
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Study on the incorporation of various elements into the Keggin lacunary-type phosphomolybdate [PMo9O34]9− and subsequent purification of the polyoxometalates by nanofiltration†
Jan-Christian
Raabe
 a,
Tobias
Esser
a,
Froze
Jameel
b,
Matthias
Stein
b,
Jakob
Albert
a and
Maximilian J.
Poller
*a
a,
Tobias
Esser
a,
Froze
Jameel
b,
Matthias
Stein
b,
Jakob
Albert
a and
Maximilian J.
Poller
*a
aInstitute of Technical and Macromolecular Chemistry, Hamburg University, Bundesstr. 45, 20146 Hamburg, Germany. E-mail: maximilian.poller@uni-hamburg.de
bMax Planck Institute for Dynamics of Complex Technical Systems, Molecular Simulations and Design Group, Sandtorstrasse 1, 39106, Magdeburg, Germany
First published on 17th July 2023
Abstract
Transition-metal substituted Keggin-type polyoxometalates (POMs) are of great interest for applications in biomedicine, material science, and catalysis. The synthesis of transition metal-substituted Keggin-type polytungstates via the formation of a lacunary structure is well established, in contrast this approach is so far unexplored for Keggin-type polymolybdates. This is because the prevailing doctrine assumes that the lacunary Keggin phosphomolybdate [PMo9O34]9− is too unstable and can only be stabilized with organic ligands such as pyridine in organic solvents. In this work, we present a reliable procedure for the synthesis of the lacunary compound [PMo9O34]9− and its application in a novel in situ approach for the synthesis of different metal substituted POMs. The method is based on generating the lacunary species in situ, where the metal-substituted POMs are produced by adding another precursor compound. We employed this method to synthesize several new specific element-substituted POMs, which we present with comprehensive characterization. The interpretation of the analytical results was complemented by DFT calculations. For the separation of by-products from synthesis, we employed a novel membrane-based nanofiltration process, that enables superior separation of alkali salts from the POM solution resulting in >99% rejection of the POM components.
Introduction
Polyoxometalates (POMs) are an inorganic substance class of polyanionic metal oxide cluster compounds with defined nanoscale sizes and geometries. In general, there are two main coordination motifs for the oxo ligand: the metal–metal bridging M–O–M and the terminal M![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O coordination motif. The history of the development of POMs starts in 1788 with Sheele, who studied the first example of Molybdenum Blue. Later in 1826, Berzelius discovered the anion [PMo12O40]3− which was isolated as an ammonium salt. One century later in 1934, Keggin presented the first solid-state structure as determined by X-ray diffraction techniques, of the anion [PW12O40]3− which is known as the Keggin-type structure.1–3 Today, POMs are in the focus of different research topics ranging from catalysis, biomedicine, nanostructure to photovoltaic applications.4,5 Furthermore, there are applications for POMs acting as proton conductors or as motifs to modify perovskite and metal organic framework (MOF) materials to use the resulting compounds in photovoltaic applications.1,6–10 Due to their unique properties, an inorganic POM structure can be combined with an organic cation. The resulting hybrid materials are interesting for nanoscience and biobased applications.6
O coordination motif. The history of the development of POMs starts in 1788 with Sheele, who studied the first example of Molybdenum Blue. Later in 1826, Berzelius discovered the anion [PMo12O40]3− which was isolated as an ammonium salt. One century later in 1934, Keggin presented the first solid-state structure as determined by X-ray diffraction techniques, of the anion [PW12O40]3− which is known as the Keggin-type structure.1–3 Today, POMs are in the focus of different research topics ranging from catalysis, biomedicine, nanostructure to photovoltaic applications.4,5 Furthermore, there are applications for POMs acting as proton conductors or as motifs to modify perovskite and metal organic framework (MOF) materials to use the resulting compounds in photovoltaic applications.1,6–10 Due to their unique properties, an inorganic POM structure can be combined with an organic cation. The resulting hybrid materials are interesting for nanoscience and biobased applications.6
The incorporation of different elements, especially transition metals, into the Keggin-type phosphomolybdate POMs (also known as heteropolyanions/heteropolyacids (HPA))11–13 is a currently very active area of research.14,15 It is an interesting field not only from a synthetic point of view, but also from a catalytic perspective; in fact a major goal of this field is the synthesis of tailor-made catalysts.15–17 By substituting the framework metal with redox active transition metals, the redox potentials and thus the redox activity of the POM can be tuned.15 Various transition metal-substituted phosphomolybdates are mentioned in literature. One of the most prominent examples is H8[PV5Mo7O40] called HPA-5 (HPA for heteropolyacid and “5” representing the number of Mo(VI) positions that are substituted by V(V)) or HPV5Mo, a POM that has been successfully established as a catalyst for the conversion of biomass to formic acid, as well as for the oxidative desulfurization of fuels.18–24 Quantum chemical calculations have shown that the incorporation of the different metals alters the shape of the POMs, where the bond distances between the oxygen atoms and upcoming metal change with respect to their atomic radii.25 These distortions in M–O bonds originate from a pseudo Jahn–Teller vibronic instability at high symmetry configurations.26
There are two possible pathways to synthesize element-substituted Keggin-type POMs: the self-assembly process,14,15,27,28 or, alternatively, via the formation of a lacunary structure. In the self-assembly process, the precursor compounds are dissolved in water in the targeted stoichiometry and then converted to the desired POM structure. This strategy has been successfully employed for the synthesis of V(V) substituted Keggin phosphomolybdates by Odyakov et al. using V2O5 as a precursor compound, resulting in structures of the type H3+x[PVxMo12−xO40] (with x = 0 to 6).14,27,28 Our own group has successfully improved this method for the synthesis of Mn(II) and mixed Mn(II)–V(V)-substituted Keggin phosphomolybdates.15
The other possibility is the lacunary route: a lacunary POM is a defect POM structure where one or more MO6 octahedra have been removed.29,30 These vacancies can then be filled with suitable multivalent transition metals, resulting in a redox substituted POM (Fig. 1). This approach has been successfully employed for Keggin and Wells–Dawson tungstates31,32 but so far has not successfully been extended to the respective molybdates. This is mainly due to the fact that phosphomolybdates are less stable than the corresponding tungstates. There are some reports of the lacunary phosphomolybdate structures dating back as far as the 1970s,33–38 however a detailed study of this compound is still missing.
 | ||
| Fig. 1 Lacunary approach used in this work. [PMo9O34]9− (LHPA-3) as main compound for further substitution by different redox-active metals of various valence. | ||
In the present study, we present a reproducible synthetic procedure and comprehensive characterization for the lacunary Keggin-type phosphomolybdate anion [PMo9O34]9− (LHPA-3, where “3” represents the number of vacancies). Furthermore, we have thoroughly explored this synthetic opportunity by successfully incorporating various metals of different valence into LHPA-3 (Fig. 1) and characterized the resulting new compounds in detail.
Previous computational studies have focused on relative stabilities of isomers of Keggin structures,39,40 the dimerization of Lindqvist and Keggin structures41 and spectra of first-row substituted Keggin structures.42 Here, the investigation is extended to phosphomolybdate POMs and substitutes thereof. The results from DFT calculations allow to make statements about relative stabilities of metal substitutions and rationalize peak positions in IR and Raman spectra.
A particular challenge in our synthetic procedure was the separation of the products from the counterions of the precursor materials (usually alkali salts). Commonly, this problem is solved by crystallisation or extractive removal of the POM using diethyl ether from the aqueous reaction solution.43 From our experience the diethyl ether purification method does not work for transition-metal substituted POMs with high degrees of substitution or with high charges. Crystallisation is usually time consuming and lowers the yield significantly due to similar solubilities of the POM and the impurities.29 For the here presented study, we have used a membrane process using nanofiltration as a novel approach to solve this problem. Esser et al. have already demonstrated that a Keggin-type POM catalyst can be separated from the reaction products using nanofiltration membranes, achieving catalyst rejections of over 99%.44 Hereby, the competitiveness of an alternative membrane system with an integrated stirrer in the membrane cell could be demonstrated and showed its potential economic advantages in the enrichment of POMs in the retentate.44 In this work, we used the same nanofiltration membrane setup for the efficient removal of accompanying metal salts from the POM solution to isolate the synthesized compounds in high yield and purity.
Results and discussion
Synthesis and characterization of the Lacunary compound
For the synthesis of the Keggin-type lacunary POM LHPA-3, we adapted the procedure described by Himeno et al.45 An aqueous solution of Na2MoO4 and Na2HPO4 was acidified using concentrated hydrochloric acid. Upon evaporation of the solvent, the product precipitated in the form of yellow crystals, some of them were suitable for single crystal X-ray diffraction (Fig. 2). | ||
| Fig. 2 Solid-state structure of LHPA-3 as determined by single crystal X-ray diffraction. The compound was crystallized in the hexagonal space group P63 (173). R1: 1.85%, wR2: 5.38%, Rint: 3.28%, GooF: 1.098. Thermal ellipsoids are drawn at 50% probability level. Colour code: purple: phosphorous, blue: molybdenum, and red: oxygen. Hydrogen atoms have not been modelled. The full crystallographic information file (cif) is available through the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service (deposition number: 2205006†). | ||
The asymmetric unit of the structure (depicted in the ESI, Fig. S40†) contains three different metal sites, aside from the phosphorus atom and 12 oxygen atoms. The oxygen atoms can be grouped into four categories: oxygens connecting the central P atom to Mo atoms (OP), bridging oxygen atoms between two Mo atoms (Ob), terminal oxygen atoms bound to a Mo atom (Ot), and oxygen atoms pointing into the lacunary position (Ol), i.e. the void which is caused by the missing MoO6 octahedra.
The comparison of selected structural features of LHPA-3 and the parent compound H3[PMo12O40] (HPMo) (Table 1) shows a significant contraction of the internal metal oxygen bonds (Mo–OP,b) and an elongation of the bonds to the terminal/lacunary oxygen atoms in LHPA-3. Generally short bond lengths are expected for the terminal (and lacunary) oxygen atoms, corresponding to their double bond character. Noteworthy is, that the oxygen atoms pointing into the lacunary void have very different bond lengths: half of them have very short bonds and are connected to a convex coordinated Mo (Ol–Mo–OP,b,t > 90°), the other half has a very long bond and is connected to a concavely coordinated Mo (Ol–Mo–OP,b,t < 90°). As expected, angles of the polyhedra deviate more strongly from the mathematically ideal angles in LHPA-3 due to the loss of the high symmetry present in the full Keggin structure.
The bulk product was further characterized by AAS/ICP-OES and thermogravimetric analysis (TGA) (Table 2).
| Compound | Targeted molecular composition | Na/K/P/Mo ratioa | Hydration waterb [mol per mol-POM] |
|---|---|---|---|
| a P and Mo were determined by ICP-OES, Na and K by AAS, the data were normalized to the target Mo content. b The content of hydration water was determined by thermogravimetric analysis (TGA). | |||
| LHPA-3 | Na9[PMo9O34] | 20.8/0/1.2/9 | 6 |
The analysis confirms the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9 P:Mo stoichiometry. Using alkali metal containing precursors and adjustment of the pH value using hydrochloric acid, undesirable salt formation is not avoidable. Therefore, the increased sodium content is expected due to the coprecipitation of the sodium chloride. Purification with the nanofiltration membrane was not possible as the conditions in the setup caused LHPA-3 to decompose, possibly forming [P2Mo18O62]6− (vide infra). The potassium content was determined, as it is a common impurity in sodium salts, but no potassium was detected. The amount of hydration water of LHPA-3 was determined as 6 mol H2O per mol LHPA-3.
:9 P:Mo stoichiometry. Using alkali metal containing precursors and adjustment of the pH value using hydrochloric acid, undesirable salt formation is not avoidable. Therefore, the increased sodium content is expected due to the coprecipitation of the sodium chloride. Purification with the nanofiltration membrane was not possible as the conditions in the setup caused LHPA-3 to decompose, possibly forming [P2Mo18O62]6− (vide infra). The potassium content was determined, as it is a common impurity in sodium salts, but no potassium was detected. The amount of hydration water of LHPA-3 was determined as 6 mol H2O per mol LHPA-3.
The material was further analysed with ATR-FT-IR spectroscopy (Fig. 3) and Raman spectroscopy (Fig. 4). In both, the IR- and the Raman spectrum, LHPA-3 exhibits several additional bands compared to HPMo, corresponding to the loss of symmetry. These additional bands were then assigned by spectral comparison with quantum chemical (DFT) calculations.
 | ||
| Fig. 3 ATR-FT-IR spectra of LHPA-3 (experimental and calculated) in comparison to HPMo. | ||
 | ||
| Fig. 4 Experimental and calculated Raman spectra of LHPA-3 (in comparison to HPMo). | ||
Fig. 3 shows the comparison of the experimental IR spectra of both Lacunary and HPMo structures with DFT calculated spectrum of the LHPA-3 lacunary complex. In the IR spectrum of HPMo, characteristic vibrational modes can be seen, which are assigned to the antisymmetric P–O (above 1000 cm−1) and to the terminal MoO vibration (above 900 cm−1) modes, as well as two types of Mo–O–Mo vibration modes: (M–O–M)vertex and (M–O–M)edge in the range of 800 to 900 cm−1 and 600 to 800 cm−1.15,48,49 LHPA-3 exhibits several additional bands in the range of the terminal MoO vibrations. This matches the observation of several different bond lengths for the terminal and lacunary O atoms, identified in the solid-state structure (vide supra).
The new peaks in the experimental IR absorption spectrum of LHPA-3 appear at roughly 850, 904, 939, 1008, and 1125 cm−1 compared to the HPMo species. In order to be able to assign these new peaks to specific vibrations, the LHPA-3 lacunary structure was optimized using DFT methods. The results from DFT calculations show that new peaks appear due to the central phosphate group. The broad peak at 1125cm−1 in the experimental spectrum can be split into two small peaks at 1138 and 1092 cm−1 and refers to the central PO bond stretches in DFT optimized LHPA-3. The peaks at 1054 and 1035 cm−1 correspond to the symmetric and anti-symmetric terminal MoO stretches, respectively, and can be related to the experimental peaks at 1060 cm−1 in both LHPA-3 and HPMo complexes. The peak at 1016 cm−1 represents the OPO asymmetric stretching and the peak at 995 cm−1 refers to a symmetric in-plane PO stretch motion. The peak at 975 cm−1 shows an out-of-plane PO bond vibrational mode. The peaks at 923 and 895 cm−1 correspond to symmetric and asymmetric M–O–M distortions along the vertex and finally the strong peak at 835 cm−1 in the DFT calculated spectrum of LHPA-3 complex refers to the M–O–M stretch along the edge.
The Raman spectra (Fig. 4) also show a noticeable difference in the bridging Mo–O–Mo vibrational bands (600 to 800 cm−1) between the two molecules and confirms the spectral assignment. Frequently, a fitted scaling factor is applied to the computed spectra in order to improve the agreement with the experiment. For our unscaled theoretical peak positions, a systematic shift of ∼50 cm−1 is considered satisfying. Furthermore, the comparison of experimental Raman spectra from solid samples with isolated molecules may contribute to the shift. Theoretical means are thus able to provide reliable spectral features in IR and Raman to support the assignment of peak positions from the experiment.
In order to gain further insights into the behaviour of LHPA-3 in solution, the compound was studied using 31P-NMR spectroscopy at pH 1 in comparison with HPMo (Fig. 5).
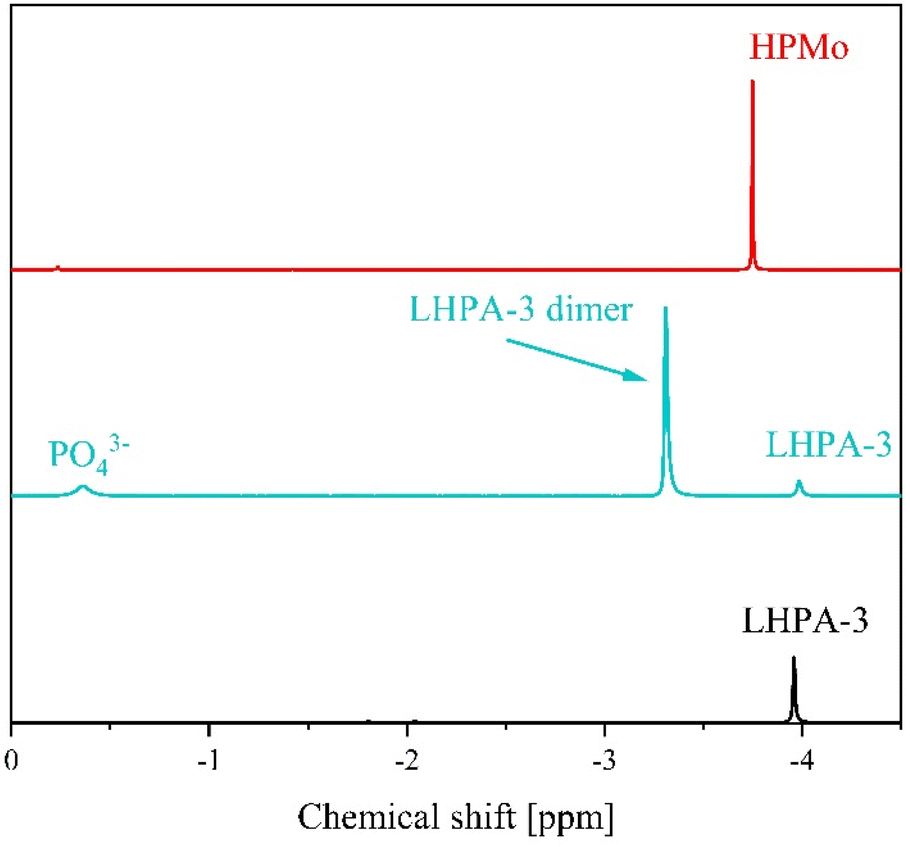 | ||
| Fig. 5 31P-NMR spectra of LHPA-3 (bottom) in comparison with HPMo (top) and dimerized LHPA-3/Wells–Dawson phosphomolybdate (middle) in a mixture of 90% H2O (pH 1, adjusted with HCl) and 10% acetone-d6. The spectra were measured at 242.9 MHz, using 85% H3PO4 as external standard. The dimerization was induced by heating the solution to 85 °C for some minutes. | ||
The 31P-NMR spectrum of LHPA-3 (Fig. 5 bottom) shows a single signal at −3.96 ppm, which is similar to the chemical shift of HPMo at −3.75 ppm (Fig. 5 top). This clearly shows that LHPA-3 remains stable in solution (at room temperature) as no impurities or dissociation products are observed. While our here presented results clearly demonstrate that LHPA-3 is sufficiently stable to obtain single crystals and to be used for further synthesis, the notion of the instability of LHPA-3 is not entirely unfounded. Our attempts to work up aqueous solutions of LHPA-3 using a rotary evaporator (80 °C, ca. 200 mbar), resulted in crystals of the mixed sodium/potassium salt of [P2Mo18O62]6−. This compound, which is a Wells–Dawson type phosphomolybdate, is formed by the condensation of two LHPA-3 units, likely induced by the elevated temperature. Since the structure of this anion has been discussed in sufficient detail by Haiyan An et al.,50 we refrain from further discussion here but provide details in the ESI (Fig. S42 and S43†). We have also verified this dimerization of LHPA-3 by briefly heating up an acidic aqueous solution of LHPA-3 to 85 °C and subsequently performing 31P-NMR spectroscopy (Fig. 5 middle). The resulting spectrum clearly shows an additional peak at −3.31 ppm, which is attributed to the dimerized Wells–Dawson species [P2Mo18O62]6−. This value is close to the literature value of −3.22 ppm.51 So, this behaviour might have led researchers to conclude that LHPA-3 is not stable enough for further use.
The Gibbs free energy of the formation of LHPA-3 was calculated using quantum chemical (DFT) calculations, where a Wells–Dawson species [P2Mo18O62]6− is formed from two LHPA-3 monomers and 6 water molecules are released in the process (Fig. 6). In an acidic medium, the terminal oxygen atoms at the vertex are most likely to be protonated. The formation of the dimerized Wells–Dawson species is thermodynamically favourable in acidic solution with a change in Gibbs free energy of −357 kJ mol−1 using an implicit solvent model.
 | ||
| Fig. 6 Calculated dimerization of LHPA-3 in acidic aqueous medium (pH 1) forming a Wells–Dawson species. | ||
UV-Vis measurements of the acidic aqueous solution of LHPA-3 (Fig. 7) show two significant peaks: 215 nm and 321 nm. After reaching the maximum from the LMCT peak at 215 nm, the intensity continues to increase towards smaller wavelengths. However, the maximum of the increase is no longer reached in the available measurement range (>200 nm), which could indicate the presence of another peak below 200 nm.
 | ||
| Fig. 7 UV Vis spectrum of LHPA 3 in aqueous, pH 1 solution with the characteristic LMCT bands. | ||
The observed signals correspond to the O→Mo(VI) ligand-to-metal-charge-transfer excitations (LMCT) of different oxygen atoms. Since for HPMo the main UV-Vis band has its maximum at 218 nm with no additional peaks at higher wavelengths,15 it can be concluded that the band at 321 nm belongs to the O→Mo(VI) LMCT of the lacunary oxygen atoms (Ol).
To complete the characterization of LHPA-3 we have performed electrochemical measurements, in detail cyclic voltammetry (CV) and square-wave voltammetry (SWV). The results are shown in the ESI Table 8 and Fig. S30 and S31.† From the obtained data we see that LHPA-3 undergoes similar redox processes as HPMo. However, the structural changes in LHPA-3 cause a shift of its potentials to higher values.
Application of the lacunary phosphomolybdate for the synthesis of various addenda element substituted Keggin-type POMs
As stated in the introduction, the significance of LHPA-3 lies in its potential as a precursor for the synthesis of POMs with further element substitutions in the metal framework. We therefore investigated this synthetic route by combining LHPA-3 with various metals of different oxidation states, namely: Co(II), Ni(II), Zn(II), Cu(II), Fe(III), In(III), Sn(IV), V(V), Nb(V), and W(VI). Most of these elements are of interest for catalytic applications due to their redox properties such as V(V)/V(IV), Fe(III)/Fe(II), Co(III)/Co(II)/Co(I), Cu(II)/Cu(I), and Sn(IV)/Sn(II).52 Other metals were chosen to explore the limits of this synthetic strategy, especially the main group elements In and Sn, as the incorporation of main group elements in POMs, although previously demonstrated, is still in its infancy.53,54A particular challenge was the choice of a suitable precursor for Nb(V) as most Nb salts are either not water soluble or precipitate as Nb2O5 in aqueous solution. Various studies report potassium hexaniobate K8[Nb6O19] (KNb) as a suitable precursor, a Lindqvist-type POM structure that can only be stabilized in strongly alkaline media (pH > 12).55–60 Since LHPA-3 is only stable in acidic solutions, KNb was first dissolved in an aqueous hydrogen peroxide solution, which was then acidified.61 The peroxide prevents the precipitation of Nb2O5, presumably via formation of a Nb(V) peroxo complex, long enough for the Nb(V) to be incorporated into LHPA-3. A detailed procedure for the synthesis, characterisation, and utilisation of K8[Nb6O19] is presented in the ESI (section 1.4).†
In a typical synthesis procedure for these metal incorporations, LHPA-3 was prepared in situ and used in aqueous solution, without previous purification. To this solution, suitable precursors or respective aqueous solutions were added, and the mixture was heated to reflux for 30 minutes. The pH of the mixture was adjusted to approx. 1.5 using hydrochloric acid. For subsequent purification, a nano-filtration membrane setup, which has previously been successfully employed for the recycling of homogenous POM catalysts,44 was used. An overview of the synthesised compounds is given in Table 4.
Purification of the newly synthesized POMs by nanofiltration
Since the above-described procedure requires the adjustment of a defined pH value, various alkali salts are present in addition to the POM. For the purification of the POMs, we have developed a nanofiltration method, which succeeds in separating salts and POM from each other.The nanofiltration system was operated in a modified diafiltration mode. Samples were taken from the permeate of each cycle to determine the salt content and catalyst loss by AAS/ICP-OES analysis. In addition, the permeates were concentrated by rotary evaporation and the resulting salt fractions were combined and analyzed by FT-IR spectroscopy as well as ICP-OES analysis. Fig. 8 shows the evolution of sodium and potassium concentrations over the four diafiltration cycles. There is a clear exponential decrease in the concentrations of sodium and potassium. The sodium concentration in the permeate already decreases from an initial value around 17.6 g L−1 to 6.1 g L−1 within one diafiltration cycle, which corresponds to a decrease by a factor about 3. After the fourth cycle, the sodium concentration in the permeate reaches a value below 0.5 g L−1. The potassium concentration shows a comparable trend.
 | ||
| Fig. 8 Development of the sodium and potassium concentrations in the permeate depending on the conducted diafiltration cycles. | ||
AAS measurements of the retentate reveal that there is still a residual sodium concentration of 7.0 g L−1, which corresponds to a difference about 6.5 g L−1, compared to the corresponding permeate. However, for each POM anion, a sufficient number of cations must remain in the retentate. In this exemplarily presented case, the anion is [PNb3Mo9O40]6−, which requires six equivalents of alkali counterions. A visual representation of the decreasing salt concentration from cycle to cycle and the purified yellow POM fraction [PNb3Mo9O40]6− is shown in Fig. S7.† To finally confirm that the isolated POM is free of chloride (and thus free of the corresponding alkali chloride salts), the POM sample was analyzed for chloride content by energy dispersive X-ray spectroscopy (EDX). As an average value over three measurements, a chloride content of 0.028 wt% was found. Thus, it can be assumed that the corresponding salts were almost completely removed by the nanofiltration process.
In order to minimize losses of the valuable products, a high selectivity is extremely important for the purification process. For this purpose, the permeate samples were analysed by AAS and ICP-OES with regard to their content of the POM components molybdenum, niobium and phosphorous. For each permeate the rejections of these components were determined as described in the corresponding section of the experimental part (ESI†). All catalyst components were rejected virtually to 100%, meaning that almost no POM anion passed the membrane (see Fig. S8†). Furthermore, the permeate sample was analyzed using powder XRD. The powder diffractogram in the ESI (Fig. S9†) shows that the permeate fraction consists mainly of sodium and potassium chloride. Essentially, the potassium content comes from the KNb precursor.
The success of the purification process can be evaluated based on the composition of the POM fraction as target product of the process. For this purpose, the stoichiometric ratios of the individual catalyst components before and after purification are shown in Table 3.
| Entrya | Process step | Stoichiometryb | ||||
|---|---|---|---|---|---|---|
| Moc | Nbc | Pc | Nad | Kd | ||
| a Experimental conditions: pre-wetted DK-series membrane, ambient temperature, 30 bar transmembrane pressure, 15 ml min−1 flow rate, 1100 rpm stirring speed. b Calculated as described in the corresponding section of the experimental part. c Determined with ICP-OES. d Determined with AAS. | ||||||
| 1 | Target value | 9.0 | 3.0 | 1.0 | 6.0 | |
| 2 | Before purification | 9.0 | 2.9 | 1.1 | 19.5 | 4.0 |
| 3 | After purification | 9.0 | 2.8 | 1.0 | 5.1 | 1.5 |
| a The content of the elements (except O) was determined by ICP-OES or AAS. The results were normalized to the targeted Mo content. Residual negative charge is assumed to be compensated by H+, as all compounds were obtained from acidic solutions. b Hydration water was determined by thermogravimetric analysis (TGA). |
|---|

|
Finally, after synthesis and purification, the elemental composition of all compounds was confirmed by ICP-OES and AAS (Table 4).
Based on the results from elemental analysis (Table 4 and ESI section 1.7†) the incorporation of most of guest elements, particularly the penta- and hexavalent metals, was successful. From the obtained ICP-OES data (yellow and red markers), limitations were found for the following elements: Cu(II), Zn(II), In(III) and Sn(IV). The limitations are limited to the late transition metals and to the main group elements, whereas the incorporation of Sn(IV) was not successful.
DFT calculations were performed in order to probe structural changes by substituting one molybdenum atom of HPMo with di- to hexavalent metal atoms (see Fig. 9). The structural changes in the POM structure upon introducing the (di- to hexavalent) metal atoms were characterized based on: (i) metal–oxygen bond lengths and (ii) distance of central phosphorous atom to the newly introduced metal atom.
 | ||
| Fig. 9 DFT optimized structures of mono-substituted Keggin phosphomolybdates. Characteristic bond distances for Ot = terminal oxygen, Ob = bridging O, OP = phosphate O are given. | ||
The metal–oxygen and phosphorus–metal distances remain almost unchanged upon substituting molybdenum with pentavalent vanadium. Due to the increase in the atomic radii, the metal–oxygen distances slightly increase from 1.8 Å to 1.97 Å and 1.99 Å for M–O1 and M–O2 upon introduction of niobium into the POM. However, the rest of the structural parameters remains almost unchanged. The similar trend was also found for tungsten substitution, thus suggesting an easy incorporation of penta- and hexavalent metals into the Keggin structure. Exceptions are the incorporation of Cu(II), Zn(II) and In(III), which where only partially successful, and the incorporation of Sn(IV), which was entirely unsuccessful. A reason for this might be that the coordination chemistry of these metal ions is dominated by p-orbitals as opposed to d-orbitals. In(III) (4d105s05p0) and Sn(IV) (4d105s05p0) are main-group elements in which the full 4d-orbitals are generally unavailable for coordination chemistry. The DFT-optimized mono-substituted Co(II), In(III) and Sn(IV) POM structures show a significant elongation of M–O and P–M bond distances. The large bond distances indicate a weak binding interaction of the newly incorporated metal into the phosphomolybdate complex. M–O bond and P–M bond distances are increasing by 0.2 Å to 0.46 Å and 0.14 Å to 0.57 Å by introducing Co(II), In(III), and Sn(IV), with In(III) showing the largest increase followed by Sn(IV) and Co(II). In comparison to In(III), the introduction of Fe(III) reduces the P–M distance by 0.24 Å (from 3.56 Å to 3.32 Å) compared to HPMo. The possible reason for the reduction might be localization of the unpaired electron on the iron atom. Zn(II) is formally a transition metal, however it also has full d-orbitals (3d104s0) and has therefore similar coordination propensities as main group elements.
The Cu(II) substituted POM exhibits a highly reactive behaviour: in contact with stainless steel, the solution turned dark blue, indicating reduction of the POM. It might therefore be possible, that a corresponding compound containing two Cu(II) ions is not thermodynamically stable. For this reason, the resulting compound, nBu4NPCu2Mo, was not purified with the membrane setup, instead the POM was selectively precipitated from aqueous solution as a tetrabutylammonium (TBA) salt, whereby the other salts remain in solution.
The integrity of the Keggin structure was confirmed by ATR-FT-IR and Raman spectroscopy (Fig. 10 and ESI Fig. S11–S20†). Vibrational spectroscopy of POMs as a method for identifying the structure type is a well-established method and is discussed in literature to a great extent.15,48,49,62
 | ||
| Fig. 10 ATR-FT-IR spectra of selected POMs, substituted with di-, tri- penta- and hexavalent metals. | ||
The IR spectra of all compounds exhibit the above described (see Fig. 3), characteristic bands for Keggin structures.
From previous investigations, it is known, that for POMs, which are substituted with elements of significant lower mass than Mo(VI), the vibration bands are shifting to lower wavenumbers in comparison to the unsubstituted HPMo.15,18,48,49 This trend is observed for the substitution with Co(II), Ni(II), Cu(II), Zn(II), Fe(III) and V(V). Interestingly, although Nb and Mo have a similar mass, the vibration bands of the Nb(V) substituted POMs show the same trend indicating that the charge/oxidation state of the metal ion also plays a role (Table 5 and ESI Table 5†).
| POM | P–O | MOt |
(M–O–M)vertex | (M–O–M)edge |
|---|---|---|---|---|
| HPMo15 | 1059 | 962 | 877 | 744 |
| NaPCoMo | 1049 | 946 | 869 | 772, 707 |
| NaPNiMo | 1047 | 942 | 869 | 783 |
| n Bu4NPCu2Mo | 1077, 1055 | 935 | 864 | 777 |
| NaPZnMo | 1075, 1060, 1045 | 939 | 900 | 775 |
| NaPFeMo | 1051 | 945 | 864 | 766 |
| NaPIn3Mo | 1058 | 956 | 867 | 770 |
| NaKPNbMo | 1058, 1034 | 944 | 859 | 741 |
| NaKPNb2Mo | 1049 | 947 | 856 | 757 |
| NaKPNb3Mo | 1047 | 944 | 857 | 760 |
| NaKPV2NbMo | 1045 | 942 | 847 | 750 |
| NaKPVNb2Mo | 1048 | 944 | 859 | 762 |
| NaPV3Mo | 1064, 1048 | 937 | 846 | 758 |
| NaPW3Mo | 1062 | 959 | 872 | 777 |
In contrast, a shift to higher wavenumbers occurs for POMs that are substituted with elements of significant higher mass. This is observed for NaPW3Mo (Table 5 and Table 5 ESI†), the higher mass of W(VI) leads to shoulder formation of the P–O band (see ESI Fig. S19 and S20†).
The Raman spectra of the substituted POMs also exhibit significant differences in comparison with the unsubstituted HPMo, indicating the successful incorporation of the elements.
In order to elucidate the structural influence of the metal substitution, a crystal of NaPV3Mo was analysed with single-crystal X-ray diffraction (Fig. 11, crystallographic information file is available through the CCDC under the deposition number 2205007†).
 | ||
Fig. 11 Solid-state structure of NaPV3Mo determined by single crystal X-ray diffraction. The compound crystallizes in the tetragonal space group P![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) 21c (114). Hydrogen atoms have not been modelled. R1: 3.03%, wR2: 7.84%, Rint: 4.49%, GooF: 1.089. Color code: turquoise: sodium, purple: phosphorous, blue: molybdenum, and red: oxygen. 21c (114). Hydrogen atoms have not been modelled. R1: 3.03%, wR2: 7.84%, Rint: 4.49%, GooF: 1.089. Color code: turquoise: sodium, purple: phosphorous, blue: molybdenum, and red: oxygen. | ||
The compound crystallized in the tetragonal space group P21c (114) and conforms to the typical Keggin structure type. The asymmetric unit contains a quarter of the molecule with three distinct metal sites, which are all partially occupied by V and Mo, therefore a substitution pattern cannot be recognised. For each Keggin unit, four Na atoms are found, the remaining charge is presumably compensated by H+, which was not modelled. The closest distance between the counter ions and the POM is to two of the bridging O atoms. Since the distance is significantly larger than the sum of the respective covalent radii (Table 6), this interaction can be considered purely ionic with no covalent/dative bond character.

The bond lengths within the POM molecule are mostly similar to the bond lengths of the unsubstituted HPMo,46 with the exception of the bond between the metals and the phosphate oxygen atoms, which is significantly shorter in NaPV3Mo although it still exceeds the sum of the respective covalent radii (Table 6). Among the bridging O atoms, two groups can be distinguished: three atoms with an average M–O–M angle of 123.4° connection, two edge-sharing MO6 octahedra, and three other atoms with an average angle of 149.7° connecting corner-sharing MO6 octahedra. These values are slightly lower than in HPMo.46
Since the primary intended application for transition metal substituted POMs is homogeneous catalysis in aqueous solution, we further studied the newly synthesized compounds in solution. For this purpose, we measured 31P- and, where applicable, 51V-NMR spectra, of the POMs. Fig. 12 shows the 31P-NMR data of selected POMs, substituted with di-, penta- and hexavalent metals in comparison with HPMo. The 31P data of NaPFeMo are only shown in the ESI in Fig. S24,† because this spectrum shows no peaks, due to the strong paramagnetism of Fe(III). Co(II) is also a paramagnetic nucleus, so the 31P signals are broadened in comparison to HPMo as shown in Fig. 12. Although the NMR samples were prepared of pure compounds, a complex pattern of signals is observed.
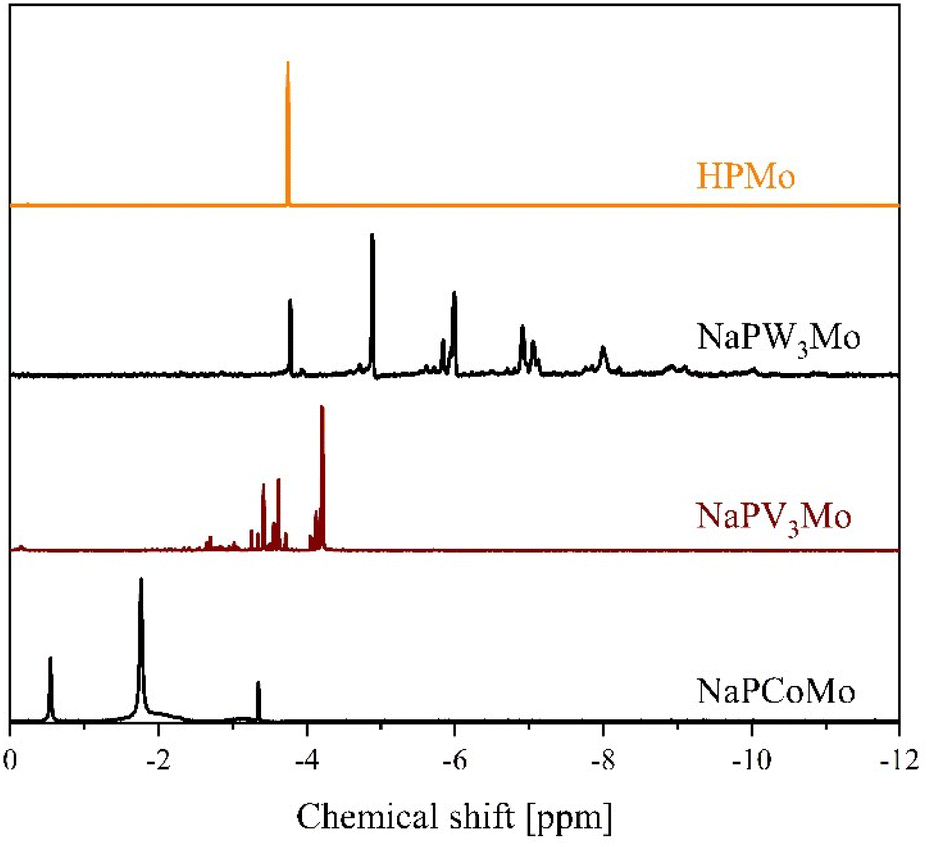 | ||
| Fig. 12 31P-NMR spectra of selected POMs substituted with di-, penta- and hexavalent elements in a mixture of 90% H2O (pH 1) and 10% D2O. The spectra were measured at 242.9 MHz, 85% H3PO4 was used as external standard. | ||
This is caused by two phenomena: the pH depended dissociation of the POMs and the isomerism (positional isomerism caused by the substitution as well as the intrinsic α/β isomerism of the Keggin structure).63–65 The dissociation, especially of substituted Keggin POMs has been described previously by several research groups.63,64 In aqueous solution, dissociation equilibria such as the ones shown in Fig. 13 cause multiple signals in the NMR spectra and lead to positional isomerisation.
 | ||
| Fig. 13 Examples of possible dissociation equilibria POMs undergo in aqueous solution. | ||
In an unsubstituted POM like HPMo, all metal positions are equivalent, therefore only one signal is observed in the 31P-NMR spectrum (ESI Fig. S20–S24†).63–67 However, as soon as a metal atom is substituted, the metal sites are not all equivalent anymore. Therefore, a second substitution can lead to several positional isomers with different substitution patterns. The number of such isomers increases drastically with the degree of substitution. While a substitution degree of two leads to only five isomers, the substitution of three metal atoms in the Keggin structure results in 13 possible isomers.65 Furthermore, there are different isomers for a Keggin-structure, most importantly the α and the β isomer. The β isomer is significantly less stable than the corresponding α isomer, so the peaks corresponding to the β isomer have a comparatively low intensity.62
As a result, the recorded spectra show a multitude of signals which are difficult to assign. In the recorded NMR-spectra (Fig. 12 and ESI Fig. S21–S25†) we find the above described phenomena confirmed in the fact that a multitude of signals is observed for all investigated compounds.
For catalytic applications of the POMs, the HOMO–LUMO gap (HOMO = highest occupied molecular orbital, LUMO = lowest unoccupied molecular orbital) is a relevant molecular property. To investigate this, UV-Vis spectra of all compounds were measured in aqueous solution (Fig. S26S29 ESI†). For the unsubstituted HPMo only one maximum is observed at a very low wavelength (218 nm), which can be assigned to the O→Mo(VI)O6 LMCT (see ESI Table 7†).15 Most of the transition-metal substituted POMs, however, show an additional smaller maximum at slightly higher wavelengths (280–350 nm, Fig. 14). This second band can be attributed to the LMCT from oxygen to the substituted transition element. Excitations from lower metal d orbitals to higher metal d orbitals are not expected to be visible, since they are known to be 1000 times less intense than LMCT transitions.68
 | ||
| Fig. 14 LMCT positions of the metal-substituted POMs. | ||
The data in Fig. 14 show the O→Mo(VI)O6 LMCT above 200 nm and the different transition metal LMCT bands about 300 nm for all elements investigated in this work, indicating that the latter transition requires less energy. The LMCTs for Nb(V) tend to be found in the 297 to 300 nm range, while the additional incorporation of vanadium causes the LMCT bands to shift to higher wavelengths. For NaPV3Mo, the LMCT for the V(V) reaches 308 nm and for the V(V) and Nb(V) mixed substituted POMs the LMCT for Nb(V) and V(V) are overlapping and cannot be distinguished from each other. It is known from literature that the V(V) LMCT protrudes into the visible region: this is the reason for the colours of POMs.15,62,69 With increasing substitution of vanadium, the V(V) LMCT increases while the Mo(VI) LMCT decreases, leading to a darker color.15 Theoretically, the LMCT bands contain two LMCTs. The LMCT at smaller wavelengths is assigned to the transition from the terminal oxygen atom to the metal VOt and the LMCT at longer wavelengths is assigned to the transition from the metal-bridging oxygen atoms V–Ob/V–Oc to the metal. This effect is often not resolved but may manifests itself by the formation of a shoulder at higher wavelengths.15,69,70
For applications in oxidation catalysis, the redox potentials of the transitions-metal substituted POMs are of great importance. Therefore, the redox potentials of the synthesized POMs were measured using cyclic voltammetry (CV) and square-wave voltammetry (SWV). Exemplary the data are shown in Fig. 15 for the three-fold substituted POMs with Nb(V) as well as V(V) in comparison with the unsubstituted HPMo. Electrochemical data of the other POMs are provided in the ESI (see ESI Fig. S30–S39 and Tables 8–10†).
 | ||
| Fig. 15 Comparison between the SWV measurements of the three-fold substituted POMs with V(V) and Nb(V) in comparison to HPMo (concentration 1 mmol L−1, scan rate 5 mV s−1 (SWV) and pH 1). Hydrochloric acid was used as supporting electrolyte. | ||
It is known from previous reports, that phosphomolybdates show several two-electron redox processes that can be tuned by incorporating different elements into the Keggin structure.15,71,72 The SWV data of the POMs containing Co(II), Ni(II), Cu(II), and Fe(III) (ESI Fig. S33 and S35†) show that these elements cause a shift of the redox potentials to higher potentials in comparison to HPMo. For nBu4NPCu2Mo (measured in acetonitrile) there are two peak potentials which are not observed for HPMo at 615 mV and 790 mV. These peak potentials might correspond to a redox process of Cu(II) to Cu(I). Comparing NaPV3Mo and NaKPNb3Mo, the former shows a pronounced redox activity while the latter seems to be redox inactive. The V(V) substituted POMs show that the V(V) lowers the peak potentials in comparison to HPMo.15 For the Nb(V)-substituted POMs, the following trend is observed: by incorporating one Nb(V), the peak potentials seem to shift initially to lower potential values, while by multiple substitution with Nb(V) the potentials partially shift back to higher values (ESI Table 10†). For the Nb(V)/V(V) mixed substituted POMs, some peak potentials are shifted to lower potentials in comparison to the pure Nb(V) substituted POMs. NaPW3Mo shows extremely low redox activity in the electrochemical measurements as it was also observed for the Nb(V) containing POMs.
Through the collected electrochemical results, it is shown, as also indicated in our previously published study, that the redox potentials – and thus also the electronic structure – of the POM can be specifically tuned by the incorporation of different metals.15 This represents an essential step forward for the synthesis of tailor-made POM catalysts and a preliminary assessment which POM catalysts might be suitable for redox catalysis.
Materials and methods
Detailed experimental procedures as well as information on the materials and devices used is provided in the ESI.†Computational details plus Cartesian coordinates of all structures can be found in the ESI.†
Conclusion
In this work, we show that the lacunary compound LHPA-3 is more stable and potentially applicable than anticipated and described in the literature. The LHPA-3 complex was characterized using various experimental spectroscopic techniques and the previously not annotated spectral peaks were assigned from quantum chemical calculations. Furthermore, DFT calculations show that in acidic medium the formation of a dimerized Wells–Dawson species is thermodynamically feasible. Using our novel in situ approach, in which LHPA-3 is formed and directly reacted by addition of other elemental precursors, a variety of element-substituted phosphomolybdates can be formed. In order to purify these compounds, we applied a novel approach using a nanofiltration membrane process, which is superior to (re-)crystallisation or extraction with diethyl ether, because it results in high purity products with no significant loss of yield. Using this synthetic approach and purification method, we successfully produced Keggin-type POMs containing Co(II), Ni(II), Zn(II), Cu(II), Fe(III), In(III), V(V), Nb(V), and W(VI). Quantum chemical calculations showed that incorporation of these metals significantly affects the structural properties of Keggin-type POMs. These new compounds were comprehensively characterised by various spectroscopic and electrochemical methods and it was found that the incorporation of these elements has a significant influence on the molecular properties (i.e. the redox potential) of the POM. Overall, our findings represent a major step forward in the development of tailor-made functional POMs for catalytic applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank the analytical service teams of the Chemistry Department at Hamburg University: Dr. Dirk Eifler and his team for elemental analysis, Dr. Frank Hoffmann for his assistance in refining our crystallographic data sets and his co-worker, Mrs. Isabelle Nevoigt for data collection of the single crystals and the powder XRD samples, and Mrs. Ute Gralla for Raman measurements. Our thanks also go to the research group of Prof. Dr. Peter Burger, in particular to Mr. Thomas Marx for lending the equipment and assisting with electrochemical measurements. Prof. Dr. Matthias Stein and Froze Jameel are grateful to the Max Planck Society of the Advancement of Science for financial support. This work is part of the Research Initiative “SmartProSys: Intelligent Process Systems for the Sustainable Production of Chemicals” funded by the Ministry for Science, Energy, Climate Protection and the Environment of the State of Saxony-Anhalt.References
- Y. Wei, Polyoxometalates: An interdisciplinary journal focused on all aspects of polyoxometalates, Polyoxometalates, 2022, 1, 9140014 CrossRef.
- J. J. Berzelius, Beitrag zur näheren Kenntnis des Molybdäns, Ann. Phys., 1826, 82, 369–392 CrossRef.
- J. F. Keggin, Structure of the Molecule of 12-Phosphotungstic Acid, Nature, 1933, 131, 908–909 CrossRef CAS.
- X. Huang, W. Cui, S. Liu, G. Liu, Y. Zhang, Z. Zhang, G. Shen, Z. Li, J. Wang and Y. Chen, One-step assembly of Pd-Keggin-polyoxometalates for catalytic benzothiadiazole generation and derived cell-imaging probe application, Chin. Chem. Lett., 2023, 34, 107692 CrossRef CAS.
- Y. Ma, F. Gao, W. Xiao, N. Li, S. Li, B. Yu and X. Chen, Two transition-metal-modified Nb/W mixed-addendum polyoxometalates for visible-light-mediated aerobic benzylic C–H oxidations, Chin. Chem. Lett., 2022, 33, 4395–4399 CrossRef CAS.
- B. Li and L. Wu, Perspective of polyoxometalate complexes on flexible assembly and integrated potentials, Polyoxometalates, 2023, 2, 9140016 CrossRef.
- Y. Wang, Y. Lu, W. Zhang, T. Dang, Y. Yang, X. Bai and S. Liu, Construction of hydrogel composites with superior proton conduction and flexibility using a new POM-based inorganic–organic hybrid, Polyoxometalates, 2022, 1, 9140005 CrossRef.
- H. Li, H. Pan, Y. Fan, Y. Bai and D. Dang, Syntheses, crystal structures, and properties of four polyoxometalate-based metal–organic frameworks based on Ag(I) and 4,4′-dipyridine-N,N′-dioxide, Polyoxometalates, 2022, 1, 9140007 CrossRef.
- S. Duan, X. Xu, W. Chen, J. Zhi and F. Li, Grain boundaries passivation of high efficiency and stable perovskite photodetector by polyoxometalate-based composite SiW 11 @ZIF-8, Polyoxometalates, 2022, 1, 9140003 CrossRef.
- Z. Xia, L. Wang, Q. Zhang, F. Li and L. Xu, Fast degradation of phenol over porphyrin-polyoxometalate composite photocatalysts under visible light, Polyoxometalates, 2022, 1, 9140001 CrossRef.
- M. T. Pope and A. Müller, Polyoxometalate Chemistry: An Old Field with New Dimensions in Several Disciplines, Angew. Chem., Int. Ed. Engl., 1991, 30, 34–48 CrossRef.
- M. T. Pope and A. Müller, Polyoxometalate Chemistry From Topology via Self-Assembly to Applications, Kluwer Academic Publishers, New York, Boston, Dordrecht, London, Moscow, 2002 Search PubMed.
- R. Dehghani, S. Aber and F. Mahdizadeh, Polyoxometalates and Their Composites as Photocatalysts for Organic Pollutants Degradation in Aqueous Media—A Review, Clean: Soil, Air, Water, 2018, 46, 1800413 Search PubMed.
- V. F. Odyakov and E. G. Zhizhina, New process for preparing aqueous solutions of Mo-V-phosphoric heteropoly acids, Russ. J. Inorg. Chem., 2009, 54, 361–367 CrossRef.
- J.-C. Raabe, J. Albert and M. J. Poller, Spectroscopic, Crystallographic, and Electrochemical Study of Different Manganese(II)–Substituted Keggin–Type Phosphomolybdates, Chem. – Eur. J., 2022, 28, 1–12 CrossRef PubMed.
- J. Albert, M. Mendt, M. Mozer and D. Voß, Explaining the role of vanadium in homogeneous glucose transformation reactions using NMR and EPR spectroscopy, Appl. Catal., A, 2019, 570, 262–270 CrossRef CAS.
- S. Pathan and A. Patel, Transition-Metal-Substituted Phosphomolybdates: Catalytic and Kinetic Study for Liquid-Phase Oxidation of Styrene, Ind. Eng. Chem. Res., 2013, 52, 11913–11919 CrossRef CAS.
- J. Albert, D. Lüders, A. Bösmann, D. M. Guldi and P. Wasserscheid, Spectroscopic and electrochemical characterization of heteropoly acids for their optimized application in selective biomass oxidation to formic acid, Green Chem., 2014, 16, 226–237 RSC.
- J. Reichert, B. Brunner, A. Jess, P. Wasserscheid and J. Albert, Biomass oxidation to formic acid in aqueous media using polyoxometalate catalysts – boosting FA selectivity by in situ extraction, Energy Environ. Sci., 2015, 8, 2985–2990 RSC.
- P. Preuster and J. Albert, Energy Technol., 2018, 6, 501–509 CrossRef CAS.
- J. Claußnitzer, B. Bertleff, W. Korth, J. Albert, P. Wasserscheid and A. Jess, Kinetics of Triphase Extractive Oxidative Desulfurization of Benzothiophene with Molecular Oxygen Catalyzed by HPA-5, Chem. Eng. Technol., 2020, 43, 465–475 CrossRef.
- B. Bertleff, J. Claußnitzer, W. Korth, P. Wasserscheid, A. Jess and J. Albert, Extraction Coupled Oxidative Desulfurization of Fuels to Sulfate and Water-Soluble Sulfur Compounds Using Polyoxometalate Catalysts and Molecular Oxygen, ACS Sustainable Chem. Eng., 2017, 5, 4110–4118 CrossRef CAS.
- J.-C. Raabe, M. Poller, D. Voß and J. Albert, H8[PV5Mo7O40] (HPA–5) – a unique polyoxometalate for acid and RedOx catalysis: synthesis, characterization, and modern applications in green chemical processes, ChemSusChem, 2023, 2013–2015 Search PubMed.
- M. J. Poller, S. Bönisch, B. Bertleff, J.-C. Raabe, A. Görling and J. Albert, Elucidating activating and deactivating effects of carboxylic acids on polyoxometalate-catalysed three-phase liquid-liquid-gas reactions, Chem. Eng. Sci., 2022, 264, 118143 CrossRef CAS.
- F. Steffler, G. F. De Lima and H. A. Duarte, The effect of the heteroatom (X=P, As, Si and Ge) on the geometrical and electronic properties of α-Keggin polyoxometalates (M=Mo, W and Nb) – A DFT investigation, J. Mol. Struct., 2020, 1213, 128159 CrossRef CAS.
- L. Yan, X. López, J. J. Carbó, R. Sniatynsky, D. C. Duncan and J. M. Poblet, On the Origin of Alternating Bond Distortions and the Emergence of Chirality in Polyoxometalate Anions, J. Am. Chem. Soc., 2008, 130, 8223–8233 CrossRef CAS PubMed.
- V. F. Odyakov, E. G. Zhizhina and R. I. Maksimovskaya, Synthesis of molybdovanadophosphoric heteropoly acid solutions having modified composition, Appl. Catal., A, 2008, 342, 126–130 CrossRef CAS.
- V. F. Odyakov and E. G. Zhizhina, A novel method of the synthesis of molybdovanadophosphoric heteropoly acid solutions, React. Kinet. Catal. Lett., 2008, 95, 21–28 CrossRef CAS.
- M. Abbessi, R. Contant, R. Thouvenot and G. Hervé, Dawson Type Heteropolyanions. 1. Multinuclear (31P, 51 V, 183 W) NMR Structural Investigations of Octadeca(molybdotungstovanado)diphosphates a-1,2,3-[P2MM'2W15062]n- (M, M′ = Mo, V, W): Syntheses of New Related Compounds, Inorg. Chem., 1991, 30, 1695–1702 CrossRef CAS.
- A. Patel, N. Narkhede, S. Singh and S. Pathan, Keggin-type lacunary and transition metal substituted polyoxometalates as heterogeneous catalysts: A recent progress, Catal. Rev. - Sci. Eng., 2016, 58, 337–370 CrossRef CAS.
- P. A. Abramov, A. A. Shmakova, M. Haouas, G. Fink, E. Cadot and M. N. Sokolov, Self-assembly of [PNbxW12-xO40]n- Keggin anions-a simple way to mixed Nb-W polyoxometalates, New J. Chem., 2016, 41, 256–262 RSC.
- D. K. Lyon, W. K. Miller, T. Novet, P. J. Domaille, E. Evitt, D. C. Johnson and R. G. Finke, Highly Oxidation Resistant Inorganic-Porphyrin Analogue Polyoxometalate Oxidation Catalysts. 1. The Synthesis and Characterization of Aqueous-Soluble Potassium Salts of α2-P2Wl7O61(Mn+·OH2)(n−10) and Organic Solvent Soluble Tetra-n-butylammonium Salts of, J. Am. Chem. Soc., 1991, 113, 7209–7221 CrossRef CAS.
- H. D'Amour, Vergleich der Heteropolyanionen [PMo9O 31(H2O)3]3−, [P2Mo18O62]6− und [P2W18O62]6−, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1976, 32, 729–740 CrossRef.
- C. Marchal-Roch, E. Ayrault, L. Lisnard, J. Marrot, F.-X. Liu and F. Sécheresse, Dimerization in Acetonitrile of [H6PMo9O34]3− into [P2Mo18O62]6−: Structural Characterization of the Tetrabutyl Ammonium Salt, J. Cluster Sci., 2006, 17, 283–290 CrossRef CAS.
- C. Li, A. Jimbo, K. Yamaguchi and K. Suzuki, A protecting group strategy to access stable lacunary polyoxomolybdates for introducing multinuclear metal clusters, Chem. Sci., 2021, 12, 1240–1244 RSC.
- C. Li, K. Yamaguchi and K. Suzuki, Synthesis of a phosphomolybdate with a tetranuclear vanadium core by installing vanadium atoms in a lacunary template using the protecting group strategy, Chem. Commun., 2021, 57, 7882–7885 RSC.
- K. Suzuki, N. Mizuno and K. Yamaguchi, New Strategy for Precise Synthesis of Polyoxometalate Catalysts with Designed Active Sites, J. Jpn. Pet. Inst., 2020, 63, 258–266 CrossRef CAS.
- C. Li, K. Yamaguchi and K. Suzuki, Ligand–Directed Approach in Polyoxometalate Synthesis: Formation of a New Divacant Lacunary Polyoxomolybdate [γ–PMo 10 O 36] 7−, Angew. Chem., 2021, 133, 7036–7040 CrossRef.
- X. López, J. M. Maestre, C. Bo and J.-M. Poblet, Electronic Properties of Polyoxometalates: A DFT Study of α/β -[XM 12 O 40 ] n - Relative Stability (M = W, Mo and X a Main Group Element), J. Am. Chem. Soc., 2001, 123, 9571–9576 CrossRef PubMed.
- P. Miró, J. M. Poblet, J. B. Ávalos and C. Bo, Towards a computational treatment of polyoxometalates in solution using QM methods and explicit solvent molecules, Can. J. Chem., 2009, 87, 1296–1301 CrossRef.
- X. López, I. A. Weinstock, C. Bo, J. P. Sarasa and J. M. Poblet, Structural Evolution in Polyoxometalates: A DFT Study of Dimerization Processes in Lindqvist and Keggin Cluster Anions, Inorg. Chem., 2006, 45, 6467–6473 CrossRef PubMed.
- S. Mir, B. Yadollahi and R. Omidyan, Theoretical comparative survey on the structure and electronic properties of first row transition metal substituted Keggin type polyoxometalates, J. Solid State Chem., 2022, 305, 122667 CrossRef CAS.
- J. M. Brégeault, M. Vennat, S. Laurent, J. Y. Piquemal, Y. Mahha, E. Briot, P. C. Bakala, A. Atlamsani and R. Thouvenot, From polyoxometalates to polyoxoperoxometalates and back again; potential applications, J. Mol. Catal. A: Chem., 2006, 250, 177–189 CrossRef.
- T. Esser, M. Huber, D. Voß and J. Albert, Development of an efficient downstream process for product separation and catalyst recycling of a homogeneous polyoxometalate catalyst by means of nanofiltration membranes and design of experiments, Chem. Eng. Res. Des., 2022, 185, 37–50 CrossRef CAS.
- S. Himeno, M. Hashimoto and T. Ueda, Formation and conversion of molybdophosphate and -arsenate complexes in aqueous solution, Inorg. Chim. Acta, 1999, 284, 237–245 CrossRef CAS.
- M.-X. Xu, S. Lin, L.-M. Xu and S.-L. Zhen, Crystal structure and properties of H 3 [PMo 12 O 40 ] · 3C 2 H 6 O, Transition Met. Chem., 2004, 29, 332–335 CrossRef CAS.
- P. Pyykkö and M. Atsumi, Molecular Single-Bond Covalent Radii for Elements 1–118, Chem. – Eur. J., 2009, 15, 186–197 CrossRef PubMed.
- J. K. Lee, J. Melsheimer, S. Berndt, G. Mestl, R. Schlögl and K. Köhler, Transient responses of the local electronic and geometric structures of vanado-molybdo-phoshate catalysts H3+nPVnMo12−nO40 in selective oxidation, Appl. Catal., A, 2001, 214, 125–148 CrossRef CAS.
- A. J. Bridgeman, Computational Study of the Vibrational Spectra ofα- andβ-Keggin Polyoxometalates, Chem. – Eur. J., 2004, 10, 2935–2941 CrossRef CAS PubMed.
- H. An, T. Xu, X. Liu and C. Jia, A series of new hybrid compounds constructed from Dawson-type phosphomolybdates and metal–organic coordination complexes, J. Coord. Chem., 2010, 63, 3028–3041 CrossRef CAS.
- L. E. Briand, G. M. Valle and H. J. Thomas, Stability of the phospho-molybdic Dawson-type ion P2Mo18O626− in aqueous media, J. Mater. Chem., 2002, 12, 299–304 RSC.
- A. F. Holleman, E. und Nils Wiberg and G. Fischer, Lehrbuch der Anorganischen Chemie, Berlin, New York, 2009 Search PubMed.
- X. Wu, T. Huang, Q. Wu and L. Xu, Synthesis and conductive performance of indium-substituted ternary heteropoly acids with Keggin structures, Dalton Trans., 2015, 45, 271–275 RSC.
- E. Rafiee, I. M. Baltork, S. Tangestaninejad, A. Azad and S. Moinee, Tin(II) Polyoxometalate as an Efficient Catalyst for the Selective Oxidation of Sulfides to Sulfoxides, Z. Naturforsch., B: J. Chem. Sci., 2006, 61, 601–606 CrossRef CAS.
- F. W. Smith, The Ion-Exchange Behaviour of Niobate and Tantalate in Alkaline Solution, University of Cape Town, 1967 Search PubMed.
- V. G. Maiorov, A. I. Nikolaev, V. K. Kopkov, V. Y. Kuznetsov and N. L. Mikhailova, Preparation of Alkaline Solutions of Niobium(V), Russ. J. Appl. Chem., 2011, 84, 1137–1140 CrossRef CAS.
- M. Nyman, T. M. Alam, F. Bonhomme, M. A. Rodriguez, C. S. Frazer and M. E. Welk, Solid-state structures and solution behavior of alkali salts of the [Nb6O19]8- Lindqvist ion, J. Cluster Sci., 2006, 17, 197–219 CrossRef CAS.
- M. Nyman, F. Bonhomme, T. M. Alam, M. A. Rodriguez, B. R. Cherry, J. L. Krumhansi, T. M. Nenoff and A. M. Sattler, A General Synthetic Procedure for Heteropolyniobates, Science, 2002, 297, 996–998 CrossRef CAS PubMed.
- D. J. Sures, P. I. Molina, P. Miró, L. N. Zakharov and M. Nyman, Cesium salts of niobo-tungstate isopolyanions with intermediate group V-group VI character, New J. Chem., 2016, 40, 928–936 RSC.
- D. J. Sures, S. K. Sahu, P. I. Molina, A. Navrotsky and M. Nyman, Distinctive Interactions of Cesium and Hexaniobate in Water, ChemistrySelect, 2016, 1, 1858–1862 CrossRef CAS.
- M. Dabbabi and M. Boyer, Syntheses et proprietes d'hexa niobo(V)-tungstates(VI), J. Inorg. Nucl. Chem., 1976, 38, 1011–1014 CrossRef CAS.
- J.-C. Raabe, J. Aceituno Cruz, J. Albert and M. J. Poller, Comparative Spectroscopic and Electrochemical Study of V(V)-Substituted Keggin-Type Phosphomolybdates and -Tungstates, Inorganics, 2023, 11, 138 CrossRef CAS.
- L. Pettersson, I. Andersson, J. H. Grate and A. Selling, Multicomponent Polyanions. 46. Characterization of the Isomeric Keggin Decamolybdodivanadophosphate Ions In Aqueous Solution by 31P and 51 V NMR, Inorg. Chem., 1994, 33, 982–993 CrossRef CAS.
- D. V. Evtuguin, C. Pascoal Neto, J. Rocha and J. D. Pedrosa de Jesus, Oxidative delignification in the presence of molybdovanadophosphate heteropolyanions: mechanism and kinetic studies, Appl. Catal., A, 1998, 167, 123–139 CrossRef CAS.
- M. T. Pope and T. F. Scully, Geometrical Isomerism Arising from Partial Substitution of Metal Atoms in Isopoly and Heteropoly Complexes. Possibilities for the Keggin Structure, Inorg. Chem., 1975, 14, 953–954 CrossRef CAS.
- K. Lee, G. Pozarnsky, O. Zarembowitch and A. McCormick, 51 V NMR of homogeneous multicomponent vanadium oxide solutions, Chem. Eng. J. Biochem. Eng. J., 1996, 64, 215–223 CrossRef CAS.
- A. Selling, I. Andersson, J. H. Grate and L. Pettersson, A Potentiometric and (31P,51 V) NMR Study of the Aqueous Molybdovanadophosphate System, Eur. J. Inorg. Chem., 2000, 2000, 1509–1521 CrossRef.
- B. Weber, Koordinationschemie, Springer Berlin Heidelberg, Berlin, Heidelberg, 2014, vol. 7 Search PubMed.
- N. K. K. Raj, A. V. Ramaswamy and P. Manikandan, Oxidation of norbornene over vanadium-substituted phosphomolybdic acid catalysts and spectroscopic investigations, J. Mol. Catal. A: Chem., 2005, 227, 37–45 CrossRef CAS.
- H. Salavati and N. Rasouli, Synthesis and characterization of supported heteropolymolybdate nanoparticles between silicate layers of Bentonite with enhanced catalytic activity for epoxidation of alkenes, Mater. Res. Bull., 2011, 46, 1853–1859 CrossRef CAS.
- D. Y. Hwang, Y. S. Ha and S. Kim, Electrode-Assisted Wacker Process: Phosphomolybdate-Mediated Oxidation of 1-Butene to Methyl Ethyl Ketone, Bull. Korean Chem. Soc., 2001, 22, 441–442 CAS.
- M. Sadakane and E. Steckhan, Electrochemical Properties of Polyoxometalates as Electrocatalysts, Chem. Rev., 1998, 98, 219–237 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2205006, 2205007, 2216946 and 2216947. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3qi00937h |
| This journal is © the Partner Organisations 2023 |