
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFluorescent detection of brown spot of tobacco caused by Alternaria alternata based on lambda exonuclease-induced DNAzyme amplification
Kai Lian†
a,
Guangyan Chen†a,
Xiaoqiang Wangb,
Wenna Zhangc,
Xihao Hud,
Hui Wangb,
Yusen Lia,
Dongmei Xia and
Ying Wang *a
*a
aCollege of Life Science, Linyi University, Linyi 276005, China. E-mail: m13854869219@163.com
bPlant Protection Research Center, Tobacco Research Institute of Chinese Academy of Agricultural Sciences, Qingdao 266101, China
cShandong (Linyi) Institute of Modern Agriculture, Zhejiang University, Linyi 276000, China
dShandong Tobacco Company Qingdao Branch, Qingdao 266101, China
First published on 9th January 2023
Abstract
A rapid, simple, and sensitive fluorescent detection method for brown spot of tobacco is established by lambda exonuclease-induced Mg2+-dependent DNAzyme amplification. It contains hybridization of the Alternaria alternata genome and HP1, digestion of the 5′-phosphorylated strand of the hybrid dsDNA by lambda exonuclease, acquisition of complete Mg2+-dependent DNAzyme, cleavage of the substrate modified with FAM and BHQ-1, and fluorescent detection. The proposed assay exhibits good sensitivity (10 pg L−1), selectivity and reproducibility. The method does not require pure DNA and expensive instruments, and can be performed within 2.5 hours. To the best of our knowledge, this is the first report of fluorescent detection of Alternaria alternata and its tobacco field samples. This method can be applied to the rapid and sensitive detection of Alternaria alternata in tobacco and its seedlings, and is particularly important for the green prevention and control of tobacco brown spot disease.
1. Introduction
Brown spot is a serious tobacco disease caused by Alternaria alternata (A. alternata), and occurs mainly at the mature stage of tobacco leaf.1 Since it was first reported in the United States in 1892, the disease has repeatedly caused huge economic losses to the tobacco industry around the world.2–4 It mainly harms leaf, stem, pedicel, and capsule, and greatly reduces the economic value of tobacco.5 Lots of methods including plate culture method, serological method, PCR method, Mass spectrometry,6 high performance liquid chromatography,7 and surface-enhanced Raman spectroscopy have been reported for detection of A. alternata.8,9 However, these detection methods are tedious, time-consuming, costly, and require precise, advanced equipment and professional detection personnel, so they are not suitable for on-site detection.10 At present, immunocolloidal gold dipstick method is commonly used for qualitative detection in field detection, and it is difficult to detect the whole population of a pathogen because of the restriction of the antigen antibody reaction method.11,12 For example, a colloidal gold test strip can be used to detect brown spot disease caused by Alternaria longus in tobacco, and the detection limit is 20 ng mL−1, but it is not suitable for detecting brown spot disease caused by other species of Alternaria alternata.13 Therefore, it is very important to establish an economical, accurate and rapid detection method for prevention and control of brown spot of tobacco.Deoxyribozyme amplifies signals through specific recognition and shear cycling, which is very attractive for efficient and rapid detection of pathogens.14–16 Deoxyribozymes are single-stranded DNA molecule with catalytic function obtained by in vitro screening technology.17,18 They are folded into specific spatial structure to catalyze specific reactions, such as RNA cutting,19,20 DNA/RNA linking,18,21 DNA phosphorylation,22 etc. Compared with traditional protein enzymes, deoxyribozymes have good chemical stability and are hardly affected by pH and temperature. They have small molecular weights and are easy to be synthesized and modified.23 Lambda exonuclease is a 5′–3′ exonuclease, which catalyze the removal of nucleotides stepwise at the 5′-phosphorylated strands of double-stranded DNA, and shows low activity to non-phosphorylated substrates, such as cleaved DNA, single-stranded DNA and double-stranded DNA. Thus, lambda exonuclease provides an alternative methods for strategies related to exonuclease-based cascade amplification.
In this study, a novel fluorescent method is established for A. alternata detection by lambda exonuclease and Mg2+-dependent DNAzyme. A species-specific sequence of A. alternata is hybridized to HP1, and 5′-phosphorylated strand of HP1 in the hybrid strand are degraded stepwise by lambda exonuclease, releasing partial sequence of Mg2+-dependent DNAzyme. This product is hybridized to P1 bearing another part of the Mg2+-dependent DNAzyme sequence to assemble the complete Mg2+-dependent DNAzyme. In the presence of Mg2+, the enzyme recognizes and cleaves the ribonucleotide of its substrate MB. FAM and BHQ-1 modified at both ends of MB are separated at room temperature, and the fluorescence signal appears.
2. Materials and methods
2.1. Chemicals
Lambda exonuclease is acquired from Thermo Fisher Scientific Ltd (Shanghai, China). 20 bp DNA ladder is purchased from TaKaRa Bio Ltd (Dalian, China). Rapid DNA Extraction Detection Kit (KG203), lnRcute lncRNA First-Strand cDNA Kit (KR202), and RNA Easy Fast Plant Tissue RNA Rapid Extraction Kit (DP452) are acquired from Tiangen Biotech Co., Ltd (Beijing, China). Ultrapure water (electrical resistance above 18.2 MΩ) used in the experiment is acquired from an ULUP-IV system (Shanghai, China). Other reagents used in this experiment are analytically pure grade and have not been further purified.Internal transcribed spacer (ITS) of the species of A. alternata were highly conservative. In order to improve the sensitivity and selectivity of the detection method, ITS sequences of A. alternata genome were acquired from NCBI, and aligned by DNAMAN software (version 7, LynnonBiosoft Co., Ltd). The GenBank IDs of these A. alternata ITS were MG601454.1, KY550233.1, NR_136120.1, KC492447.1, MN654955.1, EU520171.1, MT704965.1, OW986459.1, ON796496.1-ON796488.1, ON745551.1, ON740895.1, ON738701.1, ON738588.1. A high conserved segment (Target, Table 1) was found in the ITS region of A. alternata genome, which originally used as primer in PCR.24 So, we designed the fluorescent detection strategy to target this segment of the Alternaria alternata genome. Strains of A. alternata, Rastonia solanacearum, Phytophthora nicotianae, and tabacco samples carring A. alternata, cucumber mosaic virus (CMV), tobacco mosaic virus (TMV), 4 positive tabacco samples and 26 field tobacco samples, were kindly provided from a laboratory of Plant Protection Research Center in Tobacco Research Institute of Chinese Academy of Agricultural Sciences. Oligonucleotides were synthesized by Sangon Biotech (Shanghai) Co., Ltd and listed in Table 1.
| Name | Base sequence (5′–3′) |
|---|---|
| Target | TGCAATCAGCGTCAGTAACAAAT |
| HP1 | 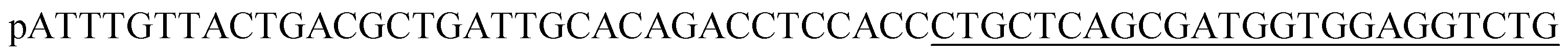 |
| P1 | 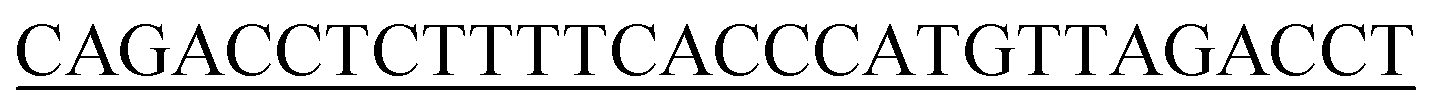 |
| MB | FAM-TGACGTAAGGTCTATrAGGAGCAGTACGTCA-BHQ-1 |
| A–F | AGGCCGGCTG CCAATTAC |
2.2. Apparatus
Fluorescence was monitored by a F4600 spectrofluorimeter (Hitachi, Tokyo, Japan). Range of emission spectra was from 500 to 650 nm and the excitation and emission wavelengths were 494 nm. The excitation and emission slit widths were set at 5 nm. NanoDrop 2000 (Thermo, New York, USA) was used for concentration measurements of genomes in the experiment. Isothermal reaction was performed in a heating block (Thermo, New York, USA). Polyacrylamide gel electrophoresis (PAGE) is performed in double board sandwich type vertical electrophoresis apparatus (DYCZ-30C, Liuyi Co., Ltd, Beijing, China). Gel pictures were captured by a WD-9413A imaging system (Liuyi Co., Ltd, Beijing, China).2.3. Genome extraction
Tobacco plants infected with A. alternata and healthy tobacco plants were put into grinding bags respectively, and then buffer solution (10 mM Tris-Cl, 1 mM EDTA pH 8.0) with twice the quality of tobacco leaf was added for grinding. The suspensions were heated in boiled water for 5 min, and cooled down in ice-water, and vortexed for 1 min. The above steps were repeated one more time. The genomic DNA was released from the cell wall of A. alternata. The result suspension was centrifuged, and the genome of A. alternata in the supernatant was used as the target, and its concentration was quantified by this method. Rapid DNA Extraction Detection Kit (KG203) was used to extract and purify genomic DNA of Alternaria alternate, Phytophthora nicotianae, and Ralstonia solancearum from their liquid medium. These pure genomes were used for sensitivity, selectivity, and reproducibility tests in this method. Total RNAs of tobacco infected by TMV or CMV were purified by RNA Easy Fast Plant Tissue RNA Rapid Extraction Kit (DP452). cDNAs of TMV and CMV were obtained by lnRcute lncRNA First-Strand cDNA Kit (KR202). Reverse transcription of TMV and CMV were done with 5× reverse transcription supermix (2 μL), primer (10 μM, 2 μL), and total plant RNA (6 μL). The reaction was performed at 42 °C for 30 min, followed by suppression of reverse transcriptase activity at 85 °C for 5 min.2.4. Procedures for A. alternata detection
Genomic DNA of A. alternata was hybridized to HP1 in 1× reaction buffer of lambda exonuclease. In the presence of lambda exonuclease, HP1/A. alternata genome hybrid was degraded stepwise from the 5-phosphorylated end of HP1 until it becomes a single-stranded DNA. It was reacted in 50 μL 1× reaction buffer (67 mM Glycine-KOH 2.5 mM MgCl2 0.01% (v/v) Triton X-100 (pH 9.4 @ 25 °C)). Hybrid product and lambda exonuclease (1U) were added into the reaction solution. It was performed at 37 °C, and then incubated at 75 °C for 10 min to inactivation of lambda exonuclease. P1 was then introduced in the solutions, and incubated for a period time at room temperature. At last, 30 μL of 1 μM MB, 10 μL of 20 mM Mg2+ and 160 μL of buffer solution (25 mM HEPES, 100 mM NaCl, pH 7.0) were introduced into the solution. The solution was incubated at room temperature for 40 min, and the fluorescence intensity was then measured by the F4600 spectrofluorimeter. To improve hybridization efficiency, samples to be hybridized were mixed and incubated at 95 °C for 1 min to eliminate their secondary structures, followed by incubation at room temperature for a period of time for subsequent reactions. DNA that need to be hybridized went through this procedure in the experiment.The results of the hybridization and digestion were analyzed by PAGE(8%). It was carried out in 1× TBE (50 mM Tris-boric acid, 1 mM EDTA). Each lane was loaded with 5 μL of sample, and electrophoresed at 120 V for 120 min. The PAGE gel is colored by silver stain. Gel picture was scanned by the WD-9413A imaging system.
2.5. PCR method
PCR systems included Taq PCR Master Mix (25 μL), Target (10 μM, 2 μL), A–F (10 μM, 2 μL), DNA template (2 μL), and H2O (19 μL). PCR condition was divided into three steps, the first step was a one minute incubation at 94 °C, and the second step was 35 thermal cycles, including denaturation at 94 °C for 30 s, annealing at 55 °C for 30 s, and extension at 72 °C for 30 s. The last step was an extension at 72 °C for 5 min. PCR products were then analyzed by 1% agarose gel electrophoresis.3. Results and discussion
3.1. Principle of the assay
The method is shown in Scheme 1, and it involves three steps, (1) hybridization and exonuclease reaction, (2) formation of complete Mg2+-dependent DNAzyme, (3) cleavage of molecular beaconing (MB) and fluorescent detection. HP1 is designed with three regions, (a) the trans-complementary sequence of part ITS gene of A. alternata, position 1st–23rd of 5′-terminal of HP1(orange in Scheme 1), (b) the stem part of HP1, position 24th–35th (grass green) and 48th–59th (light green), (c) part sequence of Mg2+-dependent DNAzyme, position 36th–47th of HP1 (light pink, sky blue and purple). The 5′-terminal hydroxyl group of HP 1 is attached to a phosphate group. P1 contains a trans-complementary sequence (1st–8th. fluorescent green) of HP1(52nd–59th), a short interval sequence (9th–12th, fluorescent green), and another part sequence of Mg2+-dependent DNAzyme (13th–28th, Indian red and yellow). The ribonucleotide-containing molecular beacon (MB) is a fluorescent-labeled oligonucleotide consisting of a loop region, a stem region, a FAM and a BHQ-1 group, which is the substrate of Mg2+-dependent DNAzyme. First, genome of A. alternata (deep sea blue) is extracted in tobacco leaves with brown spots, and hybridized with nucleotides of 1st–23rd of 5′-terminal of HP1. Lambda exonuclease can efficiently degrade the 5′-phosphorylated strand of dsDNA hybrid until it becomes a single DNA strand, that is, 1–35 nucleotides of hybrid HP1 is removed at 37 °C. Partial sequence of Mg2+-dependent DNAzyme (36th–59th of HP1) is released from the stem-loop structure of HP1. Genome of A. alternata goes free, and starts a new round of hybridization with HP1, and cleavage process in the presence of lambda exonuclease. Second, P1 is then introduced to the solution, and hybridized with the partial sequence of Mg2+-dependent DNAzyme to obtain the complete Mg2+-dependent DNAzyme, which is hybridized and nicked MB at rA site in the presence of Mg2+. As a result of this scission of the substrate, the distance between FAM and BHQ is enough to generate great fluorescence intensity, thus providing the optical readout signal for sensing of A. alternata. The Mg2+-dependent DNAzyme continuously proceeds hybridization-cleavage step of MBs, and the fluorescence signal increases significantly. However, if there is no genome of A. alternata, 5′-protrude terminal of HP1 cannot be digested by lambda exonuclease, partial sequence of Mg2+-dependent DNAzyme is still caged in the duplex structure of HP1. No intact Mg2+-dependent DNAzyme is assembled, and the ribonucleotide-containing MB is not be cleaved, and fluorescence can almost not be monitered.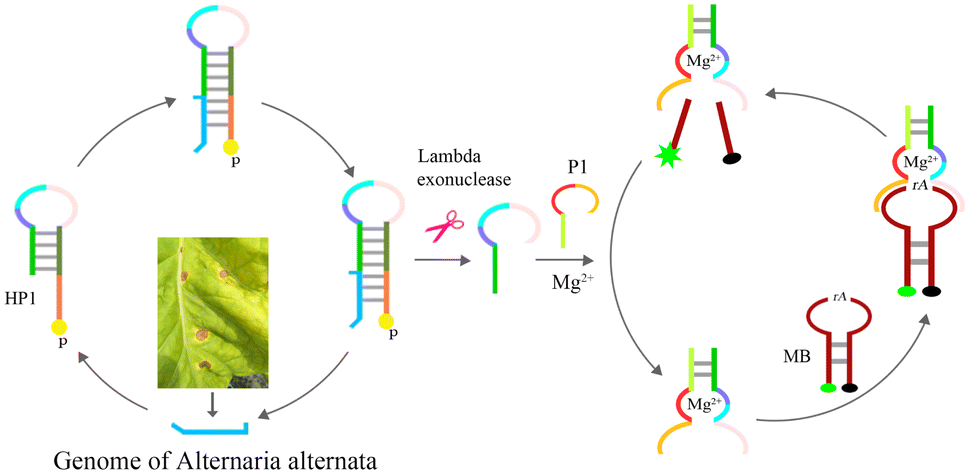 | ||
| Scheme 1 Schematic illustration of lambda exonuclease-induced DNAzyme amplification for fluorescent detection of A. alternata. Genome of A. alternata is extracted in tobacco leaves with brown spots, and hybridized with 5′-terminal of HP1, which is degraded by lambda exonuclease. Partial sequence of Mg2+-dependent DNAzyme is released from HP1, and then hybridized with P1 to obtain the complete Mg2+-dependent DNAzyme, which continuously cleaves ribonucleotide-containing molecular beacon (MB) substrate in the presence of Mg2+. FAM moves far away from BHQ, greater fluorescence intensity was detected. | ||
3.2. Feasibility of fluorescent detection of A. alternata
The genome of A. alternata contains millions of nucleotides, and the hybridization between genome of A. alternata and HP1 cannot be determined by PAGE. In order to better reflect the effect of hybridization and enzyme digestion, genomic DNA of A. alternata was replace by Target sequence. Target is a 23 nucleotide DNA sequence, which is a high conserved sequence found in ITS region of A. Alternata genome (Table 1). 20 bp DNA lander consists of 13 double-stranded DNA fragments from 20 bp–200 bp, and is used as a measure of molecular weight of double-stranded DNA in PAGE. Since HP1, Target, and HP1/Target hybrid are short single-stranded DNA or partial double-stranded DNA in the assay, the lander could only be used as a reference, and not an accurately indication of the size of these DNAs. The effect of hybridization and enzyme digestion in this assay was verified by 8% PAGE (Fig. 1A). HP1 was in lane 1, and Target was in lane 2. The 5′-terminal of HP1 and Target were hybridized and formed HP1/Target hybrid (lane 3). Lambda exonuclease catalyzed the degradation of the double-stranded portion of the HP1/Target hybrid. HP1 was degraded from the 5′-phosphorylated end until it becomes a single strand, that is, the 1st to 35th nucleotides of HP1 were successively degraded to single nucleotides. The product was a DNA sequence consisting of 24 nucleotides (positions 36–59 of HP1), bearing a partial sequence of Mg2+-dependent DNAzyme. Target (23 nucleotides) was released from HP1/Target hybrid. Since the nucleotide number of the partial sequence of Mg2+-dependent DNAzyme and Target is similar, their electrophoretic bands were overlapped (lane 4). The results show HP1 could hybridize to Target, and 1–35 nt sequence of HP1 was indeed digested by lambda exonuclease.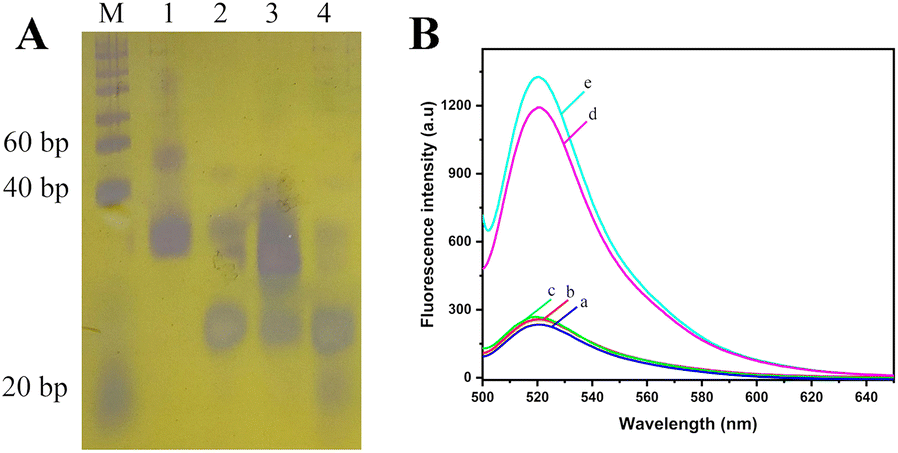 | ||
| Fig. 1 (A) Analysis of HP1/Target hybridization and lambda exonuclease digestion by 8% PAGE. M, 20 bp DNA lander, lane 1, HP1, lane 2, Target, lane 3, HP1 + Target, lane 4, HP1 + Target + lambda exonuclease. (B) Fluorescence emission spectra of solution containing only MB (a), genome DNA of healthy tobacco plant (b), and in the absence of lambda exonuclease (c), total DNA of tobacco plant infected by A. alternata (d), and genome DNA of A. alternata (e). The concentrations of HP1, P1, MB, and lambda exonuclease are 1.0 μM, 1.2 μM, 1 μM, and 1 U, respectively. | ||
In order to verify the feasibility of this method for quantitative detection of A. alternata, a series of samples were detected by this method, and the fluorescence emission spectra of each solution were obtained. As shown in Fig. 1B, no obvious fluorescence signal was detected in the solution containing only MB (curve a). The distance between FAM and BHQ was close, and the fluorescence was quenched. Similar fluorescence signals were detected in the solutions with genomic DNA of healthy tobacco plants (curve b) or reaction system without lambda exonuclease (curve c). In the presence of total DNA of tobacco plant infected by A. alternata (curve d) or genome of A. alternata (curve e), significant fluorescence signals near 520 nm were detected in the solutions. The results indicated that the proposed method could effectively monitor A. alternata, and was used for the detection of A. alternata in tobacco plants.
3.3. Optimizing the conditions for maximizing the fluorescence intensity
In this experiment, the fluorescence intensity depends on the reaction conditions of lambda exonuclease-induced DNAzyme amplification, such as digestion time by lambda exonuclease, concentration of P1, hybrid time of A. alternata genome and HP1, and the part sequence of Mg2+-dependent DNAzyme and P1. In order to obtain the best performance for quantitative detection of A. alternata, these experiment conditions were optimized respectively.Lambda exonuclease catalyzes the progressive removal of nucleotides from the 5′-terminal of double-stranded DNA. The optimal substrate for this enzyme is 5′-phosphorylated double-stranded DNA, while the degradation rate of non-phosphorylated substrates is relatively low. To reduce the cost and reaction time, digestion time of lambda exonuclease was optimized, and the concentration of A. alternata genome was 0.1 ng L−1. As shown in Fig. 2A, the fluorescence intensity increased with the reaction time from 5 to 20 min and dropped slightly beyond 20 min, indicating the completion of digestion reaction. Excessively long reaction time may lead to excessive digestion of HP1 in hybrid of A. alternata genome/HP1, thereby reducing the fluorescence intensity in the solution.25 Therefore, reaction time of 20 min was the best condition, and applied to the following experiment.
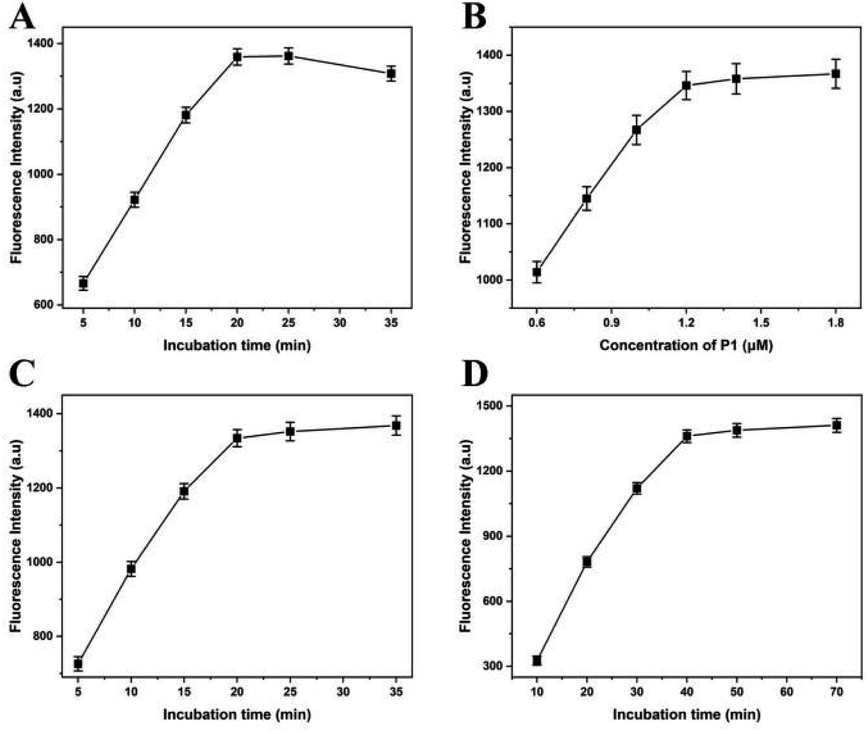 | ||
| Fig. 2 Optimization of digestion time of lambda exonuclease (A), concentration of P1 (B), hybridization time of HP1 and genome of A. alternata (C), and formation time of intact Mg2+-dependent DNAzyme (D). Concentration of A. alternata genome was 0.1 ng L−1 in the above experiments. Error bars in the figures represent the standard deviations of three experiments, and the same below. | ||
The concentration of P1 was increased from 0.6 to 1.8 μM, and the fluorescence intensity increased correspondingly. When the concentration of P1 was lower than 1.2 μM, the fluorescence intensity increased rapidly with the increase of P1 concentration.
When the concentration of P1 was greater than 1.2 μM, the fluorescence intensity increased slowly with the increase of P1 concentration (Fig. 2B). Therefore, P1 concentration of 1.2 μM was the optimal concentration, and used for the following experiments.
In order to improve the hybridization efficiency and shorten the hybridization time, genome of A. alternata and HP1 were mixed and subjected to 95 °C for 1 min to remove secondary structures, then slowly reduced to room temperature. The mixture was incubated at room temperature for 5–35 min. The results showed that the fluorescence intensity reached a plateau when the incubation time was more than 20 min (Fig. 2C). Therefore, 20 min of incubation time was optimal and used for the following experiments.
The A. alternata genome/HP1 hybrid was digested with lambda exonuclease to obtain the partial sequence of Mg2+-dependent DNAzyme, which was then mixed with P1 containing another partial sequence of Mg2+-dependent DNAzyme. After the similar heating, cooling and incubation processes, the complete Mg2+-dependent DNAzyme was acquired. As shown in Fig. 2D, fluorescence intensity increased along with the incubation time from 10 to 70 min. When the reaction time was 10 min, fluorescence signals could hardly be distinguished from the background. However, the fluorescence intensity was hardly no longer elevated once the reaction time was above 40 min. Therefore, the optimal time to form complete Mg2+-dependent DNAzyme was 40 min, which was used for the subsequent experiments.
3.4. Analytical performance of the fluorescence detection method
In order to evaluate the sensitivity of the assay, the fluorescence intensity of the solution was detected by this method when the genome concentration of A. alternata was in a range of 0–0.2 ng L−1. Greater amounts of A. alternata genome were used in the assay, and more A. alternata genome/HP1 hybrids were acquired. Lambda exonuclease digested the genome/HP1 hybrids, and greater amounts of partial sequence of Mg2+-dependent DNAzyme were obtained. The products were hybridized to more P1s, assembling more complete Mg2+-dependent DNAzyme. More MBs were nicked by Mg2+-dependent DNAzyme in the presence of Mg2+, causing a significant increase of fluorescence intensity. As shown in Fig. 3A, a dramatic increase of fluorescence intensity was acquired as the increase of A. alternata genome concentration, and the increase rate of fluorescence intensity decreased as the concentration of the genome was more than 0.1 ng L−1. The fluorescence intensity was linearly correlated with the logarithm of A. alternata genome in the concentration range of 10 pg L−1 to 0.1 ng L−1. The linear regression equation was F = −638.7 + 1040 lg(c), and the correction coefficient was 0.992 (Fig. 3B). F and c represent the fluorescence intensity and concentration of the genome of A. alternata, respectively. Based on the sum of blank response and three times standard deviation, the detection limit of the method was calculated to be 10 pg L−1. The results show that this strategy has considerable sensitivity for detecting A. alternata. Moreover, the sensitivity of the assay was also compared with that of other methods for A. alternata detection (Table 2).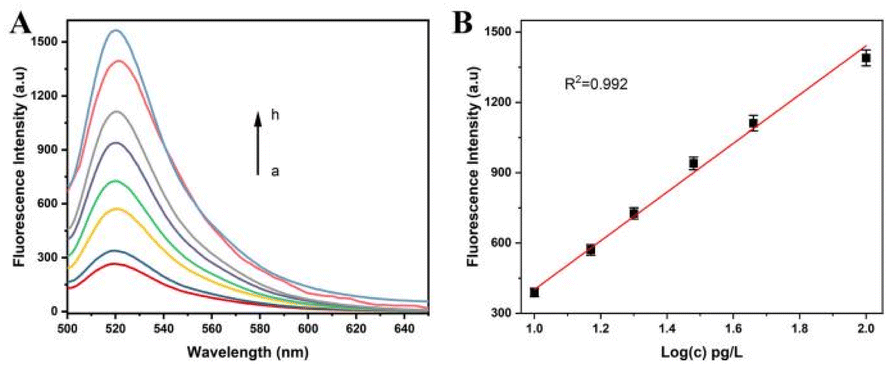 | ||
| Fig. 3 (A) Fluorescence intensity of the solutions at different genome concentrations of A. alternata. Arrow from a to h represents the genome concentrations of 0 pg L−1, 10 pg L−1, 15 pg L−1, 20 pg L−1, 30 pg L−1, 50 pg L−1, 0.1 ng L−1, and 0.2 ng L−1. (B) Linear correlation between fluorescence intensity and logarithm of A. alternata genome concentration from 10 pg L−1 to 0.1 ng L−1. Pg means picogram, and ng means nanogram. | ||
| Analytical method | Detection limit | Reference |
|---|---|---|
| Surface-enhanced Raman spectroscopy based silver nanodots array method | 1.0 × 103 cfu mL−1 | 8 |
| Colloidal gold test strips based method | 20 ng mL−1 | 26 |
| Real-time fluorescence PCR based method | 1.9 pg μL−1 | 27 |
| Loop-mediated isothermal amplification based method | 3 pg μL−1 | 28 |
| Chemiluminescence enzyme immunoassay based method | 0.068 ng mL−1 | 29 |
| PCR based method | 1.0 × 102 cfu mL−1 | 30 |
| Time-resolved fluoroimmunoassay based method | 0.78 ng mL−1 | 31 |
| This method | 10 pg L−1 |
The reproducibility of the assay was detected with 0.1 ng L−1 A. alternata genome. The intra-assay reproducibility was performed for three times, and the relative standard deviation value of three times was 2.2%. The inter-assay reproducibility test was performed for 3 consecutive days using the same amount DNA of A. alternata every time, and the relative standard deviation value was 3.8%. The results showed that the strategy had satisfied reproducibility.
3.5. Specificity of the method
Rastonia solanacearum, Phytophthora nicotianae, CMV and TMV are common pathogens in tobacco. Since co-infection of these pathogens with A. alternata is very common in tobacco fields, they are selected as the control for the specific test of this strategy. Since tobacco and pathogens contain a large amount of DNA and RNA, the specificity requirement of the method for detecting A. alternata is very high. The results show that the fluorescence intensities of the solutions bearing genomes of TMV (1), CMV (2), Rastonia solanacearum (3), Phytophthora nicotianae (4) were almost the same as that of the healthy tobacco plant (5). The fluorescence intensity were very low. However, the fluorescence intensity was high in the solution containing A. alternata genome (6) or total DNA of tobacco infected by A. alternata (7). The results show that the method had good selectivity in monitoring of A. alternata (Fig. 4).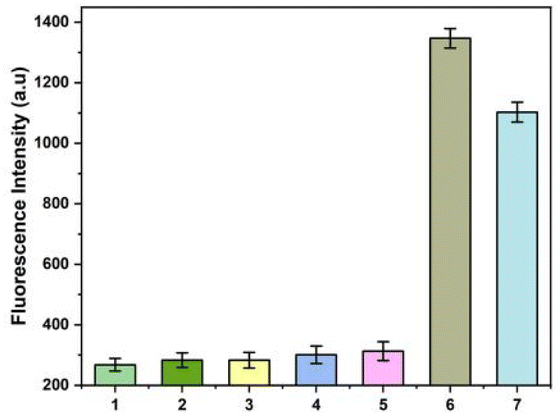 | ||
| Fig. 4 Selectivity of the proposed method. Total genome of tobacco plant infected by TMV (1), CMV (2), A. alternata (7). Genome of Rastonia solanacearum (3), genome of Phytophthora nicotianae (4), genome of healthy tobacco plant (5), and genome of A. alternata (6). Concentration of A. alternata genome was 0.1 ng L−1 in the experiments. | ||
3.6. Application of this method in tobacco field
PCR is the most common method for detecting target DNA. To verify the application of this strategy in field samples, 4 positive samples and 26 field samples were tested by this method and PCR. The detection results of both methods show that all positive samples and 11 field samples were infected by A. alternata. The results of the two methods were completely consistent, indicating that the method could be used for the detection of A. alternata in tobacco field (Table 3).| Sample | PCR | Fluorescent | ||
|---|---|---|---|---|
| Positive | Negative | Positive | Negative | |
| 4 | 4 | 0 | 4 | 0 |
| 26 | 11 | 15 | 11 | 15 |
4. Conclusions
In this study, a lambda exonuclease-induced DNAzyme signal amplification strategy for fluorescent detection of A. alternata is established. The proposed assay exhibites good sensitivity (10 pg L−1), selectivity and reproducibility. To the best of our knowledge, this is the first report of fluorimetric detection of A. alternata and its tobacco field samples. The method does not require pure DNA and expensive instruments, and can be performed within 2.5 hours. This method can be applied to the rapid and sensitive detection of A. alternata in tobacco and its seedlings, and is particularly important for the green prevention and control of tobacco brown spot disease.Author contributions
Kai Lian and Guangyan Chen, data curation, writing original draft. Wenna Zhang and Hui Wang, formal analysis, ethodology, Yusen Li, conceptualization and methodology, Xiaoqiang Wang and Xihao Hu, writing – review & editing. Dongmei Xi, funding acquisition and writing – review & editing. Ying Wang: funding acquisition, supervision, validation, writing, review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by ShanDong Students' Platform for innovation and entrepreneurship training program (S202210452017), Key Research and development project of Linyi City (2021030), Commissioned research projects of Linyi University (HX210035, HX210046), the Open Project Foundation of Shandong (Linyi) Institute of Modern Agriculture, Zhejiang University (ZDNY-2021-FWLY02012). Major science and technology projects of China Tobacco Corporation (110202101054(LS-14), 2022370200270186).References
- J. Stavely and L. Slana, Phytopathology, 1971, 61, 73–78 CrossRef.
- J. LaMondia, Plant Dis., 2001, 85, 230 Search PubMed.
- N. Tadakazu, U. Tamio and F. Hiroshi, Tetrahedron Lett., 1982, 23, 4469–4472 CrossRef.
- A. Tanaka, H. Shiotani, M. Yamamoto and T. Tsuge, Mol. Plant-Microbe Interact., 1999, 12, 691–702 CrossRef PubMed.
- Y. J. Jia, D. D. Cheng, W. B. Wang, H. Y. Gao, A. X. Liu, X. M. Li and Q. W. Meng, Physiol. Plant., 2010, 138, 164–175 CrossRef PubMed.
- R. Deshidi, S. Devari, M. Kushwaha, A. P. Gupta, R. Sharma, R. Chib, I. A. Khan, S. Jaglan and B. A. Shah, ChemistrySelect, 2017, 2, 362–368 CrossRef.
- Z. L. Xiao, Y. L. Wang, Y. D. Shen, Z. L. Xu, J. X. Dong, H. Wang, C. Situ, F. Wang, J. Y. Yang and H. T. Lei, Food Anal. Methods, 2018, 11, 635–645 CrossRef.
- T. Pan, D. Sun, H. Pu, Q. Wei, W. Xiao and Q. Wang, J. Food Eng., 2017, 215, 147–155 CrossRef CAS.
- A. Crespo Sempere, N. Estiarte, S. Marín, V. Sanchis and A. J. Ramos, Int. J. Food Microbiol., 2013, 165, 214–220 CrossRef CAS.
- O. Laczka, E. Baldrich, F. X. Munoz and F. D. Campo, Anal. Chem., 2008, 80, 7239–7247 CrossRef CAS.
- S. Nara, V. Tripathi, H. Singh and T. G. Shrivastav, Anal. Chim. Acta, 2010, 682, 66–71 CrossRef CAS PubMed.
- Y. D. Wu, M. J. Xu, Q. Q. Wang, C. X. Zhou, M. Wang, X. Q. Zhu and D. H. Zhou, Vet. Parasitol., 2017, 243, 199–203 CrossRef CAS.
- L. H. Yan Ji, X. Cai, C. Xu, Z. Fan, M. Shi, G. Tang, H. Ding and D. Yan, Tob. Sci. Technol., 2022, 55, 10–15 Search PubMed.
- Y. Wang, J. Liu and H. Zhou, Sensors, 2019, 19, 1298 CrossRef CAS PubMed.
- Y. Wang, B. Li, J. Liu and H. Zhou, Anal. Bioanal. Chem., 2019, 411, 2915–2924 CrossRef CAS.
- J. Riu and B. Giussani, TrAC, Trends Anal. Chem., 2020, 126, 115863 CrossRef CAS.
- S. C. B. Gopinath, Anal. Bioanal. Chem., 2007, 387, 171–182 CrossRef CAS PubMed.
- R. R. Breaker and G. F. Joyce, Chem. Biol., 1994, 1, 223–229 CrossRef CAS.
- A. Sreedhara, Y. Li and R. R. Breaker, J. Am. Chem. Soc., 2004, 126, 3454–3460 CrossRef CAS PubMed.
- D. R. Semlow and S. K. Silverman, J. Mol. Evol., 2005, 61, 207–215 CrossRef CAS.
- K. Schlosser, J. Gu, L. Sule and Y. Li, Nucleic Acids Res., 2008, 36, 1472–1481 CrossRef CAS.
- A. J. Camden, S. M. Walsh, S. H. Suk and S. K. Silverman, Biochemistry, 2016, 55, 2671–2676 CrossRef CAS PubMed.
- K. S. Scott, Org. Biomol. Chem., 2004, 19, 2701–2706 Search PubMed.
- X. Zhongyu, W. Xiaoqiang, S. Hong, S. Guangjun, L. Bin, C. Zhiyan, Z. Yongfeng, R. Mao, C. Haitao and Z. Shuai, Tob. Sci. Technol., 2021, 54, 11–16 Search PubMed.
- L. Liu, J. Lei, F. Gao and H. Ju, Talanta, 2013, 115, 819–822 CrossRef.
- J. Yuan, H. liwei, C. xiangjie, X. chengyue, F. ziyan, S. mowen, T. gangling, D. huimin and Y. ding, Tob. Sci. Technol., 2022, 55, 10–15 Search PubMed.
- F. J. Wang, X. L. Zhang, M. A. Hai-Bo, X. Z. Feng, J. L. Feng and P. Hua, Food Sci., 2013, 34, 170–173 Search PubMed.
- Z. Xinyue, X. Guojie, T. Huaqi, L. Yanpeng and L. Chunsheng, J. AOAC Int., 2017, 99–103 Search PubMed.
- C. Y. Yao, Z. L. Xu, H. Wang, F. Zhu, L. Luo, J. Y. Yang, Y. M. Sun, H. T. Lei, Y. X. Tian and Y. D. Shen, Food Chem., 2019, 283, 359–366 CrossRef PubMed.
- M. Pavon, I. Gonzalez, N. Pegels, R. Martín and T. García, Food Control, 2010, 21, 1745–1756 CrossRef.
- Y. Ji, L. Hu, W. Xiong, Y. Wang, F. Yang, M. Shi, H. Zhang, J. Shao, C. Lu, D. Fang, H. Deng, Z. Bian, G. Tang, S. Liu, Z. Fan and S. Liu, J. Appl. Microbiol., 2022, 132, 1250–1259 CrossRef.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |