNA also through intercalation mode using cyclic voltammetry.14 Although there are numerous DNA binding studies, none analyzed the effect of charge and size on the complex-DNA interaction, mainly on the effect of moieties.
Thus, the limitation of research on the effect of charge and size of the complex on the interaction with the DNA has instigated the synthesis, characterization and binding study of the three copper(II) tetraaza complexes (1a, 2a, 2b). The use of copper(II) tetraaza complex as a label for DNA detection was newly developed and showed to be an excellent electrochemical indicator. The electrochemical biosensor has been fabricated by immobilizing gold nanoparticles (AuNPs) onto the disposable screen-printed carbon electrode (SPCE), followed by the deposition of 3-mercaptopropionic acid (MPA). Activation of the layer was performed by self-assembled 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide/N-hydroxysuccinimide (EDC/NHS), forming an intermediate ester. 2b was then applied to obtain the DNA hybridization's electrochemical response using differential pulse voltammetry (DPV). The developed biosensor has a lower detection limit and broad linear range suitable for establishing new rapid tools for porcine detection.
2. Experimental
2.1. Chemicals and instrumentations
Analytical grade quality of reagents and solvents were used without further purification. DNA was acquired from Sigma Aldrich Company. Tris(hydroxymethyl)amino methane–HCI (Tris–HCI) buffer solution was prepared using deionized distilled water. The infrared spectra were analyzed by KBr pellets and measured on a PerkinElmer 400 FT-IR spectrophotometer. Barnstead Electrothermal Melting Point IA9100 series was used to obtain the melting point. Shimadzu UV-2450 PC spectrophotometer was used to record the electronic spectra in the 200–800 nm region. PerkinElmer LS55 spectrophotometer was used to determine the emission spectra at room temperature. 1H NMR and 13C NMR spectra were obtained on a JEOL spectrometer ECP 400 MHz in d4-MeOH and d6-DMSO as a solvent. Single Crystal X-ray Diffractometer model Bruker SMART APEX CCD was used to conduct the single-crystal X-ray study. For pH titrations, a Eutech pH 700 was used to read the pH values directly. A concentrated HCI was used to adjust the acidity. Ionic strength was kept constant by adding 50 mM NaCl. The current response was measured using differential pulse voltammetry (Autolab potentiostat). SPCE used was from Scrint Technology. The oligonucleotide sequence of each DNA is as below:Probe DNA: (5′)-CTG ATA GTA GAT TTG TGA TGA CCG TAG-(CCC-NH2)-(3′).
Complementary DNA: (3′)-GAC TAT CAT CTA AAC ACT ACT GGC ATC-(5′).
Chicken DNA: (3′)-GCG TCC ATA ATG ATA GTA GGT GGA G-(5′).
Beef DNA: (3′)-GTA TCG TTA ACG GTA TCA GGT GGA T-(5′).
2.2. Synthesis
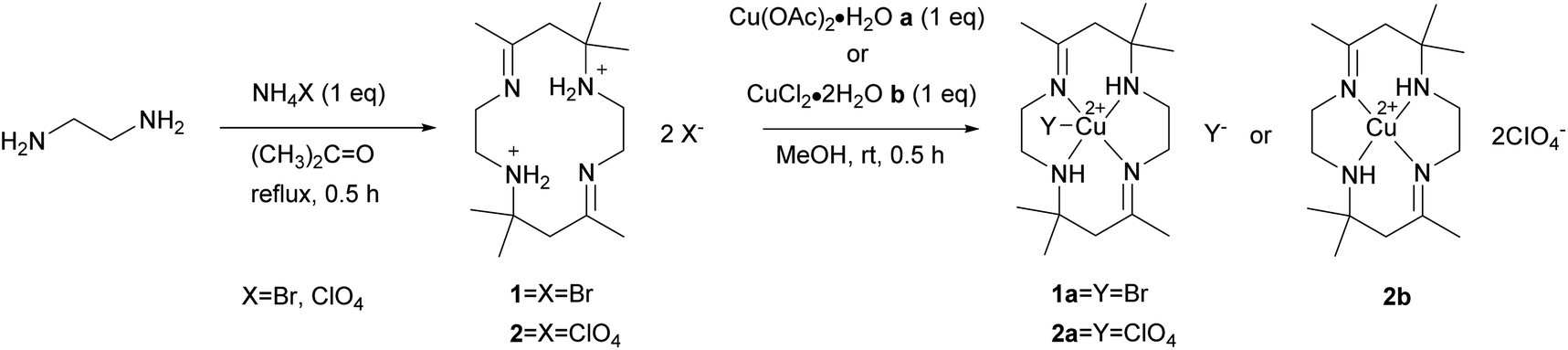 | ||
| Scheme 1 Synthesis of tetraaza ligand 1 and 2 and copper(II) tetraaza complexes 1a, 2b and 2b. | ||
[H2L]Br2 (1) Yield: 72%; m.p. 113.4–115.3 °C; UV-vis λ 246 nm, ε 333 M−1 cm−1; FT-IR (KBr) (νmax/cm−1): 3460, 3402 (N–H), 2980 (C–H), 1665 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N), 1226 (C–N). 1H-NMR (d4-MeOH, 400 MHz): 1.49 (s, 6H, (CH3)2), 2.06 (s, 3H, CH3), 2.81 (s, 2H, CH2), 3.43 (t, 2H, CH2–NH2+, J = 4.8 Hz), 3.70 (s, 2H, CH2–NC); 13C-NMR (d4-MeOH, 400 MHz): 20.4 (CH3)2, 23.3 (CH3), 41.2 (C(CH3)–CH2–C(CH3)2), 43.7 (CH2–NH2+), 46.9 (CH2–NC), 58.4 (C–(CH3)2), 175.8 (CN) ppm.
N), 1226 (C–N). 1H-NMR (d4-MeOH, 400 MHz): 1.49 (s, 6H, (CH3)2), 2.06 (s, 3H, CH3), 2.81 (s, 2H, CH2), 3.43 (t, 2H, CH2–NH2+, J = 4.8 Hz), 3.70 (s, 2H, CH2–NC); 13C-NMR (d4-MeOH, 400 MHz): 20.4 (CH3)2, 23.3 (CH3), 41.2 (C(CH3)–CH2–C(CH3)2), 43.7 (CH2–NH2+), 46.9 (CH2–NC), 58.4 (C–(CH3)2), 175.8 (CN) ppm.
[H2L](CIO4)2 (2) Yield: 76%; m.p. 175.8–176.7 °C; UV-vis λ 238 nm, ε 403 M−1 cm−1; FT-IR (KBr) (νmax/cm−1): 3466, 3407 (N–H), 2981 (C–H), 1667 (CN), 1114 (C–N). 1H-NMR (d6-DMSO, 400 MHz): 1.33 (s, 6H, (CH3)2), 1.92 (s, 3H, CH3), 2.65 (s, 2H, CH2), 3.39 (m, 2H, CH2–NH2+), 3.39 (m, 2H, CH2–NC), 8.56 (s, 2H, NH2+); 13C-NMR (d6-DMSO, 400 MHz): 21.5 (CH3)2, 24.5 (CH3), 41.3 (C(CH3)–CH2–C(CH3)2), 43.6 (CH2–NH2+), 47.3 (CH2–NC), 58.2 (C–(CH3)2), 174.7 (CN) ppm.
[Cu(II)LBr]Br (1a): Yield: 86%; m.p. 190.3–190.8 °C; UV-vis λ 257 nm, ε 9444 M−1 cm−1; λ 504 nm, ε 194 M−1 cm−1; FT-IR (KBr) (νmax/cm−1): 3217 (N–H), 2968 (C–H), 1669 (CN), 1163 (C–N). The data were in accordance with the literature.16
[Cu(II)L(CIO4)](CIO4) (2a) and [Cu(II)L](CIO4)2 (2b). Complex isomer 2a was obtained by reaction of ligand 2 (48.1 mg, 0.1 mmol) and a metal salt, Cu(OAc)2·H2O (20 mg, 0.1 mmol), in methanol. The mixture was continuously mixed for 30 minutes, producing a purple solution. The purple mixture was filtered and allowed to evaporate at room temperature until the reddish-orange crystal of 2a was obtained. The crystals were filtered and rinsed several times using ethanol, followed by drying in a desiccator. The reaction was repeated using metal salt, CuCl2·2H2O (17 mg, 0.1 mmol), to yield a complex of isomer 2b.
[Cu(II)L(CIO4)](CIO4) (2a): Yield: 53%; m.p. 229.7–230.7 °C; UV-vis λ 261 nm, ε 6183 M−1 cm−1; λ 506 nm, ε 122 M−1 cm−1; FT-IR (KBr) (νmax/cm−1): 3213 (N–H), 2982 (C–H), 1670 (CN), 1054 (C–N).
[Cu(II)L](CIO4)2 (2b): Yield: 37%; m.p. 198.9–200.4 °C; UV-vis λ 259 nm, ε 5262 M−1 cm−1; λ 506 nm, ε 117 M−1 cm−1; FT-IR (KBr) (νmax/cm−1): 3205 (N–H), 2985 (C–H), 1663 (CN), 1058 (C–N).
2.3. DNA binding experiments
The complexes binding studies towards the calf thymus (CT-DNA) were performed in Tris–HCI buffer (5 mM Tris, 50 mM NaCl, pH 7.1) at ambient temperature. CT-DNA absorbance at 260 nm and 280 nm were recorded, and the ratio from the absorbance value was divided to examine the purity. The 1.8–1.9 ratio indicates that CT-DNA was free from protein.17 In order to calculate the concentration of CT-DNA, the molar absorption coefficient of 6600 M−1 cm−1 at 260 nm was used.18
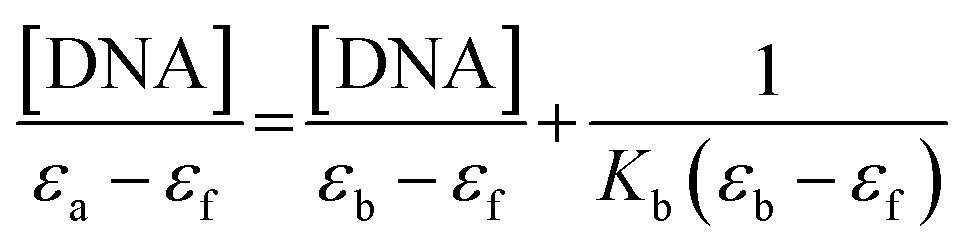 | (1) |
 | (2) |
2.4. Fabrication of sensor
15 μL of gold nanoparticles, AuNPs was dropped onto screen-printed carbon electrode, SPCE and was left to react overnight. The modified electrode was then deposited with 10 μL of 3-mercaptopropionic acid, MPA (1 mM) and incubated for 3 hours in dark conditions to form covalent bonds Au–S, followed by washing with ethanol to remove excess MPA and allowed to dry. 10 μL of the coupling reagent N-hydroxysuccinimide:1-ethyl-3-(3-dimethylaminopropyl)carbodiimide, NHS: EDC (400 mM![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100 mM) was placed onto the modified electrode and incubated for 1 hour. The modified electrode was then rinsed with distilled water before dropping 10 μL of 5 μM probe DNA and was allowed to react overnight. The modified electrode was washed using a buffer to remove the unbound aminated probe DNA. The 10 μL mixture of 30 μM 2b complex and between 1 × 10−13 M to 1 × 10−8 M of complementary DNA was placed onto the modified electrode to allow the hybridization process for 1 hour. Then, the modified electrode was soaked in a buffer for 1 minute to remove excess complementary DNA. The DNA biosensor response was evaluated by the DPV technique. The electrochemical behaviour of the modified SPCE (AuNPs/MPA/NHS:EDC) was characterized using DPV in 0.5 mM potassium ferricyanide solution, [Fe(CN)6]3−/4− containing 0.05 M PBS (pH 7.0) within the potential range of −0.8 V to 1.1 V at 5 mV s−1 scan rate. The additional DPV parameters were tabulated in Table S1 in the ESI.† The modified biosensor was determined using impedance analysis before and after the hybridization with complementary DNA (DNA target). The concept of the developed sensor is illustrated in Fig. 1.
:100 mM) was placed onto the modified electrode and incubated for 1 hour. The modified electrode was then rinsed with distilled water before dropping 10 μL of 5 μM probe DNA and was allowed to react overnight. The modified electrode was washed using a buffer to remove the unbound aminated probe DNA. The 10 μL mixture of 30 μM 2b complex and between 1 × 10−13 M to 1 × 10−8 M of complementary DNA was placed onto the modified electrode to allow the hybridization process for 1 hour. Then, the modified electrode was soaked in a buffer for 1 minute to remove excess complementary DNA. The DNA biosensor response was evaluated by the DPV technique. The electrochemical behaviour of the modified SPCE (AuNPs/MPA/NHS:EDC) was characterized using DPV in 0.5 mM potassium ferricyanide solution, [Fe(CN)6]3−/4− containing 0.05 M PBS (pH 7.0) within the potential range of −0.8 V to 1.1 V at 5 mV s−1 scan rate. The additional DPV parameters were tabulated in Table S1 in the ESI.† The modified biosensor was determined using impedance analysis before and after the hybridization with complementary DNA (DNA target). The concept of the developed sensor is illustrated in Fig. 1.
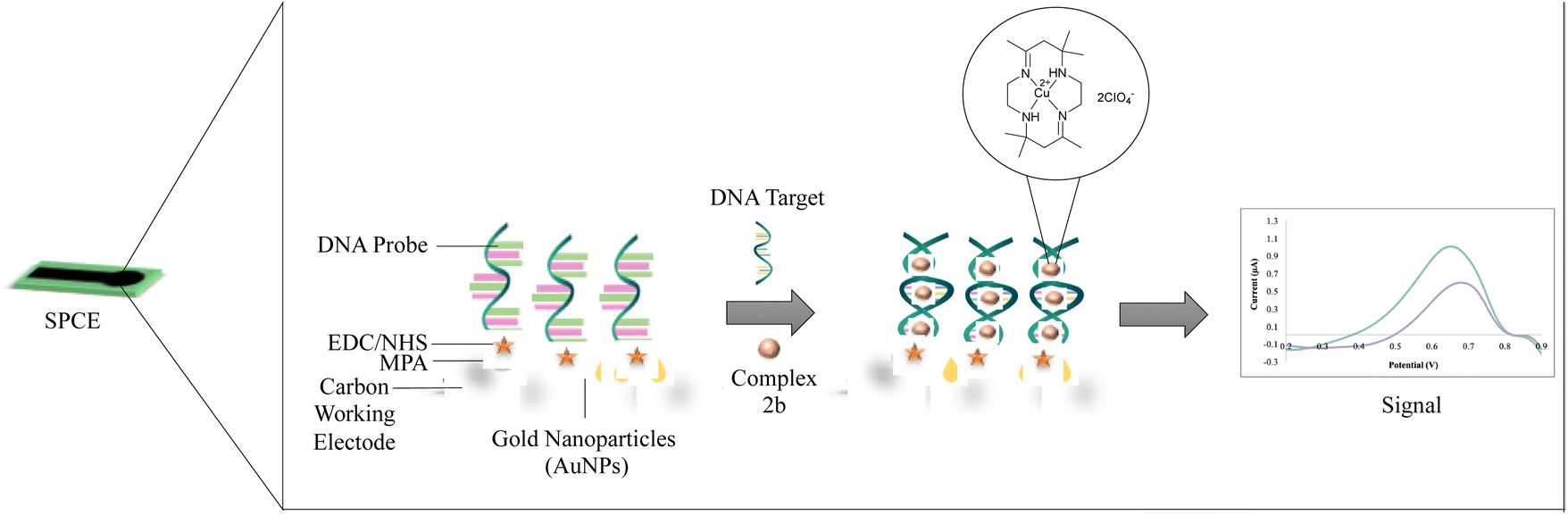 | ||
| Fig. 1 The conceptual scheme of the developed biosensor. | ||
2.5. Performance study of the fabricated porcine sensor
The porcine biosensor performance was studied in three categories: selectivity, linear range, reproducibility and shelf life. All the measurements were done in triplicates. The selectivity study analyzed the fabricated sensor's current intensity using different target DNA: complementary, probe, chicken and beef at 1 × 10−8 M DNA concentration. The linear range analysis assessed the fabricated sensor at different DNA concentrations from 1 × 10−13 M to 1 × 10−8 M. Next, the reproducibility study examined four independent sensors at complementary DNA concentrations of 1 × 10−2 μM and 1 × 10−7 μM, respectively. The shelf life experiments conducted until 23 days.2.6. Real samples preparation
The new porcine DNA biosensor was tested with raw pork, chicken and beef samples. DNA from each sample was extracted according to the Spin-Column Protocol by Qiagen DNeasy Blood and Tissue Kit. The extracted DNA was hybridized with the constructed porcine DNA biosensor with the presence of copper(II) tetraaza complex. The signal received from pork DNA was compared with chicken and beef DNA responses of the biosensor.3. Results and discussion
The macrocyclic ligand [H2L]X2, X = Br (1), CIO4 (2) reacts with metal salts in 1:1 ratio in methanol solution to give copper(II) macrocyclic complexes [Cu(II)LX]X, X = Br (1a), CIO4 (2a) and [Cu(II)L](CIO4)2 (2b) as shown in Scheme 1. The ligand and complexes were able to dissolve in common organic solvents. Spectroscopic analysis and single-crystal X-ray diffraction were used to characterize the macrocyclic ligand and copper(II) complexes.
3.1. Synthesis and characterization
The synthetic yield of complex 1a and 2a are moderate at 86% and 53%, respectively. Meanwhile, complex 2b has a low yield, 37%, because the complex exists in two isomeric forms, trans and cis. The isomer isolated under slow evaporation in this study was trans. The trans isomer adopts a conformation in which the hydrogen atoms attached to the nitrogen atoms are on the opposite side of the macrocyclic plane. The cis isomer can be separated by recrystallization in aqueous methanol solution.22The spectroscopic data of complex 1a, 2a and 2b are first discussed in this work. From the macrocyclic ligand's infrared spectrum, two bands of ν(N–H) were detected in the region 3466–3402 cm−1, corresponding to the primary amine. However, only one band of ν(N–H) at 3217–3205 cm−1 was observed for the complexes due to the secondary amine group's presence. The ν(N–H) stretching mode of the complexes undergoes a notable shift to lower frequencies than the free ligand. This suggests that the amino group coordinates with copper ions.23
The 1H NMR spectrum of macrocyclic ligand [H2L]X2, X = Br (1), CIO4 (2) showed a sharp signal δ 1.33–2.06, for which it was assigned to the methyl protons (–CH3; 9H). This signal confirmed that the cyclization between acetone and ethylene diamine has successfully occurred. The methylene protons were observed in the region δ 2.65–3.70 (–CH2; 6H). The primary amine protons were detected at δ 8.56 (–NH2; 2H) by showing a singlet signal. The 13C NMR spectra of the ligands showed the signal's characteristics to the imine group (CN). All other carbons in the macrocyclic were also observed at their appropriate positions.
The absorption spectra of the macrocyclic ligand and complexes were measured in distilled water. The absorption spectra of ligands showed one intense band at 238–246 nm assignable to π–π* transition. The π–π* transition was referred to as the azomethine CN chromophore of the ligand. A new broad peak was observed for the complexes at 504–506 nm, referring to the d–d transition. The presence of d–d transition peaks confirmed the formation of the complexes.
The X-ray molecular structures of complexes 1a, 2a and 2b have been reported earlier by other literature using different reactants and methods.24–27 Complex 1a has monoclinic crystal system with space group P21/c, a = 17.8403(16) Å, b = 15.4797(13) Å, c = 17.2018(14) Å, β = 112.037(2)°, Z = 12 and V = 4403.4(7) Å3. The tetraaza ligand coordinated to the copper atom through all four nitrogen atoms. One bromo atom was also coordinated to the metal centre copper. Hence, the complex possesses square pyramidal geometry. Another bromine ion acts as a counter anion of the monocation complex.
Complex 2a has monoclinic crystal system with space group P21/c, a = 10.4803(11) Å, b = 16.9627(17) Å, c = 13.8527(15) Å, β = 105.145(3)°, Z = 8 and V = 2377.1(4) Å3. The copper metal centre and one perchlorate ion were coordinated to all four nitrogen atoms, making the complex possess square pyramidal geometry. The other perchlorate anion stays as the counter anion of the complex.
For the complex 2b, the crystal system is monoclinic with the space group P21/c, a = 10.3221(7) Å, b = 10.6267(7) Å, c = 11.0213(7) Å, β = 111.8020(18)°, Z = 4 and V = 1122.45(13) Å3. Only the copper metal centre coordinated to all four nitrogen atoms. Hence, the complex possesses a square planar geometry. The two perchlorate anions balance the complex by having a +2 charge.
3.2. DNA binding studies
| Complex | Absorption Study | Luminescence study | |||
|---|---|---|---|---|---|
| Hypochromism (%) | Red or blue shift (nm) | Binding constant Kb (M−1) | Fluorescence decreasing factor | Stern volmer quenching constant, Ksv (M−1) | |
| 1a | 3 | 1 | 2.24 × 104 | 0.85 | 2.74 × 104 |
| 2a | 5 | 1 | 1.53 × 104 | 0.79 | 2.54 × 104 |
| 2b | 6 | 1 | 5.02 × 104 | 0.84 | 4.01 × 104 |
The small hypochromism and slight red or blueshift proposed that the complexes interact electrostatically with DNA. Electrostatic interactions involve the external binding between positively charged complexes with negatively charged DNA phosphates. The changes in spectral characteristics observed were in agreement with the tetraaza copper complex, [CuL1]2+, where L1 = 1,4,8,11-tetraazacyclotetradecane and hexaaza copper complex [CuL2]2+, where L2 = 3,10-bis(2-hydroxyethyl)-1,3,5,8,10,12-hexaazacyclotetradecane that are reported to bind electrostatically with DNA.28
The DNA binding strength can be determined from the intrinsic binding constant Kb for 1a, 2a and 2b complexes were 2.24 × 104 M−1, 1.53 × 104 M−1 and 5.02 × 104 M−1, respectively. For comparison, the complexes that interact with DNA electrostatically have intrinsic binding constant Kb in the range of 2.5 × 104 M−1 to 8.5 × 104 M−1.29–33 These complexes' binding strengths at magnitude 104 are identical to complexes 1a, 2a, and 2b. Hence, the complexes clearly bind to DNA by electrostatic mode. The complexes' binding strengths were arranged as followed 2b > 1a > 2a. The differences in the binding strength might be because of the dissimilar charge and counter ion of metal complexes. It can be explained by Coulomb's law which states that the electrostatic attraction force is directly proportional to the magnitude of positively and negatively charged ions and inversely proportional to the square value of the distance between the ionic charge. For complex 2b, the complex carries a +2 charge, thus making the strength of the electrostatic attraction between the complex and DNA higher than the other complexes. Meanwhile, for 1a and 2a, both complexes carry +1 charges. However, complex 1a has higher binding strength than complex 2a due to the smaller size of the monoatomic bromide anion ligand compared to the polyatomic perchlorate anion ligand. The small size of the anion ligand allowed the complex to interact more strongly with the DNA.
The Stern Volmer quenching constant was applied to analyse synthesised complexes' quenching strength. The value of the quenching constant for 1a, 2a and 2b complexes were 2.74 × 104 M−1, 2.54 × 104 M−1 and 4.01 × 104 M−1, respectively, as illustrated in Table 1. Complex 2b had the highest Ksv value. It can be concluded that the quenching ability of complex 2b was the strongest compared to other complexes. The complex's higher quenching ability implies a higher DNA binding ability towards the complex. In this case, complex 2b favourably interacts with DNA compared to complex 1a and 2a. The magnitude of Stern Volmer quenching constant for complexes 1a, 2a and 2b were similar to the non-intercalator compounds, which interact electrostatically with DNA.30,32,36,37
Fig. 2(a) illustrates the changes in DNA relative viscosity after adding complexes 1a, 2a, and 2b. As the complexes' concentration increased, DNA solutions' viscosity displayed no significant changes, less than 0.05 (η/η0)1/3. The findings indicated that complexes 1a, 2a, and 2b interact with DNA electrostatically. These behaviours were similar to the [Ru(bpy)3]2+ (bpy = bipyridine), a known complex that has an electrostatic binding mode when interacting with DNA. Notably, complex 2b interacts strongly with DNA compared to complexes 1a and 2a because the extent of DNA viscosity changes was the biggest. These results and the spectroscopic results mentioned above were consistent with each other.
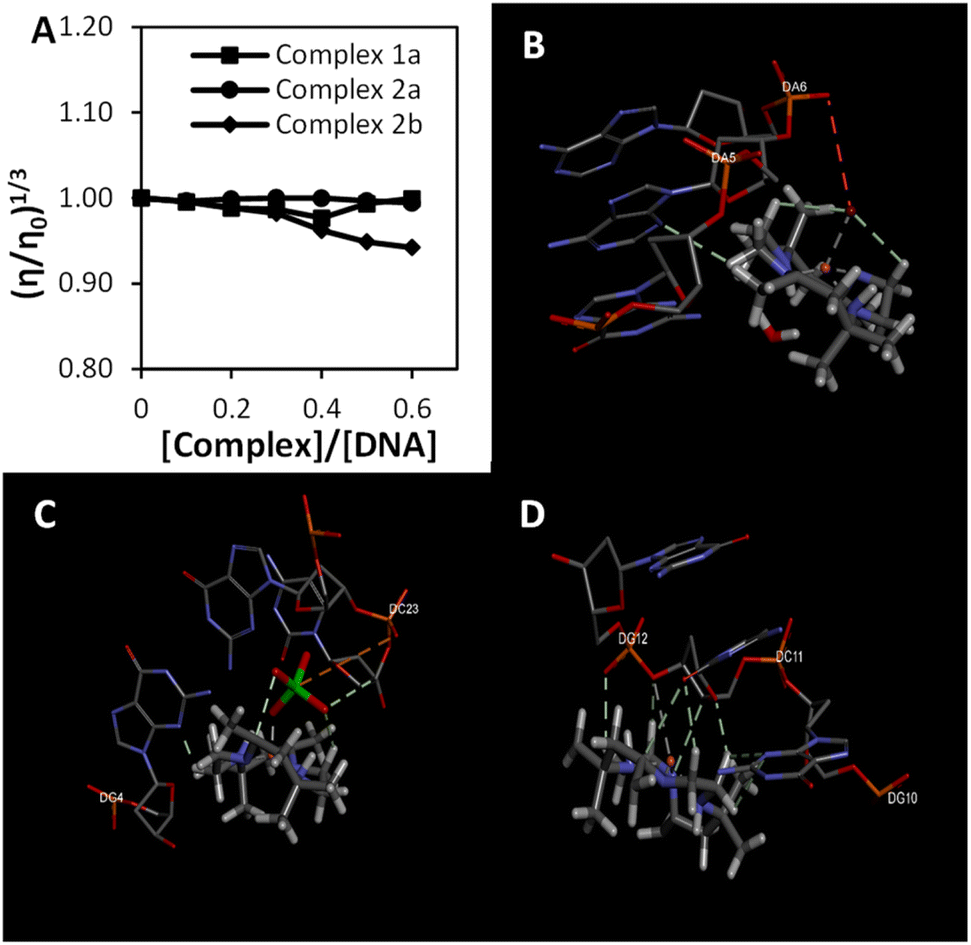 | ||
| Fig. 2 The binding study evaluations using viscosity and computational study (a) effects of the increasing amount of 1a, 2a, and 2b complexes on the relative viscosity of CT-DNA (b–d) molecular docked structures of the complex with DNA dodecamer duplex of sequence d(CGCGAATTCGCG)2 (PDB 1D: 1BNA). (b) Complex 1a, (c) complex 2a, (d) complex 2b. | ||
3.3. Electrochemical behaviour of modified SPCE in [Fe(CN)6]3−/4−
The layer of modification on SPCE was analyzed using DPV in potassium ferricyanide solution (Fe(CN)63−/4−) 0.5 mM within the range of −0.8 V to 1.1 V at a 5 mV s−1 scan rate (Fig. 3(a)). This characterization was conducted to study the electron transfer between each layer of the DNA biosensor and the electrode.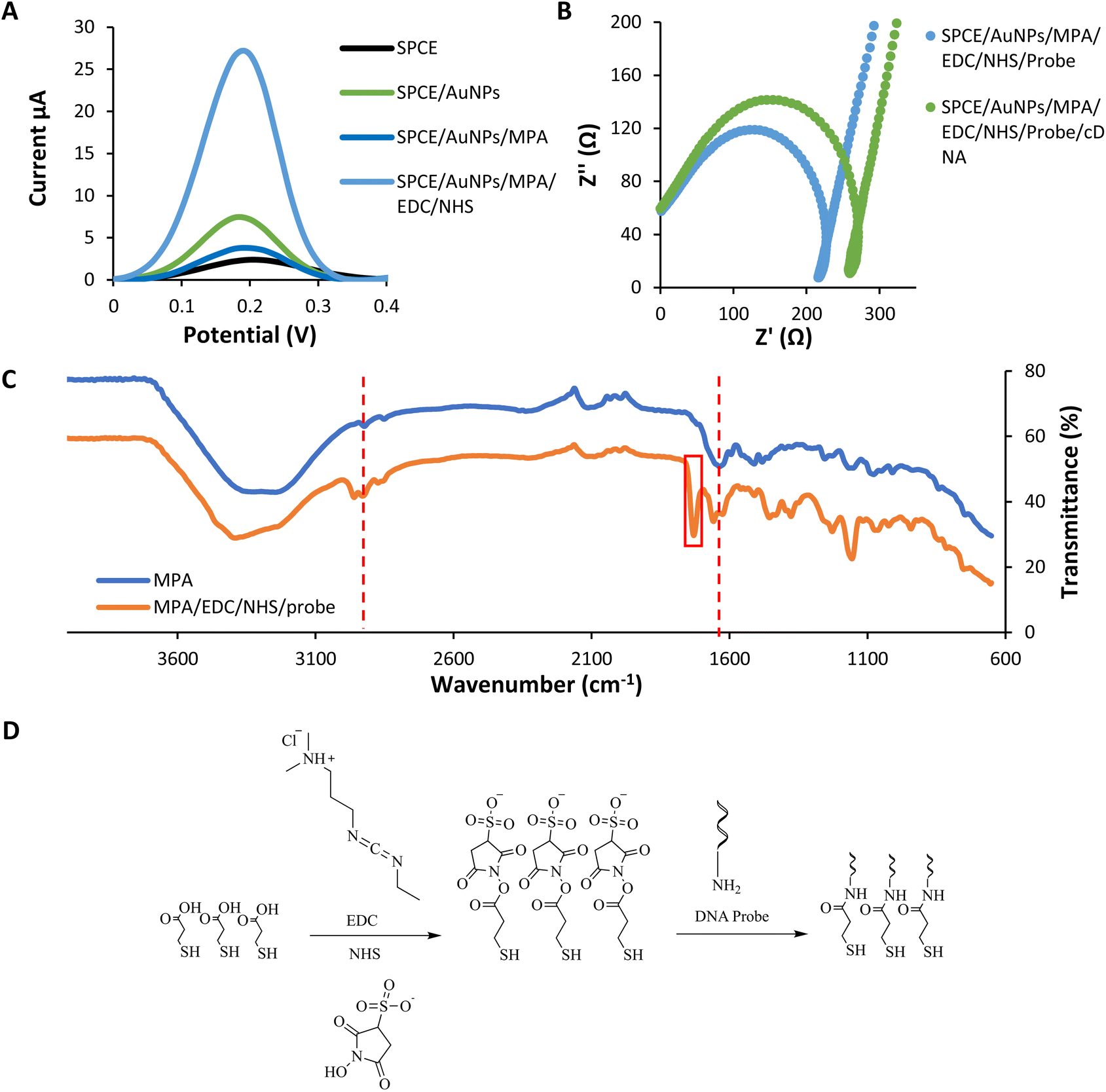 | ||
| Fig. 3 Layers analysis of the modified SPCE (a) the differential pulse voltammogram response of each self-assembled monolayer (b) the impedance spectroscopy of before and after DNA hybridization in potassium ferricyanide solution (Fe(CN)63−/4−) 0.5 mM at 5 mV s−1 scan rate (c) the infrared spectrum of the modified biosensor (d) the reaction scheme of MPA/EDC/NHS/Probe. | ||
The immobilization of the gold nanoparticles (AuNPs) enhances the current peak compared to the bare SPCE due to the gold's conductive properties.42 Gold nanoparticles have high electron transfer performance on the modified electrode. In contrast, the peak later decreases dramatically after the assembling of MPA onto the gold layer. It is because of the interruption of electron transfer caused by the MPA layers.42 MPA formed a dense layer on the gold surface and could be ascribed to the strong chain between MPA. The immobilization of EDC–NHS induces the current peak higher compared to the previous layer. The surface of –COOH groups of MPA are activated by EDC and converted into active esters intermediates. NHS converts the intermediates of EDC into a more stable active ester. This intermediate product showed good electrochemical activity.43 According to Trammell et al., the intermediate product could delocalize electrons, thus enhancing the electron tunnelling and electrons' movement.44 All these data indicated that the fabrication of each assembled monolayer was successful.
Besides DPV analysis, the characterization of every single layer was analyzed using the impedance technique (Fig. 3(b)). In impedance analysis, we are focusing on before and after DNA hybridization. Based on the Nyquist plot, the resistance charge transfer value (RCT) increased due to increased negative charge from the DNA backbone after the hybridization process.45 The negative charge repelling between DNA and potassium ferricyanide solution cause a DNA blocking effect. This effect prevents electron transfer through the electrode surface.46 The RCT value increased due to the DNA blocking effect.47,48
3.4. Layers analysis using infrared
From the infrared analysis (Fig. 3(c and d)), the assembly of the MPA layer was confirmed by the appearance of medium C–H stretching and strong CO acid stretching at 2923 cm−1 and 1635 cm−1. The successful functionalization of the DNA probe onto the layer was determined by the presence of a new vibrational peak of CO amide at 1729 cm−1 corresponds to the formation of a covalent bond between MPA and the aminated probe.49 The functionalization happens with the use of coupling agent EDC and NHS. Another important vibrational peak indicating the DNA probe's functionalization was the strong CO amide stretching of the oligonucleotides at 1658 cm−1.50
3.5. Analytical performance
The designed biosensor was fabricated based on an MPA self-assembly monolayer formation on the SPCE, modified with AuNPs. The MPA molecule consists of a thiol functional group with a high affinity toward gold. In contrast, the carboxyl group at the other end of the MPA molecule is suitable for covalently attached to DNA after the activation process of the MPA. Activation procedures involved the use of EDC and also NHS. Table S2 in ESI† shows all of the parameters optimized for fabricating a porcine DNA biosensor with slight modification from the previous studies.51This biosensor's linear concentration range based on the complex 2b redox label was 1 × 10−13 M–1 × 10−8 M with the correlation coefficient, R2 = 0.993 [Fig. 4(a)]. Within this range, the current response increases correspondingly, proving that more hybridization occurs as the concentration of target DNA increases. This electrochemical biosensor's detection limit (LOD) was 5.7 × 10−14 M calculated using LOD = [(mean blank + 3 standard deviation) − intercept]/slope.
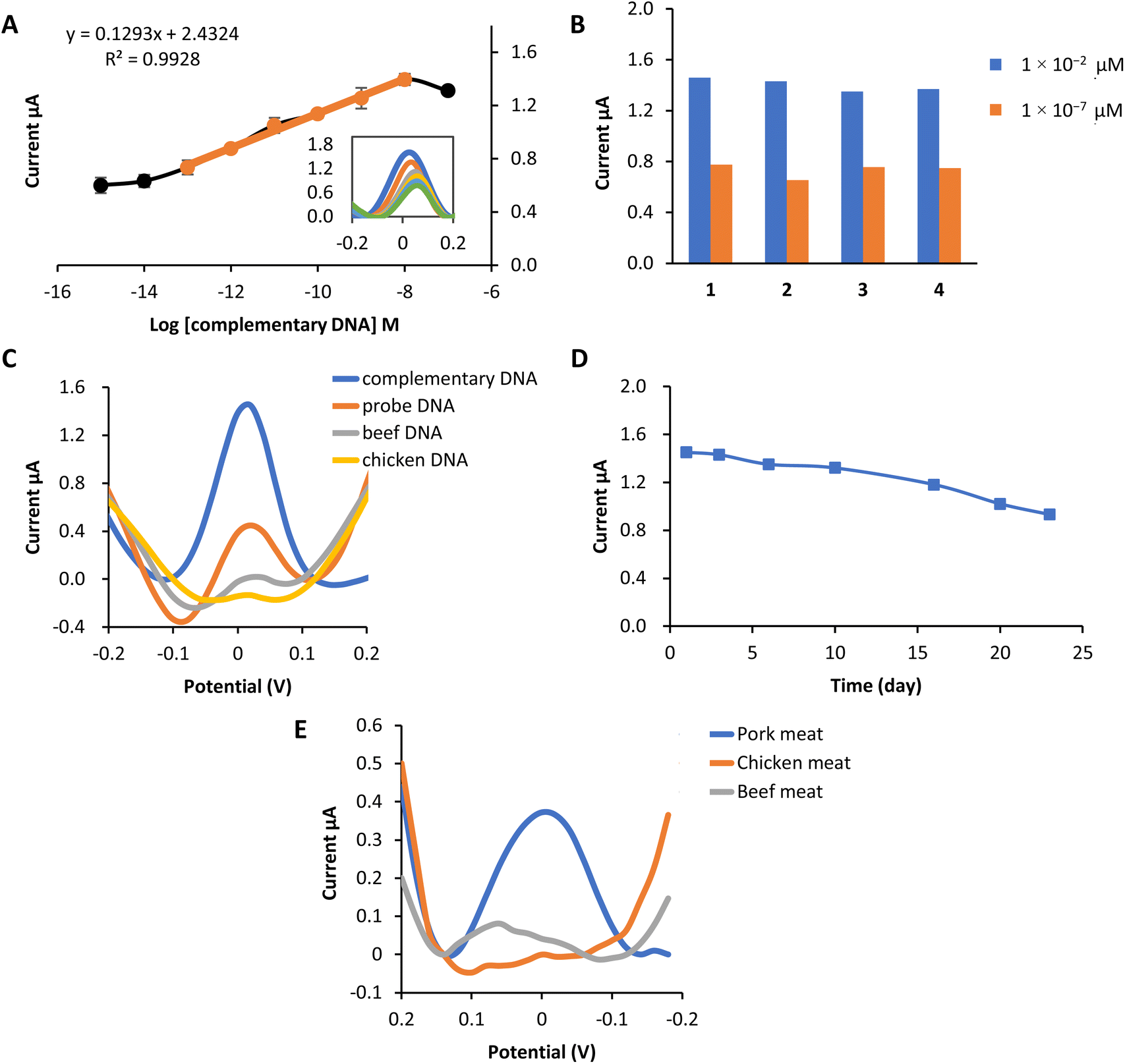 | ||
| Fig. 4 Performance of the biosensor and real sample analysis (a) the linear response range between 1 × 10−13 M–1 × 10−8 M and the dynamic range of the porcine DNA biosensor. (b) Reproducibility of the porcine DNA biosensor at concentrations of 1 × 10−2 μM and 1 × 10−7 μM. (c) Selectivity of the porcine DNA biosensor based on complex 2b as redox indicator toward the determination of probe DNA only, complementary DNA, beef DNA and chicken DNA at 1 × 10−2 μM. (d) Shelf life of the porcine DNA biosensor at the concentration of 1 × 10−2 μM store at 4 °C (e) A comparative analysis of the response of the constructed porcine DNA biosensor with extracted DNA from pork, chicken and beef meat. | ||
A reproducibility analysis was carried out using two different concentrations to assess the reusability of this electrochemical biosensor. Fig. 4(b) proved that the porcine DNA biosensor was able to provide a repeatable DPV response of 1 × 10−2 μM and 1 × 10−7 μM. The average reproducibility relative standard deviation (RSD) of each calibration point evaluated using this new DNA biosensor was estimated to be about 3.7% (n = 4) and 7.3% (n = 4), respectively.
The selectivity study of the DNA biosensor for detecting porcine DNA using copper complex 2b as the redox indicator is presented in Fig. 4(c). A selectivity study was conducted by comparing the DPV response of the DNA biosensor toward DNA probe only, complementary DNA (cDNA), beef DNA, and chicken DNA at 1 × 10−8 M (concentration). Hybridization of probe DNA showed to be profoundly specific toward complementary DNA of porcine (cDNA) as the hybridization response of complex 2b gave the highest current compared to other DNA. Peak current response at 1.45 μA indicates that complex 2b had been successfully interacting electrostatically with the phosphate backbone of immobilized probe DNA and cDNA. A significant difference can be seen with the immobilized single-stranded DNA probe before and after hybridization with other DNA. The DPV response of probe DNA only is (0.45 μA), while beef DNA and chicken DNA showed almost no biosensor response. This might be due to the ability of complex 2b to bind to the phosphate group of probe DNA only. Meanwhile, no complex 2b interacts with probe DNA after hybridization with other DNA. This is because complex 2b and the single-stranded chicken and beef are mixed first before the hybridization process, so complex 2b can bind to the DNA phosphate backbone before being deposited onto the modified electrode. However, they are washed off together because of the inability to form duplex DNA with the porcine DNA probes, hence producing practically no response.52
The lifetime of the biosensor is demonstrated in Fig. 4(d). From the study, the biosensor response remained stable from day 1 to day 16, in which the current was reduced to 81% of its initial value after being kept at 4 °C. Starting from day 20, the biosensor response decreased to 70% and below. This is due to the degradation of the biosensor system, such as gold nanoparticles (AuNPs), MPA or probe DNA from the electrode surface. Therefore, reduced the amount of probe subjected to target complementary DNA, eventually leading to the decrease in biosensor signal.
3.6. Porcine detection
The effectiveness of the constructed porcine DNA biosensor was tested with several kinds of meat. Pork, chicken and beef meat were used in this experiment. DNA from each sample was extracted using Qiagen DNeasy Blood and Tissue Kit. It is obviously shown that the DNA signal from the extracted pork meat gives the highest current value compared to chicken meat and beef meat (Fig. 4(e)). Chicken and beef meat samples give almost no response to the biosensor. This newly porcine DNA biosensor has excellent selectivity towards pork meat extracted DNA.3.7. Comparison on the performance of the reported electrochemical porcine biosensor
The comparison of the constructed biosensor performances with previously reported electrochemical porcine DNA biosensors using different DNA matrix immobilization and redox compounds is tabulated in Table 2. This comparison is essential in order to validate the developed biosensor. From the table, the limit of detection obtained in this study is the lowest and has a broad linear range. Thus, we concluded the high sensitivity of the constructed biosensor compared to other DNA ESI† and redox compounds due to the large surface area of AuNPs, which have increased the DNA binding capacity. Another essential factor that enhances the sensitivity of the constructed biosensor is the four amine groups contained in the copper(II) tetraaza complex redox compound. This compound has a very high interaction with DNA and can increase the DNA binding capacity.| Matrix immobilization | Redox compound | Linear range | LOD | References |
|---|---|---|---|---|
| SPCE/AuNPs/MPA/EDC–NHS | Copper(II) tetraaza complex | 1 × 10−13 M– 1 × 10−8 M | 5.7 × 10−14 M | This work |
| SPCE/AuNPs/NBA-NAS | Ruthenium(II) complex | 1 × 10−13 M–1 × 10−8 M | — | 53 |
| Screen printed carbon–reduced graphene oxide (SPC–RGO) electrode | Guanine | 0–10 μg mL−1 | 1.8 × 10−6 g mL−1 | 54 |
| SPCE/AuNPs | Methylene blue | 0.1–5.0 μg mL−1 | 5.8 × 10−7 g mL−1 | 55 |
| SPCE | Luminol | — | 1.0 × 10−10 g mL−1 | 56 |
| Graphene biochips | Ruthenium hexamine complex | — | 1.0 × 10−7 g mL−1 | 57 |
| Disposable electrochemical printed (dep) chips | Hoechst33258 | — | 2.0 × 10−5 g mL−1 | 58 |
| SPCE/AuNPs/SiNSs/GA | Hexaferrocenium complex | 1 × 10−12 to 1 × 10−9 M | 4.8 × 10−14 M | 59 |
4. Conclusions
In summary, the interactions between tetraaza macrocyclic complexes [Cu(II)LBr]Br (1a), [Cu(II)L(CIO4)](CIO4) (2a) and [Cu(II)L](CIO4)2 (2b) where L = 5,5,7,12,12,14-hexamethyl-1,4,8,11-tetraazacyclotetradeca-7,14-diene, with DNA were studied by spectroscopic techniques and viscosity measurements. The results collected proved that these complexes interact with DNA by electrostatic mode. The strength of binding constants, Kb of complexes, were arranged in 2b > 1a > 2a. A sensitive and selective electrochemical porcine biosensor using copper(II) tetraaza complexes 2b has been established. The developed sensor exhibits a broad linear range and good repeatability. The real sample analysis indicated that the biosensor has excellent selectivity to the extracted pork DNA. These results displayed that the copper(II) complex 2b has the potential to be used as an indicator in the prototype development of a porcine detection biosensor.Author contributions
Noraisyah Abdul Kadir Jilani: writing-original draft, formal analysis, data curation, methodology, validation, investigation. Emma Izzati Zakariah: writing-original draft, data curation. Eda Yuhana Ariffin: writing-original draft, data curation, validation, investigation. Suhaila Sapari: writing-original draft. Devika Nokarajoo: formal analysis, investigation. Bohari Yamin: supervision, resources. Siti Aishah Hasbullah: writing-review & editing, supervision, resources.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors offer thanks to Ministry of Higher Education of Malaysia for the research grant, FRGS/1/2022/STG04/UKM/02/4. Special thanks to the Faculty of Science & Technology and Centre for Research and Instrumentation Management, Universiti Kebangsaan Malaysia, for making the equipment available and aiding technically.References
- Y. Erwanto, M. Z. Abidin, E. Y. P. M. Sugiyono and A. Rohman, Asian-Australas. J. Anim. Sci., 2014, 27, 1487 CrossRef CAS
.
- S. M. K. Uddin, M. M. Hossain, Z. Z. Chowdhury and M. R. B. Johan, Food Addit. Contam., Part A, 2021, 38, 1273–1288 CrossRef CAS
- J. Lunn and H. Theobald, Nutr. Bull., 2006, 31, 178–224 CrossRef
- X.-D. Pan, J. Chen, Q. Chen, B.-F. Huang and J.-L. Han, RSC Adv., 2018, 8, 11157–11162 RSC
- N. Kusnin, N. A. Yusof, J. Abdullah, S. Sabri, F. Mohammad, S. Mustafa, N. A. Ab Mutalib, S. Sato, S. Takenaka and N. A. Parmin, RSC Adv., 2020, 10, 27336–27345 RSC
- H. A. El-Boraey and O. A. El-Gammal, Spectrochim. Acta, Part A, 2015, 138, 553–562 CrossRef CAS PubMed
- N. Arshad and S. I. Farooqi, Appl. Biochem. Biotechnol., 2018, 186, 1090–1110 CrossRef CAS PubMed
- H. Goudarziafshar, S. Yousefi, Y. A. Tyula, M. Dušek and V. Eigner, RSC Adv., 2022, 12, 13580–13592 RSC
- A. Ramanavičius, A. Ramanavičienė and A. Malinauskas, Electrochim. Acta, 2006, 51, 6025–6037 CrossRef
- X. Hai, Y. Li, C. Zhu, W. Song, J. Cao and S. Bi, TrAC, Trends Anal. Chem., 2020, 133, 116098 CrossRef CAS
- Q. Liu, J. Zhang, M.-Q. Wang, D.-W. Zhang, Q.-S. Lu, Y. Huang, H.-H. Lin and X.-Q. Yu, Eur. J. Med. Chem., 2010, 45, 5302–5308 CrossRef CAS PubMed
- P. Arthi, A. Haleel, P. Srinivasan, D. Prabhu, C. Arulvasu and A. K. Rahiman, Spectrochim. Acta, Part A, 2014, 129, 400–414 CrossRef CAS PubMed
- L. Yang, X. Li, X. Li, S. Yan, Y. Ren, M. Wang, P. Liu, Y. Dong and C. Zhang, Anal. Biochem., 2016, 492, 56–62 CrossRef CAS PubMed
- G. Li, N. Liu, S. Liu and S. Zhang, Electrochim. Acta, 2008, 53, 2870–2876 CrossRef CAS
- B. M. Yamin, W. Ismail and J.-C. Daran, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2012, 68, m886–m887 CrossRef CAS PubMed
- B. M. Yamin, A. Kadir and S. F. M. Yusoff, Indones. J. Chem. Res., 2015, 2, 171–175 Search PubMed
- J. Marmur, J. Mol. Biol., 1961, 3, 208 CrossRef CAS
- M. Reichmann, S. Rice, C. Thomas and P. Doty, J. Am. Chem. Soc., 1954, 76, 3047–3053 CrossRef CAS
- A. Wolfe, G. H. Shimer Jr and T. Meehan, Biochemistry, 1987, 26, 6392–6396 CrossRef CAS PubMed
- A. Pyle, J. Rehmann, R. Meshoyrer, C. Kumar, N. Turro and J. K. Barton, J. Am. Chem. Soc., 1989, 111, 3051–3058 CrossRef CAS
- J. K. Barton, J. M. Goldberg, C. V. Kumar and N. J. Turro, J. Am. Chem. Soc., 1986, 108, 2081–2088 CrossRef CAS
- T. Lu, T. Lee, B. Liang and C. Chung, J. Inorg. Nucl. Chem., 1981, 43, 2333–2336 CrossRef CAS
- B. Vinay Kumar, H. S. Bhojya Naik, D. Girija, N. Sharath, S. M. Pradeepa, H. Joy Hoskeri and M. C. Prabhakara, Spectrochim. Acta, Part A, 2012, 94, 192–199 CrossRef CAS
- S. Feifei and S. Baiwang, J. Chem. Crystallogr., 2011, 41, 1790–1793 CrossRef
- A. Bieńko, J. Mroziński and R. Kruszyński, Pol. J. Chem., 2008, 82, 1383 Search PubMed
- T.-J. Lee, T.-H. Lu, C.-S. Chung and T.-Y. Lee, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1984, 40, 70–72 CrossRef
- N. F. Curtis and J. Wikaira, Inorg. Chim. Acta, 2013, 396, 14–16 CrossRef CAS
- J. Liu, H. Zhang, C. Chen, H. Deng, T. Lu and L. Ji, Dalton Trans., 2003, 114–119 RSC
- B. Shekhar, P. Vasantha, B. Sathish Kumar, P. Anantha Lakshmi, V. Ravi Kumar and S. Satyanarayana, Appl. Organomet. Chem., 2019, 33, e5086 Search PubMed
- C. P. Popolin, J. P. Reis, A. B. Becceneri, A. E. Graminha, M. A. Almeida, R. S. Correa, L. A. Colina-Vegas, J. Ellena, A. A. Batista and M. R. Cominetti, PLoS One, 2017, 12, e0183275 CrossRef PubMed
- K. Anuja, K. H. Reddy, D. Dhanalakshmi, K. Srinivasulu and Y. Nagamani, Int. J. Pharm. Sci. Res., 2022, 13, 977–986 CAS
- N. Shivakumara and P. M. Krishna, J. Mol. Struct., 2020, 1199, 126999 CrossRef
- L. Conti, A. Mengoni, G. E. Giacomazzo, L. Mari, M. Perfetti, C. Fagorzi, L. Sorace, B. Valtancoli and C. Giorgi, J. Inorg. Biochem., 2021, 220, 111467 CrossRef CAS PubMed
- R. Esteghamat-Panah, H. Hadadzadeh, H. Farrokhpour, M. Mortazavi and Z. Amirghofran, Inorg. Chim. Acta, 2017, 454, 184–196 CrossRef CAS
- R. Kumari, S. Banerjee, P. Roy and M. Nath, Appl. Organomet. Chem., 2020, 34, e5283 CrossRef CAS
- S. Yousuf, F. Arjmand, C. Pettinari and S. Tabassum, Inorg. Chim. Acta, 2022, 542, 121105 CrossRef CAS
- K. B. Kale, M. A. Shinde, R. H. Patil and D. P. Ottoor, Spectrochim. Acta, Part A, 2022, 264, 120329 CrossRef CAS PubMed
- A. Majumder, S. Haldar, N. Dutta, A. Das and M. Bera, ChemistrySelect, 2022, 7, e202104319 CrossRef CAS
- S.-B. Kou, K.-L. Zhou, Z.-Y. Lin, Y.-Y. Lou, J.-H. Shi and Y.-X. Liu, J. Mol. Liq., 2021, 328, 115491 CrossRef CAS
- N. Shahabadi, S. Amiri and H. Zhaleh, J. Coord. Chem., 2020, 73, 35–51 CrossRef CAS
- T. Kondori, N. Akbarzadeh-T, H. Ghaznavi, Z. Karimi, J. Shahraki, R. Sheervalilou and O. Shahraki, BioMetals, 2020, 33, 365–378 CrossRef CAS PubMed
- H.-E. Lee, Y. O. Kang and S.-H. Choi, Int. J. Electrochem. Sci., 2014, 9, 6793–6808 Search PubMed
- E. B. Bahadır and M. K. Sezgintürk, Artif. Cells, Nanomed., Biotechnol., 2016, 44, 462–470 CrossRef PubMed
- S. A. Trammell, D. S. Seferos, M. Moore, D. A. Lowy, G. C. Bazan, J. G. Kushmerick and N. Lebedev, Langmuir, 2007, 23, 942–948 CrossRef CAS
- M. S. Cha, J. K. Lee, S. H. Cho, J. G. Park, H. Y. Lee, S. H. Lim and Y. R. Yoon, J. Nanosci. Nanotechnol., 2013, 13, 5245–5249 CrossRef CAS PubMed
- L. Santiago-Rodríguez, G. Sánchez-Pomales and C. R. Cabrera, Electroanalysis, 2010, 22, 399–405 CrossRef
- B.-Y. Chang and S.-M. Park, Annu. Rev. Anal. Chem., 2010, 3, 207 CrossRef CAS PubMed
- A. I. B. Tamayo, L. S. L. Rizo, M. B. de Armas, A. A. P. Ferreira, D. Manzani, H. Yamanaka and A. M. E. Guas, Microchem. J., 2020, 153, 104462 CrossRef CAS
- M. Masteri-Farahani and N. Mosleh, J. Mater. Sci.: Mater. Electron., 2019, 30, 21170–21176 CrossRef CAS
- J. Kong and S. Yu, Acta Biochim. Biophys. Sin., 2007, 39, 549–559 CrossRef CAS PubMed
- E. I. Zakariah and S. A. Hasbullah, Logam Rutenium Agen Pengesan DNA Babi, Universiti Kebangsaan Malaysia Press, Malaysia, 2019 Search PubMed
- K. Ohara, M. Smietana and J. J. Vasseur, J. Am. Soc. Mass Spectrom., 2006, 17, 283–291 CrossRef CAS PubMed
- N. I. A. Halid, E. I. Zakariah, L. Y. Heng, N. H. Abd Karim, H. Ahmad and S. A. Hasbullah, Malaysian J. Anal. Sci., 2016, 20, 1020–1032 CrossRef
- Y. W. Hartati, T. A. Setiawati, T. Sofyatin, F. Fitrilawati, A. Anggraeni and S. Gaffar, ScienceAsia, 2020, 46, 72 CAS
- Y. W. Hartati, A. A. Suryani, M. Agustina, S. Gaffar and A. Anggraeni, Food Anal. Methods, 2019, 12, 2591–2600 CrossRef
- N. F. N. Azam, S. Roy, S. A. Lim and M. Uddin Ahmed, Food Chem., 2018, 248, 29–36 CrossRef CAS PubMed
- S. Roy, I. A. Rahman, J. H. Santos and M. U. Ahmed, Food Control, 2016, 61, 70–78 CrossRef CAS
- M. U. Ahmed, Q. Hasan, M. Mosharraf Hossain, M. Saito and E. Tamiya, Food Control, 2010, 21, 599–605 CrossRef CAS
- E. Y. Ariffin, E. I. Zakariah, F. Ruslin, M. Kassim, B. M. Yamin, L. Y. Heng and S. A. Hasbullah, Sci. Rep., 2021, 11, 1–14 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ra05701h |
| This journal is © The Royal Society of Chemistry 2023 |