
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of highly polarized [3]dendralenes and their Diels–Alder reactions†
Rastislav
Antal
a,
Monika
Staś
bc,
Stefanie M.
Perdomo
a,
Marie
Štemberová
a,
Zbyněk
Brůža
a,
Petr
Matouš
 a,
Jiří
Kratochvíl
a,
Aleš
Růžička
d,
Lubomír
Rulíšek
b,
Jiří
Kuneš
a,
Pavel
Kočovský
ae,
Erik
Andris
*b and
Milan
Pour
*a
a,
Jiří
Kratochvíl
a,
Aleš
Růžička
d,
Lubomír
Rulíšek
b,
Jiří
Kuneš
a,
Pavel
Kočovský
ae,
Erik
Andris
*b and
Milan
Pour
*a
aDepartment of Organic and Bioorganic Chemistry, Charles University, Faculty of Pharmacy in Hradec Králové, Heyrovského 1203, 500 05 Hradec Králové, Czech Republic. E-mail: pour@faf.cuni.cz
bInstitute of Organic Chemistry and Biochemistry, Czech Academy of Sciences, Flemingovo náměstí 2, 166 10 Prague 6, Czech Republic. E-mail: erik.andris@uochb.cas.cz
cFaculty of Chemistry, University of Opole, 48 Oleska Street, 45-052 Opole, Poland
dDepartment of General and Inorganic Chemistry, University of Pardubice, Faculty of Chemical Technology, Studentská 573, 532 10 Pardubice, Czech Republic
eDepartment of Organic Chemistry, Charles University, Faculty of Science, Hlavova 8, 128 43 Prague 2, Czech Republic
First published on 19th September 2023
Abstract
The diene-transmissive Diels–Alder (DTDA) reactions of dendralenes are emerging as a powerful synthetic tool. To date, these processes have been studied with non-polarized or mildly polarized species. We now present an expedient synthesis of strongly electron-deficient [3]dendralenes and demonstrate, for the first time, their DTDA reactions with electron-poor dienophiles. While the combination of two electron-poor partners is believed to be generally disfavored, DTDA reactions reported herein proceed at 100 °C with high yields and stereoselectivities. DFT calculations show that this electronically disfavored process is encouraged by a steric effect of the vinylic moiety within the dendralene core, driving the diene segment into the s-cis conformation, thereby lowering the activation energy by 2–3 kcal mol−1. While the free energy barrier is typically lower for the second cycloaddition, the two barriers become similar for dendralenes with a cyclic enone fragment, which allows a controlled stepwise addition of two different dienophiles.
Introduction
Diels–Alder (DA) addition is indisputably one of the most efficient synthetic tools, as it is capable of assembling a six-membered ring with up to 4 chiral centers in a single step.1 According to general consensus, these reactions are concerted and governed by frontier orbital interactions, where an electron-rich conjugated diene typically reacts with an electron-poor dienophile; an inverse-demand scenario, using an electron-poor diene and an electron-rich dienophile is also viable.2 On the other hand, Diels–Alder reactions of two highly electron-deficient partners (substituted with two or more EWGs)3 are rare and require harsh conditions to proceed. The first example was reported by Alder, who showed that the addition of (2E,4E)-dimethyl hexa-2,4-dienedioate to maleate and/or fumarate required heating at 170 °C for 10–15 h, giving rise to the corresponding adducts in 40–50% yield.3aAmong the potential substrates for DA reactions, dendralenes stand out as a distinct class of cross-conjugated entities.4 The interest in these intriguing hydrocarbons5,6 stems mainly from their ability to undergo two or more consecutive DA reactions in an interconnected manner (Diene Transmissive Diels–Alder Reactions, DTDA, Scheme 1). The archetypal DTDA reaction of [3]dendralene with ethylene (A) shows that the reorganization of π-electrons in a DA process results in the formation of intermediate B that can participate in another DA reaction. Consequently, this DTDA domino sequence represents a very attractive process, as it allows the formation of a minimum of four new bonds, two cycles, and numerous chiral centers.
 | ||
| Scheme 1 [3]Dendralene and its DTDA reaction with ethene. | ||
As evidenced by, e.g., the behavior of [3]dendralene (1), unsubstituted representatives suffer from enhanced reactivity and, therefore, limited stability. Thus, for example, DA dimerization of this triene affords the monocyclic tetraene 2 (Scheme 2),4–6 in which it resembles cyclopentadiene. Notably, this DA reaction takes place at the central, most sterically demanding double bond.
 | ||
| Scheme 2 Self-dimerization of [3]dendralene. | ||
It can be assumed that the reactivity of [3]dendralenes in DA reactions is controlled by charge distribution in the substrate and/or in the reaction intermediates, together with conformational factors, influencing the mutual disposition of the core double bonds. Since an absolute majority of dendralene derivatives studied to date includes compounds with non-polarized or just mildly polarized C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds,7 we set out to prepare a series of more polarized dendralenes, and address these issues by a combination of experimental and high-level theoretical approach. We expected that the attachment of polar functional groups to the dendralene scaffold would harness their reactivity both electronically and sterically. As a consequence, entirely new opportunities to achieve enhanced structural diversity of the products of DTDA sequences were likely to arise as well.
C bonds,7 we set out to prepare a series of more polarized dendralenes, and address these issues by a combination of experimental and high-level theoretical approach. We expected that the attachment of polar functional groups to the dendralene scaffold would harness their reactivity both electronically and sterically. As a consequence, entirely new opportunities to achieve enhanced structural diversity of the products of DTDA sequences were likely to arise as well.
Results and discussion
The existing knowledge of dendralene behavior has been gathered mainly from the neutral or slightly polarized species. We reasoned that variation of the electron density and polarization of the core cross-conjugated triene structure by introducing electron withdrawing (EWG) and/or electron donating (EDG) groups may have a huge impact on their reactivity, decrease the propensity for dimerization (1 → 2), and perhaps enable other reactions to be performed.Initial DFT calculations of the unsubstituted [3]dendralene (1) revealed partial negative charges at all the terminal CH2 carbons (−0.34 to −0.37), whereas the internal carbons of the vinyl groups CHCH2 exhibit smaller charges (−0.21 and −0.22, respectively) (Fig. 1). By contrast, the central carbon of the methylene group CCH2 carries yet smaller negative charge (−0.10), which renders this double bond most polarized. This finding may rationalize the previously observed8 preferential cyclopropanation of the central π bond in [3]dendralene (1) or the unusual regioselectivity of the dimerization of 1, outlined in Scheme 2.
 | ||
| Fig. 1 (a) NBO charges and (b) electrostatic potential map of [3]dendralene (1). | ||
To increase the polarization of the dendralene core, we set out to prepare novel [3]dendralenes, equipped with strongly electron-withdrawing groups (EWG) at the terminal sp2 positions of the “western butadiene unit” and with EWG or EDGs at the “eastern vinyl moiety” (Fig. 2).
 | ||
| Fig. 2 X = EWG, Y = EDG, alkyl, or aryl. | ||
Synthesis of substituted [3]dendralenes
While a number of recent syntheses have opened access to limited subseries of dendralenes,7 a general approach to substituted dendralenes was reported only in a recent paper9 by the Sherburn group (Scheme 3). Notably, the substitution pattern, reported to date, includes alkyl, alkenyl, alkynyl, aryl, and heteroaryl groups, i.e., those, the electronic effects of which are rather small or conveyed by conjugated π-systems (Ar, HetAr). Several papers7a,10 reported derivatives with one or two EWGs but compounds, in which all terminal sp2 atoms of the dendralene backbone would be decorated with typical EWGs or EDGs, have not yet been described. Needless to say that, being equipped with strongly polarizing functional groups, rather than just with alkyls, the products can be expected to offer broader synthetic variability via subsequent functional group transformations. | ||
| Scheme 3 General synthesis of dendralenes by Sherburn.9 | ||
Whereas efficient construction of these cross-conjugated alkenes calls for cross-coupling processes (Scheme 3), we felt that a more convergent synthesis, relying on propiolate additions would offer a truly attractive opportunity for the construction of the “western butadiene unit” bearing two –CO2Me groups at its termini (Fig. 2), which would present a versatile coupling partner for introducing a substituted vinylic moiety (the “eastern unit”) via a selective hydrometallation or hydroiodination (Scheme 4).
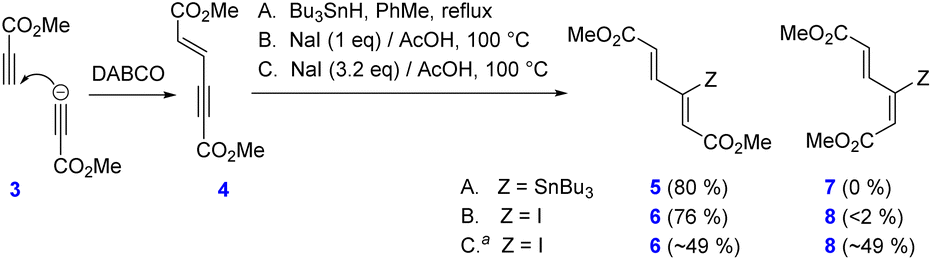 | ||
Scheme 4 Preparation of the diene coupling partners. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) NMR yield. NMR yield. | ||
Dimethyl (E)-hex-2-ene-4-ynedioate (4) was thus prepared in a nearly quantitative yield from methyl propiolate upon treatment with DABCO11 (Scheme 4). Subsequent radical hydrostannation with Bu3SnH afforded pure (2Z,4E)-3-tributylstannylhex-2,4-dienedioate 5 in 80% yield; no (2E,4E)-isomer 7 was detected. On the other hand, stereoselectivity of hydroiodination with NaI/AcOH turned out to be strongly dependent on the amount of NaI, giving rise to the almost pure isomer 6 with 1 equiv. of NaI (76% after crystallization). By contrast, using a three-fold excess of NaI resulted in the formation of a 1:1 mixture of the stereoisomers 6 and 8 in a practically quantitative yield, as revealed by the 1H NMR spectrum of the crude product.
Next, following an extensive developmental experimentation, the stannyl derivative 5 was subjected to the Pd-catalyzed cross-coupling with vinylic iodides R–I (Scheme 5). The optimized protocol required in situ generation of the highly active 14-electron catalyst12 Pd(TFP)2 from PdCl2(TFP)2 (3 mol%) on reduction with n-BuLi (6 mol%) at −78 °C, which was followed by addition of CuI (0.9 equiv.); here, the temperature must be kept below −30 °C to avoid the formation of Pd mirror on the wall of the reaction flask. Subsequent addition of the vinyl iodide R–I (1.25 equiv.) and then addition of the vinylstannane 5 (1.0 equiv.) effected the desired coupling, presumably via a Pd–Sn–Cu transmetalation.13 The resulting dendralenes 9a–n were obtained in fair to good yields, depending on the substitution pattern (Scheme 5).
 | ||
| Scheme 5 Preparation of substituted dendralenes 9a–9n; (a) L = TFP; (b) L = JohnPhos. | ||
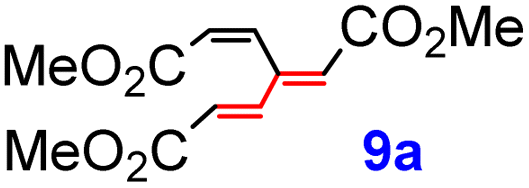
The highest yields were attained for derivatives 9a–9c, where all three terminal atoms of the dendralene core carry EWGs (type 1A). Notably, dendralene 9a has recently been isolated by Ren et al. as a natural product14 from plant material and named dracomolphesin C. There was negligible difference between derivatives 9d and 9e, in which the cyclopentenone unit is appended via its β- and α-position, respectively, and practically no difference in efficiency was observed in the formation of their cyclohexenone analogues 9f and 9g, while their acyclic counterpart 9h was obtained in a lower yield. Simple alkenyl substituents were introduced with a good yield for the acyclic (Z)-oct-2-enyl (9i, 65%) and the cyclopentenyl (9j, 53%) derivatives, while a moderate yield was attained for the cyclohex-1-enyl congener (9k, 46%). The disubstituted alkenyl moieties were also coupled efficiently (9l–9n), with a markedly higher yield for 9n (57%). Replacement of the TFP ligand15 with JohnPhos16 led to a notable yield improvement for 9l, while in the case of 9a–9c the two phosphines turned out to be comparable. Apparently, the JohnPhos-Pd complex prefers the strongest π-acid (i.e., the fumarate moiety, as in 9l), whereas weaker π-acids are favored by the TFP-Pd catalyst. All in all, the yields of this reaction seem to indicate higher dependence on steric, rather than electronic demands of the vinyl halide coupling partner.
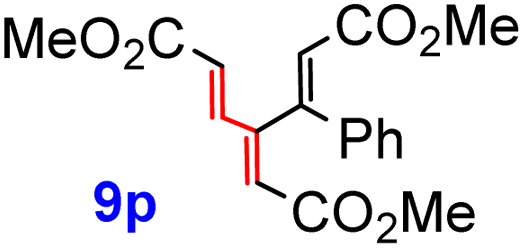
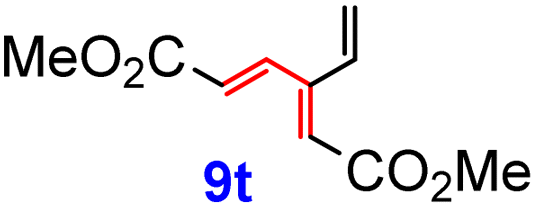
Yet another subseries was prepared from vinyl iodide 6 and the corresponding, readily available vinyl stannanes, using our protocol,17 based on a heterogeneous Pd black catalysis (Scheme 6). Since this procedure employs 6 as the vinyl halide partner, the reaction times and yields reflect the rate of transmetalation and the stability of the vinyl stannanes R-SnBu3. Using this variant, acrylate derivatives 9o and 9p were obtained in considerably higher yields than their regio- and stereoisomers 9m and 9n. Triesters 9q and 9r were also formed in good yields (78 and 57%, respectively). Cross-coupling of 6 with tributylvinylstannane furnished 9t in an excellent yield (82%), while an additional CH3 group caused a decline to 60% for 9u. The lowest yields were obtained for the vinyl ester 9s (30%), and vinyl ethers 9v (34%) and 9w (10%), presumably as a result of the limited stability of the starting (ethoxyvinyl)tributylstannanes at 70 °C. Lactone 9x was prepared previously by us18 in the same way from 6 and (E)-2-tributylstannylbut-2-en-1,4-diol. While this attractive heterogeneous protocol worked well for the coupling of β-iodoacrylate 6, all attempts to prepare the previous series 9a–9n using this procedure either failed or gave inferior yields.
 | ||
| Scheme 6 Preparation of dendralenes 9o–w; 9x was synthesized previously.18 | ||
Natural population analysis19 of DFT calculations shows that in triester 9a, the β-carbon atoms of the two acrylic moieties possess, as expected, a less negative charge (−0.12, −0.11) than the α-carbons (−0.29, −0.30) and that these two double bonds are, overall, more electron deficient than those of the vinyl groups of the parent hydrocarbon 1 (Fig. 1) as a result of the electron-withdrawing effect of the ester groups (Fig. 3). Notably, their polarization, defined as a difference of the charges at neighboring carbons, in 1 and 9a is 0.15 and 0.17, respectively. The central carbon atom in 9a practically lost its negative charge (−0.01) and the overall polarization of the central CC moiety remains the largest (0.25), similarly to 1 (0.24). The stereoisomer 9c exhibited very similar characteristics.
 | ||
| Fig. 3 NBO charges (a and c) and electrostatic potential surfaces (b and d) in [3]dendralene derivatives 9a (a and b) and 9c (c and d). | ||
Diels–Alder reactions of [3]dendralenes
With the dendralenes in hand, the DTDA reactions were explored, using triester 9a as a model compound. Given the electron-deficient nature of its double bonds, inverse electron demand DA reactions with electron-rich dienophiles could be anticipated. However, 9a failed to react with typical electron-rich dienophiles, such as 3,4-dihydro-2H-pyran, bis-trimethylsilylacetylene, cyclopentadiene and furan; only the starting materials were recovered even upon a prolonged heating at reflux in xylene in all these cases.By contrast, treatment of 9a with N-phenylmaleimide (10), one of the most reactive electron-poor, endo-selective dienophiles,20 resulted in a very slow formation of the adduct at ambient temperature (Scheme 7). Upon heating in xylene with 2.5 equiv. of 10 at 100 °C for 48 h, the reaction proceeded readily to completion, giving rise to the bis-adduct 12a as a single isomer in 78% yield (Scheme 7). Of other electron-deficient dienophiles, 9a failed to react with 1,4-benzoquinone, 1,4-naphthoquinone or cyclopentene-1,3-dione or gave complex mixtures on treatment with dimethyl fumarate or maleate. While Lewis-acid catalysis is known to accelerate DA reactions of enone-type dienophiles, it failed to promote the reaction in this case. Attempted DA addition of acroleine and cinnamaldehyde to 9a, carried out in the presence of MacMillan's secondary amines21 as organocatalysts (via the corresponding highly reactive iminium intermediates), also failed, which is in contrast with the success reported for electron-rich dendralenes.22 The isomeric triester 9c exhibited similar behavior, affording the bis-adduct 12c in high yield over a shorter period of time (using 2.5 equiv. of 10).
 | ||
| Scheme 7 DTDA reactions of 9a, 9c, 9i, and 9t; E = CO2Me. | ||
The reactivity pattern was further investigated using trienes 9i (simple alkenyl) and 9t (unfunctionalized vinyl group) that were also treated with N-phenylmaleimide (10) (2.5 equiv.) under the same conditions as those applied for 9a. Again, formation of the products of double DA addition, namely 12i and 12t was observed, both in the yields exceeding 70%; no traces of other products were detected. It is pertinent to note that when the reactions of any of the dendralenes 9a,c,i,t were carried out in the presence of just 1.1 eq. of 10, only the corresponding bis-adducts 12 were detected, together with ca. half of the unreacted starting material, showing that the second DA reaction proceeds faster than the first one. The structure of all products 12, including stereochemistry, was deduced from their NOE spectra, and was corroborated by X-ray crystallography in the case of 12i (see the ESI†).
Under the same conditions as those given in Scheme 7, the cyclopentenone derivative 9e followed the suite, giving rise to the bis-adduct 12e on reaction with maleimide (10), and so did the cyclohexenone derivative 9g, which gave the pentacycle 12g (Fig. 4). In addition, an interesting pentacyclic lactone (12x) was obtained from lactone 9x.18
 | ||
| Fig. 4 Products of DTDA reactions of 9e, 9g, and 9x with 10 (2.5 equiv., xylene, 100 °C); E = CO2Me. | ||
In sharp contrast to the reactions of 9a,c,i, and 9t (vide supra), modification of the reaction conditions for 9e and 9g (temperature decrease from 100 to 70 °C, dendralene:maleimide ratio 2:1, extended reaction time) enabled us to obtain monoadducts 11e and 11g in high yields (Scheme 8). Hence, the addition of another, different dienophile became a distinct possibility. Indeed, the latter diesters 11e and 11g readily reacted with N-phenyl-1,2,4-triazol-2,5-dione (13, vide infra) to afford the hetero bis-adducts 14e and 14g, respectively. Under similar conditions, dendralenic lactone 9x was unreactive to both dienophiles. Interestingly, when diester 9g was treated with triazole 13 first, only the corresponding bis-adduct 15g was isolated.
 | ||
| Scheme 8 Sequential reactions of 9e and 9g with two different dienophiles. | ||
Treatment of 9a with other highly reactive heterodienophiles, such as (ClSO2)NCO, (Cl3CCO)NCO, CH2C(CN)Cl, and singlet oxygen 1O2 (generated from H2O2 and Na2MoO423) were unsuccessful. On the other hand, nitrosobenzene (16) (2.5 equiv.) turned out to react at room temperature (Scheme 9), giving a rather intractable mixture, from which the bis-adduct 17a was isolated in 20% yield.24,25 More successful was the experiment with N-phenyl-1,2,4-triazol-2,5-dione (13), which afforded the bis-adduct 15a in 75% yield.
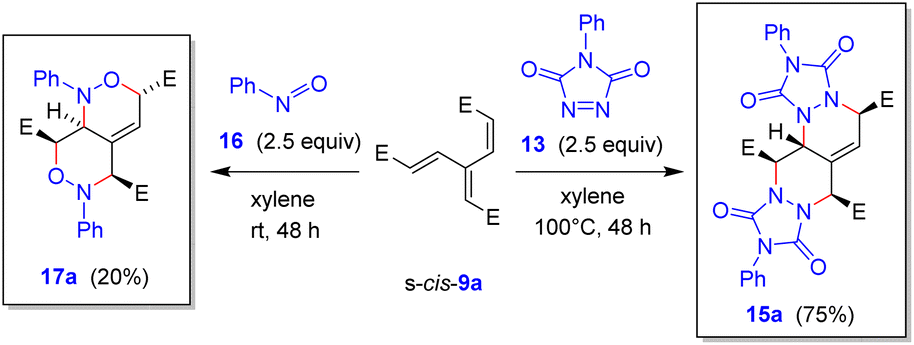 | ||
| Scheme 9 DTDA reactions of 9a with nitrosobenzene (16) and N-phenyl-1,2,4-triazol-2,5-dione (13). | ||
Formation of the tetracyclic products 12a, 12c, 12i, 12t, 15a, pentacyclic compounds 12e, 12g, 12x, and hetero adducts 14e and 14g provides evidence that it was the dienic fragment with two ester groups at the termini (Scheme 7), where the first cycloaddition must have occurred. The configuration of all adducts 12, 14 and 15 corresponds to the endo/endo selectivity for both additions, with the second dienophile approaching the corresponding intermediate 11 from the less hindered convex face (endo–anti–endo). The only exception is the configuration of 17a, whose stereochemistry indicates the direction of the second attack from the more hindered face of 11 (note, however, that it was only formed in 20% yield). In partial contrast, trienes 9o and 9r with a sterically more demanding “eastern” vinylic unit followed the same pattern, but only the monoadducts 11o and 11r were isolated (in 91% and 68% yields, respectively) (Scheme 10). On the other hand, triene 9s with the less encumbered “eastern” vinylic moiety afforded the bis-adduct 12s in 74% yield.
 | ||
| Scheme 10 DA reactions of 9o, 9r and 9s with 10; E = CO2Me. | ||
Theoretical approach to the Diels–Alder reactions of [3]dendralenes
To shed light on the behavior of the electron-deficient dendralenes in DTDA reactions, Gibbs free energies for the individual DA additions were calculated using a series of diene substrates and N-phenylmaleimide as a dienophile (Table 1). In our calculations, we employed ωB97X-D26 DFT functional, calibrated against CCSD(T)/CBS energies (for details, see ESI, section 4.2), and which has been reported to be suitable for optimization of transition state geometries.27 On average, the ωB97X-D method gives energies 3.5 kcal mol−1 higher for DA TS and 1.7 kcal mol−1 higher for the DA products than the CCSD(T)/CBS limit.| Entry | Diene | DP | ΔGt→c | ΔG‡endo | ΔG‡exo | ΔGendo | ΔGexo |
|---|---|---|---|---|---|---|---|
| a Gibbs free energies in kcal mol−1 at 298.15 K and 1 atm. Calculations were carried out with ωB97X-D/6-311+G(2d,p)//6-31+G(d,p) level of theory for xylene as the solvent. Reaction enthalpies can be found in Table S6.† | |||||||
| 1 |

|
10 | 3.1 | 27.5 | 30.4 | −31.3 | |
| 2 |
|
10 | −1.0 | 24.3 | 27.5 | −35.8 | |
| 3 |

|
10 | — | 23.7 | 26.8 | −16.1 | −15.9 |
| 4 |

|
10 | −0.3 | 24.0 | 26.7 | −36.3 | |
| 5 |
|
10 | — | 23.7 | 25.8 | −18.1 | −17.9 |
| 6 |

|
10 | — | 23.7 | 26.6 | −17.0 | −16.5 |
| 7 |

|
10 | 2.5 | 27.9 | 30.1 | −33.4 | |
| 8 |

|
10 | 1.8 | 24.6 | 28.8 | −33.9 | |
| 9 |

|
10 | −1.7 | 24.4 | 27.1 | −36.5 | |
| 10 |

|
10 | 2.8 | 28.4 | 31.4 | −26.7 | −28.1 |
| 11 |

|
10 | 5.1 | 32.4 | 34.6 | −30.1 | −28.6 |
| 12 |

|
10 | 2.7 | 30.2 | 32.7 | −22.3 | −23.7 |
| 13 |
|
10 | 2.2 | 28.3 | 30.9 | −23.1 | −27.5 |
| 14 |

|
10 | −3.2 | 27.6 | 29.8 | −26.2 | −32.0 |
| 15 |
|
10 | 2.4 | 28.3 | 30.1 | −24.6 | −30.8 |
| 16 |
|
10 | −0.4 | 28.0 | 30.4 | −24.8 | −30.2 |
| 17 |

|
10 | 2.4 | 28.2 | 30.7 | −23.3 | −29.0 |
| 18 | 9g | 13 | 2.4 | 23.6 | 33.2 | −31.6 | −31.7 |
To elucidate the effect of the electronic structure, we set out to compare the reactivity of various substituted [3]dendralenes with their 1,3-butadiene analogues (Table 1 lists Gibbs free energies; enthalpies for the reaction as orbital energies for the dienes can be found in Table S6†). In line with the Alder endo rule28 and our experiments (Schemes 7–10), calculations predict the preference for the endo pathway (Table 1).
The calculated barrier for the reaction of unsubstituted 1,3-butadiene with N-phenylmaleimide (10) was found to be 27.5 kcal mol−1 (Table 1, entry 1); according to the literature, the reaction requires 24 h at 20 °C in benzene, to give the product in 91% yield.20 On the other hand, the barrier for the exo pathway would be by ∼3 kcal mol−1 higher (30.4 kcal mol−1, entry 1). The reaction of 10 with the unsubstituted [3]dendralene 1 has a notably lower barrier of 24.3 kcal mol−1 (Table 1, entry 2) than that of butadiene and is known to take place at room temperature in toluene within 48 h with 83% yield.29 Hence, according to the calculations, the addition of a vinyl group (as in 1) reduces the energy barrier by about 3 kcal mol−1 compared to 1,3-butadiene. This result might, a priori, be attributed to the change of the electronics of the system, which would make the molecule more reactive. However, the same increase in reactivity can be observed by constraining the diene moiety in the s-cis conformation, as in cyclopentadiene, whose activation barrier is calculated to be 23.7 kcal mol−1 (Table 1, entry 3). Experimentally, the reaction between cyclopentadiene and 10 takes place at 20 °C in benzene within 15 min to afford the adduct in 98% yield.20 Interestingly, since the energy of the s-cis butadiene is by 3.1 kcal mol−1 higher than that of its most stable s-trans rotamer (Table 1, entry 1), the hypothetical s-cis butadiene would exhibit the same reactivity with a barrier of 27.5–3.1 = 24.4 kcal mol−1.
To find out whether the steric or electronic effects are responsible for the enhanced reactivity of [3]dendralenes, further calculations were performed. The presence of the vinyl group lowers the ΔG‡ (Table 1; compare entries 1 and 2) and a similar effect was observed for an ester group appended to that position (Table 1, entry 4), which may seem to support the electronics argument. However, introduction of a vinyl or ester group to cyclopentadiene (Table 1, entries 5 and 6) was found not to affect the calculated barrier, despite significantly changing the HOMO orbital energies of the dienophiles (from −8.2 eV for cyclopentadiene to −8.0 and −8.6 eV, respectively, see Table S6†). Furthermore, the barrier for 2-cyanobuta-1,3-diene (27.9 kcal mol−1; Table 1, entry 7), in which the rod-shape CN group presents much less steric influence, was found to be almost equal to the barrier of 1,3-butadiene itself (Table 1, entry 1), despite the conjugation effect and the −0.7 eV change in the HOMO orbital energy (Table S6,† entries 1 and 7). On the other hand, for ethyl and t-butyl group as a substituent, the barrier was found to decrease by ∼3 kcal mol−1 (24.6 and 24.4 kcal mol−1; entries 8 and 9) with respect to the 1,3-butadiene transition state (27.5 kcal mol−1; Table 1, entry 1). The common factor for these two examples is the stabilization of the s-cis conformation (by 1.3 and 4.8 kcal mol−1, respectively, compared to butadiene), which should lower the energy barrier for the DA reaction. Therefore, we can conclude that the vinylic group stabilizes the s-cis conformation of the diene moiety, required for the DA reaction to occur, which decreases the energy barrier for [3]dendralene.
The enhancing effect of the vinyl group is mitigated by the electron-withdrawing ester groups in the butadiene segment at 1,4-positions. Their presence results in an increase of the energy barrier by 1–3 kcal mol−1 (compare entry 1 with 10 and 12 in Table 1). Furthermore, configuration of the “eastern” double bond has a significant impact on the activation barrier. As shown above, the transition state energy for methyl (E)-penta-2,4-dienoate is by ∼4 kcal mol−1 lower than that for its (Z)-isomer (compare entries 10 and 11). These results take account for the way of formation of the addition products 12a,c,e,i,t: the first DA addition occurs always at the site of the (E,E)-dienoate, since otherwise one of the ester groups would get into a steric clash. In conclusion, for substituted [3]dendralenes, the deactivating electronic effect exercised by the ester groups is partially offset by stabilizing the s-cis conformation owing to the steric effect of the vinyl group.
The full two-step DTDA reaction pathway, experimentally examined for 9a, 9c, 9p, and 9t, was also investigated theoretically and compared with the reactivity of unsubstituted [3]dendralene (1) (Table 2, entry 1). The calculated first energy barrier for these substrates is ca. 28 kcal mol−1 (Table 2, entries 2–5; the transition states corresponding to entry 2 are shown in Fig. 5). This rather high value may explain the required high temperature and long reaction time in comparison with the unsubstituted [3]dendralene 1 (24.3 kcal mol−1; Table 2, entry 1). The second energy barriers for [3]dendralenes 1 and 9t are 23.4 and 20.7 kcal mol−1, respectively (entries 1 and 5). The relative energy for TS2 is lower than that associated with TS1 by 0.9 kcal mol−1 for 1 and 7.3 kcal mol−1 for 9t, which apparently prevents the detection of the mono-adducts during the reaction. The larger decrease of the barrier for 9t presumably originates from the absence of EWGs in the position R4 and stabilization of the s-cis conformation. On the other hand, the reaction of 9o and 9r stops at the stage of the mono-adduct. In that case, for 9p, serving as their model, the relative energies of TS for both steps are practically identical (TS1 = 28.3 and TS2 = 28.2 kcal mol−1, respectively; entry 4). The relatively high TS2 barrier here is influenced by the presence of the EWG and an impaired access to the newly formed diene system. These results also provide a rationale for the experimentally observed endo selectivity. For all these structures, the TS1 energy barrier is lower for the endo form. In the second step, the dienophile approaches the diene from a less sterically hindered convex side and formation of the endo isomer is favored again.
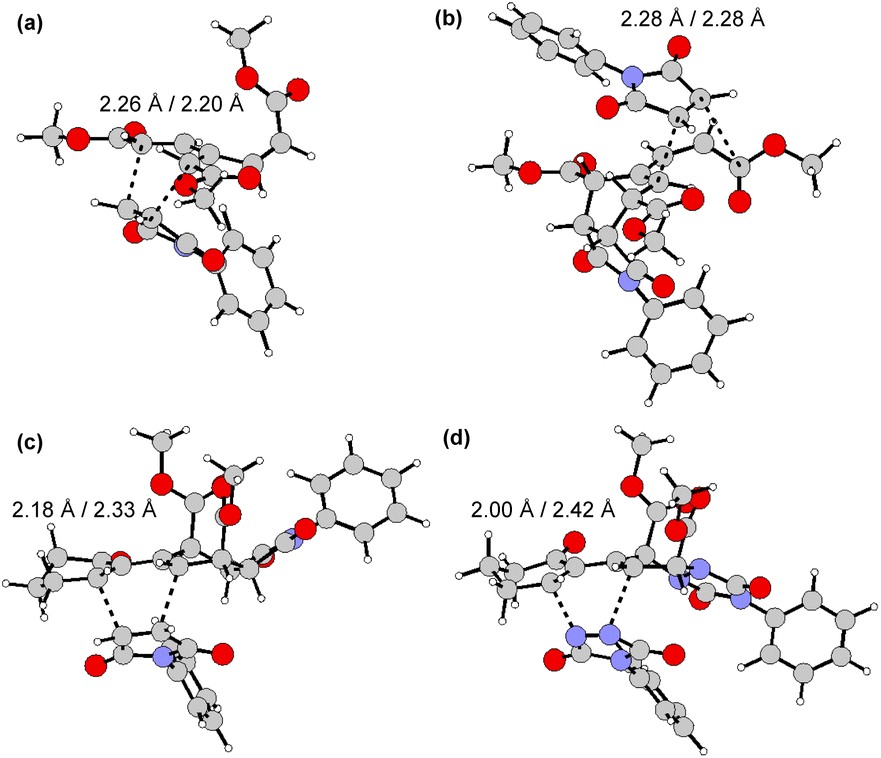 | ||
| Fig. 5 endo transition state of the first (a) and second (b) step of the reaction of 9a with 10 and for the second step of reaction of 9g with 10 (c) and 13 (d). | ||
|
|
|
||||||
|---|---|---|---|---|---|---|---|
| Entry | Diene | DP | ΔG‡TS1 | ΔG1st addition | ΔG‡TS2b,c |
ΔG2nd additionc |
ΔΔGTS2–TS1 |
| a Gibbs free energies in kcal mol−1 at 298.15 K and 1 atm. Calculations were carried out with ωB97X-D/6-311+G(2d,p)/6//6-31+G(d,p) level of theory in xylene. b Values are given for the preferred endo TS, values for exo TS can be found in Table 1 for the first addition, or in brackets for the second addition. c Free energy relative to endo-(11 + dienophile) system. | |||||||
| 1 | 1 | 10 | 24.3 | −35.8 | 23.4 (27.8) | −32.4 (−31.3) | −0.9 |
| 2 | 9a | 10 | 28.3 | −23.1 | 23.0 (31.1) | −34.7 (−28.9) | −5.3 |
| 3 | 9c | 10 | 27.6 | −26.2 | 22.2 (29.0) | −29.6 (−24.9) | −5.4 |
| 4 | 9p | 10 | 28.3 | −24.6 | 28.2 (33.8) | −24.3 (−20.6) | −0.1 |
| 5 | 9t | 10 | 28.0 | −24.8 | 20.7 (27.9) | −34.8 (−28.6) | −7.3 |
| 6 | 9g | 10 | 28.2 | −23.3 | 27.5 (30.1) | −24.5 (−26.5) | −0.7 |
| 7 | 9g | 13 | 23.6 | −31.6 | 19.8 (29.4) | −31.6 (−31.7) | −3.8 |


We also investigated the reactions of 9g with maleimide 10, where the product of the first addition can be isolated (vide supra) and then can undergo a second addition in a controlled manner with the same (10) or different (13) dienophile.
For comparison, we studied the reaction of 9g with triazole 13, which could not be stopped at the first step. Comparison of the reaction barriers of the first step and the barriers in the second step (Table 2, entries 6 and 7) shows that the first barrier is lower for 13 compared to 10 by 4.6 kcal mol−1. However, while the second barrier for 10 is only marginally lower than that for the first addition (by 0.7 kcal mol−1), in the case of 13 it is considerably lower (by 3.8 kcal mol−1). The reason for the barrier of the second step in reaction with 10 not being substantially lower (unlike for 9a, 9c, and 9t), is probably the steric clash between the keto group of the cyclohexenone ring with one of the ester groups (Fig. 6), which drives the diene system out of planarity. The substantial difference in barriers for both steps found for dienophiles 10 and 13 might originate from the nature of the corresponding transition states, which are more symmetrical in the case of 10 than in 13, as revealed by the distance of the reacting atoms (dotted lines) (Fig. 5): the more symmetric TS imposes larger constraints on both the bonds being formed, which in turn leads to stronger ketone/ester steric clash. Furthermore, the adducts with 13 seem to be less sterically congested than their counterparts arising from 10.
 | ||
| Fig. 6 Steric clash between carbonyl oxygen and hydrogen in the s-cis conformation of 11g, which is present in the TS of the second DA reaction step. | ||
Finally, we also theoretically studied the reactions between 9a with various dienophiles (Table 3 and Table S7†). Unlike maleimide, other dienophiles, namely cyclopentadiene, furan, cyclopent-4-ene-1,3-dione, 1,4-benzoquinone, 1,4-naphthoquinone, and 3,4-dihydro-2H-pyran, exhibit calculated barriers of about or more than ca. 30 kcal mol−1 or higher respectively (Table 3, entries 3–8). Therefore, it is not surprising that these reactions do not proceed.
| Entry | Dienophile | ΔG‡endo | ΔG‡exo | ΔGendo | ΔGexo | Reaction yield |
|---|---|---|---|---|---|---|
| a Gibbs free energies in kcal mol−1 at 298.15 K and 1 atm. Calculations were carried out with ωB97X-D/6-311+G(2d,p)/6//6-31+G(d,p) level of theory in xylene. b 100 °C, 48 h, bis-adduct (12a). c rt, 48 h, bis-adduct (17a). Reaction enthalpies can be found in Table S7.† | ||||||
| 1 |

|
28.3 | 30.9 | −23.1 | −27.5 | 78%b |
| 2 |

|
27.8 | 36.0 | −6.7 | −5.3 | 20%c |
| 3 |

|
30.5 | 33.7 | −16.5 | −18.7 | No reaction |
| 4 |

|
38.0 | 40.0 | −4.7 | −6.4 | No reaction |
| 5 |
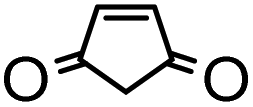
|
32.6 | 32.8 | −14.4 | −19.1 | No reaction |
| 6 |

|
30.3 | 32.0 | −17.9 | −21.9 | No reaction |
| 7 |

|
29.7 | 32.3 | −17.8 | −21.0 | No reaction |
| 8 |

|
37.7 | 37.5 | −17.4 | −21.8 | No reaction |
Conclusions
In summary, a representative set of [3]dendralenes 9a–x, decorated with substituents at each terminal sp2 carbon of the butadiene moiety, was prepared in two steps from the readily available enyne dicarboxylate 4. Two complementary cross-coupling protocols were employed, including homogeneous catalysis by a PdP2 (5 → 9a–n) and heterogeneous catalysis by Pd black (6 → 9o–x), respectively.The electron-deficient [3]dendralenes 9a–x thus prepared were subjected to a Diels–Alder reaction with the heterocyclic dienophiles 10 and 13. Contrary to the general belief that the combination of two electron-poor partners is disfavored, these reactions did proceed (at 100 °C), giving rise to the bis-adducts 12 and 15 as a result of the highly endo/endo stereoselective diene transmissive Diels–Alder reactions (DTDA), occurring in two consecutive steps (9 → 11 → 12 or 15). The second step was found to be faster than the first one, both experimentally and computationally. However, this scenario became reversed in dendralenes 9e and 9g with an appended enone fragment: here, the reaction could be stopped after the first DA addition for the first time, affording mono-adducts 11e and 11g. This finding opened the pathway towards sequential additions of two different dienophiles, giving rise to the corresponding adducts 14e and 14g.
Calculations revealed that this electronically disfavored process is encouraged by a steric effect of the vinylic appendix (as part of the dendralene core), which drives the diene moiety into the s-cis conformation, thus lowering the activation energy by 2–3 kcal mol−1. The same effect is also operating in the second step, which is actually faster, so that the intermediate 11 cannot be isolated in most cases. On the other hand, when the substitution pattern does not allow the molecule to assume the s-cis conformation required for the second step, as in the case of 9o,r, the reaction stops at the stage of the mono-adduct 11o,r. According to our calculations, carried out for 9a, 9c, 9p, and 9t, the first addition requires the highest activation energy of the whole DTDA sequence (∼28 kcal mol−1), which is only by less than 4 kcal mol−1 higher than that for the bare [3]dendralene 1. By contrast, dendralenes decorated with simple, sterically non-demanding alkyls (methyl) but lacking electron-withdrawing substituents, react in DA processes at lower temperatures.4–6
From the synthetic standpoint, this approach employing functionalized starting [3]dendralenes, which generates DTDA products with up to five cycles (as in 12e,g,x, 14e,g, and 15g) and up to eight chiral centers (12) in one pot, offers entirely new opportunities for manipulation of the structures, thereby broadening the synthetic potential of this methodology in a highly atom-economic way.
Author contributions
R.A., S.M.P., M.Š., Z.B. and J.Kr. designed and synthesized the compounds, M.S., L.R. and E.A. designed and performed computational experiments, A.R. and J.K. conducted structural analysis, P.M. and P.K. prepared the manuscript, M.P. supervised the project and prepared the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Czech Science Foundation (project no. 22-19209S). Graduate studentships (R. A., S. M. P., M. Š.) were provided by Charles University (projects no. 1348119 and SVV 260 661). Computational resources were supplied by the project “e-Infrastruktura CZ” (e-INFRA CZ LM2018140) supported by the Ministry of Education, Youth and Sports of the Czech Republic. Calculations have been carried out using resources provided by Wroclaw Centre for Networking and Supercomputing (https://wcss.pl).References
- (a) O. Diels and K. Alder, Synthesen in der hydroaromatischen Reihe, Justus Liebigs Ann. Chem., 1928, 460, 98–122 CrossRef CAS; (b) K. C. Nicolaou, S. A. Snyder, T. Montagnon and G. Vassilikogiannakis, The Diels-Alder reaction in total synthesis, Angew. Chem., Int. Ed., 2002, 41, 1668–1698 CrossRef CAS.
- (a) R. B. Woodward and R. Hoffmann, The conservation of orbital symmetry, Verlag Chemie, Weinheim/Bergstr., 1970 Search PubMed; (b) I. Fleming, Frontier orbitals and organic chemical reactions, Wiley, London, 1978 Search PubMed.
- (a) K. Alder, H. H. Molls and R. Reeber, Darstellung Und Stereochemie Der 1.2.3.4-Tetracarbonsauren Des Cyclopentans Und Cyclohexans, Liebigs Ann. Chem., 1958, 611, 7–32 CrossRef CAS; (b) G. J. Bodwell and Z. L. Pi, Electron deficient dienes I. Normal and inverse electron demand Diels-Alder reactions of the same carbon skeleton, Tetrahedron Lett., 1997, 38, 309–312 CrossRef CAS.
- H. Hopf, The Dendralenes - a Neglected Group of Highly Unsaturated-Hydrocarbons, Angew. Chem., Int. Ed. Engl., 1984, 23, 948–960 CrossRef.
- For more recent reviews, see. (a) H. Hopf, Dendralenes: The Breakthrough, Angew. Chem., Int. Ed., 2001, 40, 705–707 CrossRef CAS; (b) H. Hopf and M. S. Sherburn, Dendralenes Branch Out: Cross-Conjugated Oligoenes Allow the Rapid Generation of Molecular Complexity, Angew. Chem., Int. Ed., 2012, 51, 2298–2338 CrossRef CAS PubMed.
- H. Hopf and M. S. Sherburn, Cross Conjugation: Modern Dendralene, Radialene and Fulvene Chemistry, John Wiley & Sons, Hoboken, NJ, 2016 Search PubMed.
- For selected examples of synthesis of dendralenes, see the following. From allenes: (a) H. G. Wang, B. Beiring, D. G. Yu, K. D. Collins and F. Glorius, [3]Dendralene Synthesis: Rhodium(III)-Catalyzed Alkenyl C-H Activation and Coupling Reaction with Allenyl Carbinol Carbonate, Angew. Chem., Int. Ed., 2013, 52, 12430–12434 CrossRef CAS PubMed. From alkynes: (b) E. Rivera-Chao and M. Fañanás-Mastral, Synthesis of Stereodefined Borylated Dendralenes through Copper-Catalyzed Allylboration of Alkynes, Angew. Chem., Int. Ed., 2018, 57, 9945–9949 CrossRef CAS PubMed. Through Suzuki–Miyaura coupling: (c) C. H. Oh and Y. M. Lim, Double Suzuki reactions of organoboronic acids with 1,n-dibromides, Bull. Korean Chem. Soc., 2002, 23, 663–664 CrossRef CAS. Through Stille coupling: (d) E. J. Lindeboom, A. C. Willis, M. N. Paddon-Row and M. S. Sherburn, Computational and Synthetic Studies with Tetravinylethylenes, J. Org. Chem., 2014, 79, 11496–11507 CrossRef CAS PubMed; (e) N. A. Miller, A. C. Willis and M. S. Sherburn, Formal total synthesis of triptolide, Chem. Commun., 2008, 1226–1228 RSC; (f) N. A. Miller, A. C. Willis, M. N. Paddon-Row and M. S. Sherburn, Chiral dendralenes for rapid access to enantiomerically pure polycycles, Angew. Chem., Int. Ed., 2007, 46, 937–940 CrossRef CAS PubMed.
- R. R. Kostikov and A. P. Molchanov, Reaction of Dichlorocarbene with 2-Vinyl-1,3-Butadiene, J. Org. Chem. USSR (English Translation), 1975, 11, 438–449 Search PubMed.
- J. George, J. S. Ward and M. S. Sherburn, A general synthesis of dendralenes, Chem. Sci., 2019, 10, 9969–9973 RSC.
- (a) For examples published before 2012, see 5b; ; (b) Y. A. Qiu, D. Posevins and J. E. Bäckvall, Selective Palladium-Catalyzed Allenic C-H Bond Oxidation for the Synthesis of [3]Dendralenes, Angew. Chem., Int. Ed., 2017, 56, 13112–13116 CrossRef CAS PubMed; (c) S. Li, B. Hou and J. Wang, Palladium-Catalyzed Oxidative Coupling of the Allenic C-H Bond with alpha-Diazo Esters: Synthesis of [3]Dendralenes, J. Org. Chem., 2021, 86, 5371–5379 CrossRef CAS PubMed; (d) G. S. Naidu, R. Singh, M. Kumar and S. K. Ghosh, Tuning the Stability and the Reactivity of Substituted [3]Dendralenes for Quick Access to Diverse Copiously Functionalized Fused Polycycles with Step and Atom Economy, J. Org. Chem., 2017, 82, 3648–3658 CrossRef CAS PubMed; (e) S. Naidu, R. Singh and S. K. Ghosh, [3]Dendralenes: Synthesis, Reactivity Studies and Employment in Diversity-Oriented Synthesis of Complex Polycyclic Scaffolds, Synlett, 2018, 29, 282–295 CrossRef.
- (a) L. H. Zhou, X. Q. Yu and L. Pu, Reactivity of a propiolate dimer with nucleophiles and an efficient synthesis of dimethyl alpha-aminoadipate, Tetrahedron Lett., 2010, 51, 425–427 CrossRef CAS; (b) F. Pünner and G. Hilt, Regioselective solvent-dependent benzannulation of conjugated enynes, Chem. Commun., 2012, 48, 3617–3619 RSC.
- For the preparation of PdP2 species, see the following: (a) E. Negishi, T. Takahashi and K. Akiyoshi, Bis(Triphenylphosphine)Palladium - Its Generation, Characterization, and Reactions, J. Chem. Soc., Chem. Commun., 1986, 1338–1339 RSC. For synthetic applications, see, e.g.: (b) M. Pour and E. Negishi, An efficient and selective synthesis of nakienone A involving a novel protocol for alpha-alkenylation of ketones via palladium-catalyzed alkenyl-alkenyl coupling, Tetrahedron Lett., 1997, 38, 525–528 CrossRef CAS; (c) B. A. Anderson, L. M. Becke, R. N. Booher, M. E. Flaugh, N. K. Harn, T. J. Kress, D. L. Varie and J. P. Wepsiec, Application of palladium(0)-catalyzed processes to the synthesis of oxazole-containing partial ergot alkaloids, J. Org. Chem., 1997, 62, 8634–8639 CrossRef CAS; (d) J. Pavlík, I. Šnajdr, J. Kuneš, M. Špulák and M. Pour, A Short Entry to alpha-Substituted gamma-Alkylidene Pentenolides. Synthesis and Preliminary Biological Evaluation of Novel Gelastatin Analogues, J. Org. Chem., 2009, 74, 703–709 CrossRef PubMed.
- (a) F. Bellina, A. Carpita, M. Desantis and R. Rossi, Synthesis of Variously 2-Substituted Alkyl (Z)-2-Alkenoate and (E)-2-Alkenoate and (Z)-Alpha-Ylidene-Gamma-Butyrolactone and (E)-Alpha-Ylidene-Gamma-Butyrolactone Via Palladium-Mediated Cross-Coupling Reactions between Organostannanes and Organic Halides, Tetrahedron, 1994, 50, 12029–12046 CrossRef CAS; (b) A. L. Casado and P. Espinet, Quantitative evaluation of the factors contributing to the “copper effect” in the Stille reaction, Organometallics, 2003, 22, 1305–1309 CrossRef CAS; (c) M. H. Pérez-Temprano, J. A. Casares and P. Espinet, Bimetallic Catalysis using Transition and Group 11 Metals: An Emerging Tool for C?C Coupling and Other Reactions, Chem. – Eur. J., 2012, 18, 1864–1884 CrossRef PubMed.
- H. Zhang, L. Xu, X. Liu, J. Fan, T. Wang, T. Shen, S. Wang and D. Ren, Dracomolphesin A–E, five 3,4-seco-phenylpropanoids with Nrf2 inducing activity from Dracocephalum moldavica, Chin. Chem. Lett., 2020, 31, 1259–1262 CrossRef CAS.
- For development of the Pd(TFP)4 catalyst, see the following: (a) V. Farina and B. Krishnan, Large Rate Accelerations in the Stille Reaction with Tri-2-Furylphosphine and Triphenylarsine as Palladium Ligands - Mechanistic and Synthetic Implications, J. Am. Chem. Soc., 1991, 113, 9585–9595 CrossRef CAS; (b) V. Farina, S. Kapadia, B. Krishnan, C. J. Wang and L. S. Liebeskind, On the Nature of the Copper Effect in the Stille Cross-Coupling, J. Org. Chem., 1994, 59, 5905–5911 CrossRef CAS; (c) V. Farina, New perspectives in the cross-coupling reactions of organostannanes, Pure Appl. Chem., 1996, 68, 73–78 CrossRef CAS. For recent synthetic applications, see, e.g.: (d) K. C. Nicolaou, P. G. Bulger and D. Sarlah, Palladium-catalyzed cross-coupling reactions in total synthesis, Angew. Chem., Int. Ed., 2005, 44, 4442–4489 CrossRef CAS PubMed; (e) A. Steven and L. E. Overman, Total synthesis of complex cyclotryptamine alkaloids: Stereocontrolled construction of quaternary carbon stereocenters, Angew. Chem., Int. Ed., 2007, 46, 5488–5508 CrossRef CAS PubMed; (f) C. I. Stathakis, S. Bernhardt, V. Quint and P. Knochel, Improved Air-Stable Solid Aromatic and Heterocyclic Zinc Reagents by Highly Selective Metalations for Negishi Cross-Couplings, Angew. Chem., Int. Ed., 2012, 51, 9428–9432 CrossRef CAS PubMed; (g) L. Klier, T. Bresser, T. A. Nigst, K. Karaghiosoff and P. Knochel, Lewis Acid-Triggered Selective Zincation of Chromones, Quinolones, and Thiochromones: Application to the Preparation of Natural Flavones and Isoflavones, J. Am. Chem. Soc., 2012, 134, 13584–13587 CrossRef CAS PubMed; (h) G. Valot, C. S. Regens, D. P. O'Malley, E. Godineau, H. Takikawa and A. Fürstner, Total Synthesis of Amphidinolide F, Angew. Chem., Int. Ed., 2013, 52, 9534–9538 CrossRef CAS PubMed; (i) Y. Yang, T. J. L. Mustard, P. H. Y. Cheong and S. L. Buchwald, Palladium-Catalyzed Completely Linear-Selective Negishi Cross-Coupling of Allylzinc Halides with Aryl and Vinyl Electrophiles, Angew. Chem., Int. Ed., 2013, 52, 14098–14102 CrossRef CAS PubMed; (j) D. Haas, J. M. Hammann, R. Greiner and P. Knochel, Recent Developments in Negishi Cross-Coupling Reactions, ACS Catal., 2016, 6, 1540–1552 CrossRef CAS; (k) M. A. Miller, B. Askevold, H. Mikula, R. H. Kohler, D. Pirovich and R. Weissleder, Nano-palladium is a cellular catalyst for in vivo Chemistry, Nat. Commun., 2017, 8, 15906 CrossRef CAS PubMed.
- (a) J. P. Wolfe, R. A. Singer, B. H. Yang and S. L. Buchwald, Highly Active Palladium Catalysts for Suzuki Coupling Reactions, J. Am. Chem. Soc., 1999, 121, 9550–9561 CrossRef CAS; (b) A. J. Kendall, L. N. Zakharov and D. R. Tyler, Steric and Electronic Influences of Buchwald-Type Alkyl-JohnPhos Ligands, Inorg. Chem., 2016, 55, 3079–3090 CrossRef CAS PubMed.
- J. Kratochvíl, Z. Novák, M. Ghavre, L. Nováková, A. Růžička, J. Kuneš and M. Pour, Fully Substituted Pyranones via Quasi-Heterogeneous Genuinely Ligand-Free Migita-Stille Coupling of lodoacrylates, Org. Lett., 2015, 17, 520–523 CrossRef PubMed.
- For the preparation of 9w, see the following: Z. Brůža, J. Kratochvíl, J. N. Harvey, L. Rulíšek, L. Nováková, J. Maříková, J. Kuneš, P. Kočovský and M. Pour, A New Insight into the Stereoelectronic Control of the Pd-0-Catalyzed Allylic Substitution: Application for the Synthesis of Multisubstituted Pyran-2-ones via an Unusual 1,3-Transposition, Chem. – Eur. J., 2019, 25, 8053–8060 CrossRef PubMed.
- A. E. Reed, R. B. Weinstock and F. Weinhold, Natural-Population Analysis, J. Chem. Phys., 1985, 83, 735–746 CrossRef CAS.
- W. J. Lording, T. Fallon, M. S. Sherburn and M. N. Paddon-Row, The simplest Diels-Alder reactions are not endo-selective, Chem. Sci., 2020, 11, 11915–11926 RSC.
- K. A. Ahrendt, C. J. Borths and D. W. C. MacMillan, New strategies for organic catalysis: The first highly enantioselective organocatalytic Diels-Alder reaction, J. Am. Chem. Soc., 2000, 122, 4243–4244 CrossRef CAS.
- N. J. Green, A. L. Lawrence, G. Bojase, A. C. Willis, M. N. Paddon-Row and M. S. Sherburn, Domino Cycloaddition Organocascades of Dendralenes, Angew. Chem., Int. Ed., 2013, 52, 8333–8336 CrossRef CAS PubMed.
- W. Nardello, S. Bogaert, P. L. Alsters and J. M. Aubry, Singlet oxygen generation from H2O2/MoO42−: peroxidation of hydrophobic substrates in pure organic solvents, Tetrahedron Lett., 2002, 43, 8731–8734 CrossRef.
- Increasing the load of 16 to 5 equiv. had no effect on the yield.
- R. M. Wang, G. Bojase, A. C. Willis, M. N. Paddon-Row and M. S. Sherburn, Nitroso-Dienophile Additions to Dendralenes: A Short Synthesis of Branched Aminosugars, Org. Lett., 2012, 14, 5652–5655 CrossRef CAS PubMed.
- J. D. Chai and M. Head-Gordon, Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- M. Linder and T. Brinck, On the method-dependence of transition state asynchronicity in Diels-Alder reactions, Phys. Chem. Chem. Phys., 2013, 15, 5108–5114 RSC.
- (a) K. Alder and G. Stein, Justus Liebigs Ann. Chem., 1934, 514, 1 CrossRef CAS; (b) K. Alder and G. Stein, Untersuchungen über den Verlauf der Diensynthese, Angew. Chem., 1937, 50, 510–519 CrossRef CAS.
- T. A. Bradford, A. D. Payne, A. C. Willis, M. N. Paddon-Row and M. S. Sherburn, Practical Synthesis and Reactivity of [3]Dendralene, J. Org. Chem., 2010, 75, 491–494 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, spectral data, and copies of NMR spectra. CCDC 2238545. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3qo01221b |
| This journal is © the Partner Organisations 2023 |