
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Oxidation of a triple carbo[5]helicene with hypervalent iodine†
Florian
Rigoulet‡
 a,
Albert
Artigas‡§
a,
Nawal
Ferdi
b,
Michel
Giorgi
b and
Yoann
Coquerel
*a
a,
Albert
Artigas‡§
a,
Nawal
Ferdi
b,
Michel
Giorgi
b and
Yoann
Coquerel
*a
aAix Marseille Université, CNRS, Centrale Marseille, ISM2, 13397 Marseille, France. E-mail: yoann.coquerel@univ-amu.fr
bAix Marseille Université, CNRS, Centrale Marseille, FSCM, 13397 Marseille, France
First published on 12th October 2023
Abstract
The reactivities of both diastereomers of hexabenzotriphenylene (HBTP), a triple carbo[5]helicene of the formula C42H24, were examined in the presence of phenyliodine diacetate (PIDA) as an oxidizing agent. The D3-symmetric diastereomer afforded two different ring rearranged ketone-containing products embedding a spirofluorene moiety. In contrast, under similar conditions, the C2-symmetric diastereomer afforded predominantly a cyclodehydrogenation product. Altogether, this shows that stereochemistry is a critical factor in the reactivity of non-planar polycyclic aromatic hydrocarbons.
Introduction
Over the past 25 years, carbohelicenes have gradually shifted from the status of laboratory curiosity to star chiral molecules. This was enabled by the progress in their synthesis and by the discovery of their intriguing (chir)optical and electronic properties, which opened new and stimulating alleys of research.1–6 Helicenes, possibly embedding main-group elements7 and/or metals8,9 in their lattice, are now used to develop optoelectronic devices such as organic solar cells (OCs),10 organic field-effect transistors (OFETs),11 and circularly polarized organic light-emitting diodes (CP-OLEDs).12–15 Despite the current enormous interest in preparing functionalized helicenes or helicenoid compounds, examples of selective functionalization of pristine carbohelicenes having no directing or activating groups are extremely rare.16 One can mention the two-fold oxidation of carbo[6]helicene with chromic acid,17 as well as its bromination, nitration and acetylation (Scheme 1a).18 The regioselective borylation reaction of carbo[5]helicene was reported through an Ir-catalyzed C–H functionalization (Scheme 1b).19 Lately, the electrochemical methoxylation of carbo[n]helicenes (n = 6–9) was realized in 61–85% yield.20 The critical factor for the functionalization of helicenes is the regioselectivity. Because of the lack of site-specific methods to functionalize carbohelicenes, the community has instead relied on the synthesis of pre-functionalized analogues, incorporating alkoxyl groups, bromine atoms, and alkynes as substituents at various positions.16 | ||
| Scheme 1 Background and results of this work. PIDA = phenyliodine diacetate, TCE = 1,1,2,2-tetrachloroethane, HFIP = 1,1,1,3,3,3-hexafluoroisopropanol, and DCM = dichloromethane. | ||
Multihelicenes built only by the fusion of benzene-like six-membered rings belong to a peculiar class of nanographenes21,22 that have promising chiroptical properties.23 After the pioneering works by Clar (1959), Laarhoven (1971) and Martin (1972), who first synthesized double [n]helicenes with n = 5–7, the field of multihelicenes remained dormant for decades.24–28 It was not until the end of the 20th century that Pascal and Guitián reported the synthesis of both diastereomers (D3- and C2-symmetric) of hexabenzotriphenylene (HBTP), a triple [5]helicene fused in a single six-membered ring (Scheme 1c).29–31 It then took another decade for the successful synthesis of additional multihelicenes. Over the past ten years, a plethora of pristine multihelicene hydrocarbons with increasing molecular contortion and diverse photophysical and chiroptical properties have been reported.32–43
Despite the bloom of multihelicenes in contemporaneous organic synthesis, there have been no successful attempts reported for their functionalization. Recently, some regioselective acyloxylation reactions of polycyclic aromatic hydrocarbons (PAH) with hypervalent iodine compounds were reported.44 Among the examples examined in this study, only one was a non-planar PAH, namely corannulene, which could be acyloxylated with 30% yield. Inspired by this work, we examined the reactivity of HBTP under comparable conditions, with the idea to prepare poly(acyloxylated) HBTP derivatives [HBTP(OAc)n] (Scheme 1c). In contrast to our hypothesis, three alternative oxidation products were obtained from these experiments, with a net difference of reactivity between the D3 and C2 diastereomers of HBTP.
Results
In an early experiment aiming at the synthesis of the (poly)acyloxylated products of type HBTP(OAc)n, D3-HBTP was reacted with excess (10 equiv.) phenyliodine diacetate (PIDA) in the presence of hexafluoroisopropanol (HFIP) under blue light irradiation.44 Under these conditions, a complex mixture of unidentified products was obtained, from which the formation of the desired product(s) could not be evidenced. Using 5 equiv. of PIDA in the absence of blue light led to a cleaner reaction, with two major products formed in reasonable amounts (Table 1, entry 1). Isolation and full characterization of these two products, including single-crystal XRD analyses (Fig. 1), revealed that two ketones 1 (chiral) and 2 (achiral) formed, in 13% and 22% yield, respectively. In a control experiment in the absence of HFIP (entry 2), no conversion of D3-HBTP occurred, emphasizing its crucial role in reactivity as previously observed.44 Reducing the amount of the oxidizing agent PIDA to 2.5 equiv. allowed us to produce the chiral ketone 1 with better selectivity, with 30% yield for 1 and 11% yield for 2 (entry 3). Using 1.1 equiv. of PIDA increased the selectivity for 1, isolated in 48% yield (entry 4). In a separate experiment, it was attempted to convert the chiral ketone 1 into the achiral ketone 2 using 1.1 equiv. of PIDA in HFIP for 12 h, which resulted in only 10% yield of 2 with poor conversion (89% of 1 recovered). This experiment indicates that 1 can convert into 2 under the reaction conditions, but with low efficiency. It is plausible that several competing pathways are involved in the formation of 2 from D3-HBTP. | ||
| Fig. 1 Monocrystalline solid-state structures of 1 (a) and 2 (b). H atoms are omitted for clarity. Ellipsoids drawn at the 50% probability level. | ||
| Entry | HBTP | PIDA (equiv.) | Yield (%) 1/2/C2-3 |
|---|---|---|---|
| Reaction conditions: all reactions were performed in a 10 mL tubular flask using 20 mg (0.038 mmol) of HBTP in 0.8 mL of 1,1,2,2-tetrachloroethane, 0.8 mL of HFIP, and 0.2 mL of dichloromethane. Yields account for the isolated homogeneous material.a Reaction performed without HFIP. | |||
| 1 | D 3 | 5.0 | 13/22/0 |
| 2a | D 3 | 5.0 | No reaction |
| 3 | D 3 | 2.5 | 30/11/0 |
| 4 | D 3 | 1.1 | 48/8/0 |
| 5 | C 2 | 5.0 | 8/0/0 |
| 6 | C 2 | 2.5 | 8/0/28 |
| 7 | C 2 | 1.1 | 10/0/19 |
When examining the reactivity of C2-HBTP under identical conditions, the outcome was found to be dramatically different. Using 5 equiv. of PIDA produced 8% of the chiral ketone 1 (entry 5) together with a complex mixture of unidentified products containing no trace of the achiral ketone 2 or the initially targeted (poly)acyloxylated products of type HBTP(OAc)n. Decreasing the amount of PIDA to 2.5 equiv. and 1.1 equiv. did not allow the enhancement of the yield of 1 as for D3-HBTP, but instead a new product C2-3 was formed predominantly, in 28% and 19% yields depending on the conditions (entries 6 and 7). Remarkably, the reactivity of HBTP with PIDA dramatically depends on the relative stereochemistry of the multihelicene substrate: the chiral ketone 1 could form from both diastereomers of HBTP, albeit with different efficiency, while the formation of ketone 2 could only be evidenced from D3-HBTP and the formation of the double helicene C2-3 could only be evidenced from C2-HBTP. This illustrates for the first time that pristine multihelicene hydrocarbons are amenable for chemical transformations, and importantly their reactivity is a function of their stereochemical arrangement.
Mechanistically, the formation of the chiral ketone 1 from D3-HBTP is supposed to be initiated by the formation of an electron donor–acceptor complex between the arene and electron-deficient PIDA, followed by a one-electron oxidation to produce the corresponding aryl radical cation and an acetate anion (Scheme 2).44 Nucleophilic addition of the acetate anion to the core ring of the aryl radical cation would produce a neutral radical amenable for a strain-releasing ring rearrangement to form the spirofluorene moiety. The resulting radical would eliminate an acyl radical to form the carbonyl group in 1. The formation of ketone 2 is more complex as it involves the additional fragmentation of the core six-membered ring, followed by a cyclization step to forge the ketone-containing six-membered ring in 2. As indicated by the experimental observations, there are possibly several competing pathways leading to the formation of 2 from D3-HBTP, involving or not involving ketone 1 as an intermediate and/or a carbenoid-like intermediate. Finally, the formation of C2-3 from C2-HBTP can be inferred by the C–C bond formation at a fjord region from the radical cation formed by the one-electron oxidation of C2-HBTP (Scholl-type reaction). The shorter C–C distance between the reactive atoms in C2-HBTP, 2.91 Å for the [5]helicene unit aligned with the C2 axis vs. 3.02 Å for D3-HBTP, may explain why C2-3 was not formed from D3-HBTP.
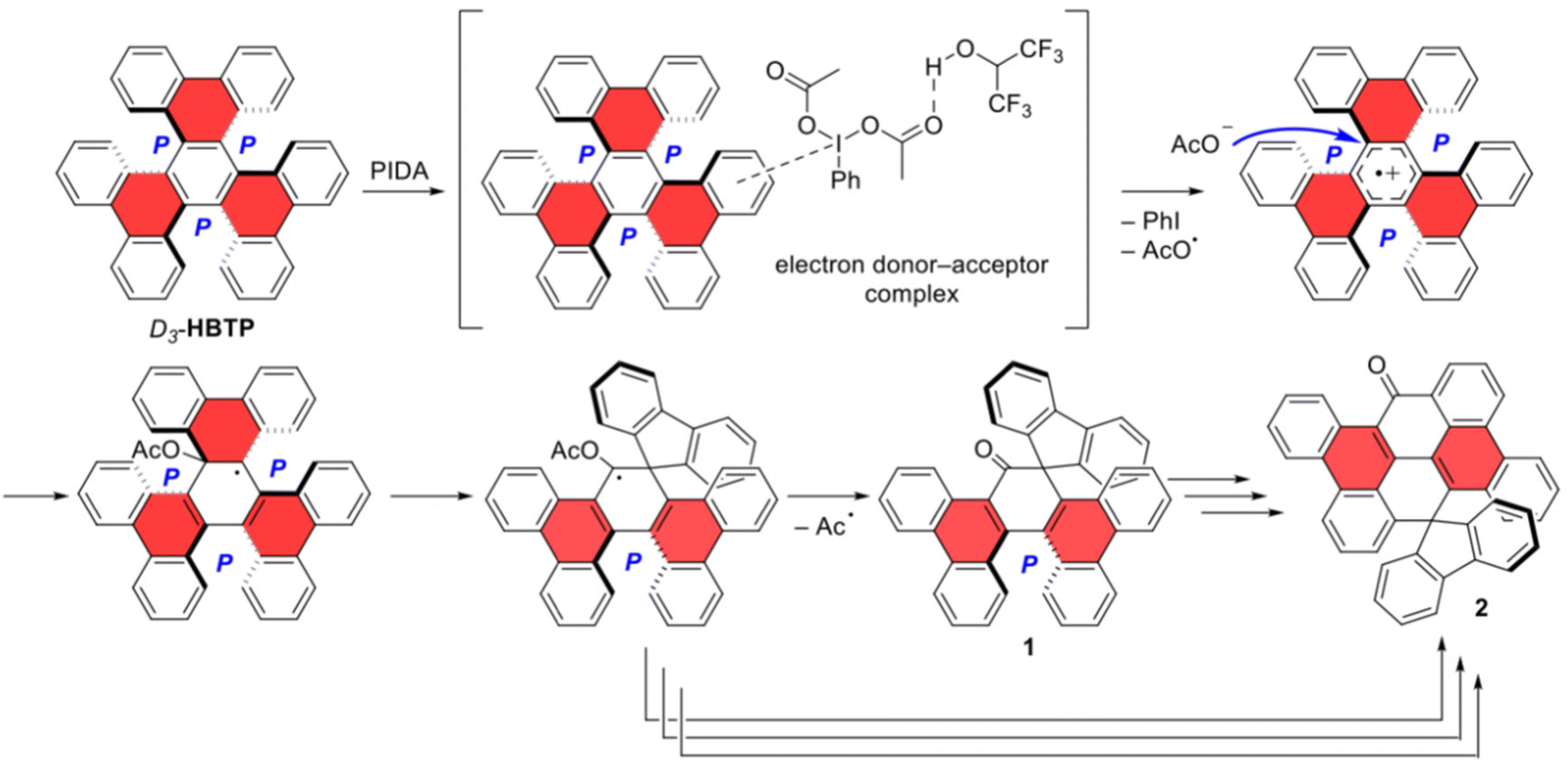 | ||
| Scheme 2 Mechanistic hypotheses. | ||
The conformational behaviours of both the chiral ketone 1 and the chiral double [5]helicene C2-3 were examined in silico by DFT methods (Fig. 2). The barrier to enantiomerization of ketone 1 was computed at 132.1 kJ mol−1, enough to consider this compound as configurationally stable at 298 K. However, despite some efforts, the separation of both enantiomers by preparative chiral HPLC has not yet been successful to date. The enantiomerization of the chiral diastereomer of C2-3 was found to be a two-step process through the thermodynamically disfavored (by 12.0 kJ mol−1) meso-diastereomer, with a barrier computed at 91.2 kJ mol−1. Because this barrier is relatively low, smaller than the barrier to the enantiomerization of pristine carbo[5]helicene,45 the resolution of the enantiomers of C2-3 was not attempted.
 | ||
| Fig. 2 Gibbs free energy profiles of the enantiomerisation and diastereomerisation processes in 1 (a) and 3 (b) computed at the ωB97XD/def2-TZVP//ωB97XD/def2-SVP level of theory at 298 K. | ||
The optical properties of products 1, 2 and C2-3 were examined (Fig. 3). While 1 and 2 have a single absorbance maximum at 253 nm and 249 nm, respectively, C2-3 shows two maxima at 239 nm and 357 nm. Ketones 1 and 2 are not fluorescent. However, C2-3 is, with a maximum fluorescence at 471 nm, which is slightly blue-shifted in comparison with D3-HBTP (483 nm),46 and the photoluminescence quantum yield was determined at ca. 0.04, typically in the range observed for pristine mutlihelicenes.23
 | ||
| Fig. 3 Normalized absorption (plain lines) and emission (dotted line) spectra in CH2Cl2 (ca. 1 × 10−5 M, λEX = 355 nm) at 298 K. | ||
The effect of the partial planarization in C2-3 relative to D3-HBTP on π-electron delocalization was evaluated using 3D isotropic magnetic shielding (IMS) maps47 and electron density of delocalized bond (EDDB(r)) analyses48,49 (Fig. 4). A comparative analysis of the 3D IMS maps indicates that electron delocalization is more intense in C2-3 relative to D3-HBTP, as visualized from the larger dark blue areas in the map of C2-3, especially at the central six-membered ring. Because of the chiral nature of C2-3 and D3-HBTP, both faces of each ring must be considered for a complete analysis. The interactive 3D IMS maps of C2-3 and D3-HBTP are available for the best appreciation of this (for vtk files, see the ESI†). The EDDBH(r) analyses provided comparable outputs for D3-HBTP and C2-3, with however a slightly better ring-to-ring delocalization in C2-3 visualized by smaller discontinuities of its EDDB(r) isosurface. Accordingly, the fraction of delocalized electrons |e| is computed and found to be greater in C2-3 relative to D3-HBTP.
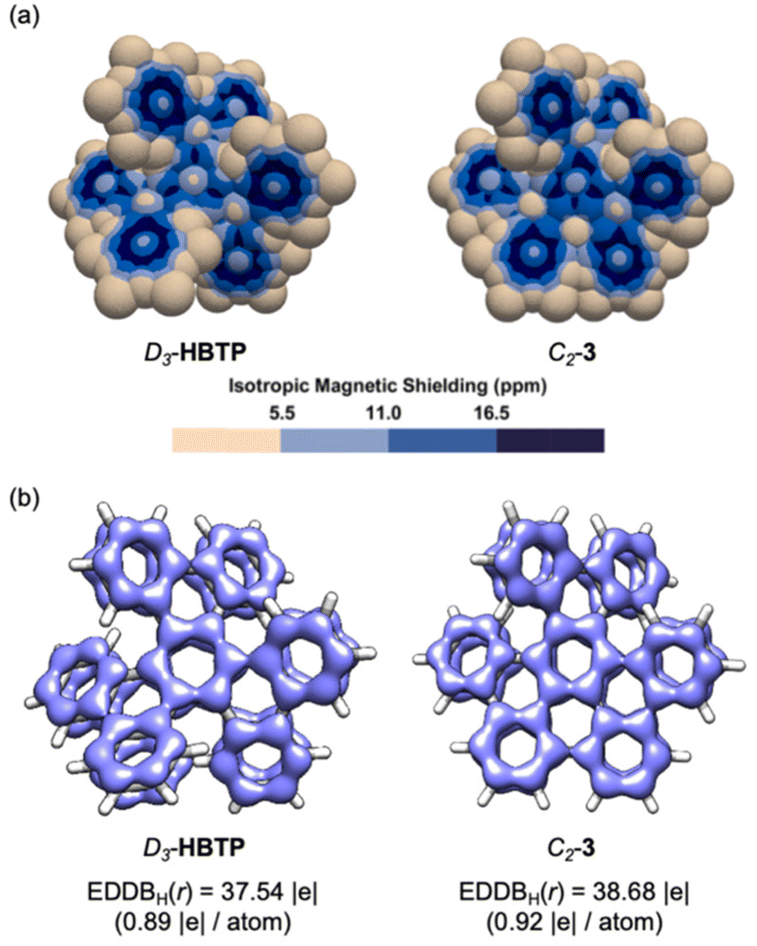 | ||
| Fig. 4 3D IMS maps (a) and EDDBH(r) analyses (b) of D3-HBTP and C2-3. EDDB(r) isovalue = 0.025. | ||
Conclusions
It is demonstrated that under oxidative conditions, using phenyliodine diacetate (PIDA) and hexafluoroisopropanol (HFIP), the two diastereomers of hexabenzotriphenylene (HBTP) reacted differently: the D3-symmetric diastereomer afforded oxidized rearranged ketone products, while the C2-symmetric diastereomer afforded predominantly a cyclodehydrogenation product. From these results, stereochemistry is considered a critical factor in the reactivity of pristine non-planar polycyclic aromatic hydrocarbons (PAH), something that has not been realized until now.Author contributions
Florian Rigoulet performed investigations – synthesis and photophysics and writing – review & editing. Albert Artigas performed investigations – synthesis and calculations and writing – review & editing. Nawal Ferdi performed investigations – structural. Michel Giorgi performed investigations – structural and writing – review & editing. Yoann Coquerel performed conceptualisation, methodology, writing – original draft, supervision, project administration and funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the French Agence Nationale de la Recherche – ANR (ANR-19-CE07-0041). The institutional financial support from Aix-Marseille University, Centrale Marseille and the CNRS is acknowledged. A. A. acknowledges the Spanish Ministerio de Universidades and the EU for a Margarita Salas grant (REQ2021_A_02). We thank the Centre Régional de Compétences en Modélisation Moléculaire (AMU) for computing facilities.References
- Y. Shen and C.-F. Chen, Helicenes: Synthesis and Application, Chem. Rev., 2012, 112, 1463–1535 CrossRef CAS PubMed.
- M. Gingras, One hundred years of helicene chemistry. Part 1: non-stereoselective syntheses of carbohelicenes, Chem. Soc. Rev., 2013, 42, 968–1006 RSC.
- M. Gingras, G. Félix and R. Peresutti, One hundred years of helicene chemistry. Part 2: stereoselective syntheses and chiral separations of carbohelicenes, Chem. Soc. Rev., 2013, 42, 1007–1050 RSC.
- M. Gingras, One hundred years of helicene chemistry. Part 3: applications and properties of carbohelicenes, Chem. Soc. Rev., 2013, 42, 1051–1095 RSC.
- C.-F. Chen and Y. Shen, Helicene chemistry. From synthesis to applications, Springer Berlin, Heidelberg, 2016 Search PubMed.
- Helicenes: Synthesis, Properties and Applications, ed. J. Crassous, I. G. Stará and I. Starý, Wiley-VCH, 1st edn, 2022 Search PubMed.
- K. Dhbaibi, L. Favereau and J. Crassous, Enantioenriched Helicenes and Helicenoids Containing Main-Group Elements (B, Si, N, P), Chem. Rev., 2019, 119, 8846–8953 CrossRef CAS PubMed.
- N. Saleh, C. Shen and J. Crassous, Helicene-based transition metal complexes: synthesis, properties and applications, Chem. Sci., 2014, 5, 3680–3694 RSC.
- E. S. Gauthier, R. Rodríguez and J. Crassous, Metal-based multihelicenic architectures, Angew. Chem., Int. Ed., 2020, 59, 1433–7851 Search PubMed.
- P. Josse, L. Favereau, C. Shen, S. Dabos-Seignon, P. Blanchard, C. Cabanetos and J. Crassous, Enantiopure versus Racemic Naphthalimide End-Capped Helicenic Non-fullerene Electron Acceptors: Impact on Organic Photovoltaics Performance, Chem. – Eur. J., 2017, 23, 6277–6281 CrossRef CAS PubMed.
- Y. Yang, B. Rice, X. Shi, J. R. Brandt, R. Correa Da Costa, G. J. Hedley, D.-M. Smilgies, J. M. Frost, I. D. W. Samuel, A. Otero-de-la-Roza, E. R. Johnson, K. E. Jelfs, J. Nelson, A. J. Campbell and M. J. Fuchter, Emergent Properties of an Organic Semiconductor Driven by its Molecular Chirality, ACS Nano, 2017, 11, 8329–8338 CrossRef CAS PubMed . See correction: Y. Yang, B. Rice, X. Shi, J. R. Brandt, R. Correa Da Costa, G. J. Hedley, D.-M. Smilgies, J. M. Frost, I. D. W. Samuel, A. Otero-de-la-Roza, E. R. Johnson, K. E. Jelfs, J. Nelson, A. J. Campbell and M. J. Fuchter, Emergent Properties of an Organic Semiconductor Driven by its Molecular Chirality, ACS Nano, 2018, 12, 6343 CrossRef PubMed.
- S. Jhulki, A. K. Mishra, T. J. Chow and J. N. Moorthy, Helicenes as All-in-One Organic Materials for Application in OLEDs: Synthesis and Diverse Applications of Carbo- and Aza[5]helical Diamines, Chem. – Eur. J., 2016, 22, 9375–9386 CrossRef CAS PubMed.
- L. Wan, J. Wade, X. Shi, S. Xu, M. J. Fuchter and A. J. Campbell, Highly Efficient Inverted Circularly Polarized Organic Light-Emitting Diodes, ACS Appl. Mater. Interfaces, 2020, 12, 39471–39478 CrossRef CAS PubMed . See correction: L. Wan, J. Wade, X. Shi, S. Xu, M. J. Fuchter and A. J. Campbell, Highly Efficient Inverted Circularly Polarized Organic Light-Emitting Diodes, ACS Appl. Mater. Interfaces, 2022, 14, 27523 CrossRef PubMed.
- D.-W. Zhang, M. Li and C.-F. Chen, Recent advances in circularly polarized electroluminescence based on organic light-emitting diodes, Chem. Soc. Rev., 2020, 49, 1331–1343 RSC.
- J. R. Brandt, F. Salerno and M. J. Fuchter, The added value of small-molecule chirality in technological applications, Nat. Rev. Chem., 2017, 1, 0045 CrossRef CAS.
- M. Jakubec and J. Storch, Recent Advances in Functionalizations of Helicene Backbone, J. Org. Chem., 2020, 85, 13415–13428 CrossRef CAS PubMed.
- J. W. Diesveld, J. H. Borkent and W. H. Laarhoven, The oxidation of hexahelicenes. Synthesis of hexahelicene-5,6-quinone, Recl. Trav. Chim. Pays-Bas, 1980, 99, 391–394 CrossRef CAS.
- P. M. op den Brouw and W. H. Laarhoven, Addition and substitution reactions of hexahelicene: Bromination, nitration and acetylation, Recl. Trav. Chim. Pays-Bas, 1978, 97, 265–268 CrossRef CAS.
- R. P. Kaiser, J. Ulč, I. Císařová and D. Nečas, Direct regioselective C–H borylation of [5]helicene, RSC Adv., 2018, 8, 580–583 RSC.
- N. Yang, C. Shen, G. Zhang, F. Gan, Y. Ding, J. Crassous and H. Qiu, Helicity-modulated remote C-H functionalization, Sci. Adv., 2023, 9, eadg6680 CrossRef CAS PubMed.
- C. Li, Y. Yang and Q. Miao, Recent Progress in Chemistry of Multiple Helicenes, Chem. – Asian J., 2018, 13, 884–894 CrossRef CAS PubMed.
- K. Kato, Y. Segawa and K. Itami, Symmetric Multiple Carbohelicenes, Synlett, 2019, 30, 370–377 CrossRef CAS.
- T. Mori, Chiroptical Properties of Symmetric Double, Triple, and Multiple Helicenes, Chem. Rev., 2021, 121, 2373–2412 CrossRef CAS PubMed.
- E. Clar, C. T. Ironside and M. Zander, The Electronic Interaction between Benzenoid Rings in Condensed Aromatic Hydrocarbons. 1:12-2:3-4:5-6:7-8:9-10:11-Hexa-benzocoronene, 1:2-3:4-5:6-10:11-Tetrabenzoanthanthrene, and 4:5-6:7-11:12-13:14-Tetrabenzoperopyrene, J. Chem. Soc., 1959, 142–147 RSC.
- W. H. Laarhoven and Th. J. H. M. Cuppen, Synthesis of a double helicene, rac. and meso diphenanthro 3,4-c;3′4-1 chrysene, Tetrahedron Lett., 1971, 12, 163–164 CrossRef.
- R. H. Martin, Ch. Eyndels and N. Defay, Studies in the helicene series. Determination of the structure and of the dl configuration of a double helicene by INDOR and ROE experiments. Part XVIII, Tetrahedron Lett., 1972, 13, 2731–2734 CrossRef.
- R. H. Martin, Ch. Eyndels and N. Defay, Double helicenes: diphenanthro[4,3-a; 3′,4′-o]picene and benzo[s]diphenanthro[4,3-a; 3′,4′-o]picene, Tetrahedron, 1974, 30, 3339–3342 CrossRef CAS.
- W. H. Laarhoven, Th. J. H. M. Cuppen and R. J. F. Nivard, Photodehydrocyclizations of stilbene-like compounds-XI: Synthesis and racemization of the double helicene diphenanthro[4.3-a; 3′.4′-o]picene, Tetrahedron, 1974, 30, 3343–3347 CrossRef CAS.
- L. Barnett, D. M. Ho, K. K. Baldridge and R. A. Pascal, The Structure of Hexabenzotriphenylene and the Problem of Overcrowded “D3h” Polycyclic Aromatic Compounds, J. Am. Chem. Soc., 1999, 121, 727–733 CrossRef CAS.
- D. Peña, D. Pérez, E. Guitián and L. Castedo, Synthesis of Hexabenzotriphenylene and Other Strained Polycyclic Aromatic Hydrocarbons by Palladium-Catalyzed Cyclotrimerization of Arynes, Org. Lett., 1999, 1, 1555–1557 CrossRef.
- D. Peña, A. Cobas, D. Pérez, E. Guitián and L. Castedo, Kinetic Control in the Palladium-Catalyzed Synthesis of C2-Symmetric Hexabenzotriphenylene. A Conformational Study, Org. Lett., 2000, 2, 1629–1632 CrossRef PubMed.
- S. Xiao, S. J. Kang, Y. Wu, S. Ahn, J. B. Kim, Y.-L. Loo, T. Siegrist, M. L. Steigerwald, H. Li and C. Nuckolls, Supersized contorted aromatics, Chem. Sci., 2013, 4, 2018–2023 RSC.
- H. Kashihara, T. Asada and K. Kamikawa, Synthesis of a Double Helicene by a Palladium-Catalyzed Cross-Coupling Reaction: Structure and Physical Properties, Chem. – Eur. J., 2015, 21, 6523–6527 CrossRef CAS PubMed.
- T. Fujikawa, Y. Segawa and K. Itami, Synthesis, Structures, and Properties of π–Extended Double Helicene: A Combination of Planar and Nonplanar π–Systems, J. Am. Chem. Soc., 2015, 137, 7763–7768 CrossRef CAS PubMed.
- Y. Hu, X.-Y. Wang, P.-X. Peng, X.-C. Wang, X.-Y. Cao, X. Feng, K. Müllen and A. Narita, Benzo-Fused Double [7]Carbohelicene: Synthesis, Structures, and Physicochemical Properties, Angew. Chem., Int. Ed., 2017, 56, 3374–3378 CrossRef CAS PubMed.
- T. Hosokawa, Y. Takahashi, T. Matsushima, S. Watanabe, S. Kikkawa, I. Azumaya, A. Tsurusaki and K. Kamikawa, Synthesis, Structures, and Properties of Hexapole Helicenes: Assembling Six [5]Helicene Substructures into Highly Twisted Aromatic Systems, J. Am. Chem. Soc., 2017, 139, 18512–18521 CrossRef CAS PubMed.
- V. Berezhnaia, M. Roy, N. Vanthuyne, M. Villa, J.-V. Naubron, J. Rodriguez, Y. Coquerel and M. Gingras, Chiral Nanographene Propeller Embedding Six Enantiomerically Stable [5]Helicene Units, J. Am. Chem. Soc., 2017, 139, 18508–18511 CrossRef CAS PubMed.
- K. Kato, Y. Segawa, L. T. Scott and K. Itami, A Quintuple [6]Helicene with a Corannulene Core as a C5-Symmetric Propeller-Shaped π-System, Angew. Chem., Int. Ed., 2018, 57, 1337–1341 CrossRef CAS PubMed.
- Y. Zhu, X. Guo, Y. Li and J. Wang, Fusing of Seven HBCs toward a Green Nanographene Propeller, J. Am. Chem. Soc., 2019, 141, 5511–5517 CrossRef CAS PubMed.
- F. Zhang, E. Michail, F. Saal, A. Krause and P. Ravat, Stereospecific Synthesis and Photophysical Properties of Propeller-Shaped C90H48 PAH, Chem. – Eur. J., 2019, 25, 16241–16245 CrossRef CAS PubMed.
- M. Roy, V. Berezhnaia, M. Villa, N. Vanthuyne, M. Giorgi, J. Naubron, S. Poyer, V. Monnier, L. Charles, Y. Carissan, D. Hagebaum-Reignier, J. Rodriguez, M. Gingras and Y. Coquerel, Stereoselective Syntheses, Structures, and Properties of Extremely Distorted Chiral Nanographenes Embedding Hextuple Helicenes, Angew. Chem., Int. Ed., 2020, 59, 3264–3271 CrossRef CAS PubMed.
- H.-C. Huang, Y.-C. Hsieh, P.-L. Lee, C.-C. Lin, Y.-S. Ho, W.-K. Shao, C.-T. Hsieh, M.-J. Cheng and Y.-T. Wu, Highly Distorted Multiple Helicenes: Syntheses, Structural Analyses, and Properties, J. Am. Chem. Soc., 2023, 145, 10304–10313 CrossRef CAS PubMed.
- A. Artigas, F. Rigoulet, M. Giorgi, D. Hagebaum-Reignier, Y. Carissan and Y. Coquerel, Overcrowded Triply Fused Carbo[7]helicene, J. Am. Chem. Soc., 2023, 145, 15084–15087 CrossRef CAS PubMed.
- Y. Sakakibara, K. Murakami and K. Itami, C–H Acyloxylation of Polycyclic Aromatic Hydrocarbons, Org. Lett., 2022, 24, 602–607 CrossRef CAS PubMed.
- Ch. Goedicke and H. Stegemeyer, Resolution and racemization of pentahelicene, Tetrahedron Lett., 1970, 11, 937–940 CrossRef.
- H. Tanaka, Y. Kato, M. Fujiki, Y. Inoue and T. Mori, Combined Experimental and Theoretical Study on Circular Dichroism and Circularly Polarized Luminescence of Configurationally Robust D3-Symmetric Triple Pentahelicene, J. Phys. Chem. A, 2018, 122, 7378–7384 CrossRef CAS PubMed.
- A. Artigas, D. Hagebaum-Reignier, Y. Carissan and Y. Coquerel, Visualizing electron delocalization in contorted polycyclic aromatic hydrocarbons, Chem. Sci., 2021, 12, 13092–13100 RSC.
- D. W. Szczepanik, M. Andrzejak, J. Dominikowska, B. Pawełek, T. M. Krygowski, H. Szatylowicz and M. Solà, The electron density of delocalized bonds (EDDB) applied for quantifying aromaticity, Phys. Chem. Chem. Phys., 2017, 19, 28970–28981 RSC.
- M. Solà, A. I. Boldyrev, M. K. Cyrański, T. M. Krygowski and G. Merino, Aromaticity and Antiaromaticity: Concepts and Application, Wiley, 1st edn, 2022, ch. 8, pp. 145–164 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic procedures, characterization, copies of NMR spectra, crystal structures, and details of the theoretical calculations. CCDC 2285157 (for 2) and 2285158 (for 1). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3qo01439h |
| ‡ These authors contributed equally. |
| § Present address: Universitat de Girona, Facultat de Cieǹcies, Campus Montilivi, Carrer de Maria Aurel̀ia Capmanyi Farneś, 69, 17003 Girona, Catalunya, Spain. |
| This journal is © the Partner Organisations 2023 |