
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Thermodynamics of the self-assembly of N-annulated perylene bisimides in water. Disentangling the enthalpic and entropic contributions†‡
Manuel A.
Martínez§
 a,
Daniel
Aranda§
b,
Enrique
Ortí
b,
Juan
Aragó
*b and
Luis
Sánchez
*a
a,
Daniel
Aranda§
b,
Enrique
Ortí
b,
Juan
Aragó
*b and
Luis
Sánchez
*a
aDepartamento de Química Orgánica, Facultad de Ciencias Químicas, Universidad Complutense de Madrid, Ciudad Universitaria s/n, 28040 Madrid, Spain. E-mail: lusamar@ucm.es
bInstituto de Ciencia Molecular (ICMol), Universidad de Valencia, 46980 Paterna, Spain. E-mail: juan.arago@uv.es
First published on 27th February 2023
Abstract
We report on the self-assembling features of the amphiphilic N-annulated perylene bisimides (N-PBIs) 1–4 in water. Their self-assembly is investigated both experimentally and theoretically and is shown to be entropically driven in all cases. Importantly, the hydrophilic/hydrophobic ratio, determined by the number and flexibility of the oligo(ethylene) glycol (OEG) chains present in monomers 1–4, plays a relevant role in the exothermic/endothermic nature of the self-assembly enthalpy. Thus, the process is enthalpically favoured in compounds 1 and 2, which are end-capped with two flexible OEG chains, but enthalpically unfavoured in compounds 3 and 4, which are end-capped with bulkier and rigid phenyl groups decorated with three OEG chains. Molecular dynamic simulations including water molecules show the influence of the initial arrangement of the hydrophilic side chains and the number of water–side chain interactions on the enthalpy of the self-assembly. Thus, the π-stacking of 3 to form the aggregated species is accompanied by the rupture of a larger number of stabilizing water–side chain interactions than that computed for compound 1. The loss of these interactions determines the sign of the enthalpy contribution and, therefore, the global stability of the aggregated species in aqueous media.
Introduction
Complexity, adaptability, and robustness are key features of self-assembled biological systems in which water plays a pivotal role.1 Supramolecular self-assembly in aqueous media yields biocompatible materials and aqua-plastics that contain water as a key component to achieve strong non-covalent interactions.2 The common feature of all these natural or synthetic self-assembled structures is the presence of amphiphilic molecules, consisting of hydrophilic and hydrophobic fragments that are soluble in polar and apolar environments and, as a result, form highly organized structures. Cell membranes, in which multiple assembled components provide structural integrity to the cell and regulate exchanges with the environment or with the double helix of DNA, which contains all the genetic code of the living organisms, are paradigms of self-assembled amphiphilic systems in water.3 A large number of supramolecular ensembles attained by the self-assembly of amphiphilic systems, based either on natural motifs (e.g., peptides or lipids) or synthetic scaffolds, can be found in the literature as functional materials4 or nanoreactors.5In all these systems, the hydrophobic effect determines the thermodynamics of the self-assembly process, with dissimilar contributions to the enthalpic and entropic terms. In natural environments (i.e., in aqueous media), the self-assembly events are entropically driven. Typical examples are the self-assembly of the tobacco-mosaic virus, the formation of β-amyloids or the formation of collagen fibres.6 The supramolecular polymerization mechanism of synthetic amphiphiles, especially those with aromatic central hydrophobic cores such as hexaperibenzocoronenes,7 naphthalene (NDIs)8 and perylene diimides (PBIs),9 benzene tricarboxylic acids,10 oligophenylenes,11 phenylene ethynylenes,12 phenylene vinylenes13 and 4,4-difluoro borodipyrromethene (BODIPY) derivatives,14 has been elucidated.15 However, a detailed investigation of the enthalpic and entropic contributions has not been performed, and, in particular, the structural factors controlling these contributions in the aqueous self-assembly of many of these systems are far from clear. Thus, only a few examples of entropically driven self-assembly of artificial amphiphiles can be found in the literature.16 To the best of our knowledge, only in the case of PBIs and NDIs endowed with oligo(ethylene) glycol (OEG) side chains of different length, a detailed investigation of the thermodynamics associated with their self-assembly has been reported.17 The synergy of UV-visible and isothermal calorimetric experiments indicates that self-assembly of these amphiphilic systems in aqueous media is entropically driven and enthalpically disfavoured. These scaffolds self-assemble by strong π-stacking of the aromatic moieties, but, more especially, by gain of entropy resulting from the release of water molecules trapped in the hydration shell of the hydrophilic OEG side chains.
Herein, we report on the self-assembling features of a series of amphiphilic N-annulated PBIs (N-PBIs) that self-assemble in an entropically driven manner in aqueous solution (compounds 1–4 in Fig. 1). The hydrophilic/hydrophobic ratio in compounds 1–4 is biased by both the OEG-bearing side groups attached to the imide nitrogens and the lack (compounds 1 and 3) or presence (compounds 2 and 4) of the 4-(2-(2-(2-methoxyethoxy)ethoxy)ethoxy)-1,1′:4′,1′′-terphenyl fragment linked to the central nitrogen atom of the N-PBI core. A detailed thermodynamic analysis has been carried out by utilizing the denaturation model, which allows derivation of a complete set of thermodynamic parameters.18 In good analogy with reported amphiphilic NDIs and PBIs, the self-assembly of N-PBIs 1–4 is entropically driven. However, the self-assembly process is enthalpically disfavoured for compounds 3 and 4, whilst it is enthalpically favoured for amphiphiles 1 and 2. These findings indicate that, in the self-assembly of compounds 1–4, the nature of the hydrophilic side chains attached to the imide groups biases the enthalpic and entropic contributions, whereas the nature of the substituent attached to the nitrogen of the aromatic core exerts a negligible influence on these contributions.
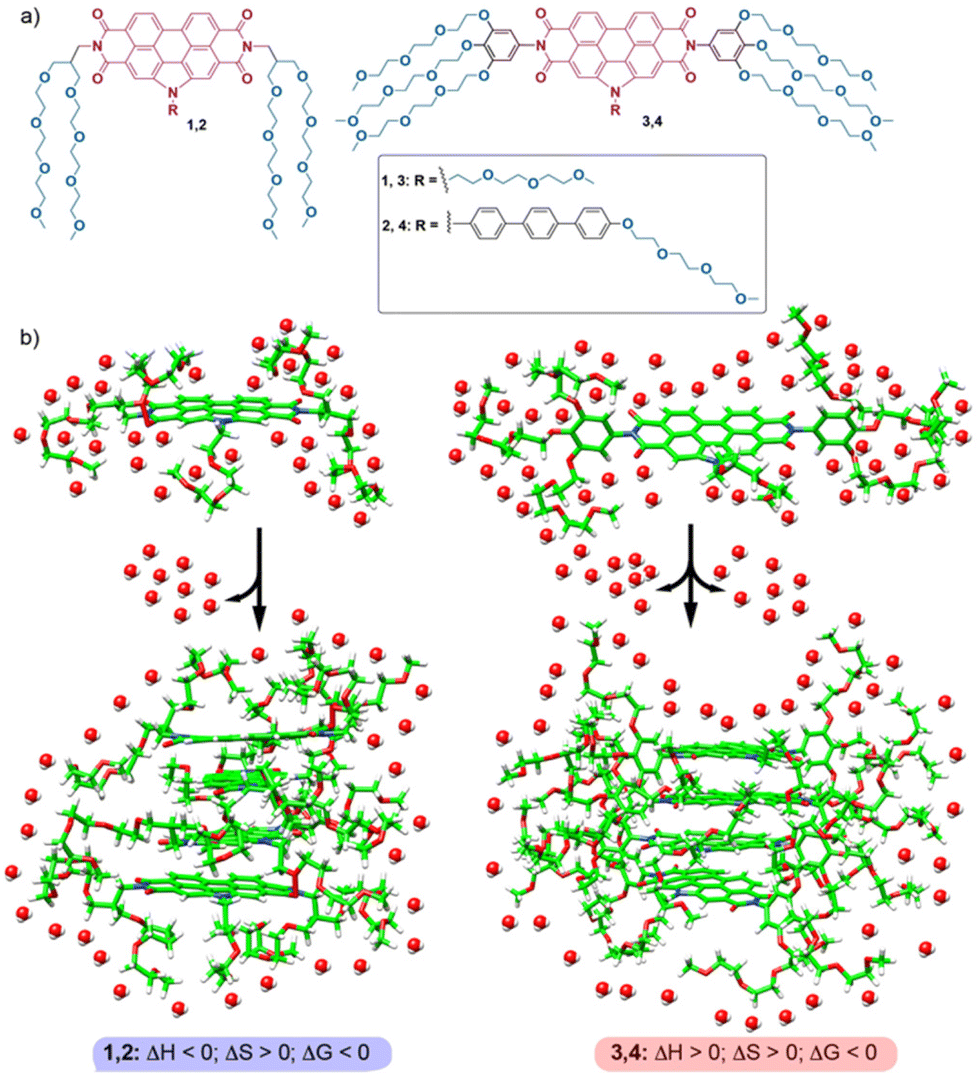 | ||
| Fig. 1 (a) Chemical structure of amphiphilic N-PBIs 1–4. (b) Schematic representation of the self-assembly of 1–4, highlighting the sign of the enthalpic and entropic terms. | ||
To shed light on the origin of the entropic and enthalpic contributions to the self-assembly of N-PBIs 1–4, molecular dynamics (MD) simulations were performed including explicit water molecules. Theoretical calculations on compounds 1 and 3 reveal that the arrangement of the side chains in the monomers plays a pivotal role in the energetics of the self-assembly. For compound 1, the proximity and flexibility of the OEG chains allows partial coating of the hydrophobic aromatic core. This, together with the small number of OEG chains, determines that compound 1 releases, upon aggregation, a lower number of water molecules non-covalently bonded to the OEG chains, and presents an enthalpically favoured process. In the case of compound 3, the rigidity of the trialkoxyphenyl unit impedes efficient coating of the central aromatic core, and, thereby, the OEG side chains are more exposed to the environment, interacting with a larger number of water molecules. The π-stacking of successive units of 3 to form the aggregated species is thus accompanied by rupture of a larger number of stabilizing water–OEG chain interactions and affords an enthalpically disfavoured process. The results presented herein contribute to our understanding of the energetics, disentangling the enthalpic and entropic contributions, of the self-assembly of amphiphilic N-PBIs in aqueous media. The comparison with previous thermodynamic data of referable amphiphilic systems also contributes to establishing structure–function relationships in entropically driven self-assembly processes, in which dissimilar back-folding of the peripheral glycol chains, with sequestration of the aromatic cores from water, plays a determinant role.
Results and discussion
Synthesis and self-assembly in solution
The synthesis of N-PBIs 2–4 was accomplished following a similar protocol to that reported previously for compound 1.19 It involves a convergent methodology in which the preparation of the hydrophilic peripheral wedges and the synthesis of the N-annulated tetraester perylene 5 are key steps (Scheme S1‡). The dendritic amino-based wedge 13 and the tetraester 5 were prepared following previously reported methodologies.19,20 The synthesis of the hydrophilic amine 8 begins with the O-alkylation of the hydroxy groups of commercial pyrogallol. The selective mononitration and the subsequent catalytic hydrogenation of the nitro group afford aniline derivative 8 in good yield.21 On the other hand, the dianhydride 12 bearing the hydrophilic terphenyl segment was prepared by a copper-assisted nucleophilic substitution of 4,4′-diiodo-1,1′-biphenyl and subsequent Suzuki cross-coupling reaction with the boronic acid 10.21a The final nucleophilic addition of amine 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 21b or 1319 in the presence of Zn(AcO)2 and imidazole yields the amphiphilic N-PBIs 2–4.
21b or 1319 in the presence of Zn(AcO)2 and imidazole yields the amphiphilic N-PBIs 2–4.
The chemical structure of the new compounds was confirmed by standard spectroscopic techniques. The N-annulated PBI cores of the four amphiphiles 1–4 show the characteristic set of resonances in their 1H NMR spectra: a singlet and a doublet at δ ∼9 and ∼8.7 ascribed to the ortho protons and an additional doublet at δ ∼8.8 corresponding to the bay protons (Fig. S1‡). These resonances are especially useful to give a first insight into the self-assembly of the aromatic units in solution. Thus, concentration-dependent 1H NMR spectra of compounds 2–4 in CDCl3 display a clear upfield shift of all the aromatic resonances upon increasing the concentration (Fig. S1‡). It is worth mentioning that compounds 3 and 4, in which the side hydrophilic segment is the trialkoxyphenyl moiety covalently attached to the imide functional group, display very broad aromatic resonances, diagnostic of efficient π-stacking. Since all the aromatic signals shield upon increasing the concentration, and considering the previous work on N-annulated PBIs,22 π-stacking of the aromatic units with the pyrrolic moieties in a parallel arrangement is a plausible self-assembly mode for these amphiphiles in CDCl3.
Our main goal is to unravel the self-assembling features of amphiphiles 1–4 in aqueous media and to disentangle the thermodynamics of the process (i.e., enthalpic and entropic contributions). Previous studies on compound 1 in polar solvents demonstrated that dioxane is a good solvent. In dioxane, the UV-vis spectrum of 1 shows the typical absorption pattern of molecularly dissolved PBI-based derivatives, with two intense, well-resolved peaks at 525 and 495 nm and a shoulder at 465 nm (Fig. 2a).19,22,23 In aqueous media, a clear hypso- and hypochromic effect is observed, with absorption maxima at 543 and 503 nm and a shoulder at 478 nm, which implies the formation of H-type aggregates. Similar changes have been observed for amphiphiles 2–4 (Fig. S3a–S5a†).
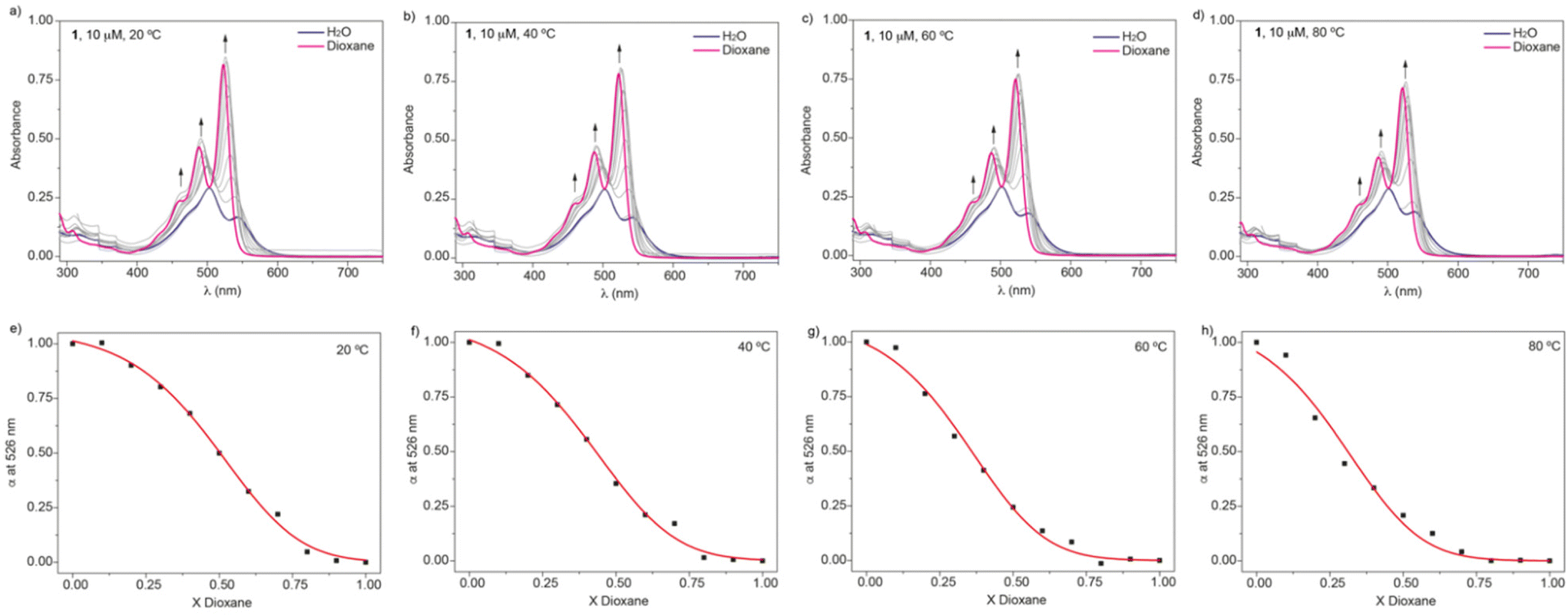 | ||
| Fig. 2 (a–d) UV-vis spectra of amphiphile 1 in water/dioxane mixtures at different temperatures and cT = 10 μM. The arrows indicate the changes in the UV-vis spectra upon adding increasing amounts of dioxane. (e–h) Denaturation curves of 1 in water/dioxane mixtures at different temperatures. Red lines depict the fit to the SD model. | ||
To derive the thermodynamic parameters associated with the self-assembly of amphiphile 1 in aqueous media, we initially used variable-temperature UV-vis experiments in water as solvent. The high stability of the aggregated species formed in water results in negligible disassembly at high temperatures. Although previous reports on amphiphilic PBIs make use of concentration-dependent UV-vis experiments or isothermal titration calorimetry to derive the thermodynamic parameters,17 we envisioned that the solvent denaturation (SD) model could be of great utility for these amphiphilic systems.18 To apply this model, increasing amounts of solutions of the investigated self-assembling unit in a good solvent are added to a solution of the same self-assembling units in a bad solvent, keeping constant the total concentration cT, which favours the disassembly of the aggregated species (Fig. 2a–d). Based on the SD model and applying eqn (1)–(3), it is possible to calculate the Gibbs free energy gain upon monomer addition (ΔG′), which depends on the molar fraction X, and it is equal to ΔG if X = 0, the parameter m, which relates the ability of the good solvent to associate with the monomer, thereby destabilizing the aggregated species, the degree of cooperativity σ, and the nucleation (Kn) and elongation (Ke) constants.
| ΔG′ = ΔG + mX | (1) |
| ΔG′ = −RTln Ke | (2) |
| σ = Kn/Ke | (3) |
In this work, the good and bad solvents are dioxane and water, respectively. Plotting the variation of the degree of aggregation α, obtained by measuring the variation of the absorbance at a specific wavelength (526 nm),24 results in sigmoidal curves that can be fitted to the SD model (Fig. 2e–h). Table 1 collects the thermodynamic parameters derived for compound 1 at 20 °C, and Table S1‡ includes the ΔG′ values, together with the m parameter and the degree of cooperativity σ for the four investigated temperatures. The σ = 1 value demonstrates the isodesmic character15a of the self-assembly process of 1 and, more importantly, the increasing values of ΔG′ display the higher stability of the aggregated species upon increasing the temperature, which is diagnostic of an entropically driven process.17
The denaturation experiments performed at four different temperatures, and considering a molar fraction of dioxane Xdioxane = 0, allow the derivation of the corresponding ΔG′ values in pristine water, which, upon applying eqn (2), yield the elongation binding constant Ke. The van't Hoff analysis affords the corresponding enthalpy (ΔH) and entropy (ΔS) changes (Fig. S2‡ and Table 1). Fig. 3 depicts the thermodynamic signature of 1 at 20 °C. The thermodynamic data indicate that the self-assembly of this amphiphile in water is enthalpically favoured (ΔH = −17.8 kJ mol−1) and entropically driven (TΔS = 19.8 kJ mol−1).
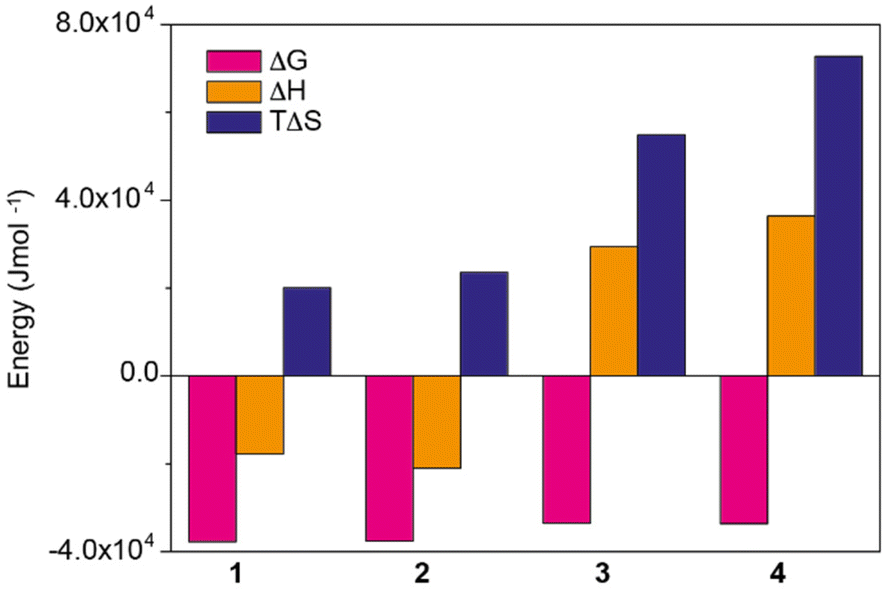 | ||
| Fig. 3 Thermodynamic contributions for the self-assembly of amphiphilic N-annulated PBIs 1–4 in water obtained by applying the SD model at 20 °C. | ||
Following this methodology, the self-assembly mechanism of amphiphiles 2–4 was also investigated and the corresponding thermodynamic parameters were derived by applying the SD model, utilizing water and dioxane as bad and good solvents, respectively (Fig. S3–S5‡). The formation of H-type aggregates from amphiphiles 2–4 in aqueous media is demonstrated by the hypso- and hypochromic effect observed in the UV-vis spectra of the aggregated species compared to the monomeric species.19 In the case of the T-shaped compounds 3 and 4, endowed with the lateral terphenyl moiety, the intense band at ∼300 nm displayed in dioxane also experiences a slight hypsochromic and a strong hypochromic effect upon addition of water, which is diagnostic of the participation of this segment in the π-stacking of the aromatic backbone, which yields the final H-type aggregates. The formation of H-type aggregates is corroborated by the strong quenching of the emissive features of the monomeric units upon aggregation (Fig. S6‡). A detailed analysis of the derived thermodynamic data indicates that the ΔG′ values increase upon increasing the temperature, implying entropically driven self-assembly processes (Tables S1–S4‡). Importantly, the increase in the Gibbs free energy (ΔΔG′) released upon aggregation is higher in amphiphiles 2 and 4, which are decorated with the lateral terphenyl units (ΔΔG′ = 13 and 17 kJ mol−1 for 2 and 4, respectively), than that calculated for 1 and 3, in which the lateral substituent is the OEG chain (ΔΔG′ = 4 and 11 kJ mol−1 for 1 and 3, respectively). These findings can be justified by considering the larger hydrophobic effect due to the larger π-surface of the T-shaped 2 and 4, in comparison to 1 and 3. It is also important to remark that the ΔG′ values are lower for amphiphiles 3 and 4, in which the imide substitution is the trialkoxyphenyl fragment, in comparison to compounds 1 and 2, which is diagnostic of their higher stability (Table 1 and Tables S1–S4‡). Inspection of the thermodynamic data extracted from the SD model also reveals that the degree of cooperativity, σ, remains similar for compounds 2 to 4, while it equals to unity for compound 1. In all cases, σ increases until reaching a value equal to unity upon increasing the temperature. These data contrast with those reported previously for referable amphiphilic NDIs, in which the self-assembly was anticooperative,17b but they are in good agreement with that reported for amphiphilic PBIs.17a
Similarly to compound 1, the values of Ke for compounds 2–4 were calculated at different temperatures and the corresponding van't Hoff analysis was performed (Fig. S2‡). To our surprise, whilst the slopes of the van't Hoff plots of compounds 1 and 2 were positive, those of compounds 3 and 4 were negative. This trend points out that in the case of amphiphiles decorated with the OEG dendron wedges (compounds 1 and 2) the self-assembly is enthalpically favoured, while the opposite is found for compounds 3 and 4, in which the OEG side chains are attached through the trialkoxyphenyl peripheral segments. As suggested by the values of ΔG′, which increase upon increasing the temperature, the self-assembly of these amphiphiles is entropically driven, with the entropy values increasing from compound 1 to compound 4 (Fig. 3). Enthalpically favoured π-stacking of PBIs and PBI-based self-assembling units is well documented due to the attractive π-stacking of the aromatic moieties.17,22 Further confirmation of this stacking is given by variable-temperature UV-vis experiments performed in apolar decalin as solvent for compounds 1 and 2 (amphiphiles 3 and 4 are insoluble in this solvent) and fitting the variation of the absorbance to the one-component equilibrium model (Fig. S7‡).25 These experiments reveal the formation of H-type aggregates upon self-assembly, and Gibbs free energies and degrees of cooperativity very similar to those obtained in aqueous media (Table S5‡).
The thermodynamic insights previously reported for the self-assembly of PBIs and NDIs in water have been justified by invoking the following arguments: (1) the hydrated OEG side chains exert a remarkable steric effect, which hinders the self-assembly, and (2) the self-assembly provokes a release of water molecules to the bulk, which results in entropically driven processes (Fig. 1b).17 The result of these two opposite effects is that the enthalpic contribution due to the π-stacking of the aromatic moieties is smaller than the loss in interaction energies ascribable to the breaking of hydrogen bonds between OEG chains and water. The final thermodynamic result for the self-assembly of these amphiphilic PBIs and NDIs is an enthalpically unfavoured and entropically driven aggregation process. Similar reasoning could be applied to the self-assembly of amphiphiles 3 and 4, but not to amphiphiles 1 and 2, for which the self-assembly is enthalpically favoured (Fig. 1b and 3).
The thermodynamic parameters derived for 1–4, and especially the ΔG values, are comparable to those reported for amphiphilic PBIs endowed with longer OEG side chains, which present values of ΔG ∼−45 kJ mol−1,17a but are more negative than those reported for the amphiphilic NDIs with OEG side chains of different length.17b The ΔG values derived for these NDIs are in the range of −20 kJ mol−1, and increase with the length of the OEG side chains.17b Importantly, and in good agreement with the findings observed for 3 and 4, the self-assembly of all these amphiphilic PBIs and NDIs, decorated with amphiphilic aromatic moieties, is entropically driven but enthalpically disfavoured.17a,b Interestingly, the NDI 2 compound reported in ref. 17c, in which OEG side chains are directly connected to the aromatic moiety, but the aromatic ring is linked to the NDI core via a methylene bridge, exhibits an enthalpically favoured self-assembly process (ΔH ∼−23 kJ mol−1) in line with the outcomes found for N-PBIs 1 and 2. This comparison between related N-PBIs, PBIs and NDIs with OEG side chains suggests that the flexibility of the side chains determines the endothermic/exothermic character of the enthalpy contribution for the self-assembly process in water; i.e., the larger capacity of OEG side chains to protect the hydrophobic π-core from water, the more favourable is the (exothermic) enthalpy-driven aggregation (see the theoretical discussion below).
Theoretical calculations
To shed light on the differences found in the enthalpic and entropic contributions to the energetics of the self-assembly of amphiphiles 1–4, we have modelled the aggregation process in water of compounds 1 and 3 (as representative examples of enthalpically favoured and unfavoured self-assemblies) by using MD simulations (see ESI for further details‡).26 Prior to examining the self-assembly process, an initial structural and energetic analysis of monomers 1 and 3 is recommended, and, in particular, an analysis of the conformational freedom of the OEG side chains in water, and of the most prominent interactions. Fig. S8‡ displays the time evolution of the minimum distance between any atom of the peripheral OEG chains at the imide position and the centroid of the central benzene ring of the N-PBI core for monomers 1 and 3. For 1, the OEG chains are flexible enough to protect part of the N-PBI core from water by CH⋯π interactions, with contacts in the range 3.0–4.5 Å along the dynamics. In contrast, the more rigid trialkoxyphenyl groups attached to the imides for 3 impede the OEG chains in efficiently reaching the π-conjugated N-PBI skeleton (core–side chain distances of ∼8.5 Å after 1 ns; Fig. S8‡). Then, the hydrophobic N-PBI skeleton of 3 is more exposed to the solvent. In terms of energetics, the chain–core, core–solvent and chain–solvent interactions (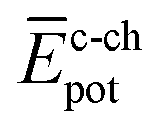 ,
, 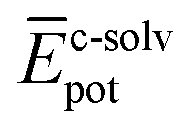 and
and 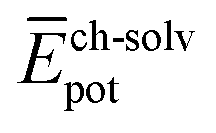 ) have also been evaluated for monomers of 1 and 3 (see ESI‡ for an extended discussion). The most important difference between 1 and 3 comes from the
) have also been evaluated for monomers of 1 and 3 (see ESI‡ for an extended discussion). The most important difference between 1 and 3 comes from the 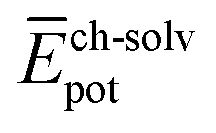 term (−831 and −1192 kJ mol−1, respectively). This is in line with the less effective wrapping of 3 by OEG chains, which are more exposed to the environment with a larger number of interacting water molecules. The self-assembly of 3, compared to 1, would therefore require the rupture of a larger number of stabilizing water–OEG chain interactions, which can be enthalpically unfavourable.
term (−831 and −1192 kJ mol−1, respectively). This is in line with the less effective wrapping of 3 by OEG chains, which are more exposed to the environment with a larger number of interacting water molecules. The self-assembly of 3, compared to 1, would therefore require the rupture of a larger number of stabilizing water–OEG chain interactions, which can be enthalpically unfavourable.
To investigate the energetics of the self-assembly, a new set of MD simulations was performed for dimers and tetramers of 1 and 3. Dimers and tetramers were initially organized in close contact (aggregated form) according to the previous MD simulations and using the same N-PBI concentration employed for the monomer by fixing the N-PBI:H2O ratio (ESI‡). Prior to determining the energetics, the structural supramolecular parameters defining the relative disposition of the molecules in the dimer were analysed along the MD trajectories (Fig. 4). These parameters are the distance between the centroids of the central N-PBI rings (d) and the twisting angle between adjacent N-PBIs (α). α is taken as the angle formed by the lines passing through the centroid of the central benzene ring and the imide nitrogen atom. On average, d ranges between 3.3 and 3.4 Å for dimer 1, in line with a typical π-stacking interaction, although larger d values are also found due to the relative sliding of adjacent N-PBIs. Dimer 3 displays a less effective π-stacking, with d values slightly larger (3.6–3.8 Å), owing to the steric hindrance of the bulky lateral chains at the imide positions. For angle α, dimer 1 shows a value centred around 45°, ranging from 40 to 60°, whereas dimer 3 presents an angle of 25° with a significant smaller fluctuation. These outcomes indicate that, compared to 1, the rotation motion of vicinal N-PBI units along the stacking axis is more impeded for 3, due to the bulkier lateral groups. As a consequence, supramolecular aggregates of 3 are more rigid and give rise to more regular helices than those of 1, as can be seen for the tetramers in Fig. S10.‡ Indeed, the lateral phenyl rings in 3 interact through π-stacking interactions, and contribute to achieving the helical arrangement. This is in line with the experimental, broad aromatic NMR signal registered for compounds 3 and 4.
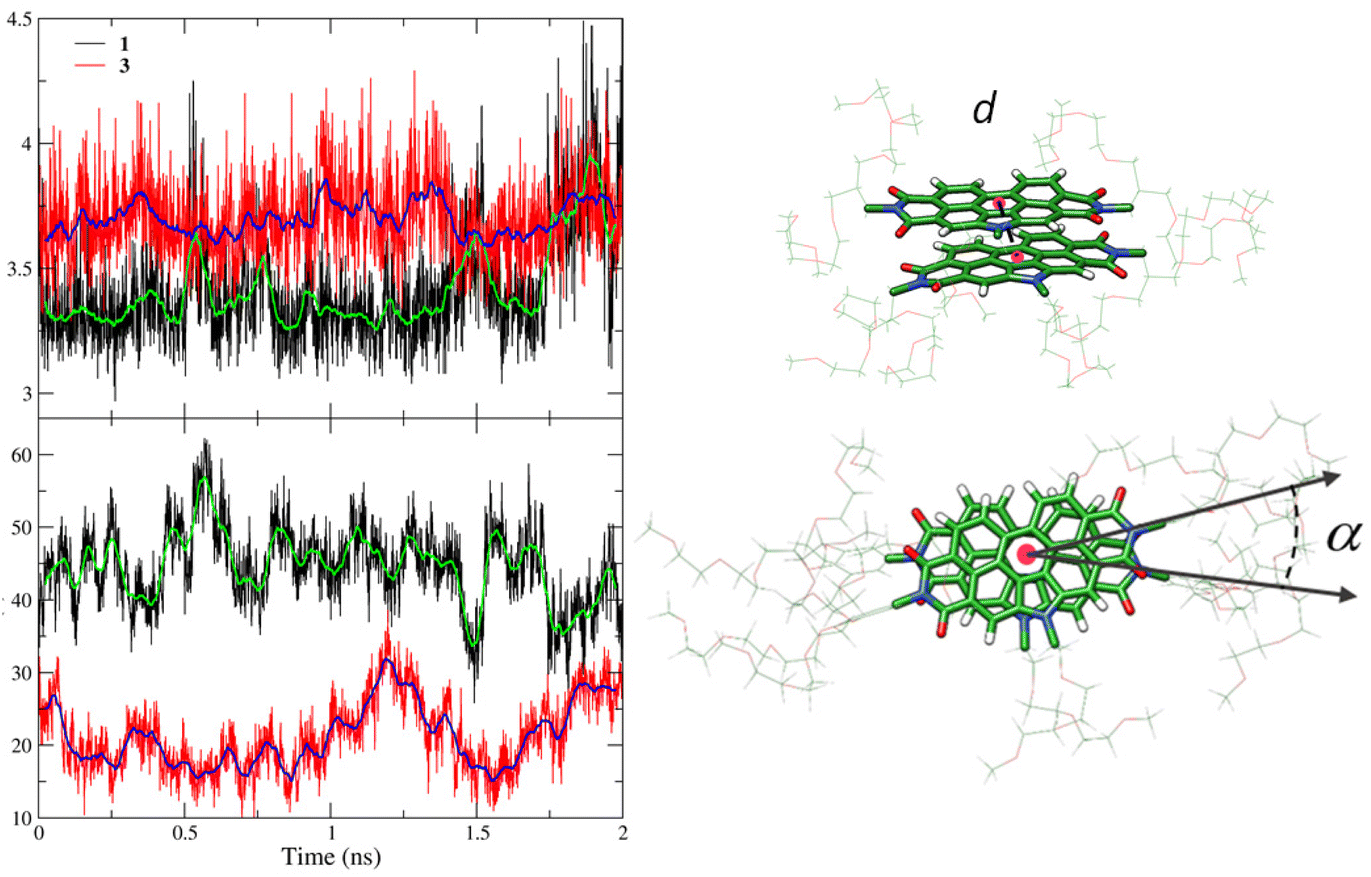 | ||
| Fig. 4 Time evolution of the selected structural parameters (d and α) computed along the MD trajectory for dimers of N-PBIs 1 and 3. Green and blue lines represent the average values in intervals of 50 points (100 fs). Distance d and angle α are defined on the right using the dimer of 1. | ||
The experimental measurements indicate that the self-assembly of 1 is enthalpically favoured, while for 3 it is enthalpically unfavoured. Table 2 shows the average 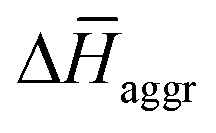 obtained from the MD simulations for dimers and tetramers of 1 and 3.
obtained from the MD simulations for dimers and tetramers of 1 and 3. 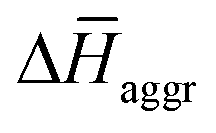 is calculated as
is calculated as 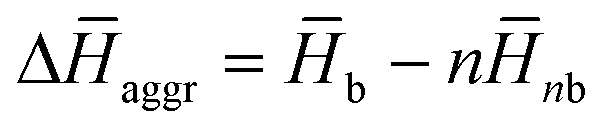 , where
, where 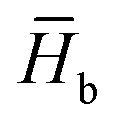 and
and 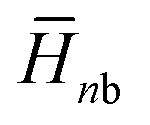 are the average enthalpy of the bonded (aggregate) and non-bonded (monomer) states, respectively, and n denotes the number of N-PBI molecules. In both cases, the experimental sign for
are the average enthalpy of the bonded (aggregate) and non-bonded (monomer) states, respectively, and n denotes the number of N-PBI molecules. In both cases, the experimental sign for 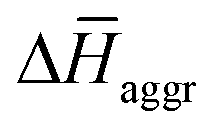 is well reproduced with a significant difference between 1 and 3. In particular, an exothermic aggregation enthalpy
is well reproduced with a significant difference between 1 and 3. In particular, an exothermic aggregation enthalpy 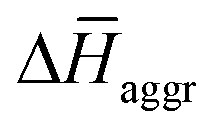 is obtained for 1, whereas an endothermic aggregation process is predicted for 3.
is obtained for 1, whereas an endothermic aggregation process is predicted for 3.
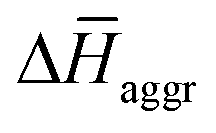 (in kJ mol−1) obtained from the MD simulations for dimers and tetramers of 1 and 3. T = 300 K, p = 1 bar
(in kJ mol−1) obtained from the MD simulations for dimers and tetramers of 1 and 3. T = 300 K, p = 1 bar
| Compound |
|
|
|---|---|---|
| 1 | −169 ± 29 | −111 ± 41 |
| 3 | 43 ± 42 | 50 ± 69 |
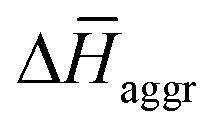
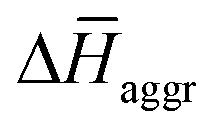
To further understand this different trend, we performed an analysis decomposing the aggregation enthalpy into several contributions. From basic thermodynamics, the enthalpy of a system is calculated by H = Ekin + Epot + pV, where Ekin and Epot, denote the kinetic and potential energy (both terms would give the internal energy), respectively, and the last term corresponds to the product between the pressure and volume. In this context, we can assume that the enthalpy changes due to the aggregation process are mainly ruled by the potential energy, and thus the kinetic energy and pV components can be omitted (see Table S8‡). Therefore, the aggregation enthalpy can be estimated as an interaction energy (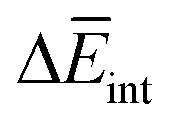 ) as
) as 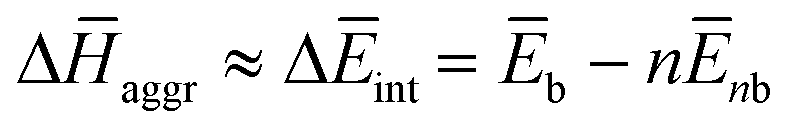 , where
, where 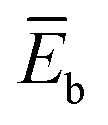 and
and 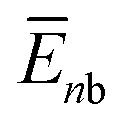 are the average potential energy contributions for the bonded (aggregate) and non-bonded (monomer) states, respectively. In the simplest picture, the average potential energy for a system (monomer, dimer, or tetramer) can be further decomposed into three main components:
are the average potential energy contributions for the bonded (aggregate) and non-bonded (monomer) states, respectively. In the simplest picture, the average potential energy for a system (monomer, dimer, or tetramer) can be further decomposed into three main components: 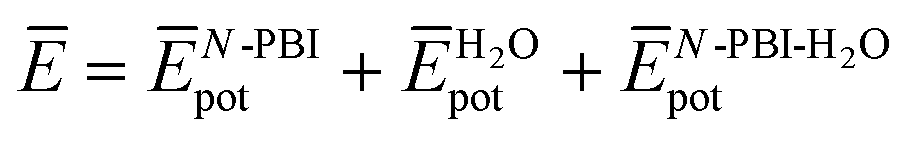 , where
, where 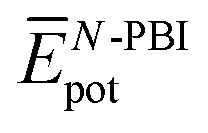 and
and 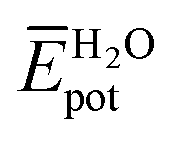 represent the average potential energy of the N-PBIs and water self-interactions, respectively, and
represent the average potential energy of the N-PBIs and water self-interactions, respectively, and 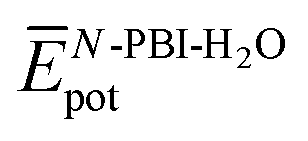 corresponds to the average potential energy due to solute–solvent interactions. In the following, we analyse the average interaction energy
corresponds to the average potential energy due to solute–solvent interactions. In the following, we analyse the average interaction energy 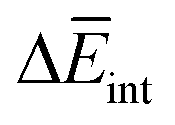 for each contribution as
for each contribution as 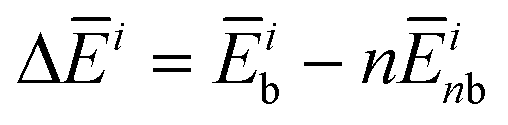 , where i denotes each subsystem (N-PBI, H2O or N-PBI–H2O).
, where i denotes each subsystem (N-PBI, H2O or N-PBI–H2O).
Table 3 shows the average interaction energy contributions estimated for dimers of 1 and 3 from the MD trajectories. For both molecules, the 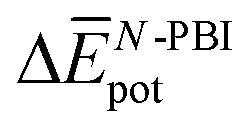 and
and 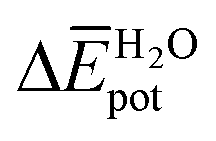 terms are negative, indicating that the N-PBI⋯N-PBI and H2O⋯H2O interactions resulting from aggregation are favourable.
terms are negative, indicating that the N-PBI⋯N-PBI and H2O⋯H2O interactions resulting from aggregation are favourable. 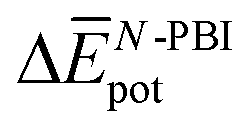 accounts for the π-stacking between the N-PBI cores and the dispersive interactions between the lateral chains.
accounts for the π-stacking between the N-PBI cores and the dispersive interactions between the lateral chains. 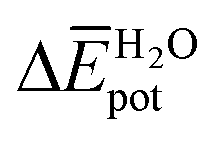 takes into account the new hydrogen bonds formed among water molecules, which were previously interacting with the N-PBI monomers. Finally,
takes into account the new hydrogen bonds formed among water molecules, which were previously interacting with the N-PBI monomers. Finally, 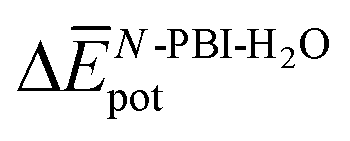 is indeed positive because of the smaller number of interactions between water and N-PBIs in the dimer with respect to the monomers in solution. It is noteworthy that for both dimers the sum
is indeed positive because of the smaller number of interactions between water and N-PBIs in the dimer with respect to the monomers in solution. It is noteworthy that for both dimers the sum 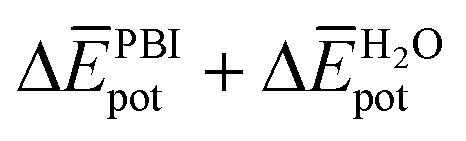 is very similar (−451 kJ mol−1 for 1 and −442 kJ mol−1 for 3). Therefore,
is very similar (−451 kJ mol−1 for 1 and −442 kJ mol−1 for 3). Therefore, 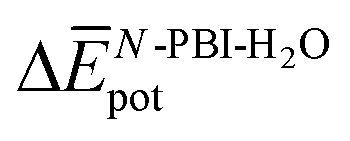 is the determining factor for the sign of
is the determining factor for the sign of 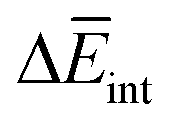 and, consequently, of
and, consequently, of 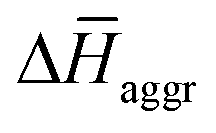 . In fact,
. In fact, 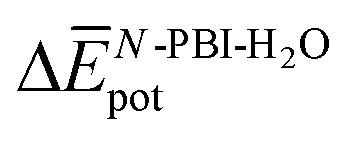 for 3 is about 1.6 times larger than that computed for 1. This is due to the larger number of OEG–water contacts (H-bonds) that are lost in 3 compared to 1, as a consequence of aggregation (Table S6‡). For instance, the average number of H-bonds lost per N-PBI monomer calculated for dimers of 1 and 3 was found to be 1.5 and 2.7, respectively. This rupture of H-bonds is even higher for tetramers (3.1 and 4.8 for 1 and 3, respectively) due to the additional hindrance effects of the non-terminal monomers on the supramolecular aggregates (see Table S9 and ESI‡ for an extended discussion).
for 3 is about 1.6 times larger than that computed for 1. This is due to the larger number of OEG–water contacts (H-bonds) that are lost in 3 compared to 1, as a consequence of aggregation (Table S6‡). For instance, the average number of H-bonds lost per N-PBI monomer calculated for dimers of 1 and 3 was found to be 1.5 and 2.7, respectively. This rupture of H-bonds is even higher for tetramers (3.1 and 4.8 for 1 and 3, respectively) due to the additional hindrance effects of the non-terminal monomers on the supramolecular aggregates (see Table S9 and ESI‡ for an extended discussion).
| Compound |
|
|
|
|
|---|---|---|---|---|
| 1 | −275 | −176 | 286 | −165 |
| 3 | −311 | −131 | 467 | 25 |
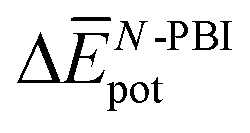
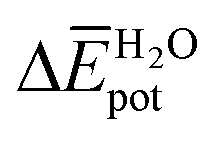
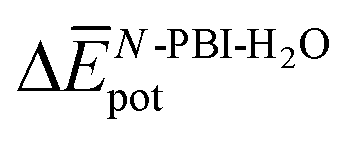
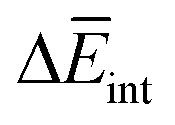
It is important to stress that the theoretical insights extracted here to rationalize the experimental thermodynamic parameters for the self-assembly of N-PBIs 1 and 3 are not only specific for these systems, but also applicable to other related PBI and NDI systems.17 MD simulations for related PBI and NDI systems show that the flexibility and, thus, the capacity of the OEG side chains to protect the hydrophobic π-conjugated core from water is key to determining the enthalpically favoured or unfavoured character of the self-assembly (see ESI for further discussion‡).
Conclusions
The synthesis and self-assembling features of a series of amphiphilic N-annulated PBIs 1–4 in water have been thoroughly investigated, both experimentally and theoretically. The thermodynamic parameters derived from the solvent denaturation method indicate that the self-assembly of the four investigated amphiphiles 1–4 is entropically driven. This feature is understood by considering the water molecules, initially bonded to the hydrophilic side chains of the monomeric species, which are released to the bulk upon aggregation. The hydrophilic/hydrophobic ratio, determined by the number and flexibility of the oligo(ethylene) glycol chains present in the monomers of compounds 1–4, plays a relevant role in the exothermic/endothermic nature of the aggregation enthalpy. Self-assembly is enthalpically favoured for compounds 1 and 2, which are decorated with flexible, hydrophilic dendron wedges attached to the imide functional groups, and enthalpically unfavoured for compounds 3 and 4, which are end-capped with rigid, hydrophilic trialkoxyphenyl rings. Molecular dynamics simulations in water show that the arrangement and flexibility of the hydrophilic side chains and the number of interactions between these chains and water molecules strongly determine the enthalpy of the self-assembly. Thus, the π-stacking of successive units of amphiphile 3 to form the aggregated species is accompanied by the rupture of a larger number of stabilizing water–side chain interactions than that computed for compound 1. The loss of these water–OEG interactions determines the sign of the enthalpy contribution and, therefore, the global stability of the aggregated species in aqueous media. The thermodynamic insights presented herein contribute to our understanding of the energetics, including enthalpy, entropy and free energy of the self-assembly of amphiphilic N-PBIs in aqueous media, and to establish structure–function relationships in entropically driven self-assembly processes in which dissimilar back-folding of the peripheral glycol chains, with sequestration of the aromatic cores from water, plays a determining role.Author contributions
M. A. Martínez: synthesis, spectroscopic characterization, supramolecular polymerization mechanisms, and writing; D. Aranda, J. Aragó, and E. Ortí: MD simulations and writing; J. Aragó, E. Ortí, and L. Sánchez: conceptualization, writing, supervision, and funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support by the MCIN/AEI of Spain (projects PID2021-128569NB-I00/AEI/10.13039/501100011033/FEDER/UE, PID2020-113512GB-I00 and CEX2019-000919-M), the Generalitat Valenciana (PROMETEO/2020/077 and MFA/2022/017) and the Comunidad de Madrid (NanoBIOCARGO, P2018/NMT-4389) is acknowledged. M. A. M. and J. A. are indebted to the MCIN/AEI for their predoctoral and Ramón-y-Cajal (RyC-2017-23500) fellowships. D. A. acknowledges the Generalitat Valenciana for his postdoctoral contract (APOSTD/2021/025).References
- (a) J. Israelachvili and H. Wennerstrçm, Role of hydration and water structure in biological and colloidal interactions, Nature, 1996, 379, 219–225 CrossRef CAS PubMed; (b) P. Ball, Water as an Active Constituent in Cell Biology, Chem. Rev., 2008, 108, 74–108 CrossRef CAS PubMed.
- (a) E. Krieg, A. Niazov-Elkan, E. Cohen, Y. Tsarfati and B. Rybtchinski, Noncovalent Aqua Materials Based on Perylene Diimides, Acc. Chem. Res., 2019, 52, 2634–2646 CrossRef CAS PubMed; (b) S. Chen, R. Costil, F. K.-C. Leung and B. L. Feringa, Self-Assembly of Photoresponsive Molecular Amphiphiles in Aqueous Media, Angew. Chem., Int. Ed., 2021, 60, 11604–11627 CrossRef CAS PubMed.
- (a) T. J. McIntosh and S. A. Simon, Long- and Short-Range Interactions between Phospholipid/Ganglioside GM1 Bilayers, Biochemistry, 1994, 33, 10477–10486 CrossRef CAS PubMed; (b) S. Matile, A. Vargas Jentzsch, J. Montenegro and A. Fin, Recent synthetic transport systems, Chem. Soc. Rev., 2011, 40, 2453–2474 RSC; (c) A. Nikoubashman and F. Schmid, The molecular Lego movie, Nat. Chem., 2011, 11, 298–300 CrossRef PubMed.
- (a) A. N. Edelbrock, T. D. Clemons, S. M. Chin, J. J. W. Roan, E. P. Bruckner, Z. Álvarez, J. F. Edelbrock, K. S. Wek and S. I. Stupp, Superstructured Biomaterials Formed by Exchange Dynamics and Host–Guest Interactions in Supramolecular Polymers, Adv. Sci., 2021, 8, 2004042 CrossRef CAS PubMed; (b) D. Straßburger, N. Stergiou, M. Urschbach, H. Yurugi, D. Spitzer, D. Schollmeyer, D. Schmitt and P. Besenius, Mannose-Decorated Multicomponent Supramolecular Polymers Trigger Effective Uptake into Antigen-Presenting Cells, ChemBioChem, 2018, 19, 912–916 CrossRef PubMed.
- M. H. Bakker, C. C. Lee, E. W. Meijer, P. Y. W. Dankers and L. Albertazzi, Multicomponent Supramolecular Polymers as a Modular Platform for Intracellular Delivery, ACS Nano, 2016, 10, 1845–1852 CrossRef CAS PubMed.
- (a) R. A. Shalabya and M. A. Lauffer, Comparison of the entropy-driven polymerization reactions of E66 and vulgare tobacco mosaic virus proteins, Arch. Biochem. Biophys., 1985, 236, 390–398 CrossRef PubMed; (b) P. Friedhoff, A. Schneider, E.-M. Mandelkow and E. Mandelkow, Rapid Assembly of Alzheimer-like Paired Helical Filaments from Microtubule-Associated Protein Tau Monitored by Fluorescence in Solution, Biochemistry, 1998, 37, 10223–10230 CrossRef CAS PubMed; (c) K. E. Kadler, Y. Hojima and D. J. Prockop, Assembly of collagen fibrils de novo by cleavage of the type I pC-collagen with procollagen C-proteinase. Assay of critical concentration demonstrates that collagen self-assembly is a classical example of an entropy-driven process, J. Biol. Chem., 1987, 262, 15696–15701 CrossRef CAS PubMed.
- (a) K. V. Rao and S. J. George, Synthesis and Controllable Self-Assembly of a Novel Coronene Bisimide Amphiphile, Org. Lett., 2010, 12, 2656–2659 CrossRef CAS PubMed; (b) J. P. Hill, W. Jin, A. Kosaka, T. Fukushima, H. Ichihara, T. Shimomura, K. Ito, T. Hashizume, N. Ishii and T. Aida, Self-Assembled Hexa-peri-hexabenzocoronene Graphitic Nanotube, Science, 2004, 304, 1481–1483 CrossRef CAS PubMed; (c) M. Yin, J. Shen, W. Pisula, M. Liang, L. Zhi and K. Müllen, Functionalization of Self-Assembled Hexa-peri-hexabenzocoronene Fibers with Peptides for Bioprobing, J. Am. Chem. Soc., 2009, 131, 14618–14619 CrossRef CAS PubMed.
- (a) M. R. Molla and S. Ghosh, Aqueous self-assembly of chromophore-conjugated amphiphiles, Phys. Chem. Chem. Phys., 2014, 16, 26672–26683 RSC; (b) A. Sikder, D. Ray, V. K. Aswal and S. Ghosh, Hydrogen-Bonding-Regulated Supramolecular Nanostructures and Impact on Multivalent Binding, Angew. Chem., Int. Ed., 2019, 58, 1606–1611 CrossRef CAS PubMed; (c) M. Al Kobaisi, S. V. Bhosale, K. Latham, A. M. Raynor and S. V. Bhosale, Functional Naphthalene Diimides: Synthesis, Properties, and Applications, Chem. Rev., 2016, 116, 11685–11796 CrossRef PubMed.
- (a) M. Sun, K. Müllen and M. Yin, Water-soluble perylenediimides: design concepts and biological applications, Chem. Soc. Rev., 2016, 45, 1513–1528 RSC; (b) D. Görl, X. Zhang, V. Stepanenko and F. Würthner, Supramolecular block copolymers by kinetically controlled co-self-assembly of planar and core-twisted perylene bisimides, Nat. Commun., 2015, 6, 7009 CrossRef PubMed; (c) M. Ogasawara, X. Lin, H. Kurata, H. Ouchi, M. Yamauchi, T. Ohba, T. Kajitani, T. Fukushima, M. Numata, R. Nogami, B. Adhikari and S. Yagai, Water-induced self-assembly of an amphiphilic perylene bisimide dyad into vesicles, fibers, coils, and rings, Mater. Chem. Front., 2018, 2, 171–179 RSC; (d) A. Ustinov, H. Weissman, E. Shirman, I. Pinkas, X. Zuo and B. Rybtchinski, Supramolecular Polymers in Aqueous Medium: Rational Design Based on Directional Hydrophobic Interactions, J. Am. Chem. Soc., 2011, 133, 16201–16211 CrossRef CAS PubMed; (e) M. Hariharan, Y. Zheng, H. Long, T. A. Zeidan, G. C. Schatz, J. Vura-Weis, M. R. Wasielewski, X. Zuo, D. M. Tiede and F. D. Lewis, Hydrophobic Dimerization and Thermal Dissociation of Perylenediimide-Linked DNA Hairpins, J. Am. Chem. Soc., 2009, 131, 5920–5929 CrossRef CAS PubMed.
- (a) N. M. Matsumoto, R. P. M. Lafleur, X. Lou, K.-C. Shih, S. P. W. Wijnands, C. Guibert, J. W. A. M. van Rosendaal, I. K. Voets, A. R. A. Palmans, Y. Lin and E. W. Meijer, Polymorphism in Benzene-1,3,5-tricarboxamide Supramolecular Assemblies in Water: A Subtle Trade-off between Structure and Dynamics, J. Am. Chem. Soc., 2018, 140, 13308–13316 CrossRef CAS PubMed; (b) M. B. Baker, L. Albertazzi, I. K. Voets, C. M. Leenders, A. R. A. Palmans, G. M. Pavan and E. W. Meijer, Consequences of chirality on the dynamics of a water-soluble supramolecular polymer, Nat. Commun., 2015, 6, 6234 CrossRef PubMed; (c) P. Besenius, G. Portale, P. H. H. Bomans, H. M. Janssen, A. R. A. Palmans and E. W. Meijer, Controlling the growth and shape of chiral supramolecular polymers in water, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 17888–17893 CrossRef CAS PubMed.
- (a) Y. Kim, T. Kim and M. Lee, From self-assembled toroids to dynamic nanotubules, Polym. Chem., 2013, 4, 1300–1308 RSC; (b) Z. Huang, S.-K. Kang, M. Banno, T. Yamaguchi, D. Lee, C. Seok, E. Yashima and M. Lee, Pulsating Tubules from Noncovalent Macrocycles, Science, 2012, 337, 1521–1526 CrossRef CAS PubMed.
- (a) C. Rest, M. J. Mayoral, K. Fucke, J. Schellheimer, V. Stepanenko and G. Fernández, Self-Assembly and (Hydro)gelation Triggered by Cooperative π–π and Unconventional C—H⋯X Hydrogen Bonding Interactions, Angew. Chem., Int. Ed., 2014, 53, 700–705 CrossRef CAS PubMed; (b) T. Rudolph, N. Kumar Allampally, G. Fernández and F. H. Schacher, Controlling Aqueous Self-Assembly Mechanisms by Hydrophobic Interactions, Chem. – Eur. J., 2014, 20, 13871–13875 CrossRef CAS PubMed.
- (a) R. Thirumalai, R. D. Mukhopadhyay, V. K. Praveen and A. Ajayaghosh, A slippery molecular assembly allows water as a self-erasable security marker, Sci. Rep., 2015, 5, 9842 CrossRef PubMed; (b) J. F. Hulvat, M. Sofos, K. Tajima and S. I. Stupp, Self-Assembly and Luminescence of Oligo(p-phenylene vinylene) Amphiphiles, J. Am. Chem. Soc., 2005, 127, 366–372 CrossRef CAS PubMed.
- I. Helmers, B. Shen, K. K. Kartha, R. Q. Albuquerque, M. Lee and G. Fernández, Impact of Positional Isomerism on Pathway Complexity in Aqueous Media, Angew. Chem., Int. Ed., 2020, 59, 5675–5682 CrossRef CAS PubMed.
- (a) T. F. A. De Greef, M. M. J. Smulders, M. Wolffs, A. P. H. J. Schenning, R. P. Sijbesma and E. W. Meijer, Supramolecular polymerization, Chem. Rev., 2009, 109, 5687–5754 CrossRef CAS PubMed; (b) M. Wehner and F. Würthner, Supramolecular polymerization through kinetic pathway control and living chain growth, Nat. Rev. Chem., 2020, 4, 38–53 CrossRef CAS.
- (a) H. Fenniri, B.-L. Deng, A. Ribbe, K. Hallenga, J. Jacob and P. Thiyagarajan, Entropically driven self-assembly of multichannel rosette nanotubes, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 6487–6492 CrossRef CAS PubMed; (b) E. Obert, M. Bellot, L. Bouteiller, F. Andrioletti, C. Lehen-Ferrenbach and F. Boue, Both Water- and Organo-Soluble Supramolecular Polymer Stabilized by Hydrogen-Bonding and Hydrophobic Interactions, J. Am. Chem. Soc., 2007, 129, 15601–15605 CrossRef CAS PubMed.
- (a) D. Görl and F. Würthner, Entropically Driven Self-Assembly of Bolaamphiphilic Perylene Dyes in Water, Angew. Chem., Int. Ed., 2016, 55, 12094–12098 CrossRef PubMed; (b) P. P. N. Syamala, B. Soberats, D. Görl, S. Gekle and F. Würthner, Thermodynamic insights into the entropically driven self-assembly of amphiphilic dyes in water, Chem. Sci., 2019, 10, 9358–9366 RSC; (c) P. P. N. Syamala and F. Würthner, Modulation of the Self-Assembly of π-Amphiphiles in Water from Enthalpy- to Entropy-Driven by Enwrapping Substituents, Chem. – Eur. J., 2020, 26, 8426–8434 CrossRef CAS PubMed.
- P. A. Korevaar, C. Schaefer, T. F. A. De Greef and E. W. Meijer, Controlling Chemical Self-Assembly by Solvent-Dependent Dynamics, J. Am. Chem. Soc., 2012, 134, 13482–13491 CrossRef CAS PubMed.
- M. A. Martínez, E. E. Greciano, J. Cuéllar, J. M. Valpuesta and L. Sánchez, Globular Aggregates Stemming from the Self-Assembly of an Amphiphilic N-Annulated Perylene Bisimide in Aqueous Media, Nanomaterials, 2021, 11, 1457 CrossRef PubMed.
- (a) R. K. Gupta, S. K. Pathak, B. Pradhan, D. S. S. Rao, S. K. Prasad and A. S. Achalkumar, Self-assembly of luminescent N-annulated perylene tetraesters into fluid columnar phases, Soft Matter, 2015, 11, 3629–3636 RSC; (b) R. K. Gupta, D. S. S. Rao, S. K. Prasad and A. S. Achalkumar, Columnar Self-Assembly of Electron-Deficient Dendronized Bay-Annulated Perylene Bisimides, Chem. – Eur. J., 2018, 24, 3566–3575 CrossRef CAS PubMed.
- (a) S. Yamaguchi, A. Fukazawa, E. Yamaguchi and C. Wang, Phosphole compounds having amino group-derived electron-donating π-conjugated units and fluorescent dyes containing them, WO2015111647A1, 2015 Search PubMed; (b) T. N. B. Truong, S. Savagatrup, I. Jeon and T. M. Swager, Modular synthesis of polymers containing 2,5-di(thiophenyl)-N-arylpyrrole, Polym. Sci., Part A: Polym. Chem., 2018, 56, 1133–1139 CrossRef CAS PubMed.
- (a) E. E. Greciano, J. Calbo, E. Ortí and L. Sánchez, N-Annulated Perylene Bisimides to Bias the Differentiation of Metastable Supramolecular Assemblies into J- and H-Aggregates, Angew. Chem., Int. Ed., 2020, 59, 17517–17524 CrossRef CAS PubMed; (b) M. A. Martínez, A. Doncel-Giménez, J. Cerdá, J. Calbo, R. Rodríguez, J. Aragó, J. Crassous, E. Ortí and L. Sánchez, Distance Matters: Biasing Mechanism, Transfer of Asymmetry, and Stereomutation in N-Annulated Perylene Bisimide Supramolecular Polymers, J. Am. Chem. Soc., 2021, 143, 13281–13291 CrossRef PubMed.
- F. Würthner, C. R. Saha-Möller, B. Fimmel, S. Ogi, P. Leowanawat and D. Schmidt, Perylene Bisimide Dye Assemblies as Archetype Functional Supramolecular Materials, Chem. Rev., 2016, 116, 962–1052 CrossRef PubMed.
- S. Ghosh, X.-Q. Li, V. Stepanenko and F. Würthner, Control of H- and J-Type π Stacking by Peripheral Alkyl Chains and Self-Sorting Phenomena in Perylene Bisimide Homo- and Heteroaggregates, Chem. – Eur. J., 2008, 14, 11343–11357 CrossRef PubMed.
- H. M. M. ten Eikelder, A. J. Markvoort, T. F. A. de Greef and P. A. J. Hilbers, An Equilibrium Model for Chiral Amplification in Supramolecular Polymers, J. Phys. Chem. B, 2012, 116, 5291–5301 CrossRef CAS PubMed.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindahl, GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers, SoftwareX, 2015, 1–2, 19–25 CrossRef.
Footnotes |
| † Dedicated to Prof. J. Font on the occasion of his 75th anniversary. |
| ‡ Electronic supplementary information (ESI) available: Experimental procedures, compound characterisation and molecular dynamics details. See DOI: https://doi.org/10.1039/d3qo00111c. |
| § These authors contributed equally to this work. |
| This journal is © the Partner Organisations 2023 |