DOI:
10.1039/D3QM00261F
(Research Article)
Mater. Chem. Front., 2023,
7, 3146-3155
Distance produces beauty? regulating the distance of Fe atomic pairs to enhance electrocatalytic CO2 reduction†
Received
14th March 2023
, Accepted 24th April 2023
First published on 9th May 2023
Abstract
Metal atom dispersed catalysts with high catalytic activity and accurate active sites are considered as promising catalyst materials for electrocatalytic CO2 reduction reactions (CO2RRs). Electronic structure regulation of active sites is a crucial means to improve the catalytic performance. Hence, Fe atom catalysts (FeN4-Dx (x = 1, 2, 3, and 4)) with pairs of Fe atoms at different distances were constructed and the effect of distance on the catalytic performance was further investigated. Based on the structural stability and the activation effect for CO2, the FeN4-D1 structure (D1 = 2.39 Å) exhibited good CO2RR selectivity and catalytic activity for producing CO. Moreover, due to the double active sites of Fe atomic pairs, the FeN4-D1 structure had superior catalytic performance and selectivity for the CO2RR to ethanol by the *CO–*CHO coupling with low reaction free energy. Combined with detailed calculations of the electronic structure, a suitable distance of Fe atomic pairs regulated the d-band center of active sites, which modified the interaction between the FeN4-D1 structure and *CO intermediates and promoted the C–C coupling process. Therefore, the FeN4-D1 structure can be used as a promising electrocatalyst for the CO2RR to C2 products and the accurate regulation method of active sites may provide new ideas for the design of efficient catalysts.
1. Introduction
Electrocatalytic CO2 reduction reaction (CO2RR) is an effective method to mitigate the greenhouse effect and promote the conversion of carbon into value-added chemicals.1–4 On the basis of high metal atom utilization rate and easily regulated active sites, atom-dispersed catalysts have broad application prospects in the CO2RR.2,5–7 Further, several means have been used to further improve the catalytic activity of the active sites, such as structural defects,8,9 introducing heteroatoms,10–12 axial coordination,13–15 bimetallic centers,16–18 and regulating the distance of the active sites.19,20 However, the distance effect of the active sites on the catalytic performance of atom-dispersed catalysts for the CO2RR is rarely reported. The double active sites of catalysts have two adsorption positions for the reaction intermediates, which may facilitate C–C coupling to proceed and C2 products to form in the CO2RR. Therefore, investigating the distance effect of the active sites on the catalytic performance of atom-dispersed catalysts is of great significance for enhancing CO2 catalytic conversion.
Generally, Cu-based materials are considered as effective catalysts for the CO2RR to multi-carbon products;21–24 however, their poor product selectivity and weak interaction with CO2 inhibit the activity for electrochemical CO2 reduction.24–26 Recently, Fe atom catalysts have been found to exhibit outstanding electrocatalytic activity in the CO2RR.5,6,12,14,27 Due to the stable adsorption interaction between CO2 molecules and Fe active sites, the reaction free energy of initial CO2 hydrogenation on the surface of Fe atom catalysts can be reduced. In addition, regulating the adsorption of key intermediates on Fe-based catalysts was an effective way to facilitate the C–C coupling process and the conversion of CO2RR to C2 products.27–29 The Fe-n-f-CNT single-atom catalyst synthesized by Hwang et al. exhibited superior electrocatalytic performance for the CO2RR to ethanol with 45% faradaic efficiency.27 Moreover, An et al. designed a hybrid Fe–B dual-atom center to facilitate C–C coupling of low free energy barrier by enhancing the spin state of Fe, which proved that Fe sites had the potential of producing C2 products in the CO2RR process.29 Considering the size effect, single-atom catalysts can exhibit unique product selectivity.7,30–32 And regulating the distance of active sites may further improve the catalytic activity.19,20 Yu et al. synthesized FeN4 single-atom catalysts with accurate structural control and found that the Fe atomic sites at a specific distance could exhibit strong oxygen reduction reaction activity by regulating the filling of occupied d orbitals of Fe sites.20 However, the electrocatalytic activity for the CO2RR on Fe atomic pair sites is rarely reported and the intrinsic mechanism of electronic structure regulation in Fe atomic pairs is not clear. Combining the superior adsorption and activity of Fe single atoms on CO2, Fe atomic pairs may also exhibit good performance in the C–C coupling process. Therefore, catalyst materials with high electrocatalytic activity and product selectivity can be designed by adjusting the electronic structure between Fe atomic pair active sites, which is of great significance to promote the development of the CO2RR.
Hence, in this work, FeN4-Dx (x = 1, 2, 3, and 4) structures of four Fe atomic pairs at different distances were designed, and the effect of distance on their electrocatalytic performance in the CO2RR was investigated by density functional theory (DFT). Firstly, the structural stability of the four catalysts was analyzed from the perspectives of geometrical structure, electronic structure, thermodynamics, and electrochemistry by calculating geometric optimization, bonding interaction, formation energy, and dissolution potential, respectively. To analyze the initial catalytic activity of the four Fe atom pairs on CO2, the adsorption and activation of CO2 were calculated and compared in detail. Subsequently, the catalytic performance of FeN4-Dx structures on the CO2RR was further investigated, including the free energy of potential limiting steps and the comparison of the CO2RR with side reactions. On the basis of double active sites, the C–C coupling process was calculated and possible C2 products in the CO2RR were analyzed. Finally, the electronic structure of the key intermediates was analyzed in detail, and the effect of distance on the electrocatalytic performance of the CO2RR was discussed.
2. Computational method
All calculations in this work were performed by spin-polarized density functional theory (DFT) in Vienna Ab initio Simulation Package (VASP) 6.1.1.33 Projector augmented wave (PAW) and Perdew–Burke–Ernzerhof (PBE) generalized gradient approximation (GGA) were used to describe the ion–electron interaction and electronic exchange–correlation energy, respectively.34 The plane wave cutoff energy was set as 450 eV. K-Point meshes of 3 × 3 × 1 and high-density 15 × 15 × 5 were used to optimize geometry structures and accurately analyze electronic structures such as Bader charge transfer, charge difference density, and density of states (DOS).35 The electronic energy and the force of catalysts converged to 1.0 × 10−5 eV per atom and 0.02 eV Å−1, respectively. The lattice parameters of FeN4-Dx structures were as follows: a = 14.76 Å, b = 12.78 Å, and α = β = γ = 90°. Besides, in order to minimize the interactions of repeated slabs, a vacuum space of 15 Å was constructed along the c-axis. And the Grimme (DFT+D3) method was performed to correct the van der Waals interactions. Considering the solvent effect of water, the VASPsol package was used to build an implicit solvent model.36
Generally, the adsorption energy (Eads) between catalysts and adsorbates can be calculated by the following formula:
where
E*a,
E*, and
Ea represent the total energy of the system, the energy of the catalyst structures, and the energy of the adsorbates, respectively.
Charge density difference (Δρ) can be used to analyze the electron transfer direction after the interaction between two fragments (A and B), which can be calculated by
And the plane-averaged electron density difference along the
z axis (Δ
ρ(
z)
avg) is described as follows:
37| | 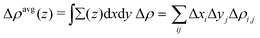 | (3) |
where the
z axis is along the
c-axis lattice of the structural model and
x/
y is the cross-section of the
x–
y plane at
z, and
i and
j are the one-point divided lattice axis and
b-axis lattice, respectively.
The calculation of reaction free energy was based on the computational hydrogen electrode (CHE) model.38 And the values of the reaction free energy difference (ΔG) can be obtained by the following formula:
| | 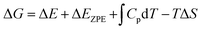 | (4) |
where Δ
E is the energy change calculated by DFT. Δ
EZPE is the zero-point energy correction.
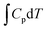
and Δ
S are the enthalpic temperature correction and the reaction entropy change at room temperature (
T = 298.15 K), respectively. Specifically, the saturated vapor pressure of 0.035, 0.053, 0.166, and 0.077 atm was employed for H
2O, HCOOH, CH
3OH, and C
2H
5OH, respectively. And VASPKIT software was used to post-process the relevant results.
39
3. Results and discussion
3.1 Structure stabilities
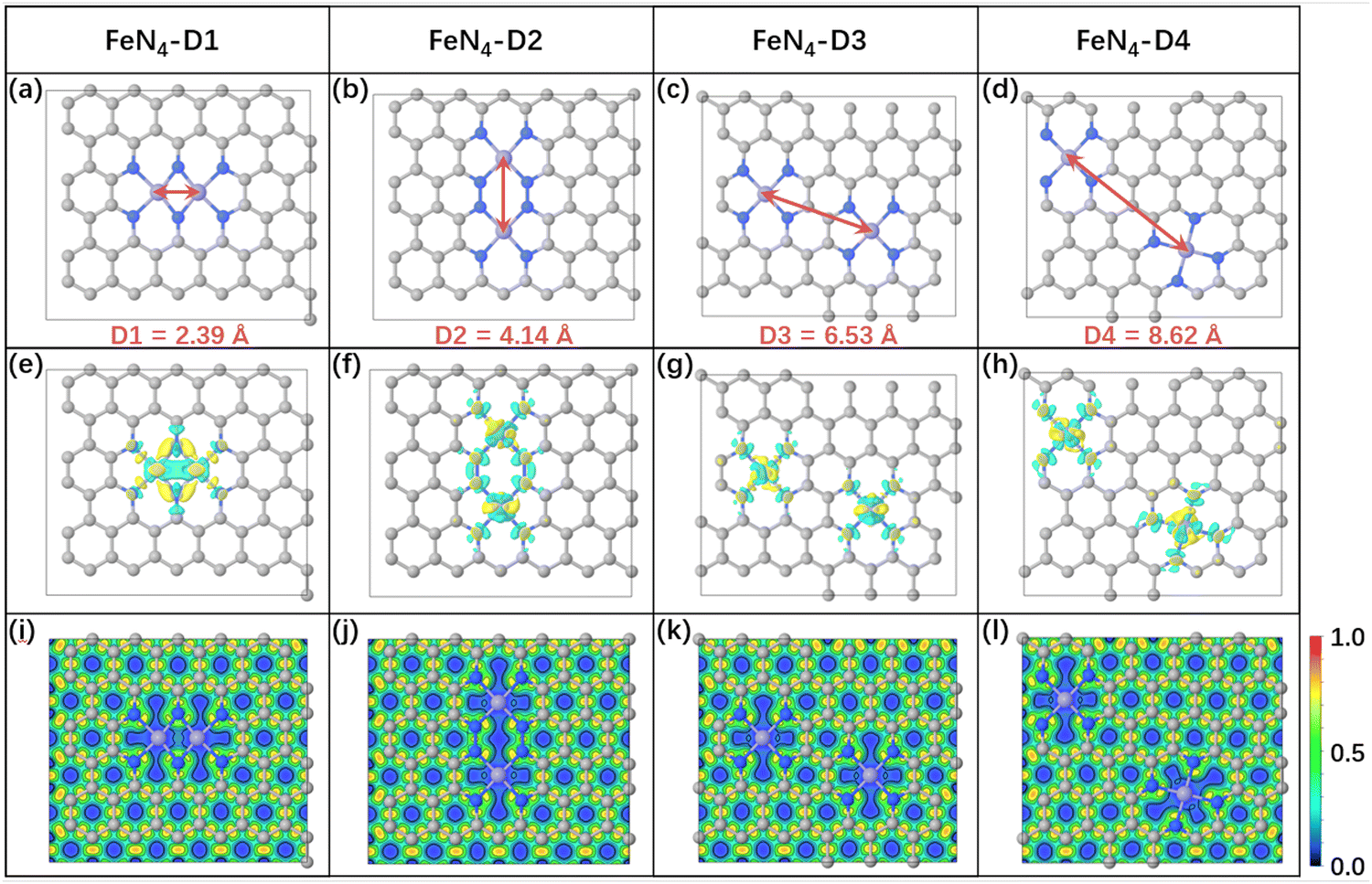
To investigate the distance effect of metal atomic pairs on electrocatalytic CO2 reduction, four typical structures (FeN4-Dx, x = 1, 2, 3, and 4) containing double FeN4 active sites were constructed in this work. And every Fe metal site was anchored by four nitrogen atoms to ensure its same coordination environment. The optimized structural models of FeN4-Dx (x = 1–4) are displayed in Fig. 1a–d, in which the distances of Fe atomic pairs were 2.39, 4.14, 6.53, and 8.62 Å, respectively. Four FeN4-Dx structures all had no obvious bending deformation in the geometry optimization. The detailed structural parameters are summarized in Table S1 (ESI†) and the Fe–N bond average lengths were between 1.87 Å and 1.91 Å. Further, charge density difference between the metal atom and nitrogen-doped carbon and corresponding Bader charge transfer indicated that Fe atomic pairs transferred electrons to adjacent nitrogen atoms, which can be anchored stably by coordinated pyridine nitrogen atoms (Table S1, ESI† and Fig. 1e–h). Based on the distance effect of the Fe atomic pairs, the magnetic moments and d-band centers of the four catalysts in Table S1 (ESI†) were different from each other, which may change the catalytic properties of Fe atom catalysts. According to the analysis of electron localization function in Fig. 1i–l, there is an obvious concentrated distribution of electrons in Fe–N bonds, indicating that Fe metal atoms have relatively strong electronic interaction with doped nitrogen. Moreover, projected density of states (PDOS) between Fe-3d and N-2p was used to analyze further their valence orbital interaction (Fig. S1, ESI†). Notably, Fe-3d orbitals in the four FeN4-Dx structures have good orbital overlapping with N-2p near the Fermi level, which indicates that the different distance Fe atomic pairs anchored by pyridine nitrogen atoms have great structural stability in the electronic structures.
 |
| | Fig. 1 The optimized structures of four different Fe atomic pairs (a–d), charge density difference between metal atoms and nitrogen-doped carbon (e–h) (the regions in yellow and cyan represent, respectively, electron accumulation and deletion. The isosurface levels are 0.01 e bohr−3) and the corresponding electron localization function (i–l). | |
Formation energy (Ef) and dissolution potential (Udiss) are used to evaluate structural stability in thermodynamics and electrochemistry, which can be calculated as follows: Ef = (Etotal − ENC − EFe × 2)/2 and Udiss = U0 − Ef/ne, respectively. Etotal, ENC, and EFe represent the energies of the total FeN4-Dx system, metal free nitrogen-doped carbon material, and Fe metal atom, respectively. U0 and n are, respectively, the standard dissolution potential of bulk metal and the number of electrons transferred during the dissolution of the metal atom. As can be seen from Fig. S2 (ESI†), FeN4-Dx structures have negative Ef values (from −9.29 eV to −7.21 eV) and positive Udiss values of 3.16–4.20 V, which ensures that FeN4-Dx structures have stable thermodynamic and electrochemical stability, respectively. Combined with previous reports,20 FeN4-Dx structures containing different distance metal atomic pairs have great potential to be synthesized and applied in electrocatalysis. Therefore, the distance effect of Fe atomic pairs might also enhance the performance of electrocatalytic CO2 reduction reaction, which can be further investigated in detail.
3.2 Initial CO2 adsorption and activation
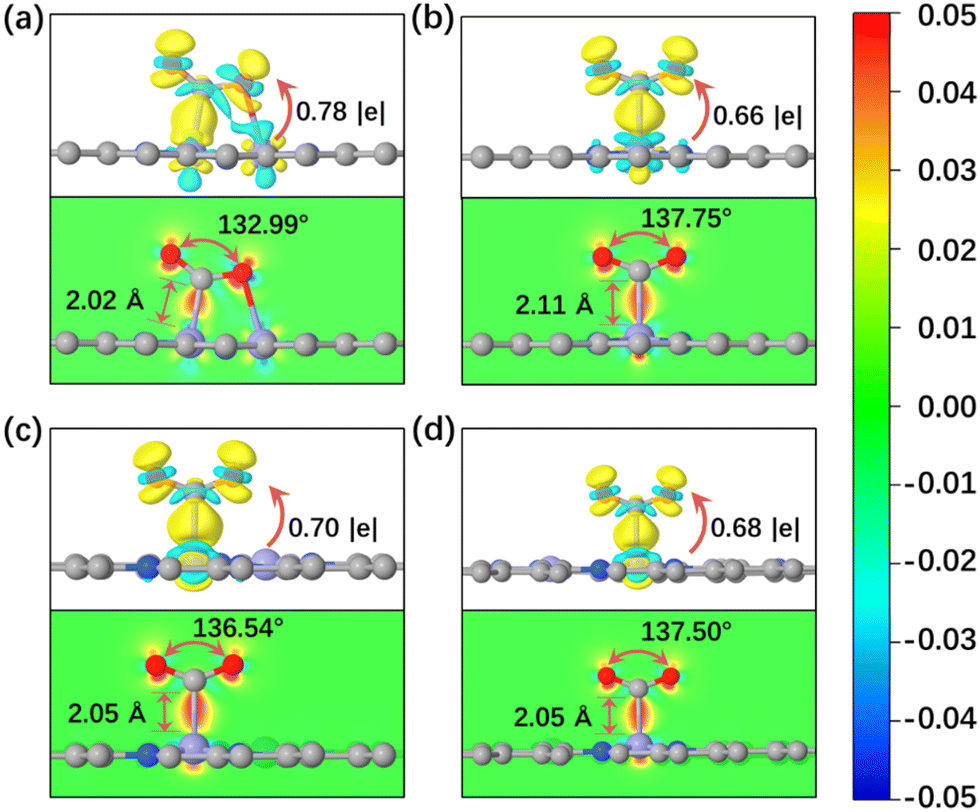
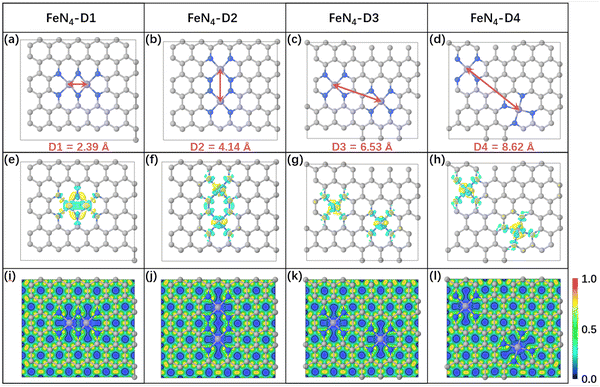
In general, the initial adsorption and activation of CO2 molecules on electrocatalysts is considered as the first step in the CO2 reduction reaction process, which is closely related to the catalytic activity of the electrocatalyst. The optimized CO2 adsorption structures and structural parameters of the four FeN4-Dx catalysts are summarized in Table S2 (ESI†). The C–Fe bond lengths between CO2 and active sites ranged from 2.02 to 2.11 Å and the bond angles of O–C–O were activated to bend (132.99°–137.75°) after CO2 adsorbed on the four FeN4-Dx structures. Obvious structural deformation indicated that CO2 molecules can be efficiently activated by Fe atomic pairs and exhibit stable chemisorption on metal centers. Furtherly, Bader charge transfer and charge density difference of CO2 adsorption structures were calculated to analyze the interaction between CO2 molecules and metal centers (Fig. 2). As shown in Fig. 2, according to the Bader charge transfer, Fe metal centers mainly acted as electron donors, while CO2 accepted most electrons to be activated. Charge density difference distribution and the corresponding profile map indicated that CO2 can be adsorbed stably on FeN4-Dx structures by strong C–Fe bond interaction. Moreover, the adsorption energies of CO2 on FeN4-Dx structures also indicated that Fe metal centers had a stable adsorption interaction with CO2 (Table S2, ESI†). Partial density of states (PDOS) in Fig. S3 (ESI†) showed the valence orbital electron interaction between the Fe 3d-orbital of active sites and CO2 2p-orbital. The orbital overlapping between Fe-3d and CO2-2p orbitals near the Fermi level further proved that the four types of Fe active sites all had good interaction with CO2 molecules. Obvious bending deformation of CO2 molecules on the four types of atomic pairs proved that different distance metal centers all had catalytic potential in the CO2RR. Specifically, CO2 was adsorbed on the bridge sites of the FeN4-D1 structure due to its close Fe pair sites. And the stable adsorption configuration showed that CO2 was more easily activated and adsorbed on FeN4-D1 than on the top sites of other FeN4-Dx structures. In addition, the values of the Bader charge transfer between CO2 molecules and FeN4-Dx structures show a similar trend with the change of CO2 bond angles (Table S2, ESI†). Therefore, the special Fe pair sites of the FeN4-D1 structure may have an advantage in promoting the initial CO2 adsorption and activation.
 |
| | Fig. 2 (a–d) Charge density difference and Bader charge transfer of CO2 adsorbed on FeN4-Dx (x = 1, 2, 3, and 4) (the regions in yellow and cyan represent, respectively, electron accumulation and deletion. The isosurface levels are 0.005 e bohr−3). | |
3.3 CO2RR to C1 products and HER side reactions
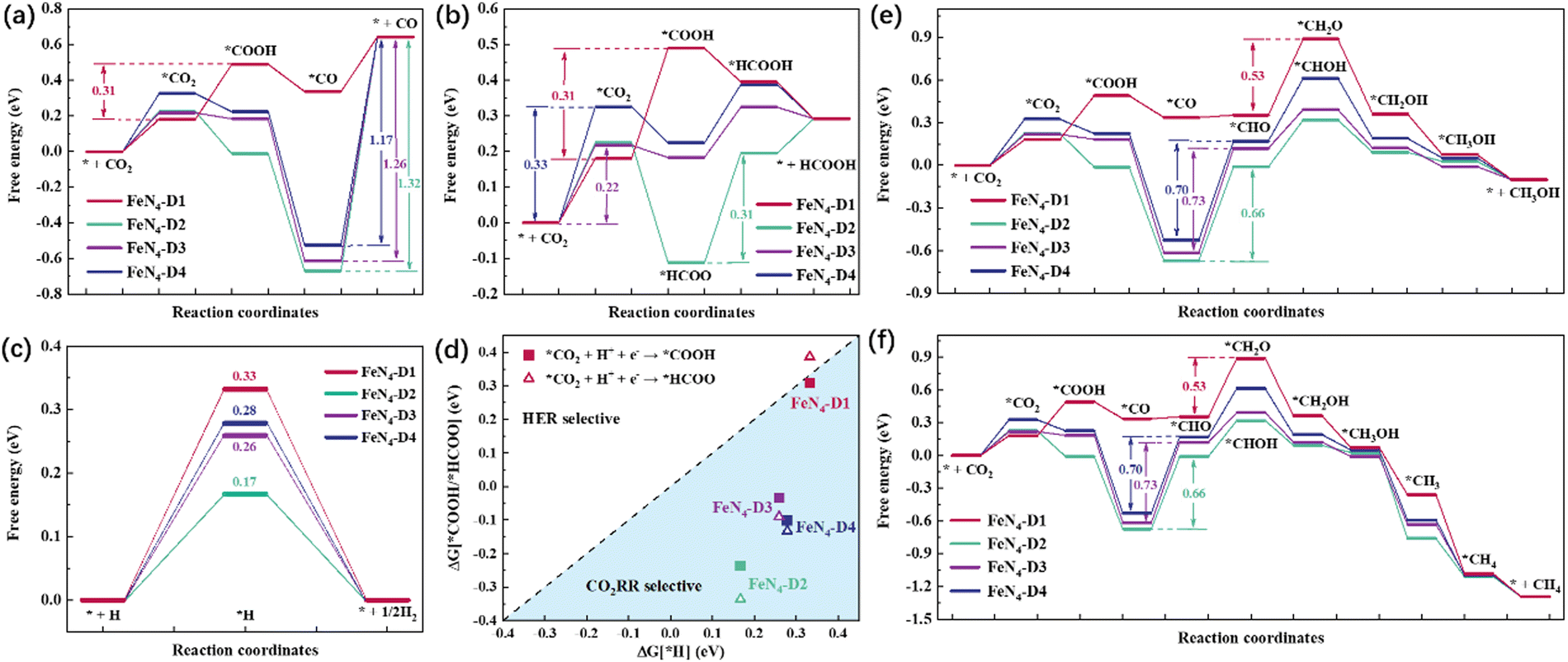
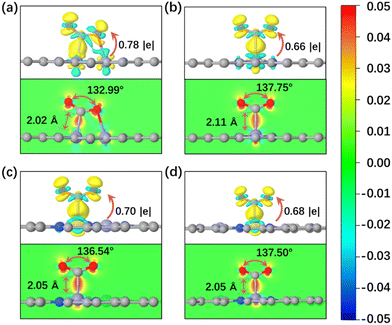
Every elementary reaction of the CO2RR after CO2 adsorption and activation is an important factor to analyze the catalytic reduction performance of electrocatalysts. Generally, the initial CO2 reduction steps can be divided into two hydrogenation reactions: hydrogen protons attacking oxygen atoms (*CO2 + H+ + e− → *COOH) and hydrogen protons attacking carbon atoms (*CO2 + H+ + e− → *HCOO). Different reduction products can be produced by different reaction pathways, depending on intermediates with relatively low reaction free energy. As for the CO product, CO2 prefers to undergo this reduction process: CO2 → *CO2 → *COOH → *CO → CO. As shown in Fig. 3a, the reaction free energies (ΔG) of the CO2RR to CO on FeN4-Dx (x = 1, 2, 3, and 4) structures were compared and the ΔG values of critical steps were labeled. Obviously, the potential limiting step (PLS) of the CO2RR to CO on FeN4-D1 was the formation of the *COOH intermediate (*CO2 + H+ + e− → *COOH, ΔG = 0.31 eV). As for FeN4-D2, FeN4-D3, and FeN4-D4 structures, the CO desorption was the PLS. The high reaction free energy of the CO desorption on FeN4-D2, FeN4-D3, and FeN4-D4 structures indicated that the Fe atomic pair sites with long distance had powerful adsorption for CO and inhibited CO desorption. The PLS ΔG value of 0.31 eV on FeN4-D1 was much lower than those on FeN4-D2 (ΔG = 1.32 eV), FeN4-D3 (ΔG = 1.26 eV), and FeN4-D4 (ΔG = 1.17 eV) structures, which showed that FeN4-D1 had the superior catalytic performance in the CO2RR to CO compared to other catalyst structures. Formic acid (HCOOH), as another two-electron reduction product, had two reaction pathways: Path 1: CO2 → *CO2 → *COOH → *HCOOH → HCOOH, and Path 2: CO2 → *CO2 → *HCOO → *HCOOH → HCOOH. By comparing the free energy of the two reaction pathways and choosing the more favorable reaction process, the free energy profile of producing HCOOH on the four catalysts was compared and analyzed. As can be seen in Fig. 3b, FeN4-D1, FeN4-D3, and FeN4-D4 structures followed Path 1 to reduce CO2 to HCOOH, while FeN4-D2 preferred to follow Path 2. And the PLS of forming HCOOH on the four catalysts was also different to some extent due to the distance difference of Fe atomic pair sites. By comparing the ΔG values of the PLS, the FeN4-D3 structure was more advantageous than other catalysts in the CO2RR to HCOOH, and its ΔG value was only 0.22 eV. Therefore, FeN4-D3 with specific distance (6.53 Å) Fe pair sites had superior catalytic performance in producing formic acid compared to other catalyst structures.
 |
| | Fig. 3 Reaction free energy profile of CO2 reduction to CO (a) and HCOOH (b). HER free energy profile (c) and the comparison between the HER and CO2RR on the four catalysts (d). Reaction free energy profile of the CO2RR to CH3OH (e) and CH4 (f) on FeN4-Dx (x = 1, 2, 3, and 4) structures. | |
It's worth noting that the hydrogen evolution reaction (HER) is considered to be the main side reaction for electrocatalytic CO2 reduction, which inhibits the CO2RR process. Fig. 3c shows the reaction free energy of the HER on the four catalysts. On the basis of the high ΔG value of 0.33 eV, the FeN4-D1 structure had a better inhibition effect on the HER than the other three catalyst structures, which was conducive to promoting the catalytic reduction of CO2. Moreover, the selectivity of FeN4-Dx (x = 1, 2, 3, and 4) structures between the HER and CO2RR was further investigated by comparing the ΔG values of first electron transfer proton coupling (Fig. 3d). The ΔG values of forming *COOH intermediates on the four catalyst structures were below the dotted line, which indicated that they all had good CO2RR selectivity. Therefore, the Fe atomic pair sites in FeN4-Dx (x = 1, 2, 3, and 4) structures were worthy to further investigate their catalytic performance in the CO2RR. Considering the stable adsorption of CO on FeN4-Dx (x = 1, 2, 3, and 4) catalysts, the multi-electron reduction products of the CO2RR, such as methanol (CH3OH) and methane (CH4), were further compared and analyzed. On the basis of reaction free energy of different intermediates in Table S3 (ESI†), the optimal reaction pathways of the CO2RR to CH3OH and CH4 on the four catalysts are summarized in Fig. 3e and f, respectively. Based on the linear scaling relations,40,41 the stable adsorption interaction between *CO intermediates and FeN4-Dx (x = 2, 3, and 4) structures resulting in the *CO hydrogenation (*CO + H+ + e− → *CHO) was still the rate-determining step of multi-electron C1 reduction products. And the ΔG values of the PLS were as follows: FeN4-D3 (ΔG = 0.73 eV) > FeN4-D4 (ΔG = 0.70 eV) > FeN4-D2 (ΔG = 0.66 eV). Specifically, the FeN4-D1 structure had a different rate-determining step with other catalysts. The hydrogenation of *CHO to *CH2O (ΔG = 0.53 eV) on the FeN4-D1 structure played the controlling role of producing CH3OH thermodynamically. It's worth noting that there were similar reaction intermediates before *CH3OH in the formation of CH3OH and CH4. Hence, the two reduction products would have the same PLS in the whole reaction process. Limiting potential (UL, V) can be obtained by the ΔG values of the PLS: UL = −ΔGmax/e. As shown in Fig. S4 (ESI†), the FeN4-D1 structure exhibited superior potential for the CO2RR to CO and multi-electron C1 reduction products, and the FeN4-D3 structure had the optimal UL values on producing HCOOH in the four catalysts. In a word, adjusting the distance of Fe atomic pair sites has an important regulatory effect on the electrocatalytic performance. Moreover, two active sites of Fe atomic pairs have the potential to promote deep CO2 reduction.
3.4 *CO intermediate analysis and C–C coupling
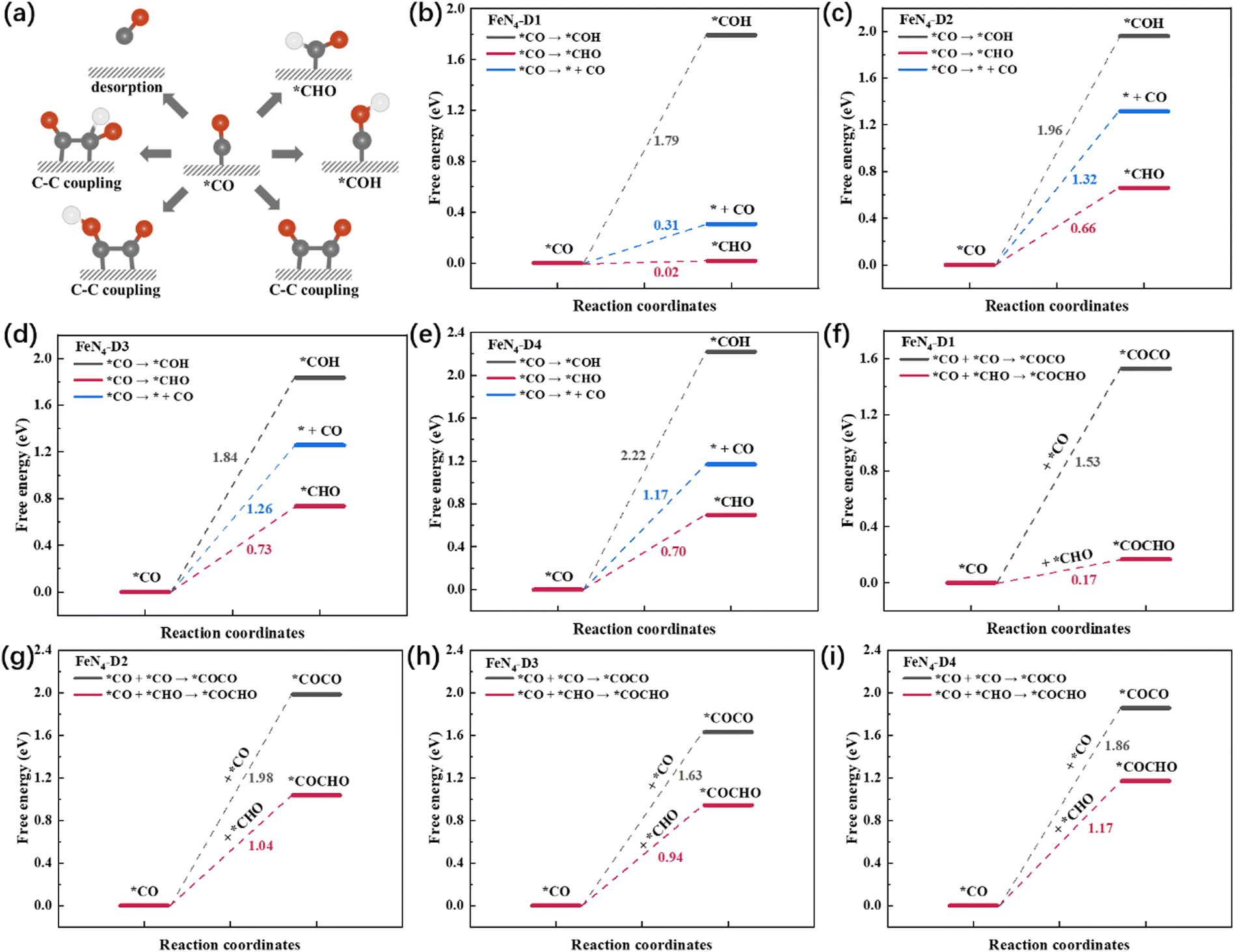
*CO was a key intermediate in the CO2RR process, participating in many possible reaction pathways such as CO desorption, two hydrogenation models, and three types of C–C coupling (Fig. 4a). In order to screen the optimal reaction pathway of *CO intermediate on the four catalysts, reaction free energies of different pathways were calculated and are compared in Fig. 4b–i. Firstly, compared with CO desorption and *CO hydrogenation to *COH, *CO intermediates were prone to form *CHO intermediates with the lowest ΔG pathway on the four FeN4-Dx (x = 1, 2, 3, and 4) structures (Fig. 4b–e). Therefore, the multi-electron reduction products might occupy a dominant position in the CO2RR process. Generally, C2 products have three types of C–C coupling: *CO–CO, *CO–*COH, and *CO–*CHO coupling. Due to the high ΔG of *CO hydrogenation to *COH intermediates, *CO–*COH coupling was not considered when comparing the optimal coupling model of the CO2RR process. As shown in Fig. 4f–i, the free energies of *CO–CO coupling and *CO–*CHO coupling on the four catalysts were calculated, respectively. Obviously, *CO–*CHO coupling exhibited the superior advantage of lower ΔG values than *CO–CO coupling, which indicated that *CO–*CHO coupling might be the main C–C coupling mode on Fe atom dispersed catalysts with Fe pair sites. What's more, the *CO–CHO coupling on the FeN4-D1 structure had the lowest free energy (ΔG = 0.17 eV) among the four catalysts, which indicated that the FeN4-D1 structure had more potential to produce C2 products in the CO2RR process. The close Fe pair sites might also play an important role in reducing the energy required for the migration of critical intermediates. The ΔG values of *CO–CHO coupling on FeN4-D2, FeN4-D3, and FeN4-D4 catalysts were, respectively, 1.04, 0.94, and 1.17 eV. Thus, the high C-C coupling free energy may be the main reason that Fe single atom catalysts have a poor catalytic performance for the CO2RR to C2 products. To sum up, precisely controlling the distance of Fe atomic pair sites may greatly improve their catalytic performance in the CO2RR.
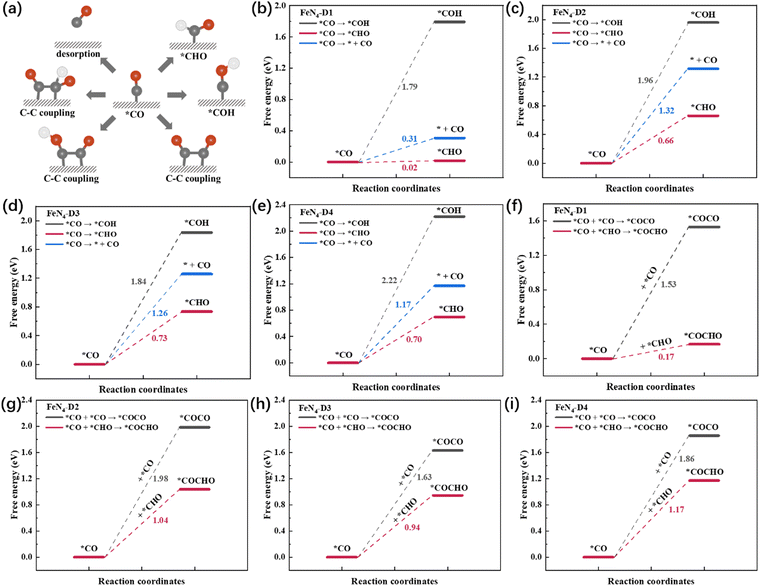 |
| | Fig. 4 (a) Schematic diagram of different reaction pathways of the *CO intermediate. (b–e) Free energy profile of CO desorption and *CO hydrogenation. (f–i) Comparison of C–C coupling on four FeN4-Dx (x = 1, 2, 3, and 4) structures. | |
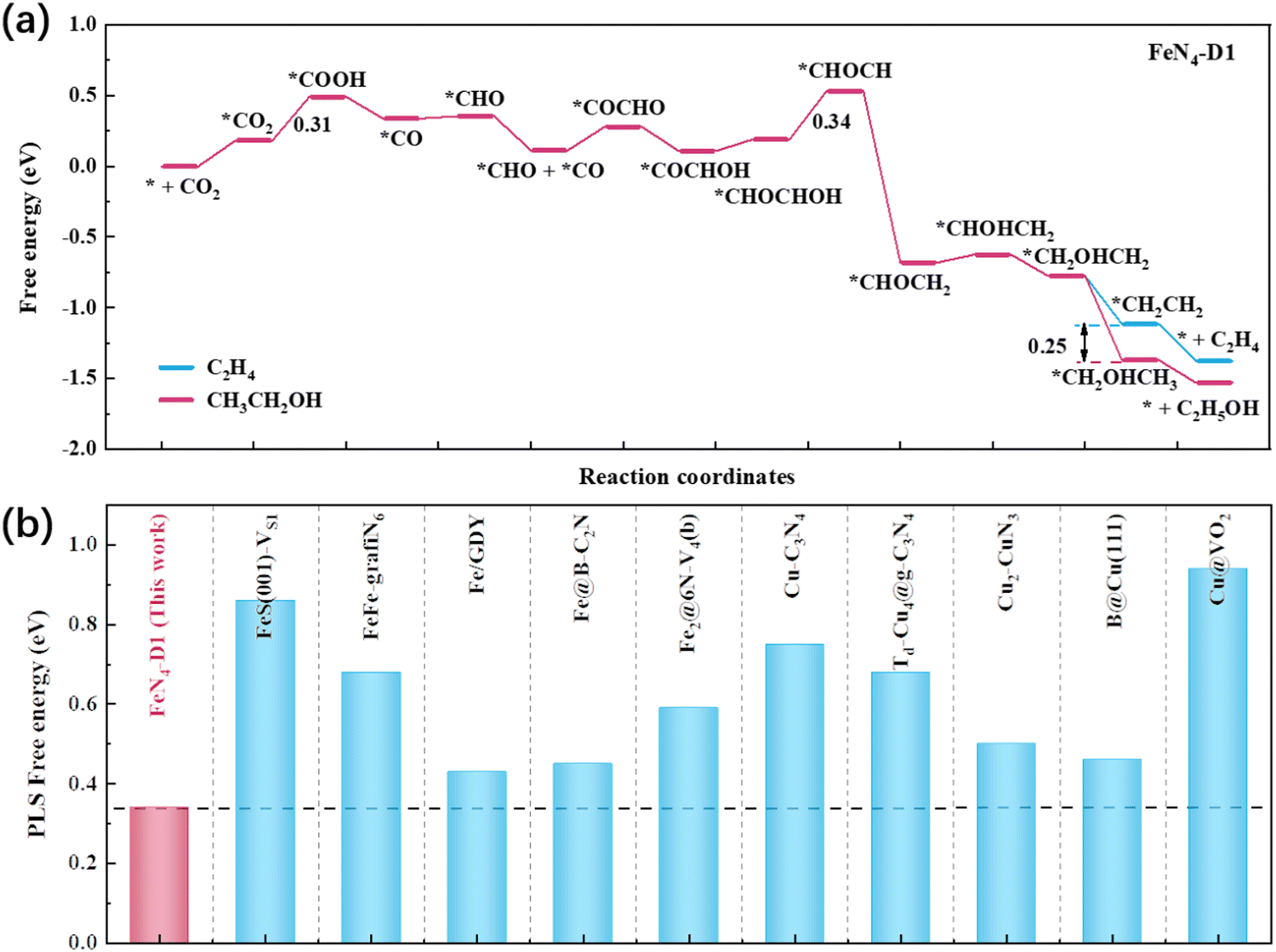
To investigate the catalytic performance of the FeN4-D1 catalyst on the CO2RR to C2 products, the reaction free energies of C2H4 and C2H5OH were calculated (Table S4, ESI†) and the optimal reaction pathways are displayed in Fig. 5a. It can be seen that the endothermic process *CHOCHOH + H+ + e− → *CHOCH + H2O (ΔG = 0.34 eV) was the key limiting step in producing C2 products. And the key ΔG values of C2H4 and C2H5OH were much lower than those of CH3OH and CH4, indicating that the FeN4-D1 structure was more inclined to deeply reduce CO2 to C2 products when *CO intermediates were formed. Moreover, the *CH2OHCH2 intermediate was a watershed between C2H4 and C2H5OH, which had two hydrogenation methods to form *CH2CH2 and *CH2OHCH3, respectively, with endothermic reaction. The Boltzmann distribution formula exp[−(ΔG)/(kBT)] can be used to estimate the selectivity of two pathways based on their free energy difference.28,42,43 *CH2OHCH3 intermediates were more readily available than *CH2CH2, with a free energy difference of about 0.25 eV. Thus, ΔG = −0.25 eV, T = 298.15 K, and the molar ratio between C2H5OH and C2H4 was about (1.68 × 104)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, which indicated that the FeN4-D1 structure showed a better product selectivity toward C2H5OH than C2H4 in the CO2RR. In order to highlight the catalytic performance of the FeN4-D1 structure on the CO2RR to C2H5OH, some reported catalysts were summarized and compared (Fig. 5b and Table S5, ESI†).23,24,28,29,44–49 Compared with usual Cu catalysts and iron-containing catalysts, the FeN4-D1 structure with specific distance Fe atomic pair sites exhibited more competitive catalytic performance for the CO2RR to ethanol. Therefore, the FeN4-D1 structure can be used as a promising electrochemical catalyst for producing ethanol.
:1, which indicated that the FeN4-D1 structure showed a better product selectivity toward C2H5OH than C2H4 in the CO2RR. In order to highlight the catalytic performance of the FeN4-D1 structure on the CO2RR to C2H5OH, some reported catalysts were summarized and compared (Fig. 5b and Table S5, ESI†).23,24,28,29,44–49 Compared with usual Cu catalysts and iron-containing catalysts, the FeN4-D1 structure with specific distance Fe atomic pair sites exhibited more competitive catalytic performance for the CO2RR to ethanol. Therefore, the FeN4-D1 structure can be used as a promising electrochemical catalyst for producing ethanol.
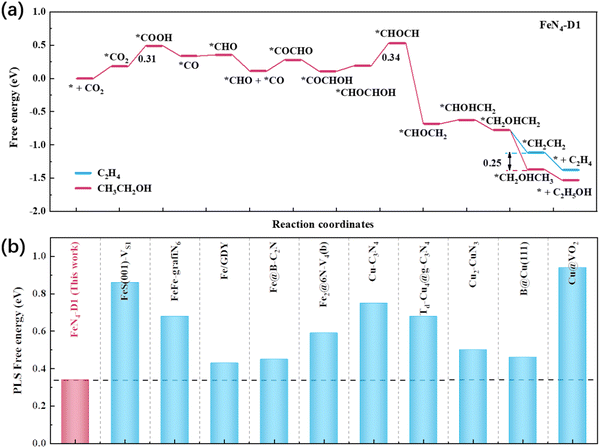 |
| | Fig. 5 (a) Reaction free energy profile of CO2 reduction to C2H4 and C2H5OH on the FeN4-D1 structure. (b) Comparison of the FeN4-D1 structure with some reported catalysts on the PLS free energy of C2H5OH. | |
3.5 Electronic structure analysis of *CO intermediates
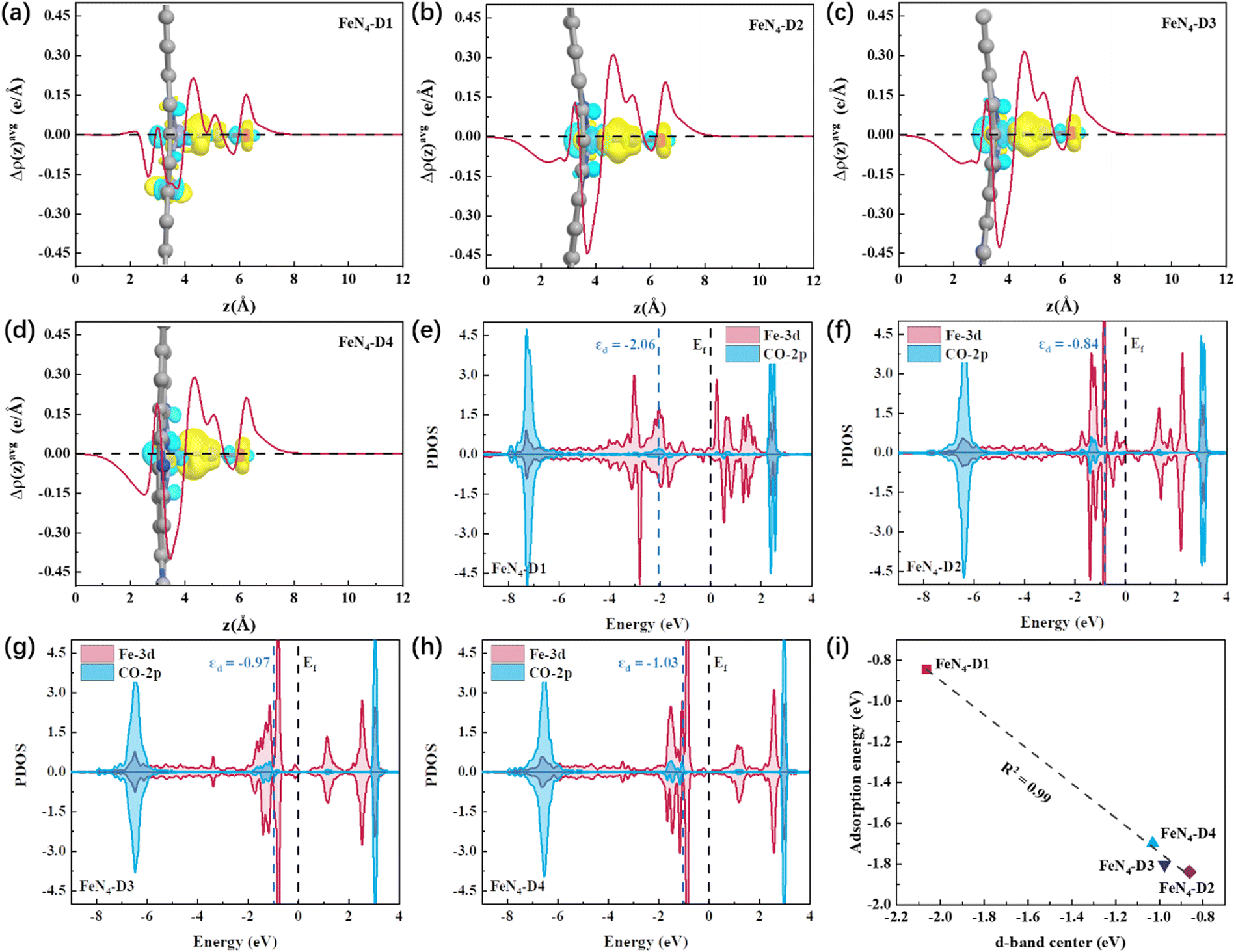
The FeN4-D1 structure had the superior catalytic performance on the CO2RR to ethanol, which can be attributed to the mild interaction between the key reaction intermediates and Fe atomic pair active sites with a specific distance (2.39 Å). *CO intermediates were important components in the C–C coupling process and played a pivotal role in the selectivity to C1 and C2 products. Therefore, the detailed electronic structures of *CO intermediates on the four different distance Fe pair sites were calculated and analyzed, and the nature of the catalytic reaction mechanism was further investigated. Fig. 6a–d shows the charge density difference between *CO intermediates and catalysts. To visualize the distribution of electron density intuitively, plane-averaged electron density difference along the z axis (Δρ(z)avg, e Å−1) was drawn together as well. It can be observed in Fig. 6a–d that a large number of electrons were concentrated on the C–Fe bonds between Fe atom active sites and *CO intermediates. Among them, the charge density concentration region of the *CO intermediate adsorbed on the FeN4-D1 structure was weaker than that of other catalysts. And the curve of z axis plane-averaged electron density difference also indicated that the *CO intermediate adsorbed on the FeN4-D1 structure had the lowest Δρ(z)avg peak (4 Å < z < 6 Å) in the four *CO intermediate adsorption configurations. Therefore, low charge density distribution of C–Fe bonds may weaken the interaction between *CO intermediates and the FeN4-D1 structure. Furtherly, partial density of states (PDOS) between the 2p-orbital of CO and 3d-orbital of adsorbed Fe sites was calculated to analyze their valence shell electron interaction (Fig. 6e–h). It was obvious that the overlapping region between CO-2p and Fe-3d orbitals of the FeN4-D1 structure below the Fermi level was the smallest among the four FeN4-Dx catalysts. Moreover, the antibonding orbitals formed by *CO with FeN4-Dx (x = 2, 3, and 4) were all higher than those formed by *CO with the FeN4-D1 catalyst, which enhanced the electronic interaction between *CO and FeN4-Dx (x = 2, 3, and 4) catalysts. The d-band center (εd) of metal active sites can serve as a descriptor to compare the adsorption interaction.50 It is worth noting that Fe active sites of the FeN4-D1 catalyst had the lowest d-band center position far away from the Fermi level. And the εd values of the four catalysts followed FeN4-D2 (εd = −0.84) > FeN4-D3 (εd = −0.97) > FeN4-D4 (εd = −1.03) > FeN4-D1 (εd = −2.06). Fig. 6i displays the relationship between the εd values and *CO intermediate adsorption energy. Obviously, the adsorption energies of *CO intermediates were linearly correlated with the d-band center of Fe active sites. The FeN4-D1 catalyst had the weak interaction with *CO intermediates based on its low d-band center. Therefore, the appropriate distance of Fe atom pairs modulated the d-band center of active sites, which adjusted the interaction between key intermediates and catalysts, and promoted the catalytic performance of the CO2RR.
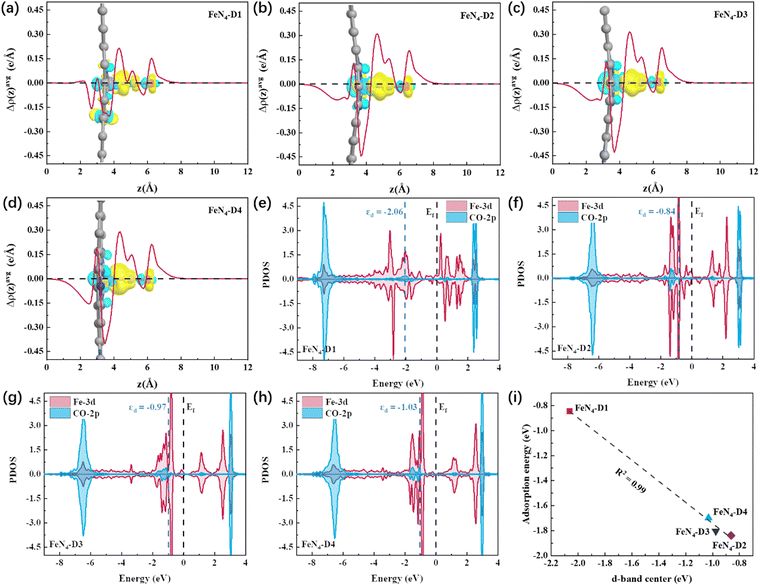 |
| | Fig. 6 (a–d) Charge density difference and the corresponding plane-averaged electron density difference along the z axis of *CO intermediates adsorbed on FeN4-Dx (x = 1, 2, 3, and 4) structures (the regions in yellow and cyan represent, respectively, electron accumulation and deletion. The isosurface levels are 0.005 e bohr−3). (e–h) Partial density of states (PDOS) between the 2p-obital of CO and 3d-orbital of adsorbed Fe sites on the four catalysts, respectively. (i) The relationship between the d-band center (εd) of adsorbed Fe sites and the adsorption energy of *CO intermediates. | |
4. Conclusions
In this work, different distance Fe atomic pairs were anchored by pyridine nitrogen to construct four FeN4-Dx (x = 1, 2, 3, and 4) structures and the distance effect on the catalytic CO2 reduction performance was further investigated. Combining charge density difference, electron localization function, and density of states between Fe atomic pair sites and coordination pyridine nitrogen, the four catalysts displayed good structural stability in electronic structure. Moreover, the negative Ef and positive Udiss indicated that the four different distance Fe atomic pairs can be stably anchored by nitrogen-containing carbon materials in thermodynamics and electrochemistry. The initial CO2 adsorption and activation was the key to reflect the activity of the catalyst. The optimized adsorption geometry and Bader charge transfer displayed that the FeN4-D1 structure had the best activation effect on CO2 molecules. Thus, the FeN4-D1 structure had good catalytic potential in the CO2RR. On the basis of the free energy profile, the FeN4-D1 structure exhibited better catalytic performance for the CO2RR to produce CO than other catalysts in C1 products. And compared with the HER side reaction, FeN4-Dx structures all showed better CO2RR selectivity, which further proved that Fe atomic pair catalysts had potential for application in electrocatalytic CO2RR. Considering the double sites of Fe atomic pair catalysts, the C–C coupling was further calculated and analyzed. The hydrogenation of the *CO intermediate was prone to form *CHO with lower reaction free energy rather than *COH. And *CO–*CHO coupling with smaller ΔG values might be the main C–C coupling method in producing C2 products. Among the four catalysts, the FeN4-D1 structure had the lowest ΔG values in the C–C coupling process, which indicated that the FeN4-D1 structure had good potential in reducing CO2 to produce C2 products. Furtherly, the free energy profile showed that the CO2RR on the FeN4-D1 structure was prone to form ethanol with a low ΔG value (0.34 eV) and high selectivity, which was superior to most reported catalysts. Charge density difference, PDOS, and d-band center of *CO intermediates on the four catalysts proved that the distance regulation of Fe atomic pairs enabled the FeN4-D1 structure to have the best catalytic performance for the CO2RR to ethanol. To sum up, the accurate distance adjustment of Fe atomic pair active sites may greatly improve the electrocatalytic CO2RR performance, and the FeN4-D1 structure would have broad application prospects in electrochemical energy conversion.
Conflicts of interest
The authors declare no competing financial interests in this paper.
Acknowledgements
This work was supported by Natural Science Foundation of Shandong Province (ZR2020KB010), and the Fundamental Research Funds for the Central Universities (22CX07010A).
References
- L. Li, X. Li, Y. Sun and Y. Xie, Rational design of electrocatalytic carbon dioxide reduction for a zero-carbon network, Chem. Soc. Rev., 2022, 51, 1234–1252 RSC.
- R. Li and D. Wang, Superiority of Dual-Atom Catalysts in Electrocatalysis: One Step Further Than Single-Atom Catalysts, Adv. Energy Mater., 2022, 12, 2103564 CrossRef CAS.
- C. Jia, K. Dastafkan, W. Ren, W. Yang and C. Zhao, Carbon-based catalysts for electrochemical CO2 reduction, Sustainable Energy Fuels, 2019, 3, 2890–2906 RSC.
- Y. Xue, Y. Guo, H. Cui and Z. Zhou, Catalyst Design for Electrochemical Reduction of CO2 to Multicarbon Products, Small Methods, 2021, 5, e2100736 CrossRef PubMed.
- X. Li, S. Xi, L. Sun, S. Dou, Z. Huang, T. Su and X. Wang, Isolated FeN4 Sites for Efficient Electrocatalytic CO2 Reduction, Adv. Sci., 2020, 7, 2001545 CrossRef CAS PubMed.
- X. Sun, Y. Tuo, C. Ye, C. Chen, Q. Lu, G. Li, P. Jiang, S. Chen, P. Zhu, M. Ma, J. Zhang, J. H. Bitter, D. Wang and Y. Li, Phosphorus Induced Electron Localization of Single Iron Sites for Boosted CO2 Electroreduction Reaction, Angew. Chem., Int. Ed., 2021, 60, 23614–23618 CrossRef CAS PubMed.
- Y. Wang, H. Su, Y. He, L. Li, S. Zhu, H. Shen, P. Xie, X. Fu, G. Zhou, C. Feng, D. Zhao, F. Xiao, X. Zhu, Y. Zeng, M. Shao, S. Chen, G. Wu, J. Zeng and C. Wang, Advanced Electrocatalysts with Single-Metal-Atom Active Sites, Chem. Rev., 2020, 120, 12217–12314 CrossRef CAS PubMed.
- Y. Zhang, L. Jiao, W. Yang, C. Xie and H. L. Jiang, Rational Fabrication of Low-Coordinate Single-Atom Ni Electrocatalysts by MOFs for Highly Selective CO2 Reduction, Angew. Chem., Int. Ed., 2021, 60, 7607–7611 CrossRef CAS PubMed.
- Q.-X. Li, D.-H. Si, W. Lin, Y.-B. Wang, H.-J. Zhu, Y.-B. Huang and R. Cao, Highly efficient electroreduction of CO2 by defect single-atomic Ni-N3 sites anchored on ordered micro-macroporous carbons, Sci. China: Chem., 2022, 65, 1584–1593 CrossRef CAS.
- D. Chen, L. H. Zhang, J. Du, H. Wang, J. Guo, J. Zhan, F. Li and F. Yu, A Tandem Strategy for Enhancing Electrochemical CO2 Reduction Activity of Single-Atom Cu-S1N3 Catalysts via Integration with Cu Nanoclusters, Angew. Chem., Int. Ed., 2021, 60, 24022–24027 CrossRef CAS PubMed.
- S. Chen, X. Li, C. W. Kao, T. Luo, K. Chen, J. Fu, C. Ma, H. Li, M. Li, T. S. Chan and M. Liu, Unveiling Proton-feeding Effect in Sulfur-doped Fe-N-C Single-Atom Catalyst for Enhanced CO2 Electroreduction, Angew. Chem., Int. Ed., 2022, 134, e202206233 Search PubMed.
- F. Pan, B. Li, E. Sarnello, S. Hwang, Y. Gang, X. Feng, X. Xiang, N. M. Adli, T. Li, D. Su, G. Wu, G. Wang and Y. Li, Boosting CO2 reduction on Fe-N-C with sulfur incorporation: Synergistic electronic and structural engineering, Nano Energy, 2020, 68, 104384 CrossRef CAS.
- H. Zhang, J. Li, S. Xi, Y. Du, X. Hai, J. Wang, H. Xu, G. Wu, J. Zhang, J. Lu and J. Wang, A Graphene-Supported Single-Atom FeN5 Catalytic Site for Efficient Electrochemical CO2 Reduction, Angew. Chem., Int. Ed., 2019, 58, 14871–14876 CrossRef CAS PubMed.
- X. Wang, Y. Pan, H. Ning, H. Wang, D. Guo, W. Wang, Z. Yang, Q. Zhao, B. Zhang, L. Zheng, J. Zhang and M. Wu, Hierarchically micro- and meso-porous Fe-N4O-doped carbon as robust electrocatalyst for CO2 reduction, Appl. Catal., B, 2020, 266, 118630 CrossRef CAS.
- L. Lin, H. Li, C. Yan, H. Li, R. Si, M. Li, J. Xiao, G. Wang and X. Bao, Synergistic Catalysis over Iron-Nitrogen Sites Anchored with Cobalt Phthalocyanine for Efficient CO2 Electroreduction, Adv. Mater., 2019, 31, e1903470 CrossRef PubMed.
- X. Wei, S. Wei, S. Cao, Y. Hu, S. Zhou, S. Liu, Z. Wang and X. Lu, Cu acting as Fe activity promoter in dual-atom Cu/Fe-NC catalyst in CO2RR to C1 products, Appl. Surf. Sci., 2021, 564, 150423 CrossRef CAS.
- J. Zhu, M. Xiao, D. Ren, R. Gao, X. Liu, Z. Zhang, D. Luo, W. Xing, D. Su, A. Yu and Z. Chen, Quasi-Covalently Coupled Ni-Cu Atomic Pair for Synergistic Electroreduction of CO2, J. Am. Chem. Soc., 2022, 144, 9661–9671 CrossRef CAS PubMed.
- W. Ren, X. Tan, W. Yang, C. Jia, S. Xu, K. Wang, S. C. Smith and C. Zhao, Isolated Diatomic Ni-Fe Metal-Nitrogen Sites for Synergistic Electroreduction of CO2, Angew. Chem., Int. Ed., 2019, 58, 6972–6976 CrossRef CAS PubMed.
- Y. Li, R. Hu, Z. Chen, X. Wan, J.-X. Shang, F.-H. Wang and J. Shui, Effect of Zn atom in Fe-N-C catalysts for electro-catalytic reactions: theoretical considerations, Nano Res., 2021, 14, 611–619 CrossRef CAS.
- Z. Jin, P. Li, Y. Meng, Z. Fang, D. Xiao and G. Yu, Understanding the inter-site distance effect in single-atom catalysts for oxygen electroreduction, Nat. Catal., 2021, 4, 615–622 CrossRef CAS.
- A. J. Garza, A. T. Bell and M. Head-Gordon, Mechanism of CO2 Reduction at Copper Surfaces: Pathways to C2 Products, ACS Catal., 2018, 8, 1490–1499 CrossRef CAS.
- C. Wang, C. Zhu, M. Zhang, Y. Geng and Z. Su, Copper Dimer Anchored in g-CN Monolayer as an Efficient Electrocatalyst for CO2 Reduction Reaction: A Computational Study, Adv. Theory Simul., 2020, 3, 2000218 CrossRef CAS.
- Y. Jiao, Y. Zheng, P. Chen, M. Jaroniec and S. Z. Qiao, Molecular Scaffolding Strategy with Synergistic Active Centers To Facilitate Electrocatalytic CO2 Reduction to Hydrocarbon/Alcohol, J. Am. Chem. Soc., 2017, 139, 18093–18100 CrossRef CAS PubMed.
- X. Su, Z. Jiang, J. Zhou, H. Liu, D. Zhou, H. Shang, X. Ni, Z. Peng, F. Yang, W. Chen, Z. Qi, D. Wang and Y. Wang, Complementary Operando Spectroscopy identification of in-situ generated metastable charge-asymmetry Cu2-CuN3 clusters for CO2 reduction to ethanol, Nat. Commun., 2022, 13, 1322 CrossRef CAS PubMed.
- M. Behrens, F. Studt, I. Kasatkin, S. Kühl, M. Hävecker, F. Abild-Pedersen, S. Zander, F. Girgsdies, P. Kurr, B.-L. Kniep, M. Tovar, R. W. Fischer, J. K. Nørskov and R. Schlögl, The Active Site of Methanol Synthesis over Cu/ZnO/Al2O3 Industrial Catalysts, Science, 2012, 336, 893–897 CrossRef CAS PubMed.
- J. A. Rodriguez, P. Liu, D. J. Stacchiola, S. D. Senanayake, M. G. White and J. G. Chen, Hydrogenation of CO2 to Methanol: Importance of Metal–Oxide and Metal–Carbide Interfaces in the Activation of CO2, ACS Catal., 2015, 5, 6696–6706 CrossRef CAS.
- K. Lakshmanan, W. H. Huang, S. A. Chala, B. W. Taklu, E. A. Moges, J. F. Lee, P. Y. Huang, Y. C. Lee, M. C. Tsai, W. N. Su and B. J. Hwang, Highly Active Oxygen Coordinated Configuration of Fe Single-Atom Catalyst toward Electrochemical Reduction of CO2 into Multi-Carbon Products, Adv. Funct. Mater., 2022, 32, 2109310 CrossRef CAS.
- X. Liu, Z. Wang, Y. Tian and J. Zhao, Graphdiyne-Supported Single Iron Atom: A Promising Electrocatalyst for Carbon Dioxide Electroreduction into Methane and Ethanol, J. Phys. Chem. C, 2020, 124, 3722–3730 CrossRef CAS.
- M. He, W. An, Y. Wang, Y. Men and S. Liu, Hybrid Metal-Boron Diatomic Site Embedded in C2N Monolayer Promotes C-C Coupling in CO2 Electroreduction, Small, 2021, 17, 2104445 CrossRef CAS PubMed.
- S. K. Kaiser, Z. Chen, D. Faust Akl, S. Mitchell and J. Perez-Ramirez, Single-Atom Catalysts across the Periodic Table, Chem. Rev., 2020, 120, 11703–11809 CrossRef CAS PubMed.
- W. Song, X. Lv, Y. Gao and L. Wang, Photocatalytic HER Performance of TiO2-supported Single Atom Catalyst Based on Electronic Regulation: A DFT Study, Chem. Res. Chin. Univ., 2021, 38, 1025–1031 CrossRef.
- Y. Song, L. Tao, Y. Zhang, J. Pan and S. Du, A DFT Investigation on the Electronic Structures and Au Adatom Assisted Hydrogenation of Graphene Nanoflake Array, Chem. Res. Chin. Univ., 2021, 37, 1110–1115 CrossRef CAS.
- G. Kresse and J. Furthmüller, Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized gradient approximation made simple, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- H. J. Monkhorst and J. D. Pack, Special points for Brillouin-zone integrations, Phys. Rev. B: Condens. Matter Mater. Phys., 1976, 13, 5188–5192 CrossRef.
- K. Mathew, R. Sundararaman, K. Letchworth-Weaver, T. A. Arias and R. G. Hennig, Implicit solvation model for density-functional study of nanocrystal surfaces and reaction pathways, J. Chem. Phys., 2014, 140, 084106 CrossRef PubMed.
- Y. Yuan, X. Gong and H. Wang, The synergistic mechanism of graphene and MoS2 for hydrogen generation: insights from density functional theory, Phys. Chem. Chem. Phys., 2015, 17, 11375–11381 RSC.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jonsson, Origin of the overpotential for oxygen reduction at a fuel-cell cathode, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef.
- V. Wang, N. Xu, J.-C. Liu, G. Tang and W.-T. Geng, VASPKIT: A user-friendly interface facilitating high-throughput computing and analysis using VASP code, Comput. Phys. Commun., 2021, 267, 108033 CrossRef CAS.
- F. Abild-Pedersen, J. Greeley, F. Studt, J. Rossmeisl, T. R. Munter, P. G. Moses, E. Skulason, T. Bligaard and J. K. Norskov, Scaling properties of adsorption energies for hydrogen-containing molecules on transition-metal surfaces, Phys. Rev. Lett., 2007, 99, 016105 CrossRef CAS PubMed.
- H. Wu and F. He, Activity Origins of Graphdiyne Based Bifunctional Atom Catalysts for Hydrogen Evolution and Water Oxidation, Chem. Res. Chin. Univ., 2021, 37, 1334–1340 CrossRef CAS.
- G. Zhu, Y. Li, H. Zhu, H. Su, S. H. Chan and Q. Sun, Curvature-Dependent Selectivity of CO2 Electrocatalytic Reduction on Cobalt Porphyrin Nanotubes, ACS Catal., 2016, 6, 6294–6301 CrossRef CAS.
- D. Jiao, Y.-j Liu, Q. Cai and J.-X. Zhao, Coordination Tunes Activity and Selectivity of Nitrogen Reduction Reaction on Single-Atom Iron Catalysts: A Computational Study, J. Mater. Chem. A, 2020, 9, 1240–1251 RSC.
- T. G. Senthamaraikannan and D.-H. Lim, CO2 Reduction to C1 and C2 Compounds on Sulfur-Deficient Mackinawite (FeS): A Density Functional Theory Study, J. Phys. Chem. C, 2022, 126, 7012–7021 CrossRef CAS.
- S. Chen, H. Yuan, S. I. Morozov, L. Ge, L. Li, L. Xu and W. A. Goddard, 3rd, Design of a Graphene Nitrene Two-Dimensional Catalyst Heterostructure Providing a Well-Defined Site Accommodating One to Three Metals, with Application to CO2 Reduction Electrocatalysis for the Two-Metal Case, J. Phys. Chem. Lett., 2020, 11, 2541–2549 CrossRef CAS PubMed.
- Y. Zhao, S. Zhou and J. Zhao, Selective C-C Coupling by Spatially Confined Dimeric Metal Centers, iScience, 2020, 23, 101051 CrossRef CAS PubMed.
- X. Bai, Q. Li, L. Shi, X. Niu, C. Ling and J. Wang, Hybrid Cu0 and Cux+ as Atomic Interfaces Promote High-Selectivity Conversion of CO2 to C2H5OH at Low Potential, Small, 2020, 16, e1901981 CrossRef PubMed.
- J.-S. Wang, G.-C. Zhao, Y.-Q. Qiu and C.-G. Liu, Strong Boron–Carbon Bonding Interaction Drives CO2 Reduction to Ethanol over the Boron-Doped Cu(111) Surface: An Insight from the First-Principles Calculations, J. Phys. Chem. C, 2020, 125, 572–582 CrossRef.
- Q. Yang, X. Liu, W. Peng, Y. Zhao, Z. Liu, M. Peng, Y.-R. Lu, T.-S. Chan, X. Xu and Y. Tan, Vanadium Oxide Integrated on Hierarchically Nanoporous Copper for Efficient Electroreduction of CO2 to Ethanol, J. Mater. Chem. A, 2021, 9, 3044–3051 RSC.
- S. Sun, X. Zhou, B. Cong, W. Hong and G. Chen, Tailoring the d-Band Centers Endows (NixFe1–x)2P Nanosheets with Efficient Oxygen Evolution Catalysis, ACS Catal., 2020, 10, 9086–9097 CrossRef CAS.
|
| This journal is © the Partner Organisations 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 and
Fangna
Dai
and
Fangna
Dai
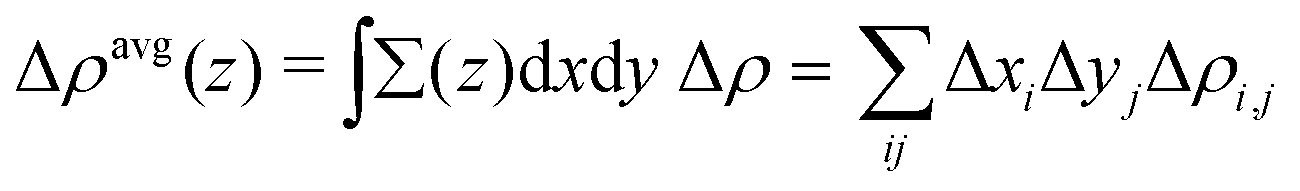
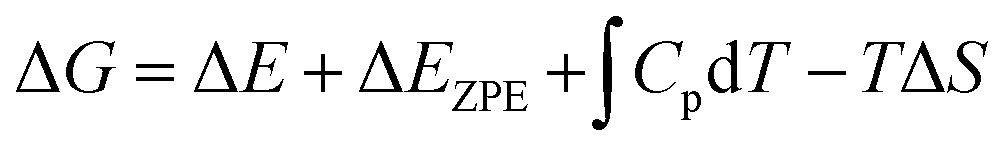
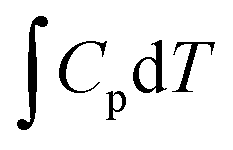 and ΔS are the enthalpic temperature correction and the reaction entropy change at room temperature (T = 298.15 K), respectively. Specifically, the saturated vapor pressure of 0.035, 0.053, 0.166, and 0.077 atm was employed for H2O, HCOOH, CH3OH, and C2H5OH, respectively. And VASPKIT software was used to post-process the relevant results.39
and ΔS are the enthalpic temperature correction and the reaction entropy change at room temperature (T = 298.15 K), respectively. Specifically, the saturated vapor pressure of 0.035, 0.053, 0.166, and 0.077 atm was employed for H2O, HCOOH, CH3OH, and C2H5OH, respectively. And VASPKIT software was used to post-process the relevant results.39