
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Aza-Wolff rearrangement of N-fluoroalkyl triazoles to ketenimines†
Anna
Kubíčková
 ab,
Athanasios
Markos
a,
Svatava
Voltrová
a,
Anežka
Marková
a,
Josef
Filgas
b,
Blanka
Klepetářová
a,
Petr
Slavíček
b and
Petr
Beier
*a
ab,
Athanasios
Markos
a,
Svatava
Voltrová
a,
Anežka
Marková
a,
Josef
Filgas
b,
Blanka
Klepetářová
a,
Petr
Slavíček
b and
Petr
Beier
*a
aInstitute of Organic Chemistry and Biochemistry of the Czech Academy of Sciences, Flemingovo nam. 2, 16610 Prague 6, Czech Republic. E-mail: beier@uochb.cas.cz
bUniversity of Chemistry and Technology, Technická 5, 166 28 Prague 6, Czech Republic
First published on 23rd May 2023
Abstract
N-Fluoroalkylated 1,2,3-triazoles underwent a microwave-heating-assisted ring opening, nitrogen molecule elimination and concomitant group rearrangement to form isolable N-fluoroalkylketenimines. This reagent-free process is characterized by a wide scope and high efficiency and provides a new route to unexplored N-fluoroalkyl compounds. The reaction mechanism was investigated by a combination of mechanistic and computational studies. [2 + 2] cycloaddition of ketenimines with alkynes or alkenes afforded novel cyclobutenimines and cyclobutanimines, respectively. Addition of oxygen-, sulfur- and nitrogen nucleophiles to ketenimines gave new N-fluoroalkyl imidates, thioimidates and amidines.
Introduction
The Wolff rearrangement of α-diazo carbonyl compounds to ketenes is one of the most significant transformations that has contributed to the growth of chemical synthesis in the 20th century (Scheme 1A).1 The ketenes generated in this way are highly useful substrates with a wide range of synthetic applications.2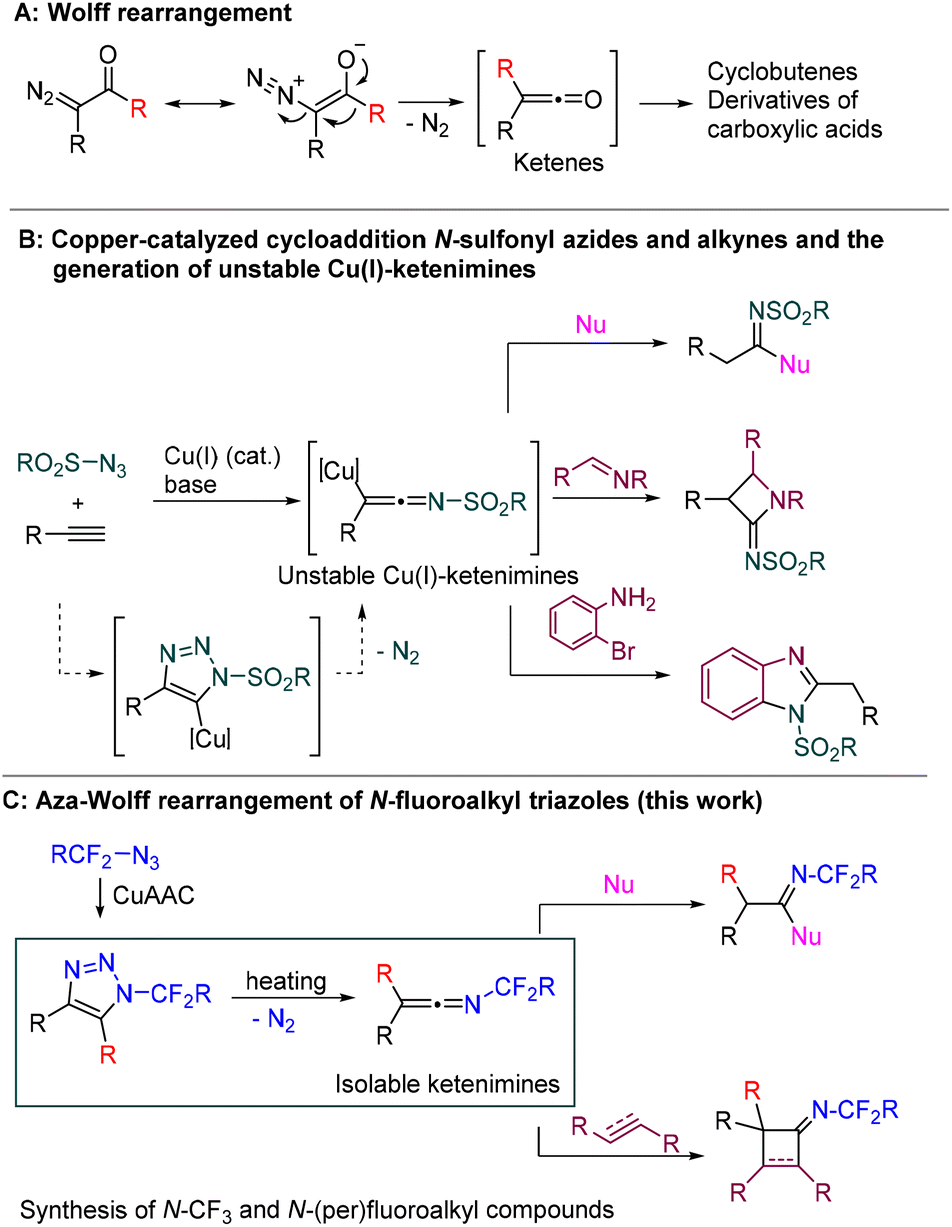 | ||
| Scheme 1 A: Wolff rearrangement. B: Generation of N-sulfonyl ketenimines and their selected reactions. C: Aza-Wolff rearrangement of N-fluoroalkyl triazoles (this work). | ||
Ketenimines, aza-analogues of ketenes, are a related class of compounds with cumulated double bonds and a broad range of applications in organic chemistry.3 Although they are generally more stable than ketenes, most ketenimines are not isolable materials and therefore need to be formed in situ as reactive intermediates. For example, highly reactive N-sulfonylketenimines are generated by copper-catalyzed azide alkyne cycloaddition (CuAAC) of N-sulfonyl azides and alkynes and subsequent spontaneous denitrogenative rearrangement of a copper-triazole intermediate (Scheme 1B, left). They can serve as three atom (C–C–N) synthons in the syntheses of amides,4–6 amidines,7 imidates,8 four-, five-, six- and seven-membered heterocycles, amino acids, and many other biologically and pharmaceutically valuable compounds (see Scheme 1B, right for selected examples).9
The formation of ketenimines by CuAAC is limited to electron-deficient sulfonyl/phosphoryl azides; alkyl or aryl azides preferably form triazole rings.10 Even N-fluoroalkyl azides with a strong electron-withdrawing N-CF2R groups react with terminal alkynes in a CuAAC fashion to form N-fluoroalkyl triazoles,11–14 substrates which have been showcased as being useful for the synthesis of N-fluoroalkyl azoles (e.g. imidazoles, pyrroles)15–18 and other fluoroalkyl-containing compounds.19–22
The incorporation of fluorinated groups into small molecules is a widely used approach to the modification of their pharmacological as well as pharmacokinetic properties.23 Whereas C-, and O-fluoroalkyl compounds have been studied extensively, N-fluoroalkyl substrates have received more attention only recently.24 That their potential is unexplored is caused mainly by the lack of efficient methods to synthesize these compounds using readily available starting materials. Introducing a CF3 group directly into nitrogen functionalities is a greatly challenging and substrate-specific process.25 However, an approach based on N-CF3 synthons has recently emerged as an alternative and atom-economical route. The Schoenebeck group introduced versatile N-CF3 carbamoyl fluorides as building blocks for the synthesis of highly attractive tertiary N-CF3 amides,26 hydrazines,27 ureas,28,29 formamides30 and other compounds.31 Synthesis of tertiary N-CF3 amides was also recently reported by Toste and Wilson from carboxylic acid derivatives and isothiocyanates in the presence of AgF.32 Another synthon, the N-CF3 nitrilium ion, was introduced by Xu and Wang for the synthesis of N-CF3 azoles and imido derivatives.33,34 Despite the great progress in recent years, most of the strategies for this are limited to N-CF3 compounds and methods for the synthesis of a wide range of N-fluoroalkyl compounds remain underdeveloped.
Here, we report a highly efficient, atom-economical, waste- and reagent-free way to synthesize isolable N-fluoroalkylketenimines by thermal aza-Wolff rearrangement of N-fluoroalkyl-1,2,3-triazoles (Scheme 1C). The synthetic utility of N-fluoroalkyl ketenimines has been shown on examples of [2 + 2] cycloadditions affording novel N-fluoroalkyl cyclobutenimines and cyclobutanimines as well as a variety of imido compounds in reactions of ketenimines with nucleophiles.
Results and discussion
While exploring the thermal stability of N-pentafluoroethyl-4-phenyl-1,2,3-triazole (1a) by differential scanning calorimetry (DSC), thermogravimetric analysis (TG) and gas chromatography with mass detection (GC/MS), we noticed decomposition at temperatures of around 140–160 °C in solution or without solvent and also decomposition in the gas phase (GC inlet temp.: 250 °C) with a mass loss of m/z 28. Detailed NMR and IR analyses of preparative reactions revealed the quantitative formation of N-(pentafluoroethyl)-2-phenylethen-1-imine (2a), which was fully characterized (Scheme 2, see the ESI†). RFA analysis ruled out any possible participation of the copper catalyst carried over from the synthesis of 1a with CuAAC. Various solvents were tested including chloroform, toluene, THF, acetone and polar protic solvents, but the best results were obtained using DCE (see the ESI† for details). Conventional heating is also possible but leads to 2a in a longer reaction time and unidentified side products.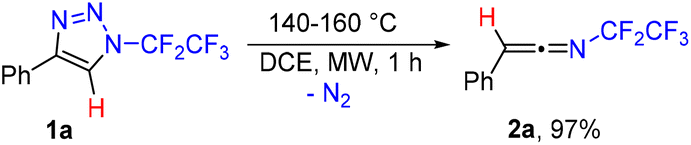 | ||
| Scheme 2 Synthesis of ketenimine 2a by the thermal decomposition of triazole 1a. | ||
Subsequent investigation revealed that the denitrogenative rearrangement of N-fluoroalkyl-1,2,3-triazoles 1 to N-fluoroalkylketenimines 2 is of a wide scope and, in a vast majority of cases, highly efficient (Table 1).
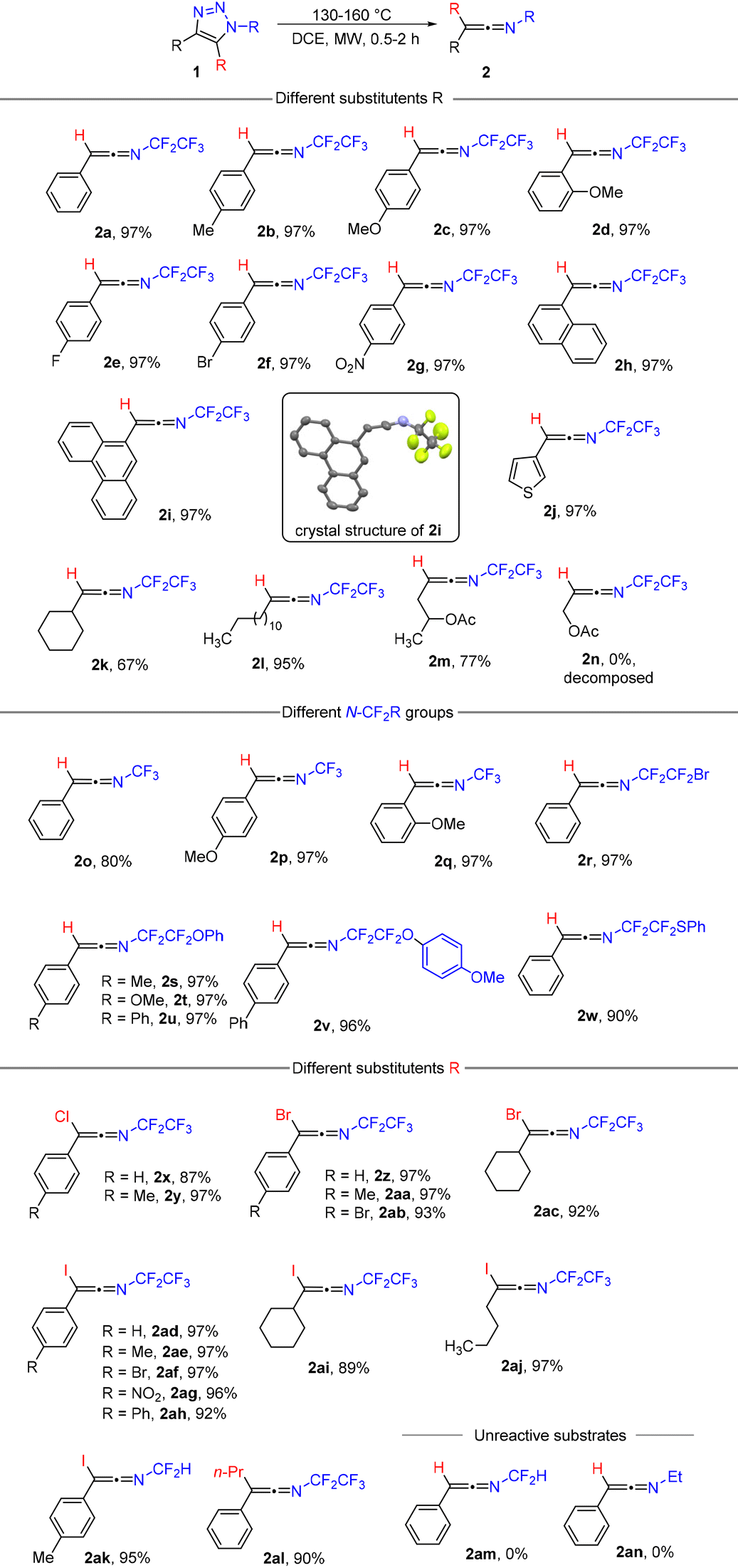
|
Triazoles with various combinations of aryl, substituted aryl, heteroaryl, alkyl, substituted alkyl or cycloalkyl groups in position four, hydrogen, Cl, Br, I or an alkyl group in position five and trifluoromethyl (–CF3), pentafluoroethyl (–CF2CF3), bromotetrafluoroethyl (–CF2CF2Br), substituted tetrafluoroethyl (–CF2CF2R) and even difluoromethyl groups (–CF2H) on the nitrogen atom all underwent the reaction and provided products 2 in high to excellent yields. Despite the robustness of the method, certain limitations were observed. Triazole 1n underwent rearrangement; however, the product was not stable (see the ESI† for details). The presence of a 5-halo substituent (iodo in particular) in triazoles 1 improved isolated yields for difficult substrates (4-alkyl, 4-cycloalkyl) compared to unsubstituted examples. This effect is strikingly strong in the case of N-difluoromethyl substrates, where the iodo product formed in high yield (2ak) and the unsubstituted one did not form at all (2am). N-Alkyl triazole (1an) was not a competent substrate in this reaction. Generally, ketenimines 2 did not require any purification; solvent removal was sufficient to obtain suitably pure samples. Their purification using silica gel column chromatography is also possible. Ketenimines 2 are mostly liquids (2i and 2v are solids; see the crystal structure in Table 1) with limited air and moisture stability as pure substances; in solution (DCE, pentane) they were stable for weeks at room temperature in an inert atmosphere.
Having demonstrated the broad scope of the reaction, we investigated the mechanism of this unprecedented transformation. First, we synthesized isotopically labelled triazoles 3–5 from 13C-phenylacetylene.35 All three triazoles underwent ring opening and rearrangement to form ketenimines 6–8, which contained the label exclusively in the central sp-hybridized carbon atom, proving that the reaction proceeds via a [1,2]-shift of the R group, similarly to the Wolff rearrangement (Scheme 3).
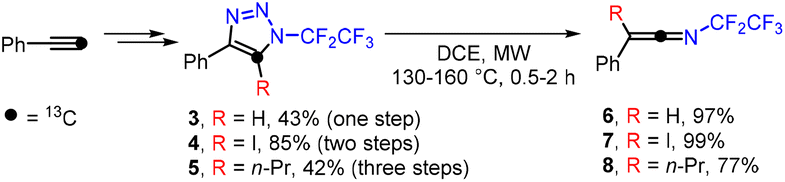 | ||
| Scheme 3 Synthesis of 13C-labelled triazoles 3–5 and ketenimines 6–8. | ||
As the Wolff rearrangement can proceed via a concerted or step-wise mechanism involving carbenes,36 we performed ab initio calculations. We optimized the structures with the multi-reference CASSCF(4,4)/6-31+g* method, as it was able to localize open-shell systems such as carbene and nitrene (unlike, e.g., density functional approaches). Single point energies were calculated using the coupled cluster CCSD(T) method with the aug-cc-pVDZ basis set. The differences in thermal corrections were found to be on the order of 10−4 eV (at the DFT/BMK/6-31+g* level), so entropic effects do not play any role here. The effect of the solvent was also found to be negligible. The lowest energy pathway involved the expected diazo intermediate and final ketenimine product (Fig. 1A, blue box). This result is consistent with a concerted Wolff rearrangement, which also requires an s-cis configuration of the diazo and heteroatom groups.
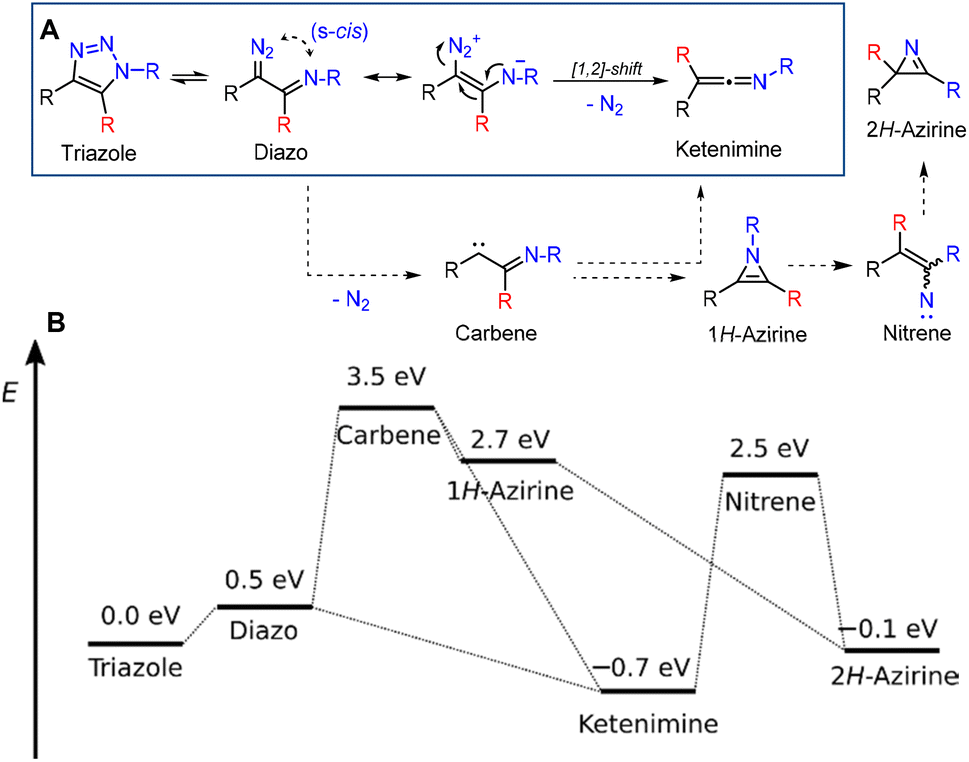 | ||
| Fig. 1 A: Reactive species considered in the ab initio calculation of the reaction mechanism (R = Me, R = H, R = CF3). B: Calculated energies of the structures (related to the starting triazole). The geometries were optimized at the CASSCF(4,4)/6-31+g* level and the energies were calculated using the CCSD(T)/aug-cc-pVDZ method. | ||
Carbene and its hypothetical rearrangement products such as 1H-azirine, and nitrene species were found to be very unstable (see the ESI† for more details), too high in energy and are not involved in the reaction mechanism (Fig. 1B).
Next, the synthetic utility of the prepared ketenimines in [2 + 2] cycloadditions and in additions of nucleophiles was explored. Thermal cycloaddition of phenylacetylene proceeded with expected regioselectivity and produced cyclobutenimines 9 in good yields as E/Z mixtures with moderate to high stereoselectivities towards E-isomers (Scheme 4). Ketenimines with halogen substituents (R = Cl, Br, I) were unreactive, but the alkyl group (R = n-Pr) was tolerated well. Internal alkyne diphenylacetylene proved to be a competent substrate in cycloaddition under microwave heating affording cyclobutenimines 10 in good yields and exclusively as E-isomers. The crystal structure of cyclobutenimine (E)-10b is presented in Scheme 4. On the other hand, the electron acceptor terminal or internal alkynes (ethyl propiolate, diethyl acetylenedicarboxylate) exhibited no reactivity even under heating.
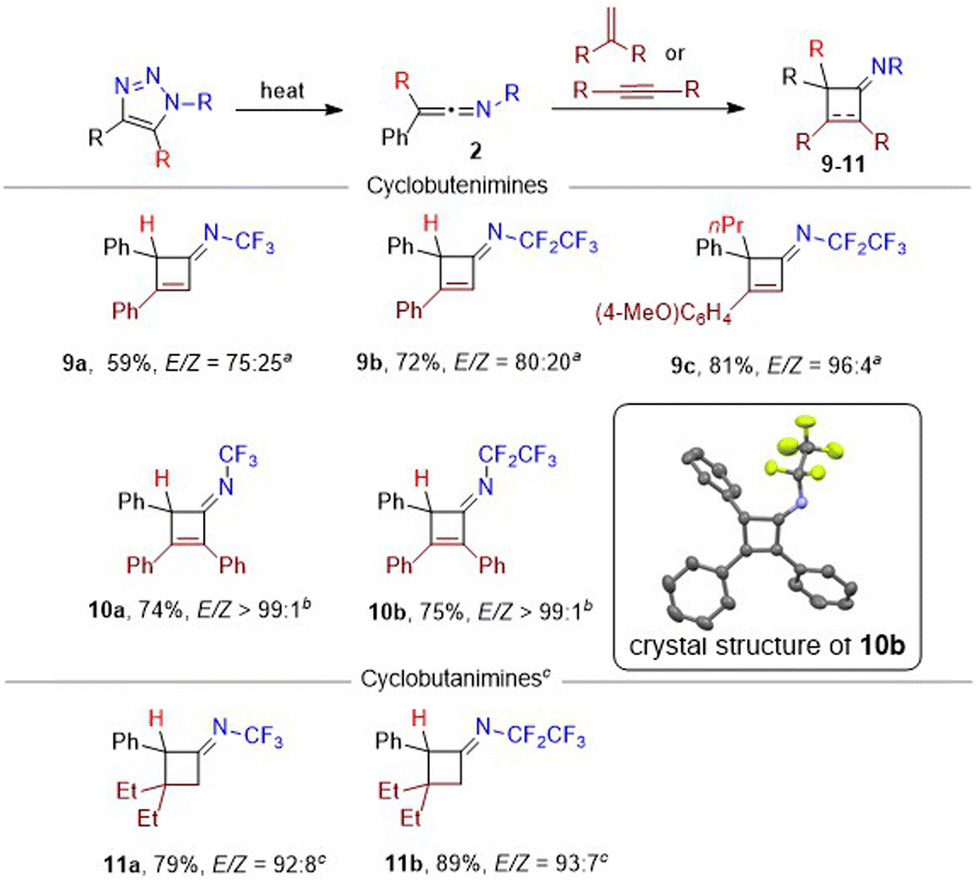 | ||
Scheme 4 [2 + 2] cycloadditions of ketenimines 2 with alkynes and alkenes. All yields were calculated from triazoles 1. Only (E)-9–11 are shown. Conditions: a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) phenylacetylene (2 equiv.), DCE, 100 °C (for R = H) or 170 °C (for R = n-Pr), 1 h; bdiphenylacetylene (2 equiv.), DCE, MW, 165–170 °C, 1–2 h; c2-ethylbut-1-ene (3 equiv.), DCE, 40 °C, 12 h. phenylacetylene (2 equiv.), DCE, 100 °C (for R = H) or 170 °C (for R = n-Pr), 1 h; bdiphenylacetylene (2 equiv.), DCE, MW, 165–170 °C, 1–2 h; c2-ethylbut-1-ene (3 equiv.), DCE, 40 °C, 12 h. | ||
Cycloaddition of ketenimines with terminal alkenes, such as styrene (60 °C) or electron-rich ethyl vinyl ether (room temperature) proceeded well; however, the products (cyclobutanimines) were unstable. With disubstituted alkene (2-ethylbut-1-ene) cyclobutanimines 11 formed with high efficiency, regio- and stereoselectivity (Scheme 4). Internal alkenes (cis- or trans-stilbene), were unlike internal alkynes unreactive, even upon prolonged heating. The E/Z selectivity is governed by the sterical factors. Cyclobutane and cyclobutene imines are rare compounds and their N-fluoroalkyl derivatives were never synthesized before.
Finally, the reactivity of ketenimines 2 with N-, O- and S-centred nucleophiles was investigated (Scheme 5). Fast reactions were observed with an excess of ethanol and ethanethiol at room temperature to afford iminoesters 12 and thioimidates 13. In the cases of 12b and 13b, dehydrofluorinated side-product 12b′ and 13b′ formed in a small amount, and instead of thioimidate 13c, imidoyl fluoride 13c′ was isolated. All imino products formed in E-configuration. With primary amines, the addition took place, but the products decomposed and a complex reaction mixture was formed; however, quantitative formation of amidines 14 was observed using secondary amines as nucleophiles. Stable products of hydrolysis N-trifluoroacetylamidines 15 were obtained from N-pentafluoroethyl derivatives, using column chromatography. The crystal structure of amidine 15f was determined (Scheme 5). Very recently, related N-CF3 amidines, imidates and thioimidates were prepared by the reaction of PhI(CF3)Cl with nitriles, followed by the addition of nucleophiles to the intermediate N-CF3 nitriliums.33
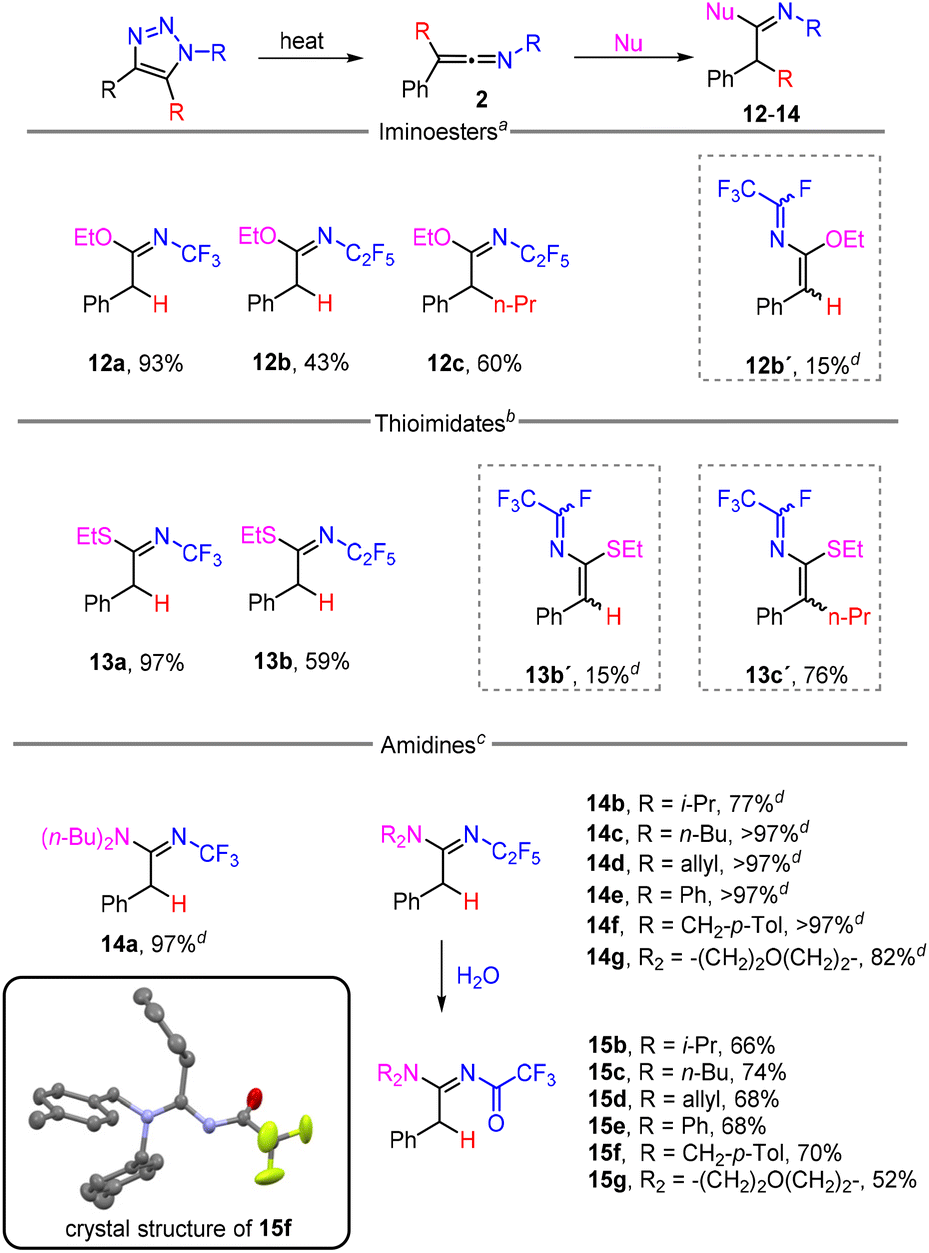 | ||
| Scheme 5 Addition of heteroatom nucleophiles to ketenimines 2. All yields were calculated from triazoles 1. Conditions: aalcohol (3 equiv.), DCE, 25 °C, 30 min (14 h for 12c); bthiol (3 equiv.), DCE, 25 °C, 30 min (14 h for 13c′); camine (1 equiv.), DCE, 25 °C, 30 min. d19F NMR yield. | ||
Conclusions
Structurally diverse N-(per)fluoroalkyl-1,2,3-triazoles rearranged under microwave heating to novel and surprisingly stable N-(per)fluoroalkyl ketenimines, which were isolated and fully characterized by spectroscopic methods and in one case also by X-ray diffraction. The mechanism, supported by control experiments, an isotopic labelling study and ab initio calculations, proceeds via triazole ring opening, nitrogen elimination and group rearrangement (1,2-shift). Ketenimines reacted with alkynes or alkenes in [2 + 2] cycloaddition reactions under mild conditions to yield novel cyclobutenimines or cyclobutanimines, respectively. The addition of alcohols, thiols, or secondary amine nucleophiles to ketenimines afforded imidates, thioimidates, or amidines, respectively. Thus, N-fluoroalkyl-1,2,3-triazoles have been demonstrated to be useful precursors of N-fluoroalkylated ketenimines, which served as valuable building blocks in the expansion of the chemical space of accessible N-fluoroalkyl compounds. Further transformations of these new N-fluoroalkyl building blocks are currently being investigated in our laboratory.Author contributions
A. Kubíčková, A. Markos, S. Voltrová, A. Marková conceived the idea, performed the experiments and partially wrote the manuscript. J. Filgas and P. Slavíček performed calculations and partially wrote the manuscript. B. Klepetářová measured X-ray diffraction and solved crystal structures. P. Beier conceived the idea and wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Czech Academy of Sciences (Research Plan RVO: 61388963) and by the Czech Science Foundation (23-04659S and 23-07066A). We thank Dr Radek Pohl (IOCB Prague) for advanced NMR experiments and Dr Lucie Bednárová (IOCB Prague) for IR experiments.References
- A. Ford, H. Miel, A. Ring, C. N. Slattery, A. R. Maguire and M. A. McKervey, Modern Organic Synthesis with α-Diazocarbonyl Compounds, Chem. Rev., 2015, 115, 9981–10080 CrossRef CAS PubMed.
- A. D. Allen and T. T. Tidwell, Ketenes and Other Cumulenes as Reactive Intermediates, Chem. Rev., 2013, 113, 7287–7342 CrossRef CAS PubMed.
- P. Lu and Y. Wang, The thriving chemistry of ketenimines, Chem. Soc. Rev., 2012, 41, 5687–5705 RSC.
- S. H. Cho, E. J. Yoo, I. Bae and S. Chang, Copper-Catalyzed Hydrative Amide Synthesis with Terminal Alkyne, Sulfonyl Azide, and Water, J. Am. Chem. Soc., 2005, 127, 16046–16047 CrossRef CAS PubMed.
- M. P. Cassidy, J. Raushel and V. V. Fokin, Practical Synthesis of Amides from In Situ Generated Copper(I) Acetylides and Sulfonyl Azides, Angew. Chem., Int. Ed., 2006, 45, 3154–3157 CrossRef CAS PubMed.
- S. H. Cho and S. Chang, Rate-Accelerated Nonconventional Amide Synthesis in Water: A Practical Catalytic Aldol-Surrogate Reaction, Angew. Chem., Int. Ed., 2007, 46, 1897–1900 CrossRef CAS PubMed.
- I. Bae, H. Han and S. Chang, Highly Efficient One-Pot Synthesis of N-Sulfonylamidines by Cu-Catalyzed Three-Component Coupling of Sulfonyl Azide, Alkyne, and Amine, J. Am. Chem. Soc., 2005, 127, 2038–2039 CrossRef CAS PubMed.
- E. J. Yoo, I. Bae, S. H. Cho, H. Han and S. Chang, A Facile Access to N-Sulfonylimidates and Their Synthetic Utility for the Transformation to Amidines and Amides, Org. Lett., 2006, 8, 1347–1350 CrossRef CAS PubMed.
- M. Alajarin, M. Marin-Luna and A. Vidal, Recent Highlights in Ketenimine Chemistry, Eur. J. Org. Chem., 2012, 5637–5653 CrossRef CAS.
- J. E. Hein and V. V. Fokin, Copper-catalyzed azide–alkyne cycloaddition (CuAAC) and beyond: new reactivity of copper(I) acetylides, Chem. Soc. Rev., 2010, 39, 1302–1315 RSC.
- S. Bahadorikhalili, M. Divar, T. Damghani, F. Moeini, S. Ghassamipour, A. Iraji, M. A. Miller, B. Larijani and M. Mahdavi, N-sulfonyl ketenimine as a versatile intermediate for the synthesis of heteroatom containing compounds, J. Organomet. Chem., 2021, 939, 121773 CrossRef CAS.
- Z. E. Blastik, S. Voltrová, V. Matoušek, B. Jurásek, D. W. Manley, B. Klepetářová and P. Beier, Azidoperfluoroalkanes: Synthesis and Application in Copper(I)-Catalyzed Azide–Alkyne Cycloaddition, Angew. Chem., Int. Ed., 2017, 56, 346–349 CrossRef CAS PubMed.
- S. Voltrová, I. Putovný, V. Matoušek, B. Klepetářová and P. Beier, Reintroducing Azidodifluoromethane: Synthesis, Stability and [3+2] Cycloadditions, Eur. J. Org. Chem., 2018, 5087–5090 CrossRef.
- S. Voltrova, M. Muselli, J. Filgas, V. Matoušek, B. Klepetářová and P. Beier, Synthesis of tetrafluoroethylene- and tetrafluoroethyl-containing azides and their 1,3-dipolar cycloaddition as synthetic application, Org. Biomol. Chem., 2017, 15, 4962–4965 RSC.
- D. Tichý, V. Koštál, V. Motornov, I. Klimánková and P. Beier, Preparation of 1-Azido-2-Bromo-1,1,2,2-Tetrafluoroethane and Its Use in the Synthesis of N-Fluoroalkylated Nitrogen Heterocycles, J. Org. Chem., 2020, 85, 11482–11489 CrossRef PubMed.
- V. Motornov, A. Markos and P. Beier, A rhodium-catalyzed transannulation of N-(per)fluoroalkyl-1,2,3-triazoles under microwave conditions – a general route to N-(per)fluoroalkyl-substituted five-membered heterocycles, Chem. Commun., 2018, 54, 3258–3261 RSC.
- V. Motornov and P. Beier, Chemoselective Aza-[4+3]-annulation of N-Perfluoroalkyl-1,2,3-triazoles with 1,3-Dienes: Access to N-Perfluoroalkyl-Substituted Azepines, J. Org. Chem., 2018, 83, 15195–15201 CrossRef CAS PubMed.
- O. Bakhanovich, V. Khutorianskyi, V. Motornov and P. Beier, Synthesis of N-perfluoroalkyl-3,4-disubstituted pyrroles by rhodium-catalyzed transannulation of N-fluoroalkyl-1,2,3-triazoles with terminal alkynes, Beilstein J. Org. Chem., 2021, 17, 504–510 CrossRef CAS PubMed.
- V. Motornov, V. Košťál, A. Markos, D. Täffner and P. Beier, General approach to 2-fluoroalkyl 1,3-azoles via the tandem ring opening and defluorinative annulation of N-fluoroalkyl-1,2,3-triazoles, Org. Chem. Front., 2019, 6, 3776–3780 RSC.
- A. Markos, L. Janecký, B. Klepetářová, R. Pohl and P. Beier, Stereoselective Synthesis of (Z)-β-Enamido Fluorides from N-Fluoroalkyl- and N-Sulfonyl-1,2,3-triazoles, Org. Lett., 2021, 23, 4224–4227 CrossRef CAS PubMed.
- A. Markos, L. Janecký, T. Chvojka, T. Martinek, H. Martinez-Seara, B. Klepetářová and P. Beier, Haloalkenyl Imidoyl Halides as Multifacial Substrates in the Stereoselective Synthesis of N–Alkenyl Compounds, Adv. Synth. Catal., 2021, 363, 3258–3266 CrossRef CAS.
- A. Markos, S. Voltrová, V. Motornov, D. Tichý, B. Klepetářová and P. Beier, Stereoselective Synthesis of (Z)-β-Enamido Triflates and Fluorosulfonates from N-Fluoroalkylated Triazoles, Chem. Eur. J., 2019, 25, 7640–7644 CrossRef CAS PubMed.
- P. A. Champagne, J. Desroches, J.-D. Hamel, M. Vandamme and J.-F. Paquin, Monofluorination of Organic Compounds: 10 Years of Innovation, Chem. Rev., 2015, 115, 9073–9174 CrossRef CAS PubMed.
- S. Schiesser, R. J. Cox and W. Czechtizky, The powerful symbiosis between synthetic and medicinal chemistry, Future Med. Chem., 2021, 13, 941–944 CrossRef CAS PubMed.
- S. Liu, Y. Huang, J. Wang, F.-L. Qing and X.-H. Xu, General Synthesis of N-Trifluoromethyl Compounds with N-Trifluoromethyl Hydroxylamine Reagents, J. Am. Chem. Soc., 2022, 144, 1962–1970 CrossRef CAS PubMed.
- T. Scattolin, S. Bouayad-Gervais and F. Schoenebeck, Straightforward access to N-trifluoromethyl amides, carbamates, thiocarbamates and ureas, Nature, 2019, 573, 102–107 CrossRef CAS PubMed.
- S. Bouayad-Gervais, T. Scattolin and F. Schoenebeck, N-Trifluoromethyl Hydrazines, Indoles and Their Derivatives, Angew. Chem., Int. Ed., 2020, 59, 11908–11912 CrossRef CAS PubMed.
- A. Turksoy, S. Bouayad-Gervais and F. Schoenebeck, N-CF3 Imidazolidin-2-one Derivatives via Photocatalytic and Silver-Catalyzed Cyclizations, Chem. Eur. J., 2022, 28, e202201435 CrossRef CAS PubMed.
- S. Bouayad-Gervais, C. D.-T. Nielsen, A. Turksoy, T. Sperger, K. Deckers and F. Schoenebeck, Access to Cyclic N-Trifluoromethyl Ureas through Photocatalytic Activation of Carbamoyl Azides, J. Am. Chem. Soc., 2022, 144, 6100–6106 CrossRef CAS PubMed.
- F. G. Zivkovic, C. D.-T. Nielsen and F. Schoenebeck, Access to N-CF3 Formamides by Reduction of N-CF3 Carbamoyl Fluorides, Angew. Chem., Int. Ed., 2022, 61, e202213829 CrossRef CAS PubMed.
- C. D.-T. Nielsen, F. G. Zivkovic and F. Schoenebeck, Synthesis of N-CF3 Alkynamides and Derivatives Enabled by Ni-Catalyzed Alkynylation of N-CF3 Carbamoyl Fluorides, J. Am. Chem. Soc., 2021, 143, 13029–13033 CrossRef CAS PubMed.
- J. Liu, M. F. L. Parker, S. Wang, R. R. Flavell, F. D. Toste and D. M. Wilson, Synthesis of N-trifluoromethyl amides from carboxylic acids, Chem, 2021, 7, 2245–2255 CAS.
- R. Z. Zhang, W. Huang, R. X. Zhang, C. Xu and M. Wang, Synthesis of N-CF3 Amidines/Imidates/Thioimidates via N-CF3 Nitrilium Ions, Org. Lett., 2022, 24, 2393–2398 CrossRef CAS PubMed.
- R. Z. Zhang, R. X. Zhang, S. Wang, C. Xu, W. Guan and M. Wang, An N-Trifluoromethylation/Cyclization Strategy for Accessing Diverse N-Trifluoromethyl Azoles from Nitriles and 1,3-Dipoles, Angew. Chem., Int. Ed., 2022, 61, e202110749 CAS.
- M. Zeng, L. Li and S. B. Herzon, A Highly Active and Air-Stable Ruthenium Complex for the Ambient Temperature Anti-Markovnikov Reductive Hydration of Terminal Alkynes, J. Am. Chem. Soc., 2014, 136, 7058–7067 CrossRef CAS PubMed.
- W. Kirmse, 100 Years of the Wolff Rearrangement, Eur. J. Org. Chem., 2002, 2193–2256 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2251498–2251500. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3qo00618b |
| This journal is © the Partner Organisations 2023 |